Bifunctional diatomic site catalysts supported by β12-borophene for efficient oxygen evolution and reduction reactions†
Received
19th September 2023
, Accepted 28th November 2023
First published on 29th November 2023
Abstract
Efficient bifunctional catalysts for oxygen evolution and reduction reactions (OERs/ORRs) are of great importance for sustainable and renewable clean energy, especially for metal–air batteries. Herein, we investigated β12-borophene with double-hole sites capped with 3d transition metal atoms to explore its catalyst performance for hydrogen evolution reactions (HERs), OERs and ORRs. It was found that the borophene is a good platform for diatomic site catalysts (DASCs) due to their advantage of stability over the corresponding single-atom catalysts (SACs) or clusters. The HER performance of DASCs on β12-BM was further improved compared to the SAC case. Furthermore, the supported FeNi DASC exhibited good catalytic performance for both OERs and ORRs, the overpotentials for which were 0.43 and 0.55 V, respectively, better than those of the corresponding supported Ni or Fe SAC due to synergistic effects. We herein propose a novel descriptor involving the Bader charges of coordinated atoms explicitly, behaving much better than the d-band center and integrated crystal orbital Hamilton population (-ICOHP) for DASCs. The synergistic effect of Fe–Ni pairs balanced the too strong binding of OH and further activated OH to achieve better catalytic performance. The results of this study can provide theoretical guidance for the design of efficient bifunctional electrocatalysts.
Introduction
With the increasing environmental pollution and exhaustion of fossil fuels,1 clean and sustainable energy technologies (e.g., metal–air batteries,2 solar cells3 and fuel cells4) have gained great interest. Rechargeable metal–air batteries2 have the potential to take the place of established lithium-ion batteries and even hydrogen fuel cells.5 Basic electrochemical reactions, such as oxygen evolution reactions (OERs) and oxygen reduction reactions (ORRs), play a pivotal role in energy conversion and storage technologies,6 especially in metal–air batteries. However, the low electron transfer kinetics in these reactions require electrocatalysts to speed up the reactions, while the development of efficient and stable electrocatalysts is a great challenge. Furthermore, the high cost and low-bifunctional OER/ORR activities of conventional unifunctional catalysts (e.g., Pt7 and IrO28) prevent their widespread practical application in rechargeable metal–air batteries. In this view, the development of highly active, conductive and safe bifunctional electrocatalysts is essential for rechargeable metal–air batteries.9
In the past few years, a variety of bifunctional OER/ORR catalysts have been investigated. On the experimental side, efficient bifunctionality was realized in P/N co-doped graphene frameworks (PNGF)10 with a combined ORR and OER overpotential of 0.705 V, which surpass noble metals in terms of (bi)functionality and durability. The OER and ORR overpotentials of the carbon-based Mn–N2C2 bifunctional electrocatalyst were 0.35 and 0.92 V, respectively.11 Carbon-based RuN4Cx also showed good catalytic performance for OERs and ORRs, the overpotentials for which were 0.37 and 0.83 V, respectively.12 Meanwhile, other metal-N-C-type carbon-based materials were also explored theoretically for bifunctional electrocatalysts. For example, the single-atom catalyst (SAC) Rh/Co@C3N was predicted to have comparable performance to the noble metal catalysts in terms of OERs and ORRs,13 and Rh@C2N had a lower OER overpotential (0.37 V) than that of the IrO2(110) benchmark with good ORR activity.14 The Co and Ni-based defective AlP monolayers also showed excellent OER/ORR bifunctional catalytic performance.15
In the past few decades, single-atom catalysts (SACs)16 have attracted much attention due to their unique advantages such as fully utilizing all metal atoms, uniform active sites and obvious selectivity, and stronger catalytic activity compared to conventional metal nanoparticles.17 However, with the increase in the loading of transition metal (TM) atoms, the cohesive energy of the cluster defeats the adsorption of single atoms onto the substrate. As a result, the TM atoms tend to cluster, and the catalytic performance decreased dramatically. In order to prevent metal atoms from aggregation, selecting a substrate with strong enough adsorption ability is very important. Two-dimensional (2D) nanosheets have been reported to be potential ideal platforms for designing novel OER/ORR electrocatalysts due to their excellent properties.18–20 Borophene monolayers (BMs) are novel two-dimensional materials composed of triangles and hexagonal holes,21–23 and such holes may serve as good anchoring sites for TM atoms, leading to independent dispersion of TM atoms without forming clusters. In addition, Wang et al. predicted that the single-atom catalyst Ni/β12-BM had good catalytic activity for OERs with an overpotential of only 0.40 V.24 Other studies also suggested that β12 borophene was a good SAC substrate for OERs/ORRs25 and CO2 reduction.26 In addition, recent studies have predicted that V2 hosted in the double vacancy of β12 borophene and Cr2/Mn2/Re2 hosted on pristine β12 borophene were promising catalysts for nitrogen reduction reactions.27,28
It was found that the synergistic effect between adjacent metal sites may be able to enhance the catalytic activity and induce bifunctionality.29,30 For example, Ni1/Ni2- and Fe1/Ni2-modified MXene-based diatomic site catalysts (DASCs) showed better bifunctional ORR/OER performance than that of the well-known unifunctional catalysts with low overpotentials, such as IrO2(110) for the OER and Pt(111) for the ORR.9 The cooperative bimetallic catalysts have become a powerful tool to realize high efficiency and selectivity in many catalytic reactions.31
In this study, we investigated the double-site catalysts TM1TM2/β12-BM (TM1 = Fe/Ni, TM2 = Cr–Cu) for hydrogen evolution reactions (HER) and ORRs/OERs using DFT methods, given the possible synergistic effect to achieve better catalytic performance. The OER activity of the Ni SAC may be further improved when adsorbing TM atoms on adjacent holes. While Fe sites in some 2D materials exhibit impressive oxygen reduction reaction activity.32,33 The coordination 3d transition metals are expected to modulate their local electronic environments34 to improve their catalytic activity. Our results indicated that the synergistic effect endows FeNi/β12-BM DASCs with good OER/ORR bifunctional catalytic performance. The corresponding OER and ORR overpotentials are 0.43 and 0.55 V, respectively, much lower than the corresponding values for Ni/β12-BM (0.45/0.95 V) and Fe/β12-BM (1.39/1.13 V) at the same accuracy. These results indicated that borophene monolayers are good platforms for OER/ORR bifunctional catalysts, and the synergistic effect in DASCs on borophene monolayers makes them candidate catalysts for other electrochemical reactions.
Computational methods
First-principles calculations were carried out using the Vienna Ab initio Simulation Package (VASP 6.1)35,36 with the projector augmented wave (PAW) method.37 The generalized gradient approximation of the Perdew–Burke–Ernzerhof (PBE)38 functional was used, and a cutoff energy of 500 eV was set for the plane waves. The convergence thresholds for the energy and force were 10−4 eV per cell and 0.01 eV Å−1, respectively. Spin polarization and Grimme's DFT-D3 van der Waals (vdW) corrections39 are considered throughout the calculations. A large vacuum spacing of more than 15 Å was taken to prevent mirror interactions. Supercells consisting of 3 × 4 × 1 unit cells of β12 borophene were used, and the Brillouin zones were sampled with 2π × 0.02 Å−1 spacing in the reciprocal space by the Monkhorst–Pack scheme.40 The climbing image nudged elastic band (CI-NEB) method41,42 was used to search transition states with a force convergence criterion of 0.05 eV Å−1.
The OER and ORR performance of our TM1TM2/β12-BM systems was investigated through widely used four intermediate steps43(e.g., as shown in Fig. S1 and S2 for FeNi/β12-BM), involving three intermediates (i.e., OH*, O* and OOH*). The details of the calculation methods for OERs and ORRs are described in the ESI.† The free energy change (ΔG) of each electrochemical step was calculated based on the computational hydrogen electrode (CHE) model proposed by Nørskov et al., where the chemical potential of an electron and proton pair is half of the free energy of H2.43–45 According to this method, ΔG is defined as follows:
| | | ΔG = ΔEDFT + ΔZPE − TΔS + ΔGU + ΔGpH | (1) |
where Δ
EDFT is the change in chemisorption energy from the DFT calculations,
T is the room temperature (298.15 K), and ΔZPE and Δ
S stand for the differences in zero point energy and entropy, respectively. Δ
GU = −
eU stands for the influence of the applied electrode potential
U, where
e is the elementary charge transferred. Δ
GpH represents the free energy correction of pH. The effect of
U and pH was not taken into account in our study (
U = 0, pH = 0). Based on the frequencies from VASP results, the zero point energy and vibrational entropy were calculated using the VASPKIT code.
46 A solvation energy correction of −0.3 eV was applied to OH* and OOH* intermediates, like the previous study on SAC Ni/β
12-BM.
24 The overpotentials for OERs and four-electron ORRs were calculated to evaluate the catalytic performance of our systems.
Results and discussions
1. Stability
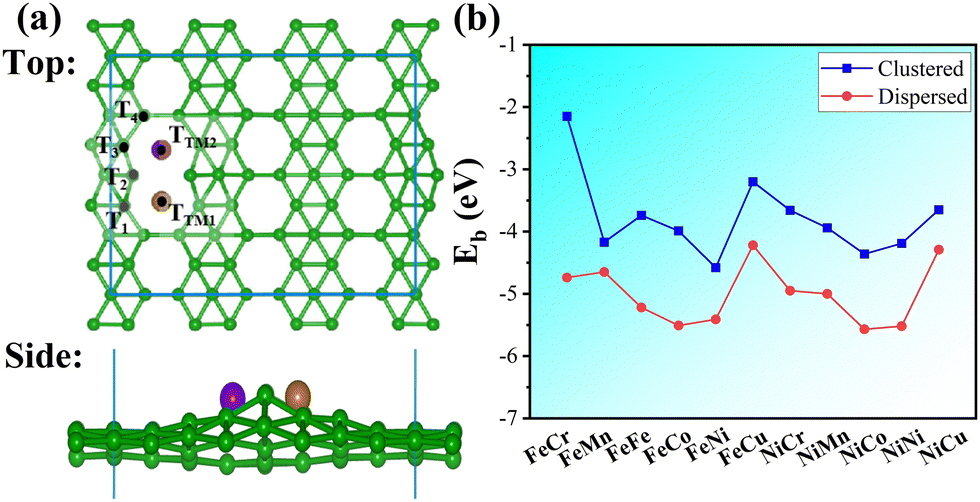
In this section, we investigated the stability of bimetallic TM1TM2/β12-BM (TM1 = Fe/Ni, TM2 = Cr–Cu) systems. The Fe and Ni atoms are very common catalytic centers for oxygen evolution and reduction reactions,47 and synergy effects are expected between Fe/Ni and other 3d TM atoms.48 The top and side views of the typical geometry of TM1TM2/β12-BM are shown in Fig. 1a. First, we investigated the possible adsorption modes (clustered or dispersed atoms) for several metal atoms, and their stabilities were evaluated using the average binding energy (Eb). For example, 2–4 Fe or Ni atoms were adsorbed on β12-BM (Fig. S3, ESI†), and their average binding energies (Ebind) were calculated as follows:| | 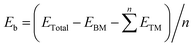 | (2) |
where ETotal, ETM and EBM stand for the energies of the total system, a single TM atom in the vacuum and borophene monolayer, respectively, and n is the number of TM atoms. Generally speaking, a more negative value of the average binding energy means a more stable state.
 |
| | Fig. 1 (a) Top and side views of typical TM1TM2/β12-BM geometry. (b) Average binding energies(Eb) for dispersed and clustered TM adatoms on β12-BM. The green, brown and purple balls represent B, TM1 and TM2 (TM1 = Fe/Ni, TM2 = Cr–Cu) atoms, respectively, and possible adsorption sites for an H atom are labeled in black. | |
It was reported that Ni/BM was a very good HER SAC,49 and hence, the adsorption of Fen and Nin (n = 2–4) clusters and their dispersed counterparts were investigated. As shown in Fig. S3 (ESI†), the dispersed Fe/Ni atoms have better stability than that of clustered Fe/Ni atoms; meanwhile, the average binding energy decreases when more Fe/Ni atoms disperse on the borophene. It indicates high loading of TM adatoms on the borophene. Furthermore, as displayed in Fig. 1b, the dispersed TM atomic pairs have better stability than their clustered counterparts. The average binding energies (Eb) of TM1TM2/β12-BM in the dispersed mode are very low (mainly smaller than −4.5 eV), which suggests high stability of TM atomic pairs on the borophene. It should be pointed that TM pairs consisting of Fe, Co or Ni atoms have better stability, facilitating their further applications.
As discussed above, the adjacent double holes of BM capped with TM atoms are stable, and the strong binding of TM atoms makes high loading of SACs/DASCs possible, though they buckle the borophene a little (Fig. 1a). The strong binding comes from the electron deficiency of the holes in borophenes and the low electronegativity of TM atoms. It tends to break the big π bonds which keep the borophenes planar50 and induces strong electrostatic interactions. However, the buckling of borophene may increase its catalytic performance, such as hydrogen evolution reactions (HERs).51
2. HER activity
High HER activity was predicted for pristine β12-BM (ΔG = 0.10 eV), and the HER performance was further improved (ΔG = 0.06 eV) when Ni SAC was adsorbed on β12-BM.49 How would the SAC perform for HERs when more TM atoms are adsorbed (e.g., diatomic catalysts) on β12-BM? To address this question, we evaluated the HER performance of several sites on TM1/β12-BM and TM1TM2/β12-BM (TM1 = Fe/Ni, TM2 = Cr–Cu), such as the top sites on TM atoms and their neighboring boron atoms (Fig. 1a). As shown in Fig. S4 (ESI†), for most sites considered on TM/β12-BM (TM = Fe/Ni) and TM1TM2/β12-BM, the calculated hydrogen adsorption free energies (ΔGH*) were very close to zero, indicating very good HER catalytic performance.52
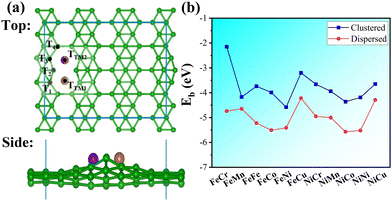
The exchange current (i0) was calculated to reflect the HER kinetics under the equilibrium potential U = 0 and pH = 0 conditions as follows:45
| |  | (3) |
where
kB is the Boltzmann constant and
T is the room temperature (298.15 K). For illustrative purposes,
k0 was set to 1.
53–55 Hence, a volcano curve was obtained to compare the HER activities of different systems (
Fig. 2a). The HER activity can be quantitatively evaluated from the position of Δ
GH* and log(
i0) with respect to the volcano peak. The closer the position to the volcano peak, the better the HER activity of the catalyst. It suggests that the HER performance is improved in most cases compared to pure β
12-BM when TM atoms were adsorbed, and several systems (
e.g., NiCu, FeFe, FeCr and FeCu) outperform the Pt(110) surface
56 and MoS
2 edges.
57
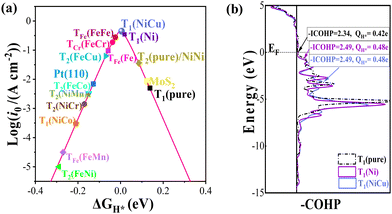 |
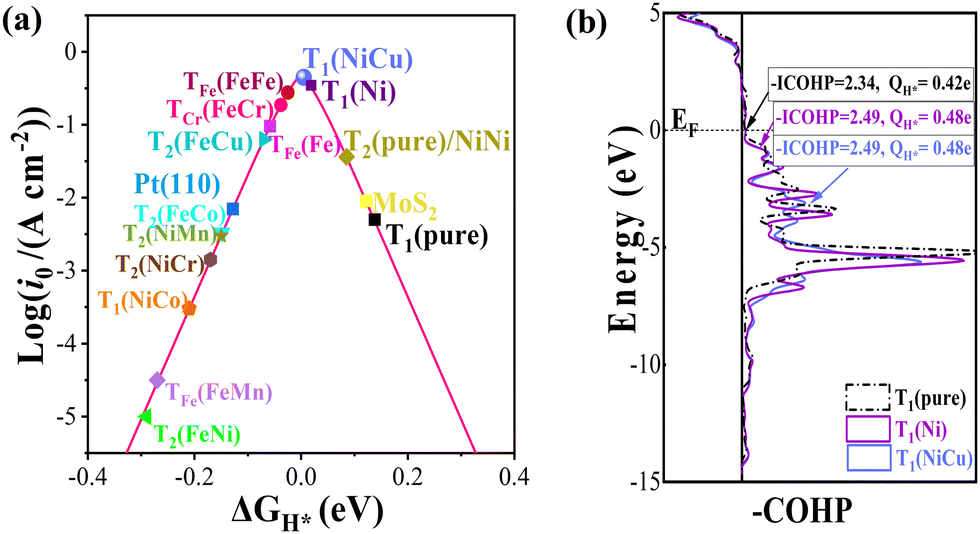
| | Fig. 2 (a) Volcano curve of exchange current (i0) as a function of the free energy of hydrogen adsorption (ΔGH*). (b) Crystal orbital Hamilton population (COHP) analysis between H* and neighbouring sites. Corresponding integrated COHP (-ICOHP) and lost electrons of H* adatoms are also listed. | |
Meanwhile, some DASCs have better HER performance than the corresponding SAC case, for example, ΔGH* of T1 site in the NiCu/β12-BM system is very close to 0 (∼0.005 eV), smaller than that of the Ni system (0.019 eV) at the same accuracy. In general, the adsorption free energy of an H adatom increases when TM atoms are adsorbed, which is beneficial for improving the HER performance. As shown in Fig. 2b, the crystal orbital Hamilton population (COHP)58 analysis suggests that the energy levels for the bonding between the H* adatom and neighbouring sites downshift, which enhances the bonding as indicated by the increased integrated COHP (-ICOHP) values. Meanwhile, more electrons transfer from H* adatoms to the BM from Bader charge analysis, indicating stronger electrostatic interactions (located at the left leg of the volcano) when more TM atoms are adsorbed.
3. OER/ORR activities
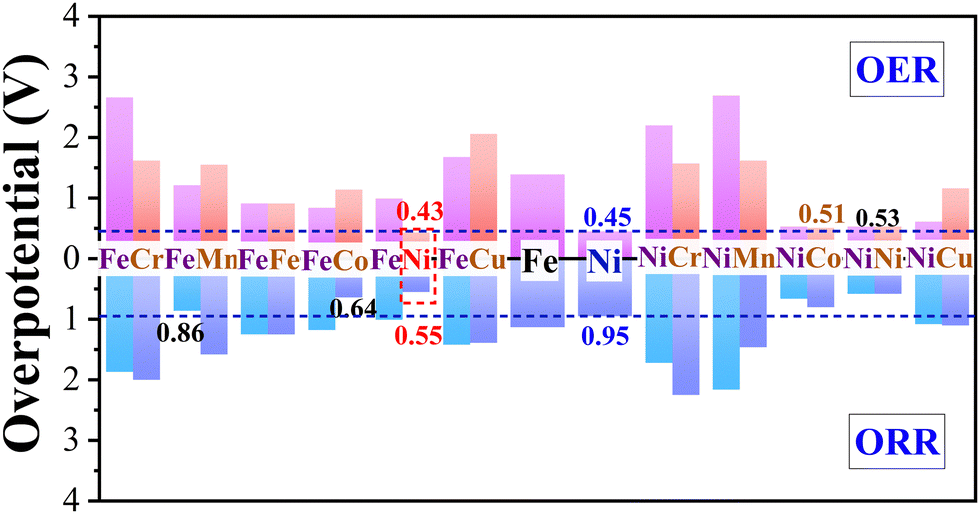
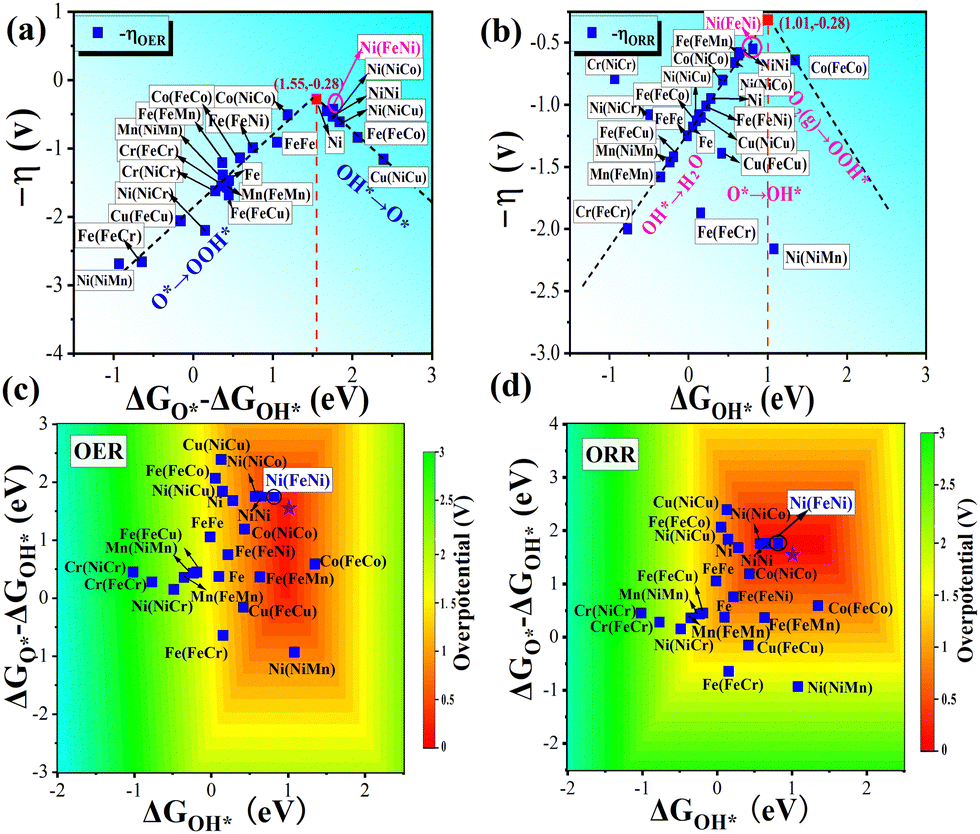
The free energies of oxygen-containing intermediates (i.e., OH*, O*, OOH*) for TM1/β12-BM and TM1TM2/β12-BM (TM1 = Fe/Ni, TM2 = Cr–Cu) were calculated and displayed in Fig. S5 (ESI†), in which OER and ORR free energy step diagrams are shown. The OER (ηOER) and 4e ORR ηORR overpotentials (η) from the potential-determining steps (PDSs) for TM1/β12-BM and TM1TM2/β12-BM (TM1 = Fe/Ni, TM2 = Cr–Cu) are displayed in Fig. 3.
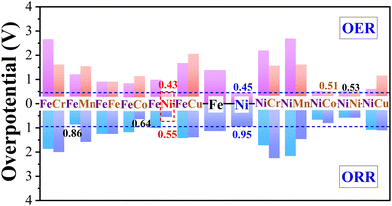 |
| | Fig. 3 Calculated OER and ORR overpotentials for TM1/β12-BM and TM1TM2/β12-BM (TM1 = Fe/Ni, TM2 = Cr–Cu) systems. The adsorption sites and corresponding OER/ORR overpotentials are labeled on the columns. The OER and ORR overpotentials for Ni/β12-BM are marked using brown dashed lines as a reference. | |
Fig. 3 shows that the OER/ORR overpotentials of the Ni(FeNi) site (i.e., abbreviation of adsorption site Ni in FeNi/β12-BM system) are 0.43/0.55 V, respectively, both lower than corresponding values of Ni/β12-BM (0.45/0.95 V) and Fe/β12-BM (1.39/1.33 V) at the same accuracy. The OER/ORR overpotentials of Ni(NiCo) and Ni(NiNi) are also very low (i.e., 0.53/0.66 V and 0.53/0.58 V, respectively). They are lower than those of previously reported MnOx (0.54/0.73 V)59 and Co/N-C-800 (1.6/0.74 V).60 The Ni(FeNi) site has comparable OER performance to RuO261 (ηORR = 0.42 V) and comparable ORR performance to Pt (ηORR = 0.45 V).43 It suggests that FeNi/β12-BM, NiNi/β12-BM and NiCo/β12-BM systems are good OER/ORR bifunctional catalysts. In brief, the synergistic effect of Fe–Ni, Ni–Ni or Ni–Co bimetallic atoms makes them behave better than single-atom catalysts in OERs and ORRs. It should be noted that the ORR overpotential of Ni/β12-BM in our study is significantly larger than that reported in a previous study25 (Table 1) using an Implicit solvation model,62 which tends to underestimate the OH* solvation energy and failed to predict the oxygen reduction onset potential on Pt(111).63 Our study suggests a method to convert unifunctional catalysts into bifunctional catalysts.
Table 1 Calculated overpotentials of OERs and ORRs for TM atoms on B38 cluster and borophenes. The references to previous data are shown in the right column. B38 is a planar boron cluster with double holes
| System |
η
OER (V) |
η
ORR (V) |
Ref. |
| Fe(FeNi)/B38 |
0.59 |
— |
72
|
| Ni(FeNi)/B38 |
0.89 |
— |
72
|
| Ni/β12-BM |
0.40 |
— |
24
|
| Ni/β12-BM |
0.38 |
0.34 |
25
|
| Ni/χ3-BM |
0.35 |
0.39 |
25
|
| Ni/β12-BM |
0.45 |
0.95 |
This work |
| NiNi/β12-BM |
0.53 |
0.58 |
| Ni(FeNi)/β12-BM |
0.43 |
0.55 |
| Ni(NiCo)/β12-BM |
0.53 |
0.66 |
4. Tendency of OER/ORR performance
According to Sabtier's principle,64 the adsorption strength of intermediates strongly affects the catalytic performance of the given catalyst. It was reported that the binding energies of intermediates (i.e., OH*, O*, OOH*) may be strongly correlated65 and the scaling relations between their free energies were found on some facets of face-centered cubic (fcc) metals and their alloys.66 To reveal the origin of the synergistic effect, the projected density of states (PDOS) with the d-band center are displayed in Fig. 4. The d-band center (εd) of the TM atom at the adsorption site was calculated as follows:| | 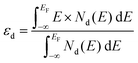 | (4) |
where EF and Nd stand for the Fermi level and PDOS of d states for TM atoms, respectively. According to the d-band center theory,67 a d-band center more close to the Fermi level usually indicates lower occupation of the antibonding states and stronger adsorption of adsorbates. However, too strong binding of adsorbates (e.g. oxygenated intermediates on TM atoms) will lead to difficult desorption of products and poor catalytic activity.
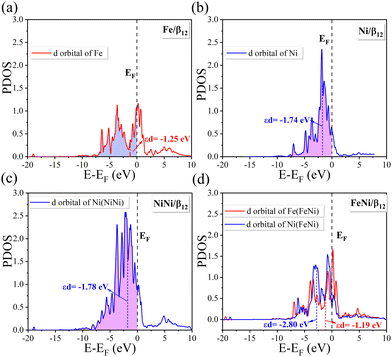 |
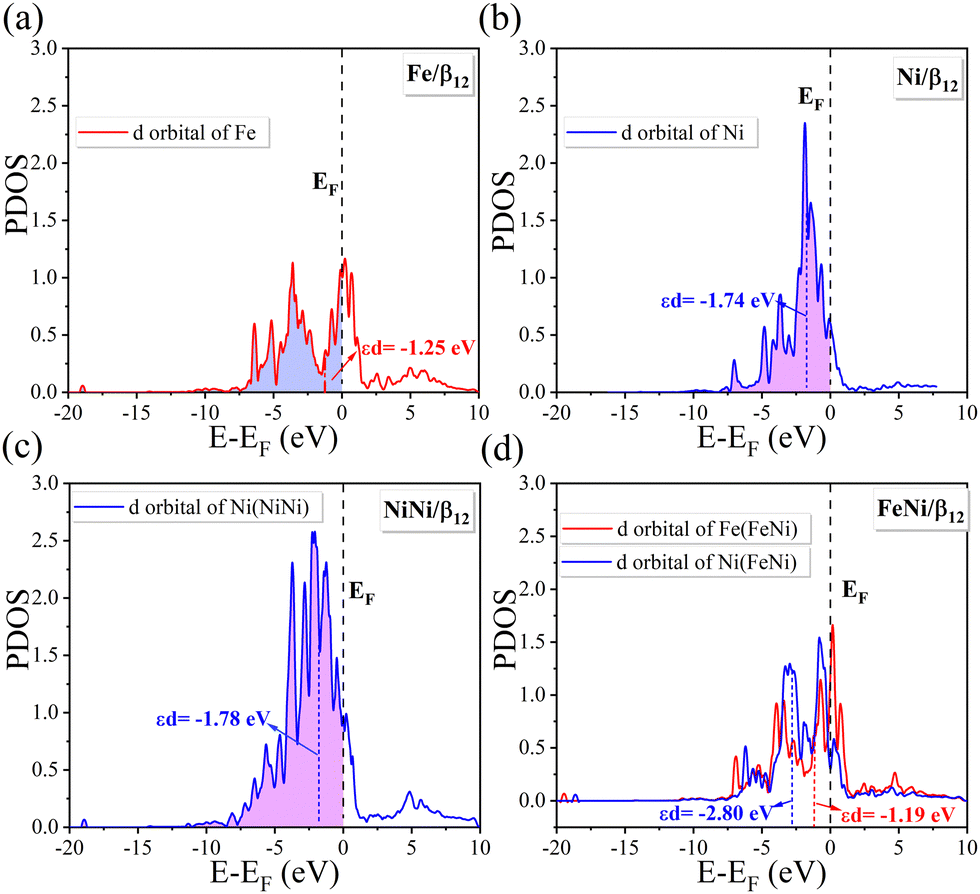
| | Fig. 4 Projected density of states (PDOS) of d-orbitals for the single (a) Fe or (b) Ni atom in OH*-TM/β12-BM systems. PDOS of d-orbitals for diatomic (c) NiNi or (d) FeNi in OH*-TM1TM2/β12-BM systems. Dotted lines denote the d-band center of the corresponding transition metal atoms. | |
As shown in Fig. 4a and b, the single Ni atom has a more negative d-band center (−1.74 eV) than that of the single Fe atom (−1.25 eV), and hence, the Ni/β12-BM exhibits weaker binding to the intermediates and, hence, has a lower free energy barrier and overpotential during the ORR and OER. For the DASC cases (in Fig. 4c, d and Fig. S6, ESI†), the Ni(FeNi) system shows the most negative d-band center of −2.80 eV, which implies the weakest binding of intermediate species and easiest desorption, leading to the highest ORR and OER activity among the DASCs. Compared to the SAC Ni/β12-BM system, the coordinated Fe/Ni/Co atom in DASC systems pushes the d-band center of the Ni atom away from the Fermi level, and thus, improves the OER/ORR catalytic performance.
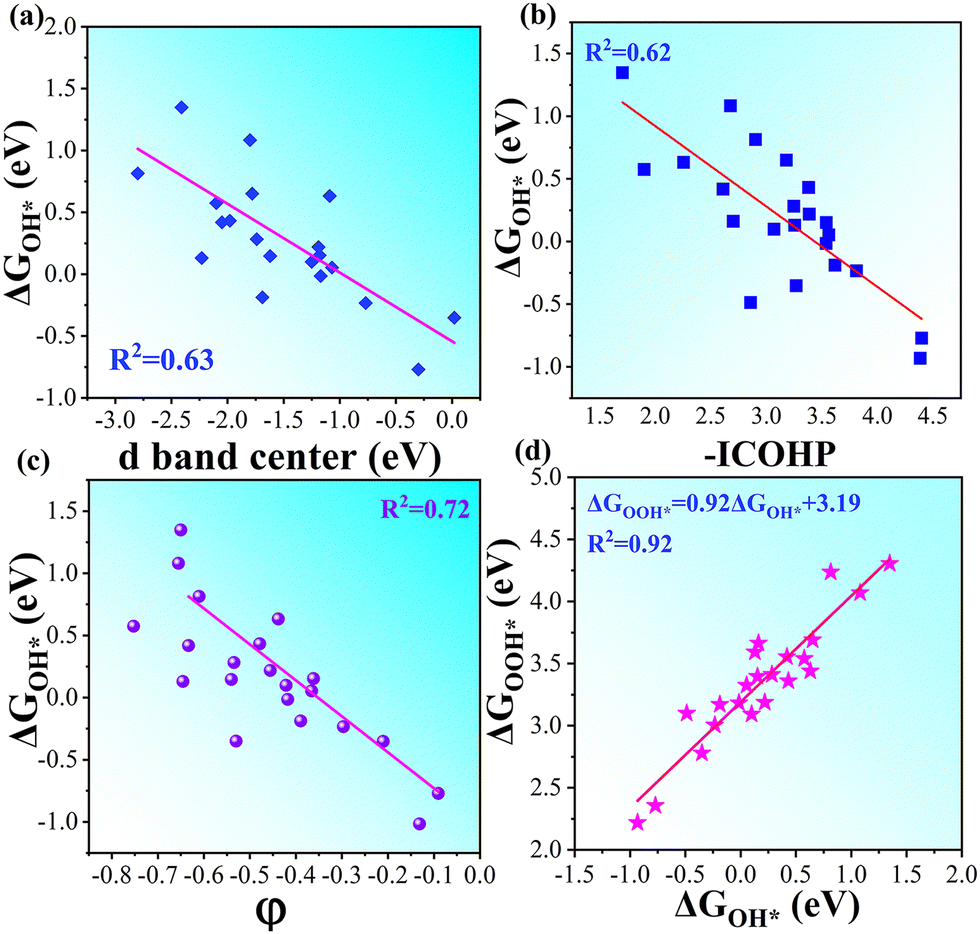
A more negative εd usually leads to a higher occupancy of antibonding states, thus weakening the adsorption, and vice versa. The -ICOHP also reflects the adsorption strength of the intermediates, and hence, it may also be a candidate descriptor for the catalytic performance68 according to Sabatier's principle.64 As shown in Fig. 5a, the d-band centers are basically linearly correlated with the free energy of OH*, and more negative d band centers suggest weaker adsorption, as discussed above. Similarly, the adsorption free energies ΔGOH* basically linearly decrease with the increase in -ICOHP. These two descriptors focus on the interaction between TM and OH*, including the effect of coordinated TM atoms implicitly; as a result, the coefficient of determination in the linear fitting is only 0.62–0.63. It suggests that the effective descriptors for the SAC24 seem to be less effective in DASCs due to the more complex environment. Inspired by previous studies,69 we tried to build a novel descriptor φ involving the Bader charges of coordinated TM and B/O atoms explicitly as follows:
| | 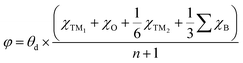 | (5) |
where
θd refers to the valence electron number of a free TM
1 atom,
χTM1 and
χTM2 stand for the Bader charges of TM
1 and TM
2 in the DASCs, respectively.
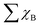
is the sum of the Bader charges of the six boron atoms around TM
1, and
χO represents the Bader charge of O, and
n + 1 is the number of B/O atoms (
n = 7 in this work) plus the TM
1 atoms.
Fig. 5c suggests that the free energies Δ
GOH* basically linearly decrease with the increase in
φ, the coefficient of determination for which is 0.72, much larger than those for the d band center and -ICOHP. Therefore, the descriptor
φ involving coordinated atoms explicitly could provide an effective and quick way to screen the catalyst for the OER and ORR.
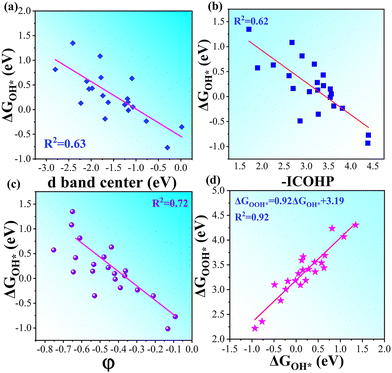 |
| | Fig. 5 (a) Linear relationship between d band centers and ΔGOH*. (b) Linear relationship between -ICOHP and ΔGOH*. (c) Linear relationship between our descriptor φ and ΔGOH* in OH*-TM1TM2/β12-BM systems. (d) Linear relationship between the absorption free energies of intermediates ΔGOH* and ΔGOH* for TM1TM2/β12-BM systems. | |
Given the possibility of linear relationship between the free energies of intermediates mentioned above, we studied the relationship between ΔGOH* and ΔGOH* in the DASC systems. As shown in Fig. 5d, ΔGOH* is linearly correlated with ΔGOH* with a coefficient of determination of 0.92, the fitted equation for which is ΔGOOH* = 0.92ΔGOH* + 3.19, while a poor linear relationship between ΔGOH* and ΔGO* is shown in Fig. S7 (ESI†). As a result, the four free energy changes can be expressed as follows:
| | 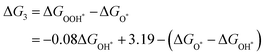 | (8) |
| | | ΔG4 = 4.92 − ΔGOOH* = 0.092ΔGOH* + 1.73 | (9) |
Therefore, the OER and ORR activities can be evaluated by Δ
GO* − Δ
GOH* and Δ
GOH*. In other words, as long as Δ
GO* and Δ
GOH* are known, the free energy of all species can be evaluated. The OER and 4e ORR process volcano diagrams are shown in
Fig. 6a and b, corresponding to the overpotentials of PDSs. According to the free energy step diagrams in Fig. S6 (ESI
†) and
Fig. 6a, the PDSs of the OER mainly occur in one step, OH* → O* or O* → OOH*, which is the right or left leg of the OER volcano displayed in
Fig. 6a, respectively. This indicates that when the adsorption of O* is too strong compared to OH*, the OER was subject to the production of OOH* (left leg). When Δ
GO* − Δ
GOH* increases (
e.g., adsorption O* of weakened compared to OH*, like that at Ni sites in
Fig. 6a), the PDS turned out to be the production of O* (right leg). The fitted boundary equations for left and right legs in
Fig. 6a can be written as follows:
| | 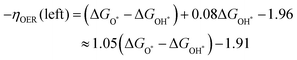 | (10) |
| | | −ηOER(right) = −1.04(ΔGO* − ΔGOH*) + 1.32 | (11) |
It shows that when Δ
GO* − Δ
GOH* arrives at 1.55, the two lines crossed at the peak (
ηminOER = 0.28), which is expected to be the optimal OER performance for these borophene-supported bimetallic catalysts.
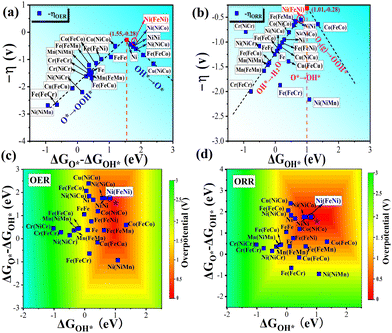 |
| | Fig. 6 Volcano plots for (a) OER overpotentials −ηOERvs. ΔGO* − ΔGOH* and (b) ORR overpotentials −ηOERvs. ΔGOH* for TM1TM2 (TM1 = Fe/Ni, TM2 = Cr–Cu)/β12-BM systems. Activity contour maps of (c) OERs and (d) ORRs for TM1TM2/β12-BM systems, and the optimal position for each map is marked with purple pentagrams. | |
As for the ORR process, the PDSs are mainly O2 → OOH* (right leg) or OH* → H2O* (left leg), as shown in Fig. 6b and Fig. S5 (ESI†). The fitted boundary equations for the ORR overpotentials can be expressed as follows:
| | | −ηORR(left) = (ΔGO* − ΔGOH*) − 1.23 ≈ 0.94ΔGOH* − 1.23 | (12) |
| | | −ηORR(right) =−1.07ΔGOH* + 0.81 | (13) |
It suggests that when Δ
GOH* arrives at 1.01, the two lines crossed at the peak (
ηminOER = 0.28), which shows the best catalytic activity for these systems. As a result, the borophene-supported bimetallic catalysts are expected to give the best bifunctional OER/ORR performance when Δ
GOH* ∼ 1.01 eV and Δ
GO* ∼ 2.56 eV.
The peaks of the volcano plots consistently show the best catalytic activity for a given catalyst system, and hence, the positions of the Ni(FeNi) system in Fig. 6a and b demonstrate its best OER and ORR performance. Good OER and ORR performance is also expected for Ni(NiCo) and Ni(NiNi) systems from the volcano plots. Based on the eqn (6)–(9), we took ΔGOH* from −2 to 2.5 eV and ΔGO* − ΔGOH* from −3 to 3 eV for OERs, and the four free energy changes were obtained. Then, the OER overpotentials were calculated using eqn (S13) (ESI†), and the ORR overpotentials were calculated in a similar way. As a result, the OER and ORR activity contour plots of the DASC systems are shown in Fig. 6c and d, and the overpotentials range from 0 to 3 V, which are represented in different colors. The optimal positions with the lowest OER and ORR overpotentials were marked with purple pentagrams, as shown in Fig. 6c and d. The Ni(FeNi) is very close to the optimal positions in both maps, indicating that it is a very promising OER/ORR bifunctional catalyst. Generally speaking, combinations between Fe, Co and Ni tend to perform better than other combinations. However, it should be pointed out that Fe, Co, and Ni may be oxidized in the OER process,70 which requires O2-poor conditions. The results of the contour plots are in good agreement with those of the free energy step diagrams and volcano plots, confirming the reliability of our calculations.
The ORR kinetic process for Ni(FeNi)/β12-BM was investigated based on the transition-state barriers (Ea) for each step (Fig. S8, ESI†). After adsorption on Ni, the  molecule tends to be hydrogenated (Ea = 0.03 eV in Fig. S8a, ESI†) rather than to dissociation (Ea = 0.15 eV in Fig. S8b, ESI†). Then, the OOH* group tends to dissociate into OH* and O* (Ea = 0.61 eV in Fig. S8c, ESI†), rather than undergoing hydrogenation (Ea = 1.1 eV in Fig. S8d, ESI†), which produces two OH* groups. Then, the O* atom reacts with a hydrogen atom (Ea = 0.59 eV in Fig. S8e, ESI†) and gives H2O after another hydrogenation (Ea = 0.87 eV in Fig. S8f, ESI†). Generally speaking, the kinetic rate-determining step (RDS) is OH → H2O, which is also the potential-determining step. It agrees with the Brønsted–Evans–Polanyi (BEP) relations,71 and confirms the reliability of the overpotentials from the potential-determining steps.
molecule tends to be hydrogenated (Ea = 0.03 eV in Fig. S8a, ESI†) rather than to dissociation (Ea = 0.15 eV in Fig. S8b, ESI†). Then, the OOH* group tends to dissociate into OH* and O* (Ea = 0.61 eV in Fig. S8c, ESI†), rather than undergoing hydrogenation (Ea = 1.1 eV in Fig. S8d, ESI†), which produces two OH* groups. Then, the O* atom reacts with a hydrogen atom (Ea = 0.59 eV in Fig. S8e, ESI†) and gives H2O after another hydrogenation (Ea = 0.87 eV in Fig. S8f, ESI†). Generally speaking, the kinetic rate-determining step (RDS) is OH → H2O, which is also the potential-determining step. It agrees with the Brønsted–Evans–Polanyi (BEP) relations,71 and confirms the reliability of the overpotentials from the potential-determining steps.
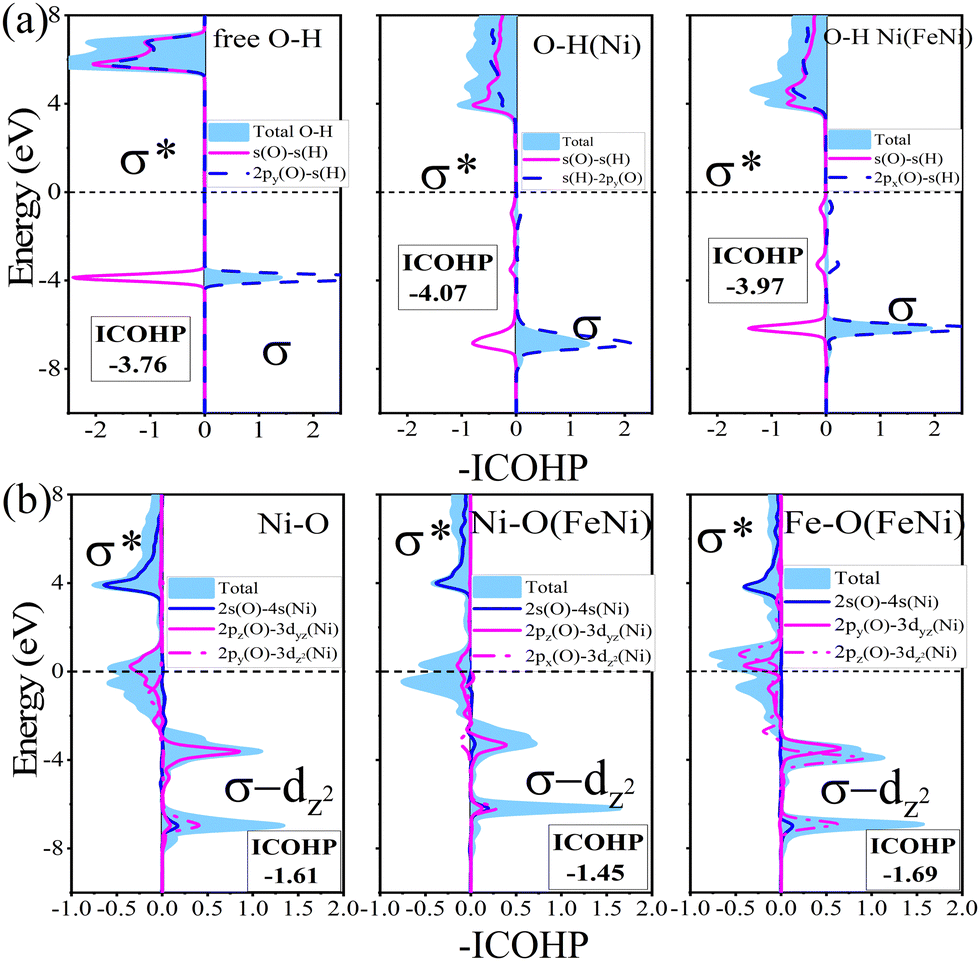
In order to further deepen the understanding of the activation of OH by Ni(FeNi) on the β12 monolayer, we analyzed the orbital hybridization using –COHP (Fig. 7a), the positive and negative values for which represent bonding and antibonding states, respectively. It can be seen that both the bonding and antibonding states of OH downshift to the Fermi level after adsorption, indicating enhanced O-H interaction, which mainly comes from the s–p hybridization (σ bond) between H and O atoms. It is confirmed by the increased -ICOHP values from 3.76 (free OH) to 4.07 (Ni adsorbed OH). In addition, as shown in Fig. 7b, there is also a bonding state in Ni–O at about −7 eV, indicating hybridization between Ni, O and H atoms. In the meanwhile, the coordinated Fe atom upshifts the Ni–O bonding states and weakens the Ni–O interaction, and activates the O–H bond (-ICOHP = 3.97). In a word, the synergistic effect of Fe–Ni pairs balances the too strong binding of OH and further activates OH to achieve better catalytic performance.
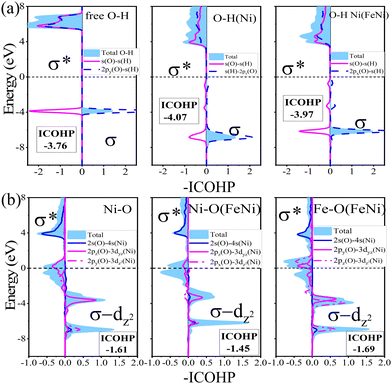 |
| | Fig. 7 (a) Crystal orbital Hamilton population (COHP) of free OH, adsorbed O–H on Ni and O–H on Ni(FeNi). (b) COHP for Ni–O, Ni–O(FeNi), and FeO(FeNi) on β12 borophene. The integral for -ICOHP ranges from minus infinity to the Fermi level. | |
Conclusions
We have systematically investigated the HER, OER and ORR performance of β12-borophene with double-hole sites capped with 3d transition metal atoms (referred to as TM1TM2/β12-BM, TM1 = Fe/Ni, TM2 = Cr–Cu) using DFT methods. It was found that the borophene is a good platform for diatomic site catalysts (DASCs) due to their advantage of stability over the corresponding single-atom catalysts (SACs) or clusters. The HER performance of DASCs on β12-BM was further improved compared to the SAC case, and ΔGOH* of NiCu DASC was smaller than 0.01 eV. Furthermore, the supported FeNi DASC exhibited good catalytic performance for both OERs and ORRs, indicating a good bifunctional catalyst. Their overpotentials were 0.43 and 0.55 V, better than those of the corresponding supported Ni or Fe SAC due to synergistic effect. Based on the scale relationship between the adsorption free energies of intermediates, the contour maps and volcano plots were established, which suggested that combinations between Fe, Co and Ni tend to perform better for the OER/ORR than other combinations. We further proposed a novel descriptor involving the Bader charges of coordinated atoms explicitly, behaving much better than the d-band center and -ICOHP for DASCs. The synergistic effect of Fe–Ni pairs balances the too strong binding of OH and further activates OH to achieve better catalytic performance. The results of this study can provide theoretical guidance for the design of efficient bifunctional electrocatalysts.
Author contributions
Jia Liu: data curation, investigation, formal analysis, visualization, validation, writing – original draft. Minjing Zhang: data curation. Si-Dian Li: project administration, funding acquisition, software, supervision. Yuewen Mu: conceptualization, methodology, formal analysis, investigation, software, project administration, supervision, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 11504213, 22373061, 21720102006 and 21803037) and the Fund for Shanxi “1331 Project” Key Innovative Research Team (1331KIRT). We greatly acknowledge the computing resources provided by the high performance computing platform of Shanxi University, Hongzhiwei Technology and National Supercomputing Center in Taiyuan.
References
- C. Sealy, Mater. Today, 2008, 11(12), 65–68 CrossRef CAS.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43(22), 7746–7786 RSC.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4(2), 145–153 CrossRef.
- M. K. Debe, Nature, 2012, 486(7401), 43–51 CrossRef CAS PubMed.
- Z. F. Huang, J. Wang, Y. Peng, C. Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7(23), 1700544 CrossRef.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 6271 CrossRef PubMed.
- V. Tripkovic, Phys. Chem. Chem. Phys., 2017, 19(43), 29381–29388 RSC.
- H. G. Sanchez Casalongue, M. L. Ng and S. Kaya,
et al.
, Angew. Chem., Int. Ed., 2014, 53(28), 7169–7172 CrossRef CAS PubMed.
- B. Wei, Z. Fu and D. Legut,
et al.
, Adv. Mater., 2021, 33(40), 2102595 CrossRef CAS PubMed.
- G. L. Chai, K. Qiu and M. Qiao,
et al.
, Energy Environ. Sci., 2017, 10(5), 1186–1195 RSC.
- H. Shang, W. Sun and R. Sui,
et al.
, Nano Lett., 2020, 20(7), 5443–5450 CrossRef CAS PubMed.
- L. Bai, Z. Duan and X. Wen,
et al., Highly dispersed ruthenium-based multifunctional electrocatalyst, ACS Catal., 2019, 9(11), 9897–9904 CrossRef CAS.
- Y. Zhou, G. Gao and J. Kang,
et al.
, J. Mater. Chem. A, 2019, 7(19), 12050–12059 RSC.
- Y. Ying, K. Fan and X. Luo,
et al.
, J. Mater. Chem. A, 2021, 9(31), 16860–16867 RSC.
- X. Liu, Y. Zhang and W. W. Wang,
et al.
, ACS Appl. Mater. Interfaces, 2021, 14(1), 1249–1259 CrossRef PubMed.
- B. Qiao, A. Wang and X. Yang,
et al.
, Nat. Chem., 2011, 3(8), 634–641 CrossRef CAS PubMed.
- X. F. Yang, A. Wang and B. Qiao,
et al.
, Acc. Chem. Res., 2013, 46(8), 1740–1748 CrossRef CAS PubMed.
- I. S. Amiinu, Z. Pu and X. Liu,
et al.
, Adv. Funct. Mater., 2017, 27(44), 1702300 CrossRef.
- R. Christensen, H. A. Hansen and C. F. Dickens,
et al.
, J. Phys. Chem. C, 2016, 120(43), 24910–24916 CrossRef CAS.
- M. Jahan, Z. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23(43), 5363–5372 CrossRef CAS.
- A. J. Mannix, X. F. Zhou and B. Kiraly,
et al.
, Science, 2015, 350(6267), 1513–1516 CrossRef CAS PubMed.
- B. Feng, J. Zhang and Q. Zhong,
et al.
, Nat. Chem., 2016, 8(6), 563–568 CrossRef CAS PubMed.
- H. Tang and S. Ismail-Beigi, Phys. Rev. Lett., 2007, 99(11), 115501 CrossRef PubMed.
- C. Ling, L. Shi and Y. Ouyang,
et al.
, Nano Lett., 2017, 17(8), 5133–5139 CrossRef CAS PubMed.
- X. Xu, R. Si and Y. Dong,
et al.
, J. Mol. Model., 2021, 27, 1–10 CrossRef PubMed.
- J. H. Liu, L. M. Yang and E. Ganz, RSC Adv., 2019, 9(47), 27710–27719 RSC.
- Y. B. Sun, Y. Q. Liu and X. Zhao,
et al.
, ChemSusChem, 2022, 15(18), e202200930 CrossRef CAS PubMed.
- L. Liu, L. Yang and H. Zhang,
et al.
, Energy Fuels, 2023, 37(13), 9682–9691 CrossRef CAS.
- J. Park and S. Hong, Chem. Soc. Rev., 2012, 41(21), 6931–6943 RSC.
- V. A. Pollard, S. A. Orr, R. McLellan, A. R. Kennedy, E. Hevia and R. E. Mulvey, Chem. Commun., 2018, 54(10), 1233–1236 RSC.
- D. Wu, B. He and Y. Wang,
et al.
, J. Phys. D: Appl. Phys., 2022, 55(20), 203001 CrossRef.
- W. Wan, Y. Zhao and S. Wei,
et al.
, Nat. Commun., 2021, 12(1), 5589 CrossRef CAS PubMed.
- K. Liu, J. Fu and Y. Lin,
et al.
, Nat. Commun., 2022, 13(1), 2075 CrossRef CAS PubMed.
- B. Zhang, X. Zheng and O. Voznyy,
et al.
, Science, 2016, 352(6283), 333–337 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, J. Phys.: Condens. Matter, 1994, 6(40), 8245 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- S. B. Fagan, R. J. Baierle and R. Mota,
et al.
, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61(15), 9994 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113(22), 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS.
- H. Niu, X. Wan, X. Wang, C. Shao, J. Robertson, Z. Zhang and Y. Guo, ACS Sustainable Chem. Eng., 2021, 9(9), 3590–3599 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl and A. Logadottir,
et al.
, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef.
- J. K. Nørskov, T. Bligaard and A. Logadottir,
et al.
, J. Electrochem. Soc., 2005, 152(3), J23 CrossRef.
- V. Wang, N. Xu and J. C. Liu,
et al.
, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- Z. Zeng, L. Y. Gan and H. B. Yang,
et al.
, Nat. Commun., 2021, 12(1), 4088 CrossRef CAS PubMed.
- Q. Deng, J. Zhao and T. Wu,
et al.
, J. Catal., 2019, 370, 378–384 CrossRef CAS.
- C. Ling, L. Shi and Y. Ouyang,
et al.
, Nanoscale, 2017, 9(2), 533–537 RSC.
- T. R. Galeev, Q. Chen and J. C. Guo,
et al.
, Phys. Chem. Chem. Phys., 2011, 13(24), 11575–11578 RSC.
- X. Qin, J. Liu and Y. Mu,
et al.
, Phys. Chem. Chem. Phys., 2022, 24(23), 14566–14572 RSC.
- B. Ding, W. J. Ong and J. Jiang,
et al.
, Appl. Surf. Sci., 2020, 500, 143987 CrossRef CAS.
- Y. N. Zhou, L. Sheng, Q. Q. Luo, W. H. Zhang and J. L. Yang, J. Phys. Chem. Lett., 2021, 12, 11652–11658 CrossRef CAS PubMed.
- J. Li, Y. N. Zhou, W. J. Tang and J. Zheng,
et al.
, Appl. Catal., B, 2021, 285, 119861 CrossRef CAS.
- Y. M. Zang, Q. Wu, W. H. Du, Y. Dai, B. B. Huang and Y. D. Ma, Phys. Rev. Mater., 2021, 5(4), 045801 CrossRef CAS.
- J. A. Santana, J. J. Mateo and Y. Ishikawa, J. Phys. Chem. C, 2010, 114(11), 4995–5002 CrossRef CAS.
- H. Wang, X. Xiao and S. Liu,
et al.
, J. Am. Chem. Soc., 2019, 141(46), 18578–18584 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132(39), 13612–13614 CrossRef CAS PubMed.
- Y. Su, Y. Zhu, H. Jiang, J. Shen, X. Yang, W. Zou, J. Chen and C. Li, Nanoscale, 2014, 6(24), 15080–15089 RSC.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver and T. A. Arias,
et al.
, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- Q. Zhang and A. Asthagiri, Catal. Today, 2019, 323, 35–43 CrossRef CAS.
- P. Sabatier, Chem. Ber., 1911, 44(3), 1984–2001 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard and C. F. Dickens,
et al.
, Science, 2017, 355(6321), eaad4998 CrossRef PubMed.
- H. A. Hansen, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. C, 2014, 118(13), 6706–6718 CrossRef CAS.
- M. Gajdoš, A. Eichler and J. Hafner, J. Phys.: Condens. Matter, 2004, 16(8), 1141 CrossRef.
- W. Zhong, Y. Qiu and H. Shen,
et al.
, J. Am. Chem. Soc., 2021, 143(11), 4405–4413 CrossRef CAS PubMed.
- Y. Zhou, L. Sheng and Q. Luo,
et al.
, J. Phys. Chem. Lett., 2021, 12(48), 11652–11658 CrossRef CAS PubMed.
- I. Yamada, A. Takamatsu, K. Asai, T. Shirakawa, H. Ohzuku, A. Seno, T. Uchimura, H. Fujii, S. Kawaguchi, K. Wada, H. Ikeno and S. Yagi, J. Phys. Chem. C, 2018, 122(49), 27885–27892 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 209, 275–278 CrossRef.
- A. Mohajeri and N. L. Dashti, J. Phys. Chem. C, 2019, 123(51), 30972–30980 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details for OER and ORR, reaction pathways of OER and ORR, Nin (n = 2–4) clusters on β12-BM, HER adsorption free energies, OER/ORR free energy step diagrams. PDOS and d-band center, linear relationship between ΔGOH* and ΔGO*, kinetic process of the FeNi DSAC, overpotentials for OER and ORR. See DOI: https://doi.org/10.1039/d3cp04543a |
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Yuewen
Mu
* and
Yuewen
Mu
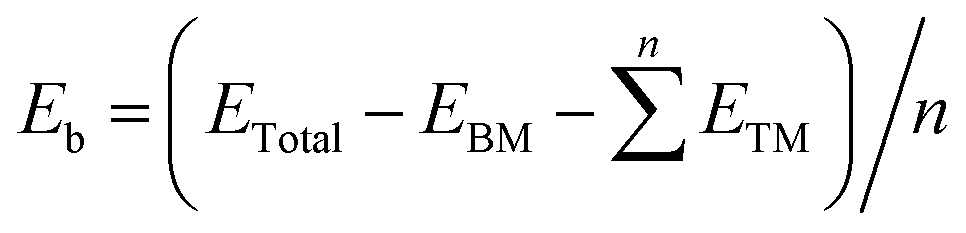
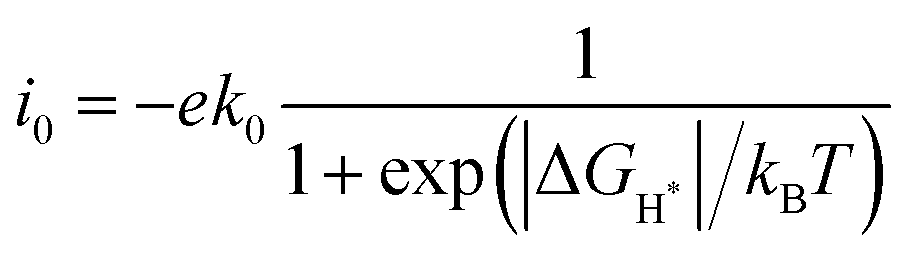
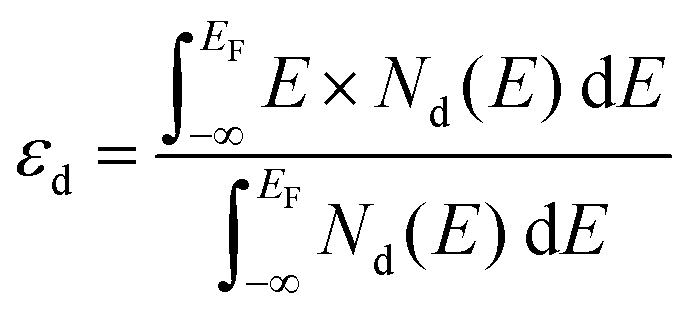
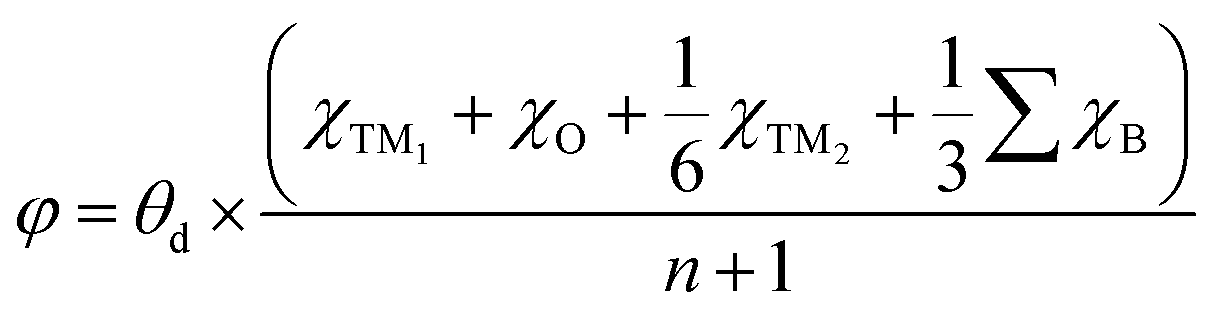
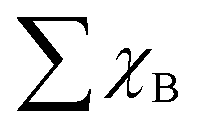 is the sum of the Bader charges of the six boron atoms around TM1, and χO represents the Bader charge of O, and n + 1 is the number of B/O atoms (n = 7 in this work) plus the TM1 atoms. Fig. 5c suggests that the free energies ΔGOH* basically linearly decrease with the increase in φ, the coefficient of determination for which is 0.72, much larger than those for the d band center and -ICOHP. Therefore, the descriptor φ involving coordinated atoms explicitly could provide an effective and quick way to screen the catalyst for the OER and ORR.
is the sum of the Bader charges of the six boron atoms around TM1, and χO represents the Bader charge of O, and n + 1 is the number of B/O atoms (n = 7 in this work) plus the TM1 atoms. Fig. 5c suggests that the free energies ΔGOH* basically linearly decrease with the increase in φ, the coefficient of determination for which is 0.72, much larger than those for the d band center and -ICOHP. Therefore, the descriptor φ involving coordinated atoms explicitly could provide an effective and quick way to screen the catalyst for the OER and ORR.
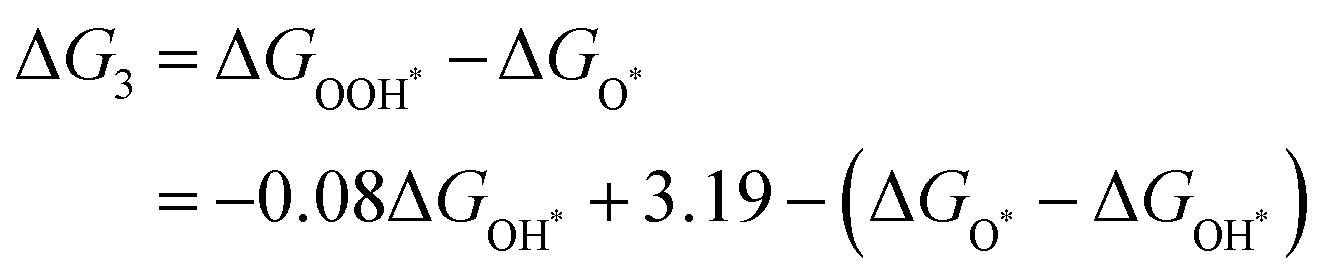
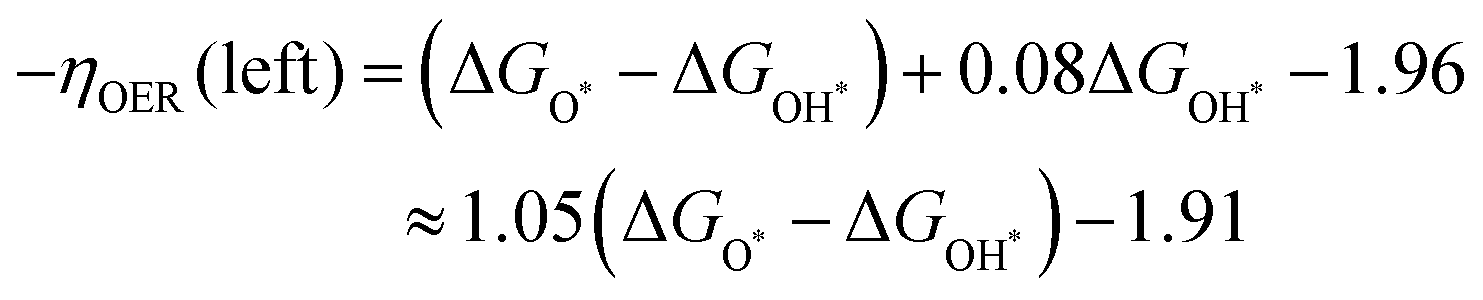
 molecule tends to be hydrogenated (Ea = 0.03 eV in Fig. S8a, ESI†) rather than to dissociation (Ea = 0.15 eV in Fig. S8b, ESI†). Then, the OOH* group tends to dissociate into OH* and O* (Ea = 0.61 eV in Fig. S8c, ESI†), rather than undergoing hydrogenation (Ea = 1.1 eV in Fig. S8d, ESI†), which produces two OH* groups. Then, the O* atom reacts with a hydrogen atom (Ea = 0.59 eV in Fig. S8e, ESI†) and gives H2O after another hydrogenation (Ea = 0.87 eV in Fig. S8f, ESI†). Generally speaking, the kinetic rate-determining step (RDS) is OH → H2O, which is also the potential-determining step. It agrees with the Brønsted–Evans–Polanyi (BEP) relations,71 and confirms the reliability of the overpotentials from the potential-determining steps.
molecule tends to be hydrogenated (Ea = 0.03 eV in Fig. S8a, ESI†) rather than to dissociation (Ea = 0.15 eV in Fig. S8b, ESI†). Then, the OOH* group tends to dissociate into OH* and O* (Ea = 0.61 eV in Fig. S8c, ESI†), rather than undergoing hydrogenation (Ea = 1.1 eV in Fig. S8d, ESI†), which produces two OH* groups. Then, the O* atom reacts with a hydrogen atom (Ea = 0.59 eV in Fig. S8e, ESI†) and gives H2O after another hydrogenation (Ea = 0.87 eV in Fig. S8f, ESI†). Generally speaking, the kinetic rate-determining step (RDS) is OH → H2O, which is also the potential-determining step. It agrees with the Brønsted–Evans–Polanyi (BEP) relations,71 and confirms the reliability of the overpotentials from the potential-determining steps.