
A dissipative particle dynamics simulation of controlled loading and responsive release of theranostic agents from reversible crosslinked triblock copolymer vesicles†
Zhikun
Wang‡
ab,
Fengting
Li‡
ab,
Li
Wang
ab,
Yueqi
Liu
ab,
Miantuo
Li
ab,
Nannan
Cui
ab,
Chunling
Li
 ab,
Shuangqing
Sun
*ab and
Songqing
Hu
*ab
ab,
Shuangqing
Sun
*ab and
Songqing
Hu
*ab
aSchool of Materials Science and Engineering, China University of Petroleum (East China), Qingdao 266580, China. E-mail: sunshuangqing@upc.edu.cn; songqinghu@upc.edu.cn
bInstitute of Advanced Materials, China University of Petroleum (East China), Qingdao 266580, China
First published on 25th November 2023
Abstract
To control the transport stability and release efficiency of loaded theranostic drugs in triblock copolymer carriers, the reversible crosslinking ability is of great significance. A molecular level exploration of such a function is needed to extend existing stabilizing and responsive dissociation mechanisms of carriers. Here, dissipative particle dynamics simulations were used to first demonstrate the formation of triblock copolymer vesicular carriers. Chemical crosslinking was used to strengthen the structural stability of the vesicle shell to avoid drug leakage. Reversible decrosslinking along with dissociation of the vesicle and release of loaded drugs were then explored. The structural, energetic and dynamical properties of the system were discussed at the molecular level. The regulation mechanism of drug release patterns was revealed by systematically exploring the effect of intra and intermolecular repulsive interactions. The results indicate that the chemical crosslinking of copolymers enhanced the compactness of the vesicle shell with a strengthened microstructure, increased binding energy, and limited chain migration, thus achieving more stable delivery of drugs. In terms of drug release, we clarified how the pairwise interactions of beads in the solution system affect the responsive dissociation of the vesicle and associated release patterns (speed and amount) of drugs. More efficient delivery and smart release of theranostic drugs are achieved using such reversible crosslinked triblock copolymer vesicles.
1 Introduction
In pursuit of higher delivery stability and targeted release accuracy of theranostic drugs for diseased tissues, scholars have conducted extensive research and exploration on the morphology and structure design of drug carriers.1–3 Vesicles self-assembled from block copolymers have shown high application potential for the theranostic need, since hydrophobic shells and hydrophilic inner cavities can be used as loading space for multiple chemical agents (e.g., anticancer drugs and fluorescent indicators) with different solvent affinities.4 In real cases, the transportation of multiple chemical agents, as well as their targeted release, in a special environment often needs an accurate regulation of the microstructure of the vesicle carriers.5 The tunable configuration and available diverse chemical groups of block copolymers provide possibility for such regulation. This is why block copolymer vesicles have become indispensable as theranostic drug carriers in pursuit of enhanced curative effects.6–8To enhance the structural stability of the block copolymer vesicles, the shell can be chemically crosslinked.9,10 The migration of copolymer segments in the crosslinked shell can be limited, thus effectively avoiding drug leakage when subjected to environmental disturbances. More intelligently, the vesicles can be crosslinked using reversible covalent bonds (e.g., imine bonds, disulfide bonds, and diselenium bonds) that are stable under normal conditions and prone to fracture in response to the tumor microenvironment.11–13 Based on this feature, the crosslinked vesicles may release loaded drugs due to reversible shell decrosslinking (i.e., dissociation), in addition to the increased shell porosity and permeability.14 Moreover, by selecting copolymer segments with an environmental response characteristic, the responsive vesicle dissociation and drug release processes can be further accelerated.15–18 The above versatility is more easily achieved in vesicles self-assembled from triblock copolymers as they have more adjustable polymer segments.4 There have been many literature reports on the construction of reversible crosslinking structures in triblock copolymer vesicles, as well as their environmentally responsive dissociation and drug release properties.12,13,19,20 However, it is often tedious in real experiments to manipulate the formation and responsive dissociation of above vesicles at the molecular level, especially to determine the parameter ranges of copolymer composition, crosslinking degree, repulsion between various polymer segments, etc., for certain drug loading or release needs.4–6 This obstacle has been encountered especially in developing novel polymeric vesicular drug delivery systems.
One popular strategy to overcome this deficiency is to explore the comprehensive effect of above parameters via computer simulations, which can scan large parameter ranges at a nanoscale based on real conditions.21–23 To date, computer simulations have been used to reveal the intra- and inter-molecular interactions in polymeric vesicular nanocontainers that drive the stable delivery and responsive release of drugs.20,24,25 Many previous reports have constructed a systematic relationship between the molecular structure, the environmental affecting factor, and the performance of polymer vesicles as drug delivery systems.26 We have previously simulated the interplay of distributions of multiple payloads within block copolymer micelles and proposed a mechanism describing the preferred locations of multiple payloads depending on their chemical properties.27 Also, we have discussed the intermolecular interactions within versatile block copolymer nanocontainers based on the architecture, composition, rigidity, etc., of copolymer segments.28–30 However, in terms of triblock copolymer vesicles with a crosslinked shell, the local structural transformation and internal driving mechanism still lack in-depth research at the molecular level. Of note for triblock copolymer systems with more selectable polymer segments is the possibility to introduce multi-functional chemical groups (e.g., crosslinking groups, responsive groups, and varied hydrophilic/hydrophobic groups).31 The central challenge is how to precisely control the component, alignment, molecular weight, etc., of these chemical groups, thus to achieve the desired stability of the crosslinked shell (for smooth drug delivery in vivo) and its responsive decrosslinking (for targeted and rapid drug release).31 A systematic and microscopic investigation of the structural, energetic, and dynamical variations of such systems is needed.
Here, we develop a simple reversible crosslinking strategy for the construction and microstructural regulation of a polymeric vesicular nanocontainer, and the exploration of the controlled loading and stimuli-triggered release of theranostic agents. We start from the self-assembly of a linear amphiphilic triblock copolymer poly(ethylene glycol)-b-poly(isobutyl methacrylate)-b-poly(N-(2-oxoprocopyl)methacrylamide) (PEG-b-PIBMA-b-POPMA) with EG as hydrophilic sites stabilizing a micelle/water interface, IBMA as hydrophobic sites loading hydrophobic fluorescent dyes, and OPMA as cross-linkable sites. 3,3′-Dithiobis(propionohydrazide) (DTP) is selected as the linker molecule that can chemically connect two OPMA groups for copolymer crosslinking,32–34 and reversibly decrosslink the copolymers through the disulfide bonds on it. The reaction between OPMA and DTP also makes the copolymer sensitive to pH conditions. In our work, the length ratio and hydrophilicity strength of different blocks are first varied to determine the range in which vesicular self-assemblies can be obtained. Then, two typical agents of hydrophilic anticancer drugs doxorubicin hydrochloride (DOX-HCl) and hydrophobic fluorescent dye Nile red are selected to be loaded in the vesicular cavity and shell, respectively. The microscopic mechanism that promotes the structural enhancement by shell crosslinking has been discussed. Furthermore, we perform decrosslinking of the shell to simulate the responsive dissociation of copolymers and release of drugs under external stimuli. Two critical aspects, i.e., crosslinking degree and intra/intermolecular interaction, are investigated with emphasis to reveal how to control the release pattern of drugs and the morphological rearrangement of the nanocontainer. The featured manipulation of such vesicular drug delivery systems provides a significant theoretical support for the design of more efficient integrated diagnosis and treatment systems.
2 Methods
2.1 Modeling and simulation details
Dissipative particle dynamics (DPD) is a coarse-grained simulation method that handles a system composed of a set of soft particles (beads).35–37 In DPD models, repeat polymer units or chemical groups are grouped into a single bead, which enables the simulation of the system at a much larger spatial scale and longer time scale than the atomistic molecular dynamics (MD) method. A soft and short ranged repulsive potential is used to describe the interactions including bond stretching between bonded beads, conservative repulsion for the excluded volume effect, dissipative forces for a viscous drag between moving beads, and random forces for stochastic impulse among beads. In this work, the initial DPD model includes randomly distributed triblock copolymers and drug molecules such as PEG-b-PIBMA-b-POPMA, DOX-HCl, and Nile red in aqueous solvents as the solution. The DPD modeling of these molecules based on their chemical structures is illustrated in Fig. 1. Three repeat units of PEG are grouped into one hydrophilic DPD bead (A). A repeat unit of PIBMA is grouped into one hydrophobic DPD bead (B). A repeat unit of POPMA is grouped into one pH-responsive and crosslinkable DPD bead (C). A linear triblock copolymer AmBnCl (m, n, and l indicate the number of A, B, and C, respectively) will be built. A linker molecule of DTP is grouped into two connected DPD beads (L), which can be disconnected to simulate the decrosslinking of copolymers. In addition, a DOX-HCl molecule is grouped into four linear connected DPD beads (D), a Nile red molecule is grouped into three linear connected DPD beads (N), and 8 water molecules are grouped into one DPD bead (W). Table S1 (ESI†) provides more details about the grouping method for DPD simulations in this work.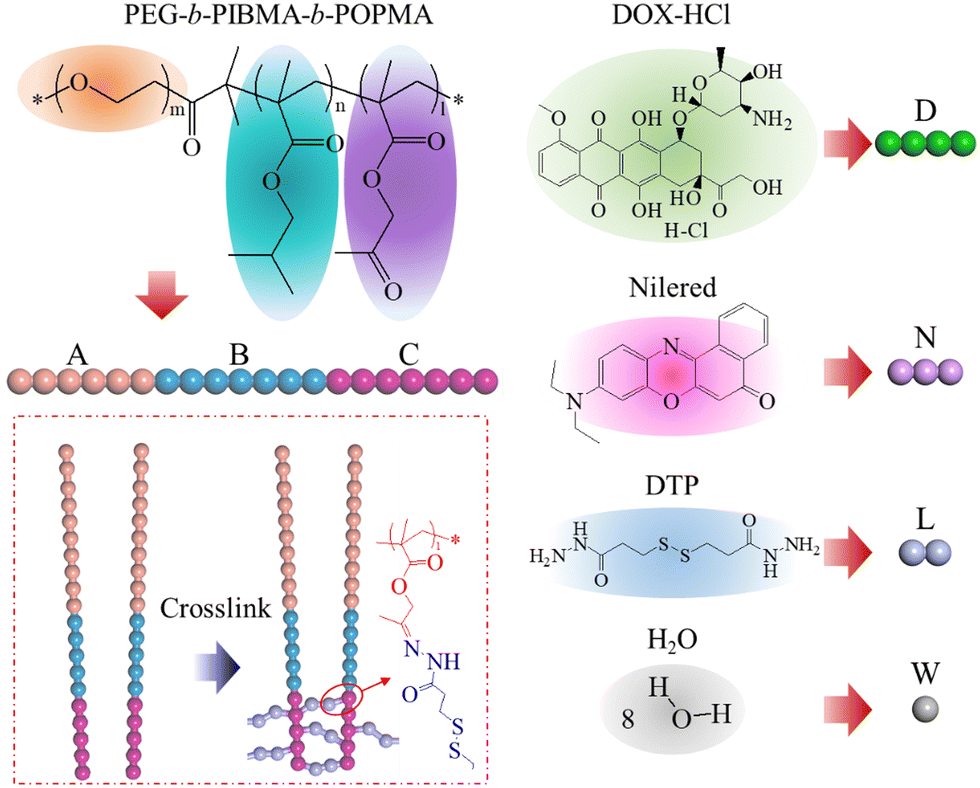 | ||
| Fig. 1 Chemical structures and coarse-grained modelling of the triblock copolymer, loaded theranostic agents, linkers and solvent molecules, as well as the chemical structure at the crosslinking site. | ||
Initial molecular and solution models were constructed using the Amorphous Cell tool in Materials Studio (Accelrys Inc.), and were exported as recognizable data files by Lammps through Perl scripts. A cubic box of size of 50rc × 50rc × 50rc was built, with a total of 375![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 beads and a number density of 3/rc3. The concentrations of the triblock copolymer, DOX-HCl, and Nile red were set to 20%, 1.5%, and 1%, respectively. The length of flexible blocks on the copolymer was set to 1 distance unit. Periodic boundary conditions were applied to all directions of the simulation box. Before conducting DPD simulations in Lammps, the critical constant aij in terms of the conservative repulsion between beads i and j in the studied system was first determined based on the hydrophilic–hydrophobic interaction theory of such copolymers and lots of previous reports.27–30,39 To match the compressibility of water, the aii value between two beads of the same type was set to 25. The aAW value between block A and water W was set to 30 to describe the hydrophilicity of A with a small repulsive effect to solvent beads. The aBW, aCW, and aNW values between water W and block B, block C, Nile red N were set to 100 to describe the hydrophobicity of these beads. The aAB, aAC, and aBC values between different blocks on the linear triblock copolymer were set to 80, 80, and 40, respectively, to ensure the self-stretching of the copolymer. The repulsive parameter between linker L and other beads was uniformly set to 40 to describe the relative compatible interactions between linkers and other beads in the system. It should be noted that such a treatment method simplifies the effect of linkers on the system, and only the utilization of its most basic functions (crosslinking and decrosslinking) is preserved. For other incompatible pairwise interactions between DOX-HCl D and block B, block C, Nile red N, as well as between Nile red N and block A, the aDB, aDC, aDN, and aNA values were set to 80. For other compatible pairwise interactions between DOX-HCl D and block A, water W, as well as between Nile red N and blocks B, C, the aAD, aDW, aBN, and aCN values were set to 25. The above settings are used for DPD simulations unless mentioned the change of values for specific study needs in the text.
000 beads and a number density of 3/rc3. The concentrations of the triblock copolymer, DOX-HCl, and Nile red were set to 20%, 1.5%, and 1%, respectively. The length of flexible blocks on the copolymer was set to 1 distance unit. Periodic boundary conditions were applied to all directions of the simulation box. Before conducting DPD simulations in Lammps, the critical constant aij in terms of the conservative repulsion between beads i and j in the studied system was first determined based on the hydrophilic–hydrophobic interaction theory of such copolymers and lots of previous reports.27–30,39 To match the compressibility of water, the aii value between two beads of the same type was set to 25. The aAW value between block A and water W was set to 30 to describe the hydrophilicity of A with a small repulsive effect to solvent beads. The aBW, aCW, and aNW values between water W and block B, block C, Nile red N were set to 100 to describe the hydrophobicity of these beads. The aAB, aAC, and aBC values between different blocks on the linear triblock copolymer were set to 80, 80, and 40, respectively, to ensure the self-stretching of the copolymer. The repulsive parameter between linker L and other beads was uniformly set to 40 to describe the relative compatible interactions between linkers and other beads in the system. It should be noted that such a treatment method simplifies the effect of linkers on the system, and only the utilization of its most basic functions (crosslinking and decrosslinking) is preserved. For other incompatible pairwise interactions between DOX-HCl D and block B, block C, Nile red N, as well as between Nile red N and block A, the aDB, aDC, aDN, and aNA values were set to 80. For other compatible pairwise interactions between DOX-HCl D and block A, water W, as well as between Nile red N and blocks B, C, the aAD, aDW, aBN, and aCN values were set to 25. The above settings are used for DPD simulations unless mentioned the change of values for specific study needs in the text.
Moreover, the dissipative parameter γ describing the viscous dragging force between each pair of moving beads was set to 4.5. The stiffness constant C describing the strength of bond stretching was set to −4. The cutoff distance rc, mass m of all beads, simulation temperature T, and thermal energy kBT were all set to united unit 1. The time step was set to Δt = 0.05τ = 0.05rc(m/kBT)1/2. To start the simulation, a Gaussian distribution method was performed to generate initial velocities for all beads in the system. For different study needs (e.g., self-assembly process and drug release process), the production simulations lasted from 1.0 × 106 to 3.0 × 106 time steps to ensure that equilibrium systems can be obtained.
2.2 Crosslinking and decrosslinking procedures
Based on the vesicle models obtained through self-assembly simulations, a nearest neighbor bonding principle28,38 was used for the construction of the crosslinked vesicle. All beads C on the copolymers were treated as possible crosslinking points, and an extra linker molecule containing two bonded L beads was used to crosslink adjacent hydrophobic blocks (beads C), by a Crosslinking Perl Script in Materials Studio (appended in the ESI†). The script conducted a truncation radius search program that was used to ensure that the newly formed chemical bonds will not cause large internal stress and geometric deformation. Specifically, three cyclic dynamic processes were performed: (1) monitor distances between all possible bead pairs of C and detect the closest ones; (2) if the distance of the closest pair is within a threshold rth, insert a linker molecule between the selected bead C pair and connect them using chemical bonds; (3) perform a 1000 step DPD simulation to remove internal stresses and geometric distortions; (4) repeat above processes until no more bead pairs can be found within rth, and then a larger distance rth + 1 is used to increase the threshold. It should be noted that the crosslinking between pairs of beads in the same molecule has been avoided. The script shows that only one crosslinking bond can be formed on the same bead C. The box size was modified according to the additional number of inserted linker beads, i.e., to ensure a number density of 3/rc3 of the DPD model. The crosslinking process stops when a desired crosslink degree was obtained. Fig. 1 provides a simple explanation on the crosslinking of two triblock copolymers and the corresponding real chemical structure at the crosslinking site.Moreover, to simulate the responsive dissociation process of a crosslinked vesicle, a decrosslinking process which is much simpler than the above crosslinking process was conducted. To simulate a dissociation degree, we only need to randomly delete a certain number of linker molecules, as well as modifying the box size to maintain the system density unchanged. The model at a specific decrosslinking state will be equilibrated by a dynamic DPD simulation, based on which further responsive dissociation simulations under external stimuli will be conducted. The release speed and accumulated release amount of loaded drugs, as well as the evolution of the vesicle dissociation morphology, in relation to the simulation time will be studied.
3 Results and discussion
3.1 Designing polymer vesicular structures
We first probe the formation of vesicles from the self-assembly of triblock copolymers. The self-assembly was carried out in a neutral solvent, during which the responsive block C was treated as an equivalent hydrophobic block as B. In this case, the hydrophilicity of block A is of great significance for the self-assembly morphology of copolymers.27,39 Here, we consider two aAW values, 25 and 30, representing fully compatible and slightly repulsive interactions between block A and solvent, respectively. Fig. 2 summarizes the clustering behavior of AmBnCl as a function of block length ratio m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :n:l at the two aAW values, for which the simulation models are provided in Fig. S1 and S2 in the ESI.† From Fig. 2(a) (aAW = 25), the self-assembly morphology of AmBnCl develops from a large vesicle to a core–shell micelle with a decreasing size, along with the increase of m. The phase diagram is separated into two parts by a dividing line near m = 4. From the cluster number and AmBnCl number per cluster indicated by the plots, a gradual decomposing process induced by the hydrophilic–hydrophobic interaction of the system is presented. Different from this mechanism, Fig. 2(b) (aAW = 30) indicates the evolution of the AmBnCl self-assembly morphology from a multi-compartment (m ≤ 2) to a vesicle (3 ≤ m ≤ 10), porous vesicle (11 ≤ m ≤ 12), and bicontinuous micelle (m ≥ 13) along with the increase of m. The cluster number and AmBnCl number per cluster keep unchanged in the range of m. Here, the hydrophilic–hydrophobic interaction does not affect the overall clustering behavior, but leads to a large variation in the local morphology. The above two conditions represent the possible variation of external stimuli (solvent exchange and pH variation) which may induce the morphological evolution of the self-assembly structure. The obtained vesicular structures are focused for the follow-up study. Two block length ratios of m:n:l at 4:8:8 and 10:5:5 (noted as A4B8C8 and A10B5C5 at aAW = 30) are chosen to construct two representative vesicles.
:n:l at the two aAW values, for which the simulation models are provided in Fig. S1 and S2 in the ESI.† From Fig. 2(a) (aAW = 25), the self-assembly morphology of AmBnCl develops from a large vesicle to a core–shell micelle with a decreasing size, along with the increase of m. The phase diagram is separated into two parts by a dividing line near m = 4. From the cluster number and AmBnCl number per cluster indicated by the plots, a gradual decomposing process induced by the hydrophilic–hydrophobic interaction of the system is presented. Different from this mechanism, Fig. 2(b) (aAW = 30) indicates the evolution of the AmBnCl self-assembly morphology from a multi-compartment (m ≤ 2) to a vesicle (3 ≤ m ≤ 10), porous vesicle (11 ≤ m ≤ 12), and bicontinuous micelle (m ≥ 13) along with the increase of m. The cluster number and AmBnCl number per cluster keep unchanged in the range of m. Here, the hydrophilic–hydrophobic interaction does not affect the overall clustering behavior, but leads to a large variation in the local morphology. The above two conditions represent the possible variation of external stimuli (solvent exchange and pH variation) which may induce the morphological evolution of the self-assembly structure. The obtained vesicular structures are focused for the follow-up study. Two block length ratios of m:n:l at 4:8:8 and 10:5:5 (noted as A4B8C8 and A10B5C5 at aAW = 30) are chosen to construct two representative vesicles.
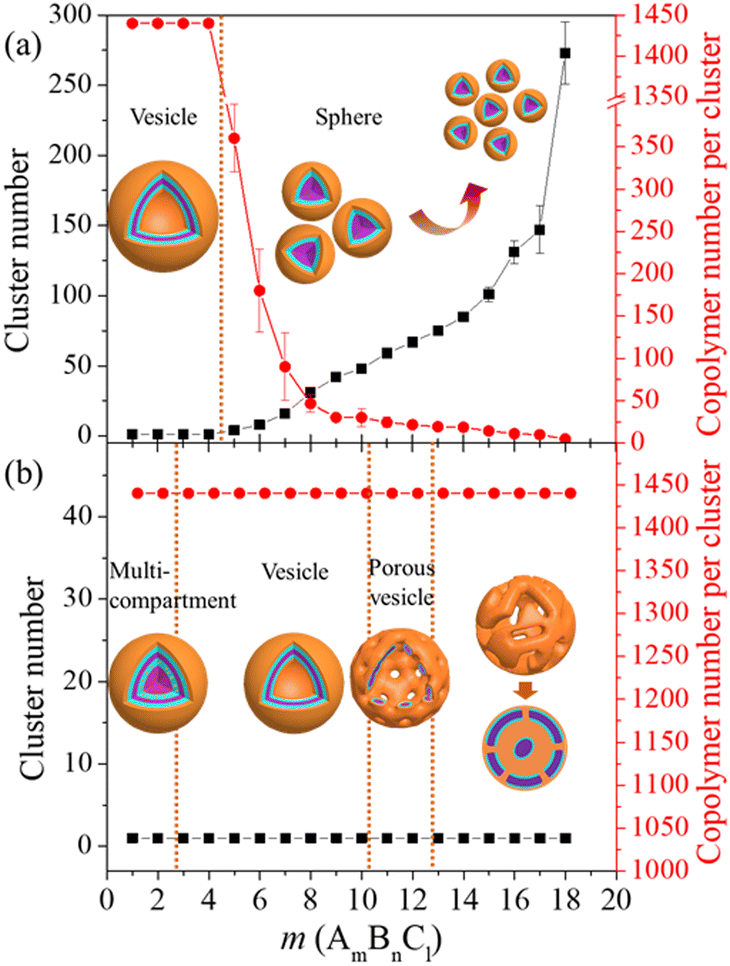 | ||
| Fig. 2 Self-assembly of AmBnCl and the evolution of clustering behavior (i.e., cluster number and copolymer number per cluster) as a function of the length increase of hydrophilic block A (m varies from 1 to 18, m + n + l = 20, n equals to the integral part of (20 − m)/2). (a) and (b) Systems with aAW equal to 25 and 30, respectively. | ||
Next, we load the A4B8C8 and A10B5C5 vesicular structures with both hydrophobic bead N (Nile red) and hydrophilic bead D (DOX-HCl), and probe the equilibrium properties of structure, energy, and dynamics in the systems. Both vesicles contain the same volume fraction of copolymer molecules or drug molecules. Fig. 3(a) and (b) indicate the cross-sectional views and the radial density profiles of split copolymers and drug beads. Obvious stratification behavior can be seen within the two vesicles. The N and D beads are distributed within the hydrophobic layer B and the hydrophilic core C regions, respectively, driven by hydrophilic/hydrophobic interactions. From A4B8C8 to A10B5C5, the thicknesses (equal to half peak width) of hydrophilic A, responsive B, and hydrophobic C layers experience clear changes, i.e., layer A increases from 2.3 to 2.5 distance unit, layer B decreases from 2.4 to 1.5 distance unit, and layer C decreases from 3.2 to 2.4 distance unit. A significant influence of the above structural change of the vesicle is on its loading capacity to N and D beads. Fig. 3(c) exhibits the interaction potential energy, E, for bead pairs of ABC-N, ABC-D, and ABC-W, respectively. The decreased EABC-N value (from 0.045 to 0.043) denotes weakened binding ability to N beads, the increased EABC-D value (from 0.009 to 0.019) indicates more favorable for loading of D beads, while the increased EABC-W value (from 0.117 to 0.129) states that the water solubility of the vesicle is enhanced. Fig. 3(d) provides the calculated dynamical fluctuation radius, Δr, of ABC, N, and D molecules, respectively. The increased ΔrABC (from 4.124 to 5.665) is induced by the above-mentioned water solubility enhancement of the vesicle, and the increased ΔrN (from 6.888 to 8.035) is due to the weakening binding capacity of the hydrophobic layer to Nile red, while the increased ΔrD (from 4.046 to 4.680) is because of the enhanced affinity of the confined space to DOX-HCl.
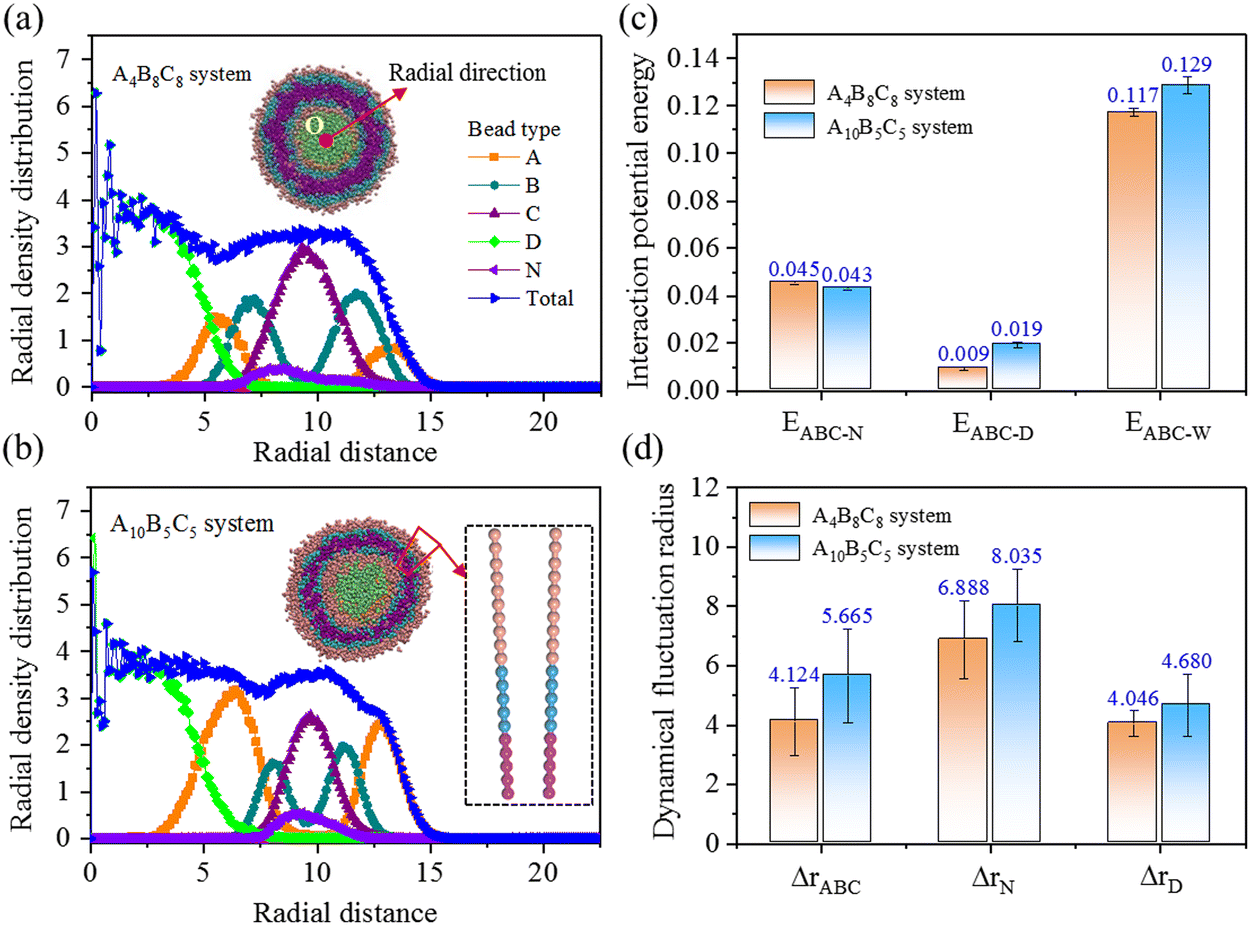 | ||
| Fig. 3 Properties of the structure, energy, and dynamics of uncrosslinked A4B8C8 and A10B5C5 polymer vesicles loaded with N in the shell and D in the core. (a) and (b) Radial density distributions of total and separate beads. (c) Interaction potential energies for bead pairs of ABC-N, ABC-D, and ABC-W, respectively. (d) Dynamical fluctuation radii of ABC, N, and D molecules. | ||
3.2 Structural enhancement by shell crosslinking
Chemical crosslinking is performed on the vesicle shell to enhance the stability of drug loading. As the linker molecule consists of two beads (type L), the vesicle shell experiences a certain degree of expansion during the crosslinking process. We performed frequent structural optimization and dynamical equilibration to soothe the generated internal stress and geometric deformation. Using a truncation radius of rth = 10 distance unit, we obtained the crosslinked A4B8C8 and A10B5C5 vesicles with the largest crosslinking degrees of 91% and 96%, respectively.Fig. 4(a) and (b) indicate the radial density distributions of beads in the crosslinked A4B8C8 and A10B5C5 systems. From the cross-sectional morphologies, it can be seen that the self-assembly structure of copolymers and the loading positions of N and D remain unchanged. The distribution of linker beads synchronizes with that of crosslinking block C. The thicknesses of crosslinked layer C in A4B8C8 and A10B5C5 systems are increased from 3.2 to 6.0 distance unit (improved by 87.5%) and from 2.4 to 4.0 distance unit (improved by 41.9%), respectively, and the volume expansion degrees of the two systems are calculated to be 19.2% and 6.7% (based on the radial distance of “total” density lines), respectively. Fig. 4(c) and (d) indicate the interaction potential energies and dynamical fluctuation radius of the crosslinked A4B8C8 and A10B5C5 systems, respectively. In terms of energy, compared to uncrosslinked systems, the EABC-N values in crosslinked A4B8C8 and A10B5C5 systems are decreased to proportions of 28.9% and 41.7%, respectively, while the EABC-D and EABC-W values have hardly decreased. It is inferred from this that the crosslinking structure mainly weakens the interaction between copolymers and N beads, i.e., the loading capacity for N may be reduced. In terms of dynamics, it can be seen that all the ΔrABC, ΔrN, and ΔrD values experience large reduction, indicating that the motion abilities of these beads are weakened by the formation of the crosslinking structure. This phenomenon reflects that the loading stability of the two drug molecules in the polymer vesicle is clearly improved by chemical crosslinking of the shell.
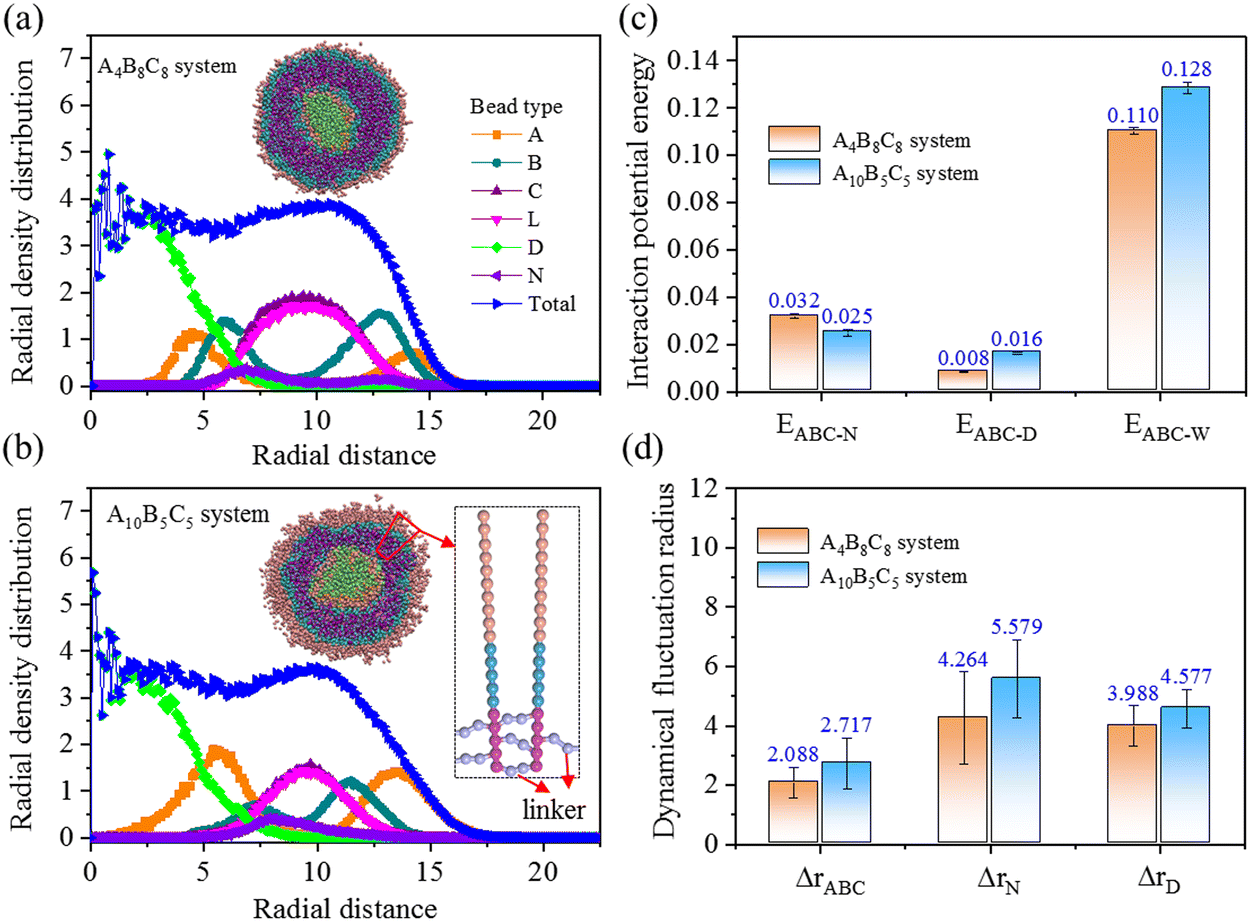 | ||
| Fig. 4 Properties of the structure, energy, and dynamics of crosslinked A4B8C8 and A10B5C5 polymer vesicles loaded with N in the shell and D in the core. (a) and (b) Radial density distributions of total and separate beads. (c) Interaction potential energies for molecule pairs of ABC-N, ABC-D, and ABC-W, respectively. (d) Dynamical fluctuation radii of ABC, N, and D molecules. | ||
3.3 Stimuli-triggered response of crosslinked vesicles
Triggered by the specific tumor microenvironment condition (e.g., low pH value), the crosslinked vesicles may experience breakage of crosslinking bonds and protonation of amino groups, which will ultimately lead to vesicle dissociation and drug release. To reveal the response mechanism of the crosslinked A4B8C8 and A10B5C5 vesicles, we simulated their responsive decrosslinking processes at low pH values and compared their differences in evolutions of morphology, drug–polymer interaction, dynamic fluctuation, and drug release properties. The interaction parameters between responsive block C and other beads under acidic conditions were also changed, i.e., the aCA, aCD and aCW values were reduced from 80, 80 and 100 to 40, respectively, to simulate the enhanced compatibility, while the aCN and aCC values were increased from 25 to 40 to simulate the enhanced incompatibility. Fig. 5 and 6 display the snapshots of equilibrated model structures of the two vesicles versus the reduction of the crosslinking degree. For the crosslinked A4B8C8 vesicle, it can be observed that the vesicle maintains the spherical morphology at crosslinking degrees of 91% and 80%, dissociates out small spherical micelles at 60% and 40%, and totally dissociates into small spherical micelles at 20% and 0%. By comparison, the crosslinked A10B5C5 vesicle maintains the spherical morphology at crosslinking degrees of 96% and 80%, experiences local dissociation at 60% and 40%, and dissociates into worm-like or small spherical micelles at 20% and 0%. The different re-assembly morphologies of the two systems can be attributed to the different length ratios of hydrophilic and hydrophobic blocks.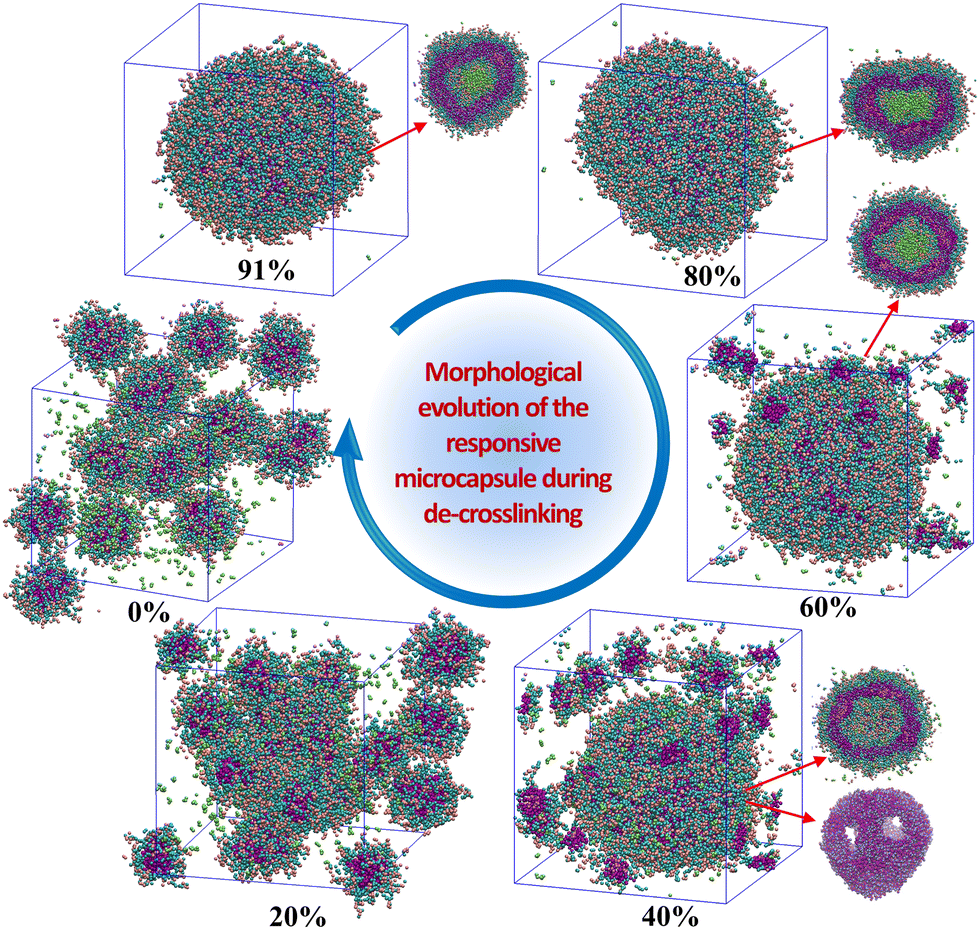 | ||
| Fig. 5 Morphological evolution of the crosslinked A4B8C8 vesicle during the responsive decrosslinking process in acidic solution. Cross-sectional views of several structures are given to show inner details. | ||
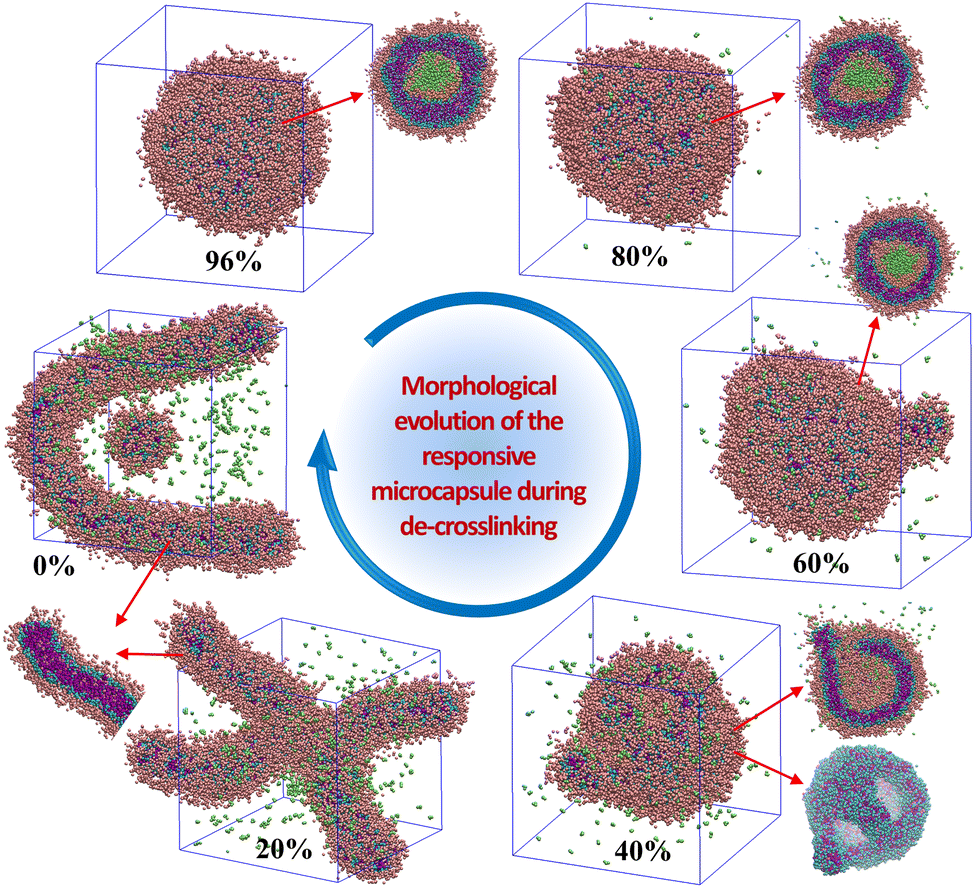 | ||
| Fig. 6 Morphological evolution of the crosslinked A10B5C5 vesicle during the responsive decrosslinking process in acidic solution. Cross-sectional views of several structures are given to show inner details. | ||
In terms of drug release, the hydrophilic D beads are observed to migrate into the solution, while the hydrophobic beads N are still encapsulated by the hydrophobic region of the re-assembled copolymer clusters. Fig. 7(a) and (b) indicate the percentages of released beads D during the decrosslinking of A4B8C8 and A10B5C5 polymer vesicles. We performed 1000000 steps of the simulation for the model at each crosslinking degree. It can be seen that with decrosslinking of the vesicles, the release rate of loaded drugs gradually increases. Crosslinking degrees at ≥60% seem to be benificial to achieve the long-term slow release of loaded drugs. While at ≤40%, rapid and complete release loaded drugs are observed during the simulation time. This release rate change induced by decrosslinking can be closely related to the morphological decomposition in Fig. 5 and 6, particularly for the transition region of 40–60% when the vesicles start to decompose into smaller copolymer clusters. By comparison, the crosslinked A4B8C8 system seems to be more sensitive to environmental stimuli and has a larger drug release rate than the crosslinked A10B5C5 system. For example, at a simulation step of 1000000, the drug release percentage of A4B8C8 increases from 5% to 12%, 29%, 100%, 100%, and 100%, while that of A10B5C5 increases from 0% to 7.5%, 26%, 88%, 100%, and 100%.
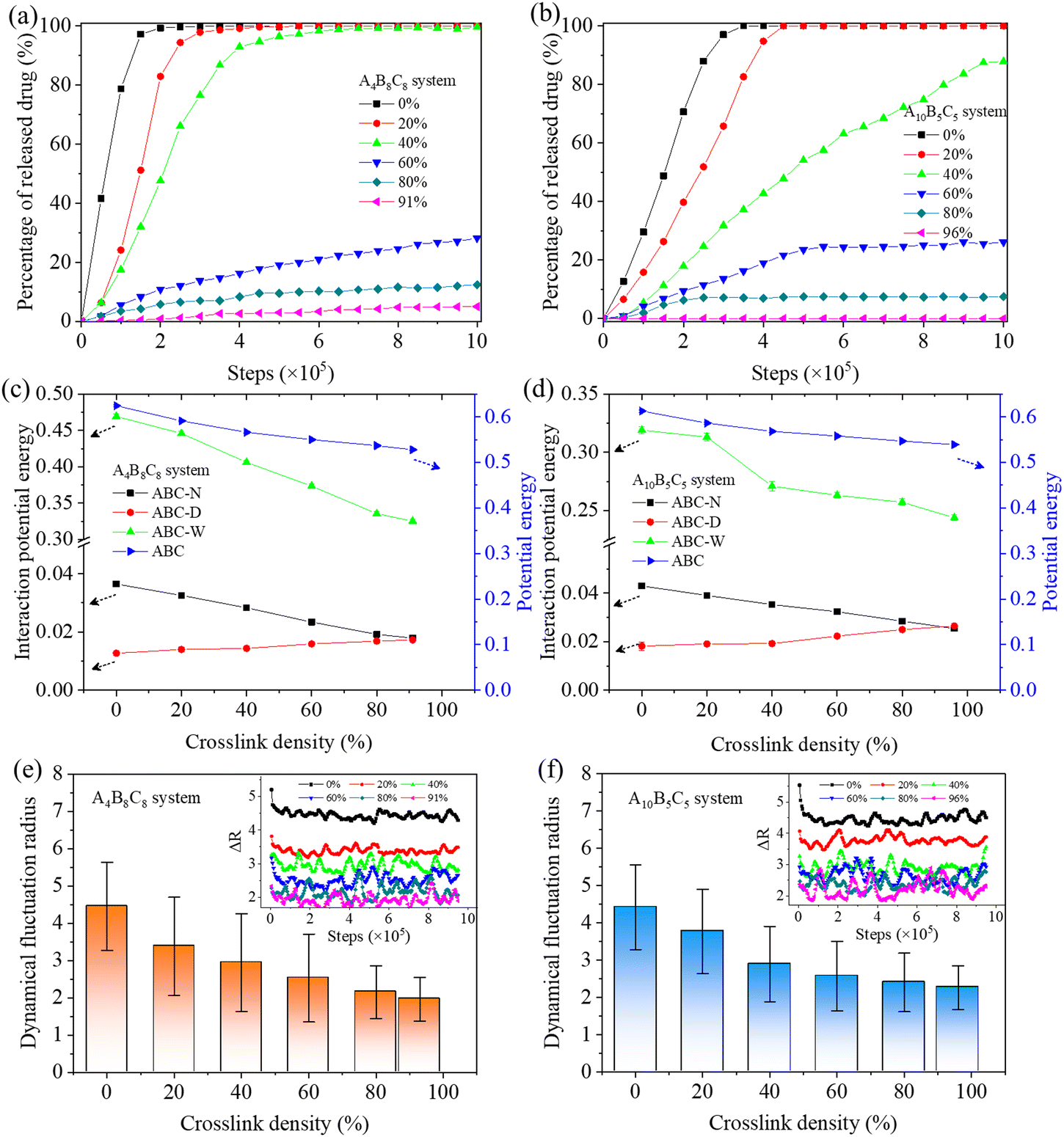 | ||
| Fig. 7 Variaitons of drug release (a) and (b), interaction potential energy (c) and (d), and dynamic fluctuation (e) and (f) properties of the A4B8C8 and A10B5C5 polymer vesicles. | ||
One important factor driving the vesicle dissociation and drug release is the varied intermolecular interaction between beads in the system under external stimuli. Fig. 7(c) and (d) indicate the calculated interaction potential energies between copolymers and Nile red, DOX-HCl, and water, respectively, and the potential energy of all copolymers, versus the decrosslinking degree. It can be seen that, along with decrosslinking of the two vesicles, the interaction potential energies between copolymers and Nile red, as well as between copolymers and water are increased, while that between copolymers and DOX-HCl is decreased. The former increased energies can be attributed to the improved interaction between the studied bead pairs since the vesicle dissociation enhances their contact probability. The latter decreased energy is due to the fact that the released DOX-HCl drugs move to the solvent and become less contacted with the copolymers. Moreover, it is observed that the total potential energy of the copolymers themselves gradually increases with the decrosslinking of vesicles, indicating that the increased contact probability between copolymers and surrounding beads enhances the interaction of them. The above evolutions of interactions promote the gradual equilibrium of copolymer reassembly morphology and achieve stable drug release.
In the meantime, we analyzed the dynamical properties of copolymers during decrosslinking of the vesicles. Fig. 7(e) and (f) provide the obtained averaged dynamical fluctuation radius (Δr) of copolymer beads at all studied crosslinking densities. The evolution processes of Δr within the range of simulation steps are also recorded. As the crosslinking density decreases, it can be seen that the Δr value of the A4B8C8 system increases from 1.97 (at 91%) to 4.45 (at 0%) with a growth degree of 126%, while that of the A10B5C5 system increases from 2.26 (at 96%) to 4.42 (at 0%) with a growth degree of 96%. Clearly, the increased Δr value indicates that the movement ability of copolymer beads becomes gradually enhanced at smaller crosslinking densities. This is mainly due to the dissociation of vesicles which greatly increases the probability of contact between the copolymer and the solution, and is beneficial to improve the self diffusion and migration of copolymers.
3.4 Effect of bead–bead interaction on drug release
Furthermore, the interaction force between different beads is also one of the important reasons for the differences in drug release properties. In experiments, the interaction force between beads may be modified by changing the type of functional groups, the property of solvents, the type of environmental stimuli, etc. Here, focusing on the 60% crosslinked A4B8C8 system, we investigated the effect of pairwise repulsive parameters on the culmulative percentage of released drugs. A series of parameter ranges have been studied in the hope of gaining a comprehensive understanding of the possible mechanisms of action. To reveal more possibilities based on the simulation system, we varied one of the interaction parameters with fixing the others. Some of the parameter settings may be unrealistic or hard to achieve in chemistry, their directionality of change patterns is our greatest concern.
Fig. 8 illustrates the drug release curves versus repulsive parameters of aAW, aBW, aCW, and aDW, which determines the repulsion between solvent W and blocks A, B, C, and drug D, respectively. For aAW (Fig. 8(a)), the culmulated release amount within the simulation time (1000000 steps) reaches up to 32% at aAW = 25 (the situation where water solubility is particularly good). When the value of aAW increases to or greater than 35, the release amount rapidly decreases below 10%, which is understandable since the increase in the hydrophobicity of block A makes it more difficult for copolymers to dissociate. For aBW (Fig. 8(b)), as the hydrophobicity of responsive block B gradually decreases (aBW drops froms 100 to 30), the drug release rate and amount will gradually increase accordingly, even close to 100% at a simulation step of 1000000 when aBW equals to 30. The decrease in aBW is equivalent to the increase in the hydrophilic block ratio, which reduces the aggregation ability of hydrophobic blocks. For aCW (Fig. 8(c)), the accumulated drug release amount does not exceed 50% within the range of investigated aCW values. The decrease of aCW represents the gradual transition of the hydrophilic–hydrophobic–hydrophobic triblock copolymer to the near hydrophilic–hydrophobic–hydrophilic triblock copolymer, and the self-assembly structure of copolymers undergoes changes accordingly. Therefore, as can be seen, the decrease of aCW first leads to a decrease and then increases the release rate and amount. For aDW (Fig. 8(d)), which represents the hydrophobicity of drug molecules, a very large value (aDW ≥ 50) effectively inhibits the release of drugs while a small value (aDW ≤ 40) promotes the release of drugs (up to 85% at aDW = 20). This result reflects the need to balance the hydrophilicity and hydrophobicity of the loaded drugs, since different drugs may experience different loadings and release mechanisms.
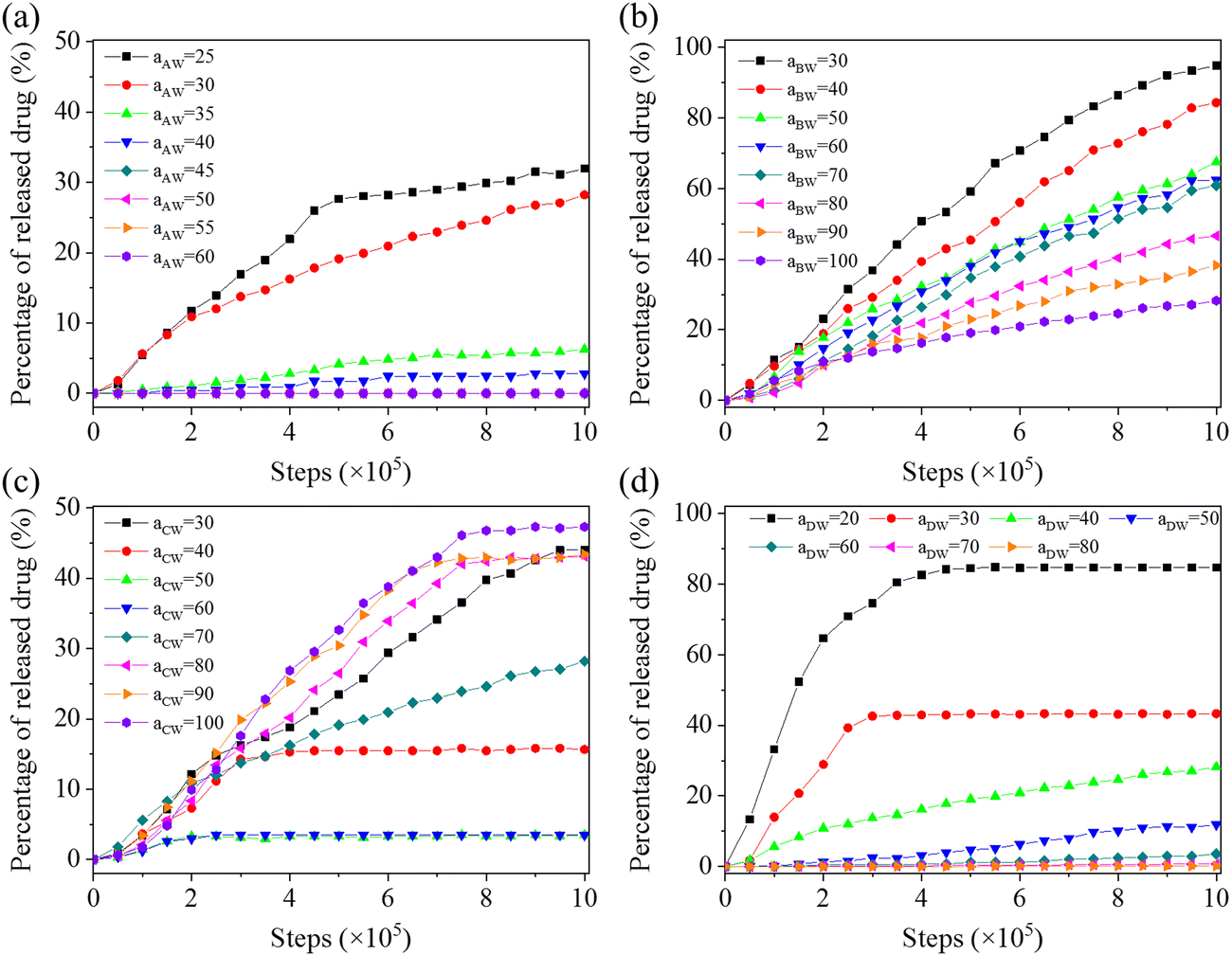 | ||
| Fig. 8 Effect of repulsive parameters between solvent W and blocks A (aAW, (a)), B (aBW, (b)), C (aCW, (c)), and drug D (aDW, (d)), respectively, on the release of drugs from the 60% crosslinked A4B8C8 polymersome. | ||
Fig. 9 illustrates the drug release profies versus the repulsive parameters of aBA, aBB, aBC, and aBD, which determine the repulsion between hydrophobic block B and copolymer blocks A, B, C, drug D, respectively. For aBA (Fig. 9(a)), which reflects the extension degree of the two connecting blocks, the increase of the value can promote the release speed and amount of drugs. We speculate that the increased entension force between blocks B and A can make the self-assembled structure more porous and becomes more easily dissociated. Similar situations can be found for aBB (Fig. 9(b)) and aBC (Fig. 9(c)), where the drug release tendency can be promoted at larger values. The impact of the extension force determined by aBC on drug release seems to be smaller than aAB, since the largest accumulated release amount only reaches 31% at a simulation step of 1000000. The self-repulsion between hydrophobic blocks B denoted by aBB exhibits a larger scope of the drug release amount, which ranges from 3% at aBB = 20 to 84% at aBB = 100 at a simulation step of 1000000. The control of aBB can be realized by, e.g., utilizing the electrostatic repulsion between charged groups. For aBD (Fig. 9(d)), which indicates the repulsion between loaded drug molecules and hydrophobic block B, it also has a limited impact on the release amount of drug molecules. The largest accumulated release amount is 31% at aBD = 25 (i.e., high compatibility between drug D and hydrophobic block B) and at a simulation step of 1000000. The increased value of aBD leads to a quick reduction of the release amount of drug molecules.
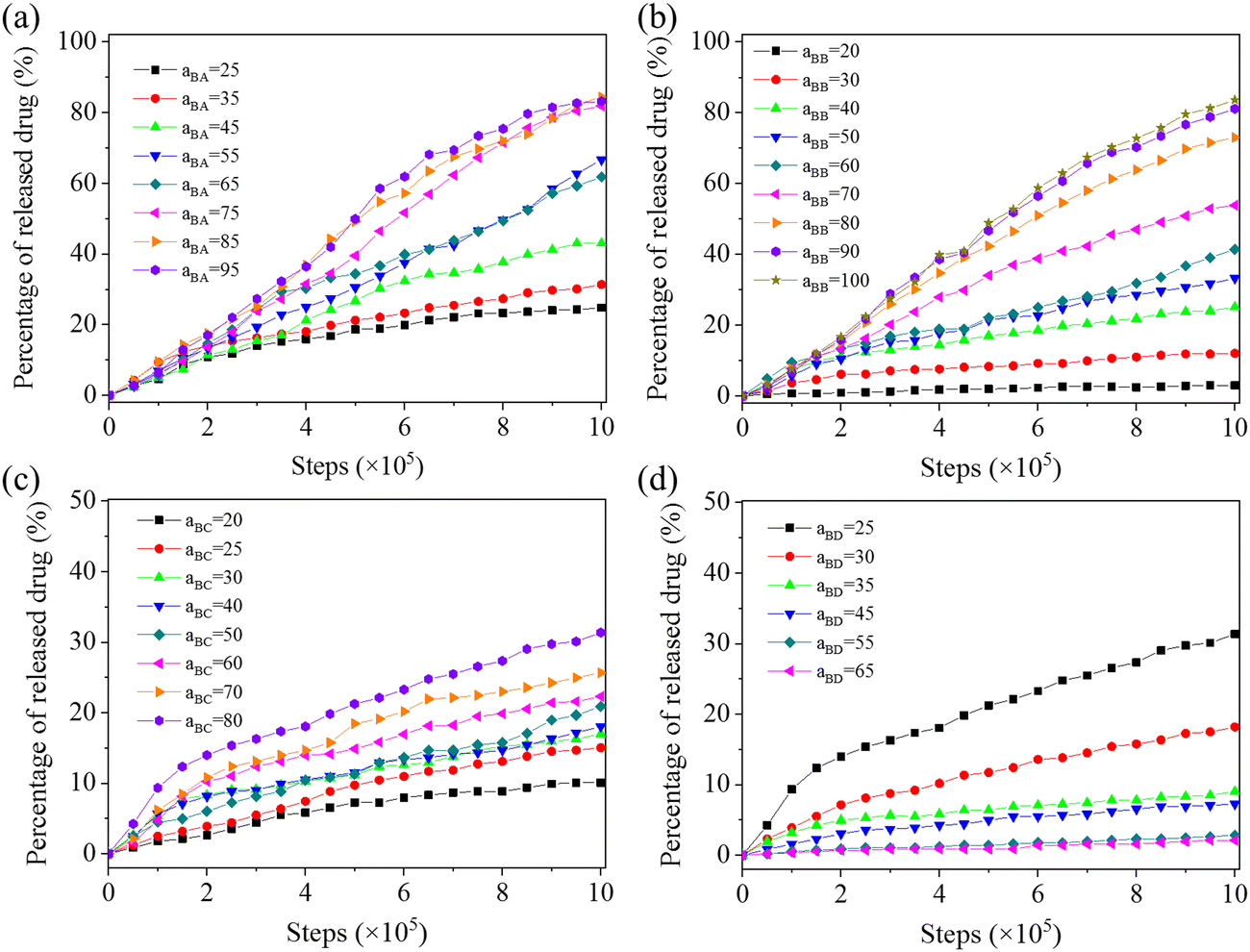 | ||
| Fig. 9 Effect of repulsive parameters between hydrophobic block B and blocks A (aBA, (a)), B (aBB, (b)), C (aBC, (c)), and drug D (aBD, (d)), respectively, on the release of drugs from the 60% crosslinked A4B8C8 polymersome. | ||
Based on above discussions, the release pattern of leaded drugs can be controlled mainly through tuning the repulsive interactions of copolymer segments or their solvent affinities. However, in real cases, it is often hard to make arbitrary settings and changes of these interactions. People need to carefully choose and synthesize block copolymers with specific structural and chemical properties, or to try control the compatibilities between copolymer blocks and solvents.40 The environmental conditions as well as forms of stimulation also have important impacts.41–43 Our findings fully exhibit the potential patterns of the change of reversible crosslinked vesicular drug delivery systems, and provide important theoretical guidance for the development and performance optimization of such systems in the experiment.
4 Summary and conclusions
In summary, the controlled loading and stimuli-triggered release of theranostic agents in the crosslinked triblock copolymer vesicle have been demonstrated by coarse-grained dissipative particle dynamics simulations. The self-assembly of triblock copolymers with different lengths of hydrophilic and hydrophobic blocks has been studied for the construction of vesicular nanocontainers. Two copolymers, i.e., A4B8C8 and A10B5C5, are chosen to form two typical vesicles with inconsistent thicknesses of hydrophobic and crosslinkable block areas in the vesicle shell. After crosslinking, the structural stability of the shell is proven to be enhanced by calculating the radial density distribution, interaction potential energy and dynamical fluctuation radius of polymers. In response to external stimuli, the responsive decrosslinking of the vesicles is further simulated, and the morphological rearrangement of dissociated copolymers is presented. The release of loaded drugs along with copolymer dissociation has been tracked. By adjusting the range of repulsive parameters between different beads, we have further elaborated on the release patterns of loaded drugs under various conditions. Our findings indicate that the crosslinking stability of the vesicle shell is the key factor to avoid drug leakage, i.e., to ensure smooth drug delivery. Small differences in the copolymer molecular structure and crosslinking degree may lead to large fluctuations in drug loading and release efficiencies. The examined large range of pairwise repulsive interation parameters of different particles in the system deepens people's understanding on how to control the release rate of drugs.Author contributions
Z. W. and F. L. planned and performed the computational work. L. W., Y. L., M. L. and N. C. assisted during the model construction and data analysis. Z. W. wrote the manuscript and prepared the figures. C. L., S. S. and S. H. supervised the research and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52204066 and 51974345), the Shandong Provincial Natural Science Foundation of China (ZR2020QB185), the Fundamental Research Funds for the Central Universities (23CX06018A), and the Taishan Scholar Program of Shandong Province in China (tsqn202211076).Notes and references
- C. Lu and M. W. Urban, Prog. Polym. Sci., 2018, 78, 24–46 CrossRef CAS.
- J. A. Finbloom, C. Huynh and X. Huang, et al. , Nat. Rev. Bioeng., 2023, 1(2), 139–152 CrossRef.
- Y. Chen, D. Qin and J. Zou, et al. , Adv. Mater., 2023, 35(17), 2207787 CrossRef CAS PubMed.
- Y. Lu, J. Lin and L. Wang, et al. , Chem. Rev., 2020, 120(9), 4111–4140 CrossRef CAS PubMed.
- Y. Dai, C. Xu and X. Sun, et al. , Chem. Soc. Rev., 2017, 46(12), 3830–3852 RSC.
- C. G. Palivan, R. Goers and A. Najer, et al. , Chem. Soc. Rev., 2016, 45(2), 377–411 RSC.
- V. Balasubramanian, B. Herranz-Blanco and P. V. Almeida, et al. , Prog. Polym. Sci., 2016, 60, 51–85 CrossRef CAS.
- X. Feng, N. Yan and J. Jin, et al. , Macromolecules, 2023, 56(6), 2560–2567 CrossRef CAS.
- J. Kim, S. Jeong and R. Korneev, et al. , Biomacromolecules, 2019, 20(6), 2430–2439 CrossRef CAS PubMed.
- D. Wang, S. Moreno and S. Boye, et al. , Macromol. Rapid Commun., 2023, 44(16), 2200885 CrossRef CAS PubMed.
- K. Zhu, G. Liu and G. Zhang, et al. , Macromolecules, 2018, 51(21), 8530–8538 CrossRef CAS.
- X. Xu, W. Yang and Q. Liang, et al. , Nano Res., 2019, 12(3), 659–667 CrossRef CAS.
- Y. Liu, M. J. van Steenbergen and Z. Zhong, et al. , Macromolecules, 2020, 53(16), 7009–7024 CrossRef CAS PubMed.
- X. Wang, J. Hu and G. Liu, et al. , J. Am. Chem. Soc., 2015, 137(48), 15262–15275 CrossRef CAS PubMed.
- S. Li, B. Xia and B. Javed, et al. , ACS Nano, 2020, 14(6), 7398–7411 CrossRef CAS PubMed.
- S. Moreno, P. Sharan and J. Engelke, et al. , Small, 2020, 16(37), 2002135 CrossRef CAS PubMed.
- O. Rifaie-Graham, N. F. B. Galensowske and C. Dean, et al. , Angew. Chem., Int. Ed., 2021, 60(2), 904–909 CrossRef CAS PubMed.
- J. Shao, S. Cao and D. S. Williams, et al. , Angew. Chem., Int. Ed., 2020, 59(39), 16918–16925 CrossRef CAS PubMed.
- W. Gu, J. An and H. Meng, et al. , Adv. Mater., 2019, 31(46), 1904742 CrossRef CAS PubMed.
- Z. Luo, Y. Li and B. Wang, et al. , Macromolecules, 2016, 49(16), 6084–6094 CrossRef CAS.
- S. Li, C. Yu and Y. Zhou, Phys. Chem. Chem. Phys., 2020, 22(43), 24934–24942 RSC.
- Y. H. Feng, X. P. Zhang and Y. Y. Hao, et al. , Colloids Surf., B, 2020, 195, 111260 CrossRef CAS PubMed.
- W. Min, D. Zhao and X. Quan, et al. , Colloids Surf., B, 2017, 152, 260–268 CrossRef CAS PubMed.
- Z. Luo and J. Jiang, J. Controlled Release, 2012, 162(1), 185–193 CrossRef CAS PubMed.
- W. Feng, L. Wang and X. Xu, et al. , Giant, 2023, 15, 100174 CrossRef CAS.
- Q. Zhang, J. Lin and L. Wang, et al. , Prog. Polym. Sci., 2017, 75, 1–30 CrossRef CAS.
- Z. Wang, J. Zhou and J. Wang, et al. , J. Colloid Interface Sci., 2022, 607, 1142–1152 CrossRef CAS PubMed.
- Z. Wang, J. Gao and V. Ustach, et al. , Langmuir, 2017, 33(29), 7288–7297 CrossRef CAS PubMed.
- Z. Wang, L. Fu and H. Zhu, et al. , Phys. Chem. Chem. Phys., 2022, 24(47), 28886–28894 RSC.
- Z. Wang, S. Sun and Q. Lyu, et al. , Macromol. Rapid Commun., 2020, 41(3), 1900561 CrossRef CAS PubMed.
- K. C. de Castro, J. C. Coco and É. M. dos Santos, et al. , J. Controlled Release, 2023, 353, 802–822 CrossRef CAS PubMed.
- N. Xu, B. Wang and Z. An, et al. , Chem. Mater., 2022, 34(10), 4732–4740 CrossRef CAS.
- H. Yang and Y. Cheng, Analyst, 2017, 142, 2654–2662 RSC.
- X. Wang, Y. Wei and D. Chen, et al. , J. Macromol. Sci., Part A: Pure Appl. Chem., 2017, 54(12), 956–966 CrossRef CAS.
- P. Español and P. Warren, Europhys. Lett., 1995, 30(4), 191 CrossRef.
- N. Thota and J. Jiang, Front. Mater., 2015, 2, 64 Search PubMed.
- R. D. Groot and P. B. Warren, J. Chem. Phys., 1997, 107(11), 4423–4435 CrossRef CAS.
- Z. Wang, Q. Lv and S. Chen, et al. , ACS Appl. Mater. Interfaces, 2016, 8(11), 7499–7508 CrossRef CAS PubMed.
- Z. Wang, S. Sun and C. Li, et al. , Soft Matter, 2017, 13(35), 5877–5887 RSC.
- S. Zehra, M. Mobin and R. Aslam, et al. , Prog. Org. Coat., 2023, 176, 107389 CrossRef CAS.
- Z. Chen, X. Li and B. Gong, et al. , ACS Appl. Mater. Interfaces, 2023, 15(1), 2067–2076 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang and Y. Wan, et al. , Prog. Polym. Sci., 2021, 116, 101386 CrossRef CAS.
- K. Yang, L. Feng and Z. Liu, Adv. Drug Delivery Rev., 2016, 105, 228–241 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04190e |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |