
Prediction of novel ground-state structures and analysis of phonon transport in two-dimensional GexSy compounds†
Asad
Ali
 and
Young-Han
Shin
*
and
Young-Han
Shin
*
Multiscale Materials Modeling Laboratory, Department of Physics, University of Ulsan, Ulsan 44610, Republic of Korea. E-mail: hoponpop@ulsan.ac.kr; Fax: +82 52 259 1693; Tel: +82 52 259 1027
First published on 30th November 2023
Abstract
We conducted this study to explore the ground-state structures of two-dimensional (2D) variable-composition GexSy compounds, driven by the polymorphic characteristics of bulk germanium sulfides and the promising thermoelectric performance of 2D GeS (Pmn21). To accomplish this, we utilized the highly successful evolutionary-algorithm-based code USPEX in conjunction with VASP for total energy calculations, leading to the discovery of three previously unexplored structures of Ge2S (P2/c), GeS (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1), and GeS2 (P21/c). These 2D materials exhibit significantly lower formation energies compared to their reported counterparts. We thoroughly scrutinized the structural stability and subsequently analyzed their electronic structures. Our analysis reveals a nearly direct band gap of 0.12/0.84 eV with the PBE/HSE06 functional for 2D Ge2S and an indirect band gap for 2D GeS and GeS2. Their semiconducting nature highlights the crucial importance of lattice thermal conductivity (κl), which we determined by solving the Boltzmann transport equation for phonons. Importantly, we predict a room temperature κl value of 6.82 W m−1 K−1 for GeS, lower than its 2D orthorhombic counterpart. In the case of GeS2, we observed an anisotropic κl value of 16.95/10.68 W m−1 K−1 along the zigzag/armchair directions at 300 K, with an in-plane anisotropy ratio of 1.59, surpassing that of 2D IV–VI compounds. We delve into detailed discussions regarding the role of lattice anharmonicity, group velocities, phonon lifetimes, and three-phonon weighted phase space in the overall thermal conductivity analysis.
m1), and GeS2 (P21/c). These 2D materials exhibit significantly lower formation energies compared to their reported counterparts. We thoroughly scrutinized the structural stability and subsequently analyzed their electronic structures. Our analysis reveals a nearly direct band gap of 0.12/0.84 eV with the PBE/HSE06 functional for 2D Ge2S and an indirect band gap for 2D GeS and GeS2. Their semiconducting nature highlights the crucial importance of lattice thermal conductivity (κl), which we determined by solving the Boltzmann transport equation for phonons. Importantly, we predict a room temperature κl value of 6.82 W m−1 K−1 for GeS, lower than its 2D orthorhombic counterpart. In the case of GeS2, we observed an anisotropic κl value of 16.95/10.68 W m−1 K−1 along the zigzag/armchair directions at 300 K, with an in-plane anisotropy ratio of 1.59, surpassing that of 2D IV–VI compounds. We delve into detailed discussions regarding the role of lattice anharmonicity, group velocities, phonon lifetimes, and three-phonon weighted phase space in the overall thermal conductivity analysis.
1 Introduction
The demand for two-dimensional (2D) materials has significantly increased due to their unique properties and potential for miniaturization in future devices. One such material is graphene, derived from graphite, which exhibits exceptional characteristics such as a high carrier mobility and lattice thermal conductivity (κl), making it suitable for efficient thermal transport.1–3 However, in the context of thermoelectric devices, the efficiency at temperature T is quantified by a dimensionless figure of merit,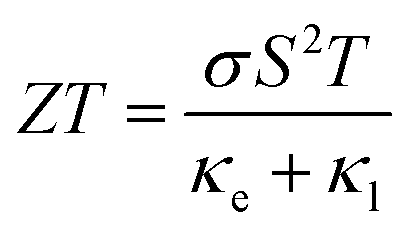 , where σ, S, κe, and κl represent the electrical conductivity, Seebeck coefficient, electronic thermal conductivity, and lattice thermal conductivity, respectively. It is worth noting that ZT is inversely proportional to the κl. Therefore, the high κl and metallic nature of intrinsic graphene pose challenges for its direct use in thermoelectric devices, and various methods like nanostructuring and doping are employed to mitigate these issues.4 Consequently, alternative materials are being explored, with 2D transition metal dichalcogenides (TMDs) gaining considerable attention. Nonetheless, most TMDs exhibit relatively high lattice thermal conductivity,5 prompting researchers to redirect their focus toward 2D IV–VI materials. These materials have emerged as a prominent research area due to their ability to achieve ultra-low lattice thermal conductivity and higher Seebeck coefficients.6
, where σ, S, κe, and κl represent the electrical conductivity, Seebeck coefficient, electronic thermal conductivity, and lattice thermal conductivity, respectively. It is worth noting that ZT is inversely proportional to the κl. Therefore, the high κl and metallic nature of intrinsic graphene pose challenges for its direct use in thermoelectric devices, and various methods like nanostructuring and doping are employed to mitigate these issues.4 Consequently, alternative materials are being explored, with 2D transition metal dichalcogenides (TMDs) gaining considerable attention. Nonetheless, most TMDs exhibit relatively high lattice thermal conductivity,5 prompting researchers to redirect their focus toward 2D IV–VI materials. These materials have emerged as a prominent research area due to their ability to achieve ultra-low lattice thermal conductivity and higher Seebeck coefficients.6
The suitable band gap, earth abundance, and non-toxic characteristics of germanium sulfides and its constituent elements make the orthorhombic (Pnma) phase of GeS the primary focus of researchers. The monolayer GeS (Pmn21) can be obtained through mechanical exfoliation from its bulk and has shown great potential for thermoelectric device applications due to its high electron mobility and ultra-low lattice thermal conductivity. Similarly, the 1T structure of 2D GeS27 and the low-symmetric (P21212) phase of two-dimensional (2D) Ge2S8 have also exhibited excellent properties, particularly as thermoelectric and anode materials. However, while the bulk counterparts of these GexSy compounds exhibit various polymorphs, such as cubic,9 orthorhombic,10 and amorphous11 phases of GeS, and monoclinic,12 orthorhombic,13 and beta14 phases of GeS2, it remains unexplored whether their monolayer forms possess unique and undiscovered structures. This intriguing possibility suggests that the monolayers of these compounds may exhibit distinct properties, thereby warranting further investigation and exploration.
In the pursuit of obtaining 2D counterparts of materials, researchers employ various experimental procedures. These methods can be of two categories: top-down and bottom-up approaches. In the first approach, a single layer is exfoliated from the bulk material, which is particularly feasible for layered materials held together by van der Waals forces such as graphene1 or 2D TMDs. Conversely, in the bottom-up approach, layers grow on a substrate from vapors of precursors through methods like physical or chemical vapor depositions.15 Bottom-up approaches are employed for synthesizing 2D sheets from non-layered bulk materials due to their high exfoliation energy. They have also enabled the synthesis of 2D structures that lack bulk counterparts. For instance, various 2D boron allotropes have been successfully prepared on substrates such as silver surfaces.16 The 2D hexagonal layer of GeSe, synthesized through chemical vapor deposition,17 shares a similar structure with our predicted 2D GeS in this work and also lacks a bulk counterpart. In parallel, density functional theory (DFT) can predict a single layer by cutting it from the layered bulk and allowing for relaxation with sufficient vacuum in the out-of-plane direction. In contrast, the non-layered bulk materials lack preferred planes for cleaving a single layer, leading to many possible 2D structures along various planes. Among these, the one with the lowest formation energy is energetically favorable, called the ground-state 2D structure of the given composition. The ground state structures of materials have particular importance due to their higher possibility of experimental realization. Another strategy involves replacing atoms in already explored monolayer lattices with the elements under investigation and searching for the most energetically favorable configuration, such as in the case of germanene and silicene.18 However, manually exploring all possible atomic arrangements can be a tedious task. Fortunately, significant advancement in computational tools for predicting ground state structures has revolutionized the field of materials science, allowing the prediction of novel 2D materials like borophenes before their experimental synthesis.16,19 Among these tools, the Universal Structure Predictor: Evolutionary Xtallography (USPEX)20,21 stands out with its high success rate among genetic-algorithm-based search codes. USPEX offers a specific 2D search capability, making it particularly suitable for identifying the lowest-energy phases in compounds with variable compositions, such as 2D CdxTey.22 By harnessing the power of USPEX, one can efficiently search for and identify the energetically stable structures of 2D GexSy compounds.
This study predicts novel ground-state structures of 2D GexSy compounds with three different compositions: Ge2S, GeS, and GeS2, that exhibit higher stability than their previously reported counterparts. Specifically, we focus on their crystal structures, electronic structure, and phonon transport properties. The 2D GeS phase demonstrates lower lattice thermal conductivity compared to its orthorhombic monolayer counterpart. We delve into the underlying mechanisms responsible for the low κl of 2D GeS, examining factors such as anharmonicity, phonon group velocity, their lifetimes, and weighted phase space for three-phonon scattering. Moreover, 2D GeS2 shows intriguing in-plane anisotropic elastic stiffness and κl, while 2D Ge2S has comparatively higher κl. The interplay of anisotropic acoustic and low-frequency optical phonon group velocity and relatively higher lifetimes to their distinguished κl in 2D GeS2 makes this study of fundamental interest.
2 Computational methodology
The search for lowest-energy structures of 2D germanium sulfides involves USPEX20,21 in conjunction with the Vienna Ab initio Simulation Package (VASP).23 Our variable-compositional search is constrained to a layer thickness of 0–6 Å and the constituent atoms in a unit cell of 2–12. In this search, a total of 80 generations are considered. The first generation contains 200 randomly created structures, while the subsequent generations have 80 structures each that evolve through natural selection and variation operators such as mutation, crossover, and symmetry operations. The relative energetic stability of different compositions and structures was determined by comparing the formation energy per atom (Ef), which is calculated using the equation: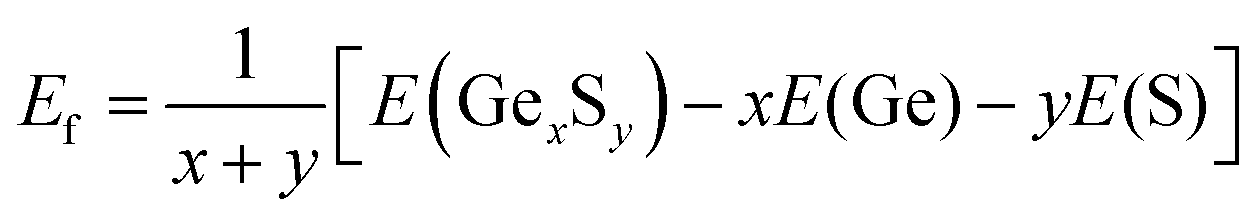 | (1) |
In this equation, E(GexSy), E(Ge), and E(S) represent the total energies of the lowest possible 2D configurations of GexSy, elemental germanium, and sulfur, respectively. The total energy of each structure is calculated using the projector-augmented wave (PAW)24 method, which is well-suited for handling core electrons. To account for the electronic exchange–correlation interaction, the generalized gradient approximation (GGA)25 with the Perdew–Burke–Ernzerhof parameterization (PBE) is employed. To maximize computational efficiency, a strategy is adopted that includes a relatively coarse k-mesh with a resolution of 2π × (0.08–0.04) Å−1, a smaller cutoff energy of 450 eV for plane wave expansion, a vacuum size of 15 Å, and exclusion of the zero damping of Grimme (DFT-D3)26 for the van der Waals corrections in the first 60 generations, followed by its inclusion in the final 20 generations.
To ensure accuracy, the resulting 2D GexSy structures undergo re-optimization using a larger cutoff energy of 540 eV, a vacuum size of 20 Å, and a denser Γ-center k-mesh with a resolution of 2π × 0.02 Å−1. The convergence criteria for energy is set to 10−8 eV and for forces to 10−3 eV Å−1. Electronic band structures are computed using both the GGA and Heyd–Scuseria–Ernzerhof (HSE06)27 methods. For ab initio molecular dynamics (AIMD) simulations, we employ the NVT ensemble with a step size of 1 fs and a total time of 10 ps at temperatures of 300 K, 600 K, and 900 K. We use the Nosé–Hoover thermostat28 to maintain a constant temperature during simulations. The Phonopy code29 is used to calculate the phonon dispersion based on the finite displacement method with supercells. We use supercells of 6 × 5 × 1 for Ge2S, 5 × 5 × 1 for GeS, and 3 × 3 × 1 for GeS2, which are the same-sized supercells as those used for AIMD simulations.
We utilized an iterative self-consistent approach implemented in the ShengBTE package30 to solve the phonon Boltzmann transport equation (PBTE) and predict the lattice thermal conductivity. This method relies on two sets of interatomic force constants (IFCs): harmonic (second-order) and anharmonic (third-order). The harmonic IFCs are obtained using the Phonopy code29 and anharmonic IFCs through the thirdorder.py python script based on the method developed by Lindsay et al.31 in combination with VASP. With these sets of IFCs, one can express the αβ component of the lattice thermal conductivity tensor as:30
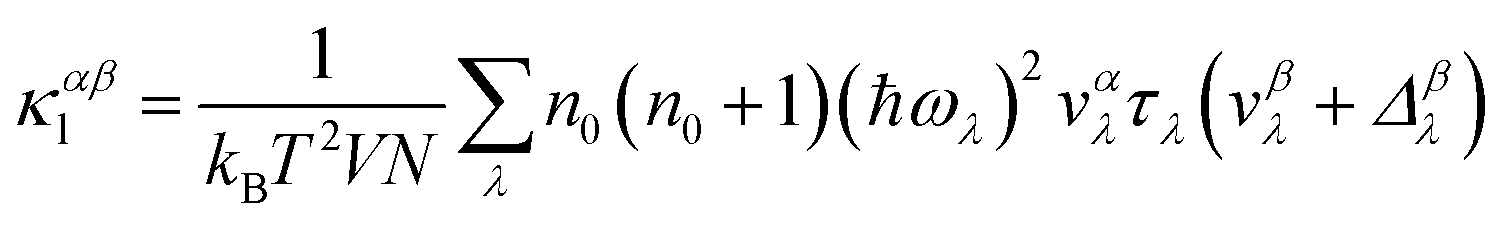 | (2) |
Here, λ(k,p) represents a phonon mode characterized by the wave vector k and branch number p. Δβλ with the dimensions of velocity is the correction to relaxation time approximation (RTA) prediction. V denotes the unit-cell volume, ωλ is the phonon frequency, N is the number of q points in the first Brillouin zone (BZ) for a Γ-centered regular grid, T represents the temperature, vαλ is the group velocity of the phonon in the α direction, τλ is the λ phonon lifetime, and n0 is the phonon occupation number given by the Bose–Einstein distribution. Here, the inverse of τλ, i.e., 1/τλ is the total scattering rate, which is determined through Matthiessen's rule given as:
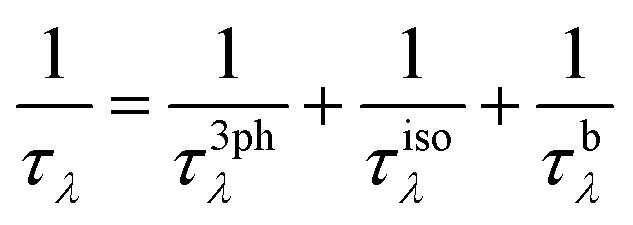 | (3) |
In this equation, 1/τ3phλ represents the three-phonon scattering rate, while 1/τisoλ and 1/τbλ denote the scattering rates associated with phonon–isotope and phonon–boundary interactions, respectively. For a detailed expression of each scattering rate, please refer to ref. 30.
The obtained κl is rescaled for 2D GexSy by multiplying a factor Lz/deff, where Lz is the total length of the unit cell along the z direction and deff is the effective thickness of the materials obtained from the corresponding bulk structure. The values of deff are 7.579, 9.068, and 7.710 in units Å for Ge2S, GeS, and GeS2, respectively (see Fig. S1 of the ESI†). The long-range interactions are included by Born effective charge tensors (see Table S1 of the ESI†) computed with the density functional perturbation theory (DFPT).32
3 Results and discussion
3.1 Structure prediction and stability
The comparative energetic stability among various structures of a material can be represented by the energy convex hull (ECH). The ECH of the 2D Ge–S system (see Fig. 1) is constructed by plotting the formation energy per atom, given by eqn (1), of a given composition ratio in all possible structures along the y-axis and composition ratio S/(Ge + S) along the x-axis. In the literature, several polymorphs of bulk GeS, such as orthorhombic α, cubic, and amorphous phases,9,11,33 have been reported due to different bonding modes. ECH construction is an efficient and powerful tool for identifying the most stable structure and composition,34 especially for materials with numerous polymorphs. These most stable structures will lie on the convex hull, such as in the Ge–S system, Ge2S (magenta), GeS (cyan), and GeS2 (dark-blue), as shown in Fig. 1. These lowest-energy 2D Ge2S, GeS, and GeS2 (discussed later) have different structures and lower formation energies than the previously reported ones. For comparison, the formation energies of the already reported 2D Ge2S,8 GeS,35 and GeS27 structures are represented in Fig. 1 by magenta, cyan, and blue triangles, respectively. The previously known 2D GexSy structures, i.e., Ge2S, GeS, and GeS2, have higher formation energies than our predicted counterpart monolayers by 85, 47, and 84 in units of meV per atom, respectively, and consequently occupy positions above the convex hull. Worth noticing from the ECH analysis is the superior energetic stability of our predicted GexSy structures, which indicates easier experimental accessibility. Therefore, they deserve detailed stability analysis, and we constrain the discussion to the structures on the convex hull only.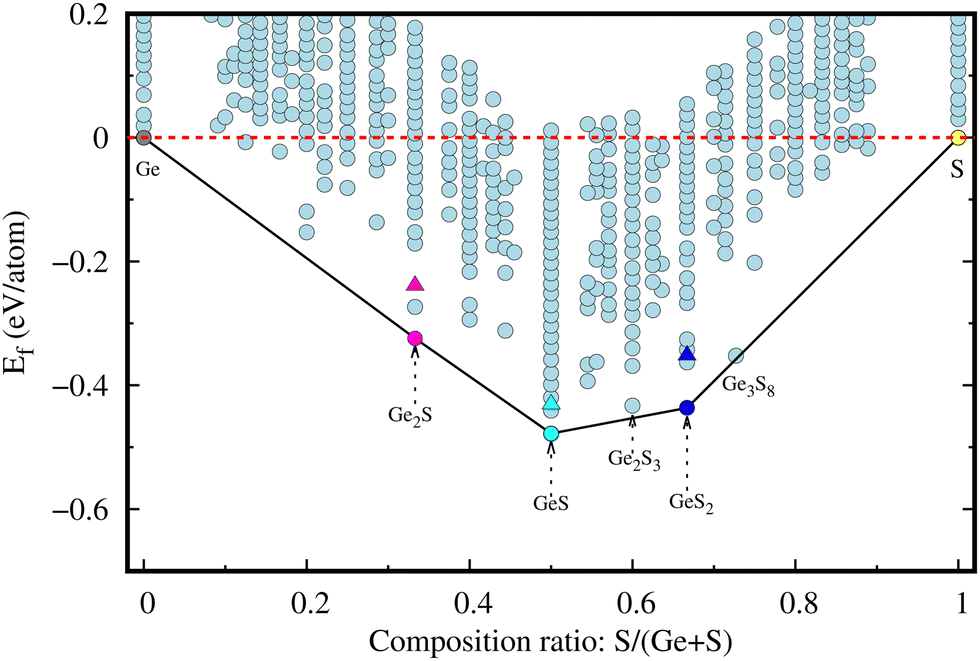 | ||
| Fig. 1 This figure shows the evolution of the energy convex hull (ECH) during the USPEX search, where the solid bounding curve indicates the final ECH of the 2D Ge–S system. The energetically most stable structures of 2D GexSy are highlighted with various colored circles, whereas those previously reported ones are represented with filled triangles. | ||
The phonon band structure, which provides critical insights into the dynamic stability of the newly predicted 2D phases of the Ge2S, GeS, and GeS2, are determined and shown in Fig. 2. These results reveal the dynamic stability of the 2D Ge2S, GeS, and GeS2, as there are no imaginary frequencies in Fig. 2(a)–(c). There are a total of 3n phonon branches, where n is the number of atoms in the primitive unit cell. The first three lowest-frequency phonon branches are called acoustic phonons, which are further classified into transverse acoustic (TA), longitudinal acoustic (LA), and z-axis acoustic (ZA). The ZA mode, also called the flexural mode, represents the out-of-plane atomic vibrations and has a parabolic wave vector dependence contrary to the TA and LA, which have linear dispersion near the Γ-point. In Fig. 2(c), the LA mode along the Γ–Y direction has softer behavior (smaller slope) than along the Γ–X direction. This difference in the phonon dispersion suggests the presence of anisotropic lattice thermal conductivity, which will be discussed later. The remaining 3n − 3 phonon branches are known as optical branches, divided into low and high-frequency bands separated by a gap. This gap is 16/41 cm−1 in Ge2S/GeS and 88 cm−1 in GeS2. The common observation from Fig. 2 is the overlapping of the low-frequency optical phonons with acoustic phonons. It is one of the indicators of the high possibility of three-phonon scattering, which will eventually decrease lattice thermal conductivity. The maximum phonon frequency (ωmax) in all predicted structures is below 400 cm−1. Specifically, ωmax is 365 cm−1 for Ge2S, smaller than its P21212 phase (561 cm−1),8 and 337 cm−1 for GeS, in the same range as the Pmn21 phase (∼330 cm−1).6ωmax is 397 cm−1 for 2D GeS2, comparable to its 1T phase (∼400 cm−1).7 Additionally, the dispersion in the high-frequency optical branches is comparatively lower in GeS2 than in Ge2S and GeS. The non-analytical correction to phonon spectra (see Fig. S8 of ESI†) exhibits a negligible effect on the dispersion of 2D Ge2S and GeS2. However, in 2D GeS, a splitting of high-frequency optical modes near the Γ-point of the BZ is observed. Despite this splitting, it has a negligible impact on the calculated lattice thermal conductivity due to their shorter lifetimes, as discussed later.
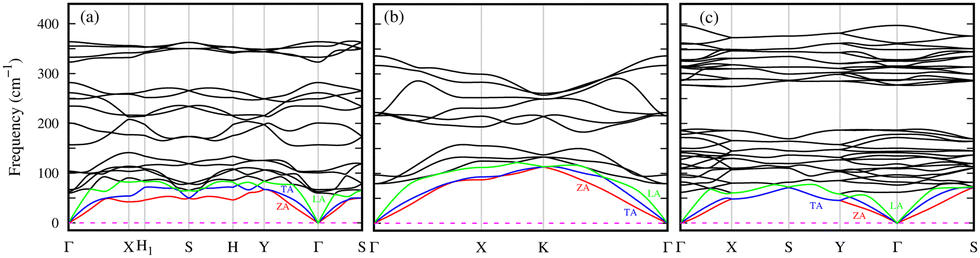 | ||
| Fig. 2 Phonon band structures of 2D (a) Ge2S, (b) GeS, and (c) GeS2. The green, blue, red, and black colors represent the longitudinal acoustic (LA), transverse acoustic (TA), flexural acoustic (ZA), and optical mode phonons, respectively. | ||
In addition to energetic and dynamic stability, the elastic stability of the predicted 2D GexSy provides more insights into their viability. The elastic stability of our predicted 2D monoclinic Ge2S (P2/c), GeS2 (P21/c), and trigonal GeS (Pm1) is confirmed using the corresponding stability criteria36 based on the elastic constants presented in Table 1. Specifically, GeS with a trigonal structure satisfies the criteria C11 > 0, C66 > 0, and C11 > |C12|. The elastic constants of GeS2 satisfy the criteria for a rectangular lattice, i.e., C11 > 0, C66 > 0, and C11 × C22 > C122. Similarly, due to its oblique structure, Ge2S satisfies the criterion det|Cij| > 0, with i, j = 1, 2, 6.
| Structure (space group) | Lattice parameters (a, b), γ (°) | Elastic constants (N m−1) C11, C22, C12, C66 | Young's moduli Yx, Yy (in N m−1) | Poisson's ratio νx, νy |
|---|---|---|---|---|
| Ge2S (P2/c) | 4.98, 5.56, 45° | 47.80, 54.68, 8.07, 27.96 C26 = 10.03, C16 = 5.84 | 46.6, 53.3 | 0.147, 0.169 |
| GeS (Pm1) |
3.63, 3.63, 120° | 85.40, 85.40, 28.46, 28.47 | 75.9, 75.9 | 0.333, 0.333 |
| GeS2 (P21/c) | 6.87, 6.17, 90° | 64.58, 27.84, 4.22, 12.56 | 63.8, 27.6 | 0.152, 0.065 |
Generally, 2D materials are more vulnerable to high temperatures compared to their bulk. Therefore, analyzing the thermal stability of predicted 2D GexSy compounds at elevated temperatures is worthwhile. For this purpose, AIMD simulations are performed to investigate the thermodynamic stability of newly predicted monolayers at different temperatures of 300, 600, and 900 K. The simulation results are shown in Fig. 3 at 300 K and Fig. S2 and S3 of the ESI† at temperatures of 600 and 900 K, respectively. These results reveal that 2D GeS and GeS2 exhibit higher structural stability because no significant changes were observed in the overall morphology or bond lengths at 300, 600, and 900 K. However, the thermally stable 2D Ge2S at 300 and 600 K becomes unstable when the temperature rises to 900 K, as evident from the broken bonds in the inset of Fig. S3(a) in the ESI.†
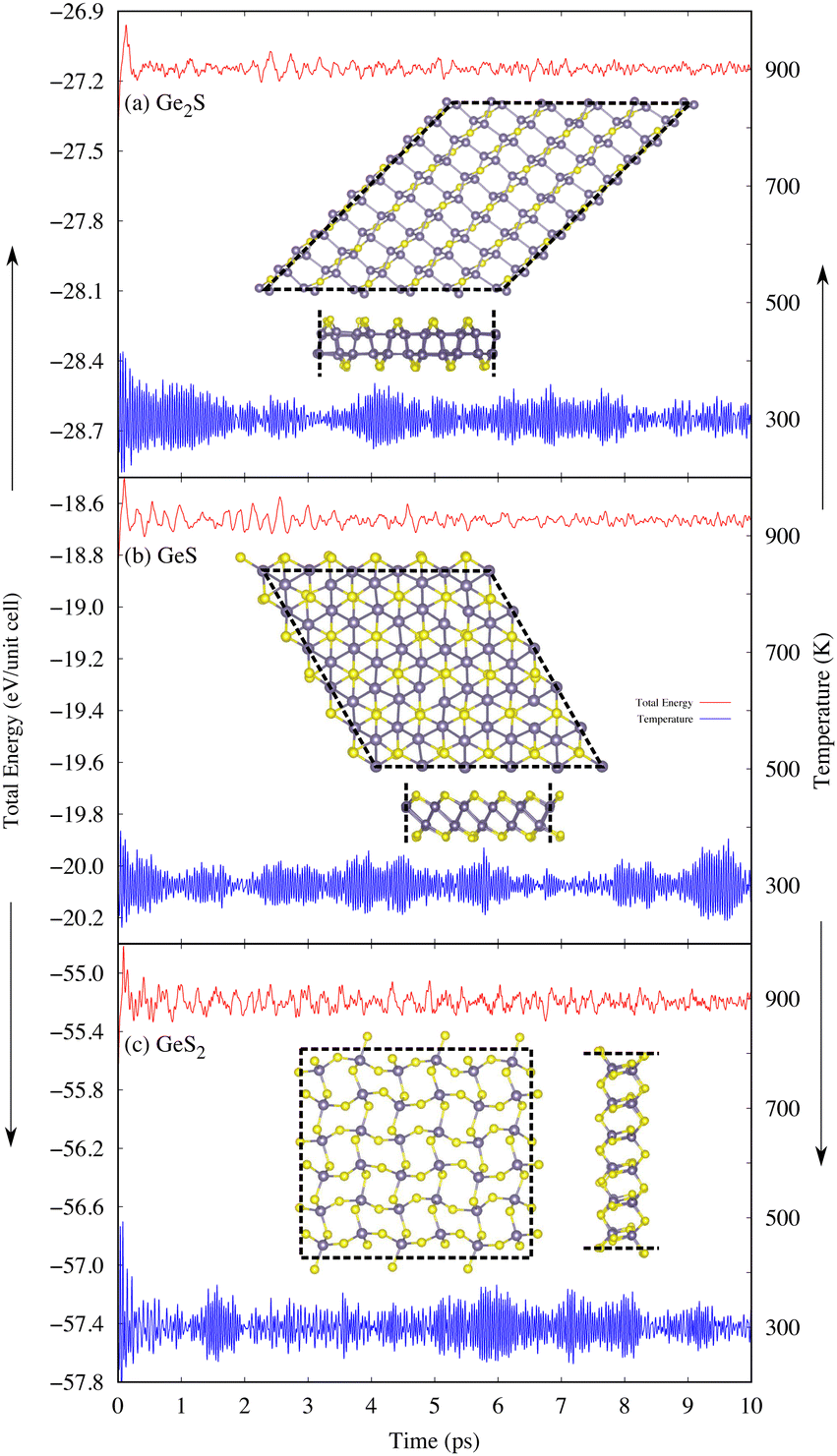 | ||
| Fig. 3 The total energy per unit cell and temperature profiles of the 2D materials (a) Ge2S, (b) GeS, and (c) GeS2 were obtained through AIMD simulations at 300 K. The simulated structures were visualized from the top and side in the insets after a simulation time of 10 ps. | ||
Under real experimental conditions, the 2D layer can interact with various molecules, including nitrogen and oxygen, which are predominant components of the air and can influence their stability. To address this concern, we assessed the stability of the predicted 2D GexSy against N2 and O2 molecules at 0 K. We conclude this section with the remarks that our predicted three compositions of 2D GexSy are energetically more favorable than previously reported ones, stable dynamically and elastically, and can withstand higher temperatures.
3.2 Structural analyses
The properties of a material, either bulk or two-dimensional, are entangled with its crystal structures.37 Therefore, before delving into the electronic structure and phonon transport properties, it is crucial to investigate the crystal structures of the low-energy lying 2D GexSy. In Fig. 4(a)–(c), we show the 2D structures of the stable compositions Ge2S, GeS, and GeS2, respectively. The top and side views of each structure in these figures will facilitate the readers understanding of the discussion in this section. The dashed lines represent the primitive unit cell in each case. Regarding symmetry, Ge2S has a monoclinic unit cell with the space group P2/c, the GeS2 unit cell has the space group P21/c, while GeS adopts a trigonal structure with the space group Pm1. Due to the low symmetry of 2D Ge2S, the views of various orientations are shown in Fig. 4(a). It composes two sublayers; the red-dashed and the solid-black rhombic lattices represent the bottom and top sublayers, respectively. The top sublayer can be obtained from the bottom by mirror symmetry along the z-axis plus a glide of 0.5b along the ![[b with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0062_20d1.gif) lattice vector, where b is the lattice constant. For clarity, we show the top view of only the bottom sublayer on the right side of Fig. 4(a). It consists of irregular (unequal bonds) parallel zigzag chains of Ge atoms in the ab-plane connected through S atoms. Each Ge atom makes four bonds, one with the S atom and three with other Ge atoms, of which two Ge belong to the same sublayer and one Ge to the neighboring sublayer. Hence, the two sublayers are attached by the Ge–Ge bond of length 2.52 Å. The Ge–Ge bond lengths in the zigzag chain are 2.47 Å and 2.51 Å. Other lattice parameters and mechanical properties, such as the elastic constants and Poisson's ratios, are tabulated in Table 1.
lattice vector, where b is the lattice constant. For clarity, we show the top view of only the bottom sublayer on the right side of Fig. 4(a). It consists of irregular (unequal bonds) parallel zigzag chains of Ge atoms in the ab-plane connected through S atoms. Each Ge atom makes four bonds, one with the S atom and three with other Ge atoms, of which two Ge belong to the same sublayer and one Ge to the neighboring sublayer. Hence, the two sublayers are attached by the Ge–Ge bond of length 2.52 Å. The Ge–Ge bond lengths in the zigzag chain are 2.47 Å and 2.51 Å. Other lattice parameters and mechanical properties, such as the elastic constants and Poisson's ratios, are tabulated in Table 1.
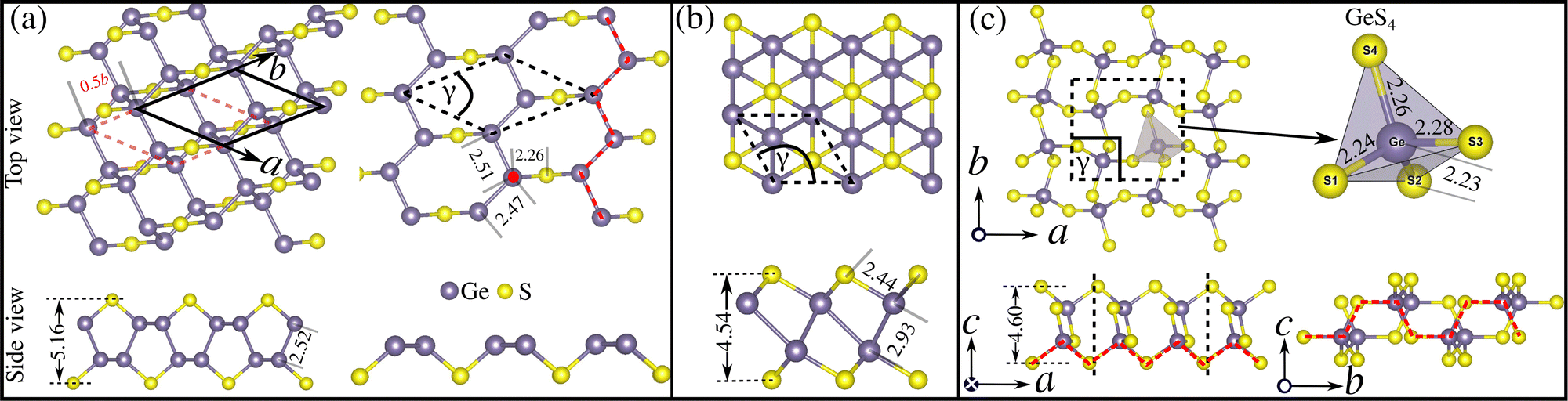 | ||
| Fig. 4 The top and side views of 2D (a) Ge2S, (b) GeS, and (c) GeS2 and the primitive unit cell are shown with dashed lines in each top view. All bond lengths and thicknesses of the layers are shown in units of angstroms (Å). The solid circle (red) in (a) represents the bond between two sublayers. The red-dashed lines in (c) represent zigzag and armchair directions. | ||
Fig. 4(b) shows the top and side views of 2D GeS. It consists of two oppositely buckled honeycomb sublayers with AB stacking. In this structure, the in-plane coordinates of the S atoms are (1/3![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) , 1/3) and of the Ge atoms are (0,0) and (1/3, 2/3), where and are the lattice vectors. For GeS, a = b = 3.63 Å is the optimized lattice constant. Within each sublayer, Ge/S atoms are bonded with three S/Ge atoms through a bond length of 2.44 Å. The two sublayers are then weakly connected by a Ge–Ge bond of length 2.93 Å. Fig. 4(c) shows that the primitive unit cell of 2D GeS2 constitutes corner-sharing distorted (unequal edges) GeS4 tetrahedra. The GeS4-tetrahedron contains the Ge atom in its center, bonded with four S atoms at the corners. All four Ge–S bonds in the GeS4 tetrahedron have different lengths, i.e., 2.23, 2.24, 2.26, and 2.28 in Å, which reduces the unit-cell symmetry from orthorhombic Pbcm (β-2D silica38) to monoclinic P21/c. The side views in Fig. 4(c) exhibit the zigzag morphology along the a-axis and armchair along the b-axis. The GeS4 tetrahedra centers align in the ac-plane and buckle in the bc-plane, which leads to a hinge-like structure along the b-axis. This arrangement in 2D GeS2 results in a rectangular lattice with lattice parameters a = 6.87 Å and b = 6.17 Å. The structural anisotropy of the lattice is reflected in its high elastic anisotropy, where the Young's modulus along the a-axis is 63.8 N m−1, which is more than twice that along the b-axis (27.6 N m−1). Like phosphorene,39,40 we expect that GeS2 will have high anisotropic lattice thermal conductivity because it is often directly proportional to the elastic modulus.41
, 1/3) and of the Ge atoms are (0,0) and (1/3, 2/3), where and are the lattice vectors. For GeS, a = b = 3.63 Å is the optimized lattice constant. Within each sublayer, Ge/S atoms are bonded with three S/Ge atoms through a bond length of 2.44 Å. The two sublayers are then weakly connected by a Ge–Ge bond of length 2.93 Å. Fig. 4(c) shows that the primitive unit cell of 2D GeS2 constitutes corner-sharing distorted (unequal edges) GeS4 tetrahedra. The GeS4-tetrahedron contains the Ge atom in its center, bonded with four S atoms at the corners. All four Ge–S bonds in the GeS4 tetrahedron have different lengths, i.e., 2.23, 2.24, 2.26, and 2.28 in Å, which reduces the unit-cell symmetry from orthorhombic Pbcm (β-2D silica38) to monoclinic P21/c. The side views in Fig. 4(c) exhibit the zigzag morphology along the a-axis and armchair along the b-axis. The GeS4 tetrahedra centers align in the ac-plane and buckle in the bc-plane, which leads to a hinge-like structure along the b-axis. This arrangement in 2D GeS2 results in a rectangular lattice with lattice parameters a = 6.87 Å and b = 6.17 Å. The structural anisotropy of the lattice is reflected in its high elastic anisotropy, where the Young's modulus along the a-axis is 63.8 N m−1, which is more than twice that along the b-axis (27.6 N m−1). Like phosphorene,39,40 we expect that GeS2 will have high anisotropic lattice thermal conductivity because it is often directly proportional to the elastic modulus.41
3.3 Electronic structure
The electronic band structure and the total density of states (DOS) with projected density of states (PDOS) for the three compositions of 2D GexSy are presented in Fig. 5. A first look shows the semiconducting nature of all these compositions. 2D Ge2S in Fig. 5(a) depicts a nearly direct and narrow band gap of 0.12/0.84 eV using the PBE/HSE06 functional. The valence band maximum (VBM) and conduction band minimum (CBM) occur along the Γ–C–V–S path. The complete k-path used for plotting this band structure is given in Fig. S4(c) of the ESI.† Interestingly, the k-points corresponding to the band edges, denoted by (C,V), do not occur along the high symmetry k-path of the first Brillouin zone (BZ). We identified this behavior of 2D Ge2S from Fig. S4(a) (ESI†), where the band edges of the high-symmetry k-path [Fig. S4(b), ESI†] do not match the total DOS. This discrepancy should be attributed to the band structure being limited to only high-symmetry k-points while the total DOS covers the whole first BZ. The k-path is revised, as shown in Fig. S4(c) of the ESI,† by including accurate band edges [C (0.333 0.384 0), V (0.37 0.425 0)] determined from the DOS with a denser k-mesh of reciprocal space resolution of 2π × 0.004 Å−1. Due to the high dispersion of the band structure at the CBM, there is a lower density of states than in the VBM. Furthermore, one can analyze the atomic orbital contributions to the band structure by the PDOS, right panel in Fig. 5(a). This implies that the VBM of 2D Ge2S is contributed by the p-orbitals of both Ge and S atoms, whereas the CBM mainly originates from the s-orbital of the Ge atom. The 2D GeS and GeS2 exhibit band structures in Fig. 5(b) and (c) with indirect band gaps. Specifically, the VBM of 2D GeS is located on both sides of the Γ-point, i.e., along Γ–X and Γ–K. The CBM is located at the X point, where the band structure is highly asymmetric, as shown in Fig. 5(b). The asymmetric conduction band around the CBM is a footprint of the electrical anisotropy. The VBM of 2D GeS includes the p and s orbitals of S and Ge atoms, respectively. Its CBM is composed of Ge-s, Ge-p, and S-p orbitals. 2D GeS2 in Fig. 5(c) has VBM at the Γ point and CBM at the Y point of the first BZ. The valence and conduction bands are flat near the band edges and have a higher density of states than the 2D Ge2S and GeS2. The p-orbital of the S atom mainly contributes to the VBM and CBM, and there is also a comparable contribution from the Ge-s orbital to the CBM. The fundamental band gaps, calculated with PBE functionals, are 0.72/1.85 eV for 2D GeS/GeS2. The dispersions of band structures with the HSE06 functional remain similar to PBE, but the fundamental band gaps open to 1.05 eV for 2D GeS and 2.92 eV for 2D GeS2. Thus, we have narrow, medium, and wide band gap 2D semiconductors: Ge2S, GeS, and GeS2, respectively.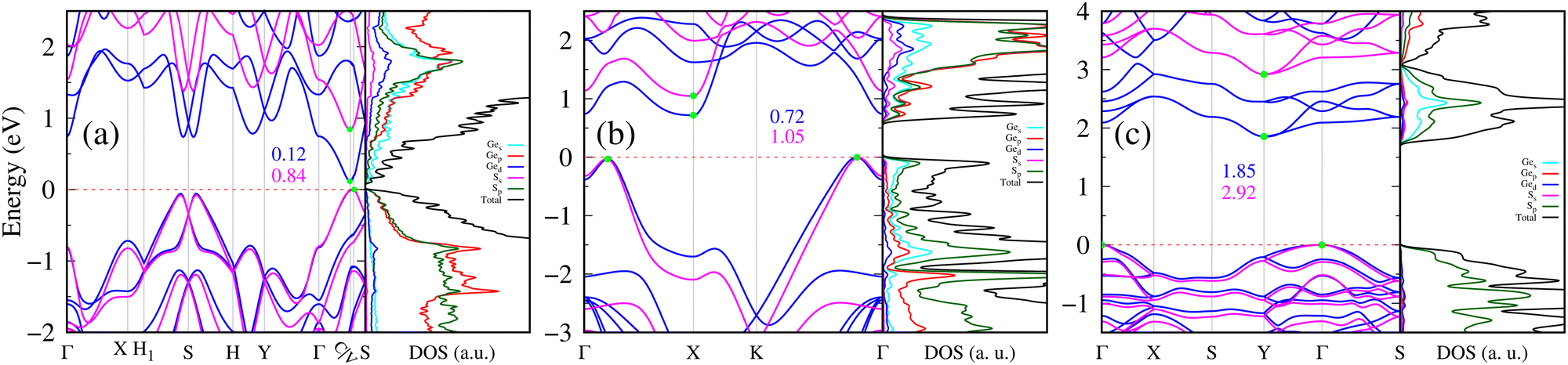 | ||
| Fig. 5 Electronic band structures using PBE (blue)/HSE06 (magenta) functionals and PBE total DOS with the corresponding projected density of states (PDOS) of Ge and S atoms are presented for 2D materials: (a) Ge2S, (b) GeS, and (c) GeS2. The zero of the energy axis is referenced to a horizontal red-dashed line that aligns with the valence band maximum (VBM). The band edges, i.e., VBM and CBM (conduction band minimum), are highlighted with solid green circles. The fundamental band gaps (in eV) are mentioned for each case. | ||
3.4 Phonon transport in 2D GexSy compounds
Lattice thermal conductivity (κl) is a key characteristic of phonon transport and is obtained by iteratively solving the linearized phonon Boltzmann transport equation (PBTE). The solution of PBTE relies on second and third-order interatomic force constants (IFCs) as inputs. The influence of four-phonon scattering is disregarded due to its minimal impact on phonon transport in materials like 2D GexSy, which lack optical-acoustic gaps in phonon dispersions42 (see Fig. 2). The third-order IFCs capture the phonon–phonon scattering processes and play a crucial role in predicting reliable thermal conductivity. A cutoff radius determines the region of interatomic interactions for third-order IFC calculations. Selecting an appropriate cutoff radius ensures the appropriate inclusion of anharmonic interactions and improves the accuracy of κl. Furthermore, solving the PBTE involves discretizing the reciprocal space into a q-point grid hence achieving an accurate κl requires a sufficiently dense q-point grid. Considering inadequate cutoff radii and q-point grids can lead to orders of variations in the reported κl values. For instance, in the case of phosphorene, different κl values in the literature can be attributed to variations in the chosen cutoff radii and q-point grids.39,40 Therefore, to avoid ambiguity in κl, we present its convergence against the cutoff radius and q-point grid in Fig. S5 of the ESI.† These figures depict an enormous variation of κl in the lower cutoff radius, which does not include sufficient nearest neighbors for interactions, and a coarse q-point grid. We achieve sufficient convergent κl by considering cutoff radii of 7.4 Å, 10.8 Å, and 6.3 Å for 2D Ge2S, GeS, and GeS2, respectively. These include up to the 20th nearest neighbors for Ge2S and 22nd for GeS and GeS2. Our q-point grid tests show that κl well converges at 60 × 60 × 1, 100 × 100 × 1, and 45 × 45 × 1 for Ge2S, GeS, and GeS2, respectively.Fig. 6(a)–(c) depict the temperature-dependent lattice thermal conductivities of the 2D Ge2S within the stable temperature range of 200–600 K, and the 2D GeS and GeS2 compounds over the temperature range of 200–900 K. Specifically, GeS has a room temperature κl of 6.82 W m−1 K−1, which is lower than monolayer MoSe2, WSe2, and phosphorene.43,44 Importantly, it is also smaller than its meta-stable orthorhombic phase (10.5/7.8 W m−1 K−1 along the zigzag/armchair directions) but higher than other IV–VI 2D compounds like GeSe (6.7/5.2 W m−1 K−1), SnS (4.7/4.4 W m−1 K−1), and SnSe (2.6/2.4 W m−1 K−1) along the respective directions.6 On the other hand, 2D Ge2S exhibits a room temperature κl of ∼29 W m−1 K−1, which is the highest among the predicted 2D GexSy compounds and closer to that of phosphorene in the zigzag direction.44 The novel stable phase of 2D GeS2 has 16.95/10.68 W m−1 K−1 along the zigzag/armchair directions at 300 K, larger than the reported value for its meta-stable 1T phase.7 The anisotropy of κl can be quantified by the ratio κaa/κbb, where κaa and κbb represent the κl values along a- and b-axes, respectively. In the case of GeS2, we designate the a-axis as the zigzag direction and the b-axis as the armchair direction (see Fig. 4(c)). For 2D GeS, this ratio is exactly one, indicating isotropic phonon transport. Similarly, for 2D Ge2S, the ratio is nearly one, implying a predominantly isotropic nature of κl. Interestingly, the in-plane anisotropy ratio κaa/κbb is 1.59 for the predicted stable 2D GeS2, which is higher than other orthorhombic 2D IV–VI compounds such as GeS (1.35), GeSe (1.29), SnS (1.07), and SnSe (1.08), indicating giant in-plane anisotropic lattice thermal conductivity. Although phosphorene exhibits a larger in-plane anisotropy ratio of 2.2144 compared to 2D GeS2, it is prone to stability issues under ambient temperatures.45 These intrinsic anisotropic materials are rare and of particular importance in nanodevice applications. Moreover, Fig. 6(a)–(c) show a consistent decrease in κl as the temperature increases, displaying a strong adherence to the 1/T relationship, as indicated by the solid fitting line in each plot. This trend is commonly observed in intrinsic 2D materials and is attributed to the dominant occurrence of three-phonon scattering via the umklapp process, which depends on temperature directly. The umklapp scattering process opposes heat flow by scattering phonons in the opposite direction, impeding their effective transport. In contrast, the phonon–phonon scattering by the normal process could not provide resistance to phonon transport.46
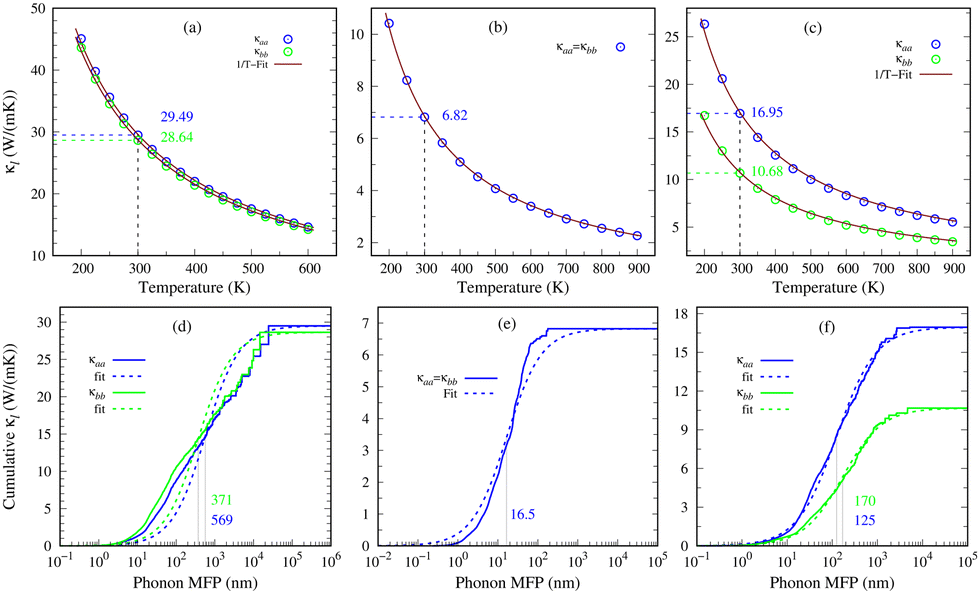 | ||
Fig. 6 The lattice thermal conductivity as a function of temperature and the corresponding 1/T fitting for 2D materials (a) Ge2S, (b) GeS, and (c) GeS2. The cumulative κl is plotted against the phonon mean free path (MFP) and fitted using the expression 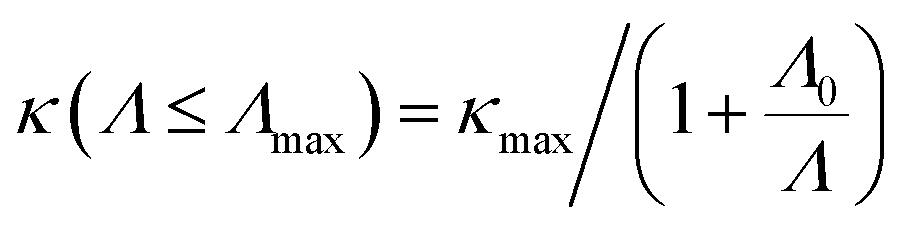 for (d) Ge2S, (e) GeS, and (f) GeS2, where the value (in nm) of the fitting parameter Λ0 along the a/b-axis is mentioned in each case. for (d) Ge2S, (e) GeS, and (f) GeS2, where the value (in nm) of the fitting parameter Λ0 along the a/b-axis is mentioned in each case. | ||
Nanostructuring is a common approach to control the κl of a material. Generally, decreasing the grain size causes a reduction in the mean free path (MFP) Λ of phonons, resulting in a lower κl while keeping the electronic conductivity unchanged. This behavior offers significant potential for enhancing the system's thermoelectric figure of merit. The cumulative κl as a function of MFP in Fig. 6(d)–(f) provides insights into the effect of grain size on the κl of 2D GexSy compounds at 300 K. To investigate the MFP range that dominantly contributes to κl, we fit the data with a logistic function 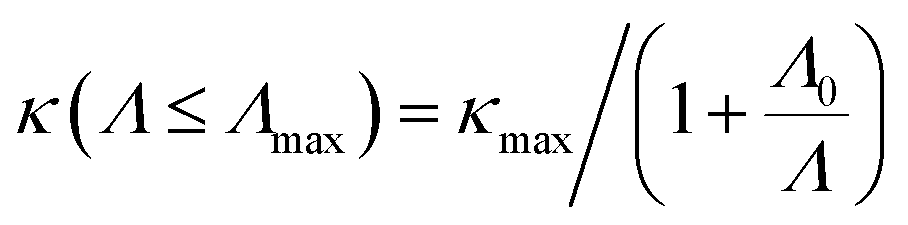 , where κmax is the ultimate lattice thermal conductivity, and Λ0 is the fitting parameter called the representative MFP at which cumulative κl reaches 50% of its maximum value. The higher values of Λ0, i.e., 569/371 nm for Ge2S and 125/170 nm for GeS2 compared to other IV–VI monolayers,47 indicate that nanostructuring will be highly effective in reducing their κl. In contrast, the low phonon MPF Λ0 of 16.5 nm for GeS, comparable to orthorhombic IV–VI monolayers, implies that it will be hard to reduce its κl with nanostructuring and other methods like alloying might be a convenient way. For a given nanostructure size of 50 nm, the κl is reduced to 5.70/7.45 W m−1 K−1 for Ge2S and 5.54/2.75 W m−1 K−1 for GeS2, which becomes competitive with κl (5.83 W m−1 K−1) of GeS. The evident anisotropy in the Λ0 for Ge2S and GeS2 along the a/b-axes implies that reducing the grain size of the system will have different impacts on the κl along the a-axis compared to the b-axis. Similar anisotropic trends in Λ0 for phosphorene allotropes are reported in previous studies.48 This anisotropy offers intriguing possibilities for tailoring the κl by providing additional flexibility for design purposes. The analysis of the cumulative κl at various temperatures within the range of 300–600 K for 2D Ge2S and 300–800 K (see Fig. S6 of the ESI†) for 2D GeS and GeS2 demonstrates a uniform decrease in Λ0 as the temperature rises.
, where κmax is the ultimate lattice thermal conductivity, and Λ0 is the fitting parameter called the representative MFP at which cumulative κl reaches 50% of its maximum value. The higher values of Λ0, i.e., 569/371 nm for Ge2S and 125/170 nm for GeS2 compared to other IV–VI monolayers,47 indicate that nanostructuring will be highly effective in reducing their κl. In contrast, the low phonon MPF Λ0 of 16.5 nm for GeS, comparable to orthorhombic IV–VI monolayers, implies that it will be hard to reduce its κl with nanostructuring and other methods like alloying might be a convenient way. For a given nanostructure size of 50 nm, the κl is reduced to 5.70/7.45 W m−1 K−1 for Ge2S and 5.54/2.75 W m−1 K−1 for GeS2, which becomes competitive with κl (5.83 W m−1 K−1) of GeS. The evident anisotropy in the Λ0 for Ge2S and GeS2 along the a/b-axes implies that reducing the grain size of the system will have different impacts on the κl along the a-axis compared to the b-axis. Similar anisotropic trends in Λ0 for phosphorene allotropes are reported in previous studies.48 This anisotropy offers intriguing possibilities for tailoring the κl by providing additional flexibility for design purposes. The analysis of the cumulative κl at various temperatures within the range of 300–600 K for 2D Ge2S and 300–800 K (see Fig. S6 of the ESI†) for 2D GeS and GeS2 demonstrates a uniform decrease in Λ0 as the temperature rises.
Understanding the mechanisms involved in heat conduction through lattice vibrations in 2D GexSy compounds requires comprehensive analysis. We calculate the phonon group velocities using the formula 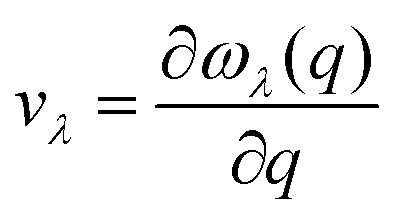 , where λ represents the mode index, ω is the frequency, and q is the wave vector of the phonon, and the results are shown in Fig. 7. Fig. 7 shows the lower group velocities, i.e., below 6 km s−1, in all 2D GexSy compared to other 2D materials like graphene, silicene,49 MoS2,50 and phosphorene,44 and the overall lower κl can be attributed to it. However, Fig. 7(b) and (e) depict comparable group velocities in the frequency range 200–300 cm−1 of the optical phonon to its acoustic branches and is higher than the optical phonon of Ge2S and GeS2. This implies that the lower group velocity alone does not guarantee the lower κl, and further investigation is required, particularly in conjunction with phonon lifetimes. The phonon lifetimes, which represent the time a phonon can exist before scattering with other lattice vibrations, for 2D GexSy compounds, are shown in Fig. 8(a)–(c). Specifically, the short phonon lifetimes of less than 10 ps in 2D GeS indicate frequent scattering events. These scattering events impede the heat transfer through the lattice, resulting in the reduced κl of GeS compared to Ge2S and GeS2.
, where λ represents the mode index, ω is the frequency, and q is the wave vector of the phonon, and the results are shown in Fig. 7. Fig. 7 shows the lower group velocities, i.e., below 6 km s−1, in all 2D GexSy compared to other 2D materials like graphene, silicene,49 MoS2,50 and phosphorene,44 and the overall lower κl can be attributed to it. However, Fig. 7(b) and (e) depict comparable group velocities in the frequency range 200–300 cm−1 of the optical phonon to its acoustic branches and is higher than the optical phonon of Ge2S and GeS2. This implies that the lower group velocity alone does not guarantee the lower κl, and further investigation is required, particularly in conjunction with phonon lifetimes. The phonon lifetimes, which represent the time a phonon can exist before scattering with other lattice vibrations, for 2D GexSy compounds, are shown in Fig. 8(a)–(c). Specifically, the short phonon lifetimes of less than 10 ps in 2D GeS indicate frequent scattering events. These scattering events impede the heat transfer through the lattice, resulting in the reduced κl of GeS compared to Ge2S and GeS2.
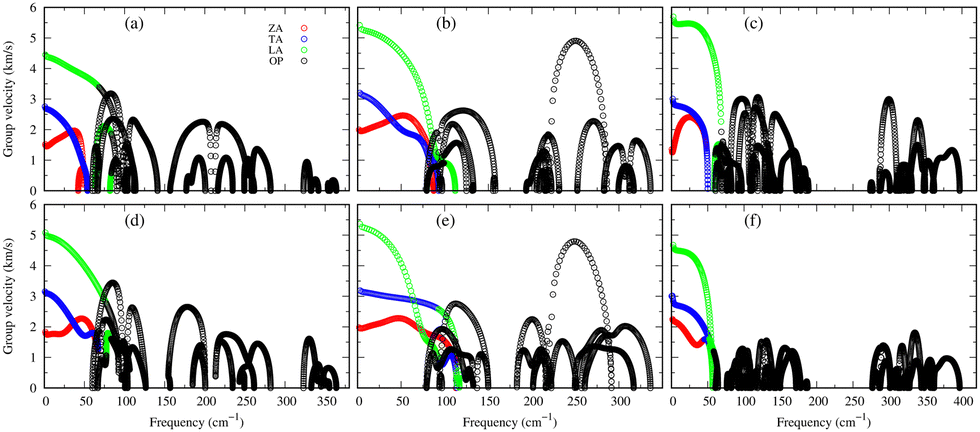 | ||
| Fig. 7 Figures (a)–(c) shows the phonon group velocity (vλ) along Γ–X for Ge2S, GeS, and GeS2, while (d) and (f) represent vλ along Γ–Y for Ge2S and GeS2, and (e) along Γ–K for GeS, respectively. | ||
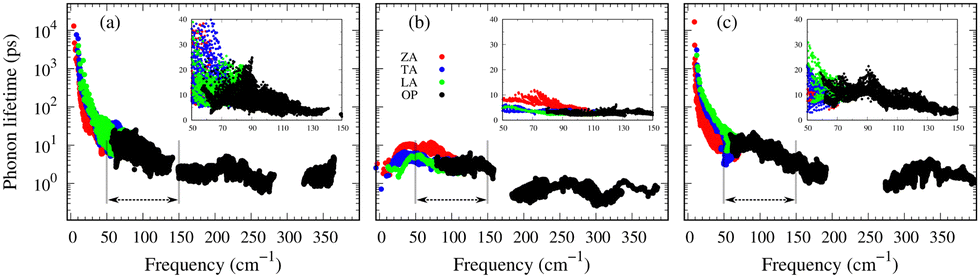 | ||
| Fig. 8 The phonon lifetimes of 2D Ge2S, GeS, and GeS2 are shown in (a), (b), and (c), respectively. The insets display the enlarged view for visual comparison in the frequency range of 50–150 cm−1. | ||
The anisotropic κl observed in 2D GeS2 can be attributed to the anisotropic group velocities of both acoustic and low-frequency optical phonons in the frequency 50–150 cm−1 range. Fig. 7(b) and (e) provide evidence of larger group velocities along the Γ–X reciprocal lattice direction compared to the Γ–Y direction in GeS2 for acoustic and low-frequency optical phonons. This anisotropy in group velocities directly translates into anisotropic κl along the respective crystallographic directions. The frequency range of 50–150 cm−1, mainly populated by optical phonons in GeS2 and Ge2S compositions, plays a crucial role due to the larger phonon lifetimes observed within this range. While heat transfer typically dominates in acoustic phonons, the acoustic-optical (low-frequency) phonon coupling effect allows for exceptions to this trend, as demonstrated in a recent study of nitride perovskite LaWN3, where 60% of the heat transfer occured through optical phonons.51 In the case of GeS2 and Ge2S, the phonon lifetimes within the mentioned frequency range are approximately 20 ps, which is four times longer than the lifetimes observed in GeS (∼5 ps), evident from the insets showing enlarged views in Fig. 8(a)–(c). This highlights the significant role played by low-frequency optical phonons in heat transport. The frequency-resolved cumulative κl analysis (see Fig. S7(c) of the ESI†) further confirms the substantial contributions of low-frequency optical phonons to the overall κl. Moreover, the thermal anisotropy in GeS2 aligns with its elastic properties. The zigzag direction exhibits a larger Young's modulus than the armchair direction, resulting in a larger κl along the zigzag direction.
Importantly, within the dominant heat-carrying frequency range (see Fig. S7 of the ESI†), the phonon lifetimes of 2D Ge2S and GeS2 are significantly larger than those of 2D GeS. However, this increase does not correspond to a proportional rise in κl. It highlights that phonon–phonon scattering cannot solely be determined by phonon lifetimes. Therefore, we investigate the lattice anharmonicity and the weighted phase space for three-phonon scattering. In Fig. 9, we display the Grüeisen parameter 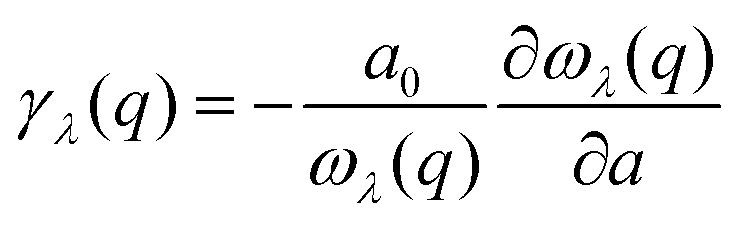 , where a0 represents the equilibrium lattice constant, serving to quantify the lattice anharmonicity of materials. Additionally, the weighted phase space W of three-phonon scattering (as defined by Wu Li52) for 2D GexSy is presented in the lower panel of Fig. 9. Both these parameters, i.e., γ and W, exhibit an inverse relationship with phonon lifetimes, providing additional insights. The large magnitude of γ and phase space W in the range of acoustic frequencies, which serve as the primary heat carriers, is evident from Fig. 9(a) and (d) and aligns with the lower κl of 2D GeS. On the other hand, a comparison of γ, W between 2D Ge2S and 2D GeS2 reveals that while the |γ|max of Ge2S (∼5) is greater than that of 2D GeS2 (∼3), the weighted phase space for Ge2S decreases significantly compared to GeS2 as the phonon frequency increases within the frequency range of 150 cm−1. Consequently, the weaker anharmonicity of 2D GeS2 is compensated by the larger three-phonon weighted phase space, resulting in a smaller κl compared to 2D Ge2S. The large W can be attributed to the higher phonon population in the lower-optical branches, as shown in Fig. 2(c), due to the greater number of atoms in the primitive unit cell of GeS2, increasing the probability of phonon–phonon scattering.53 Furthermore, the higher phase space [as observed in Fig. 9(e)] of GeS2 in the frequency range of 150 cm−1 supports our argument regarding its considerable anisotropic κl, attributed to both acoustic and low-frequency optical phonons within the 50–150 cm−1 frequency range.
, where a0 represents the equilibrium lattice constant, serving to quantify the lattice anharmonicity of materials. Additionally, the weighted phase space W of three-phonon scattering (as defined by Wu Li52) for 2D GexSy is presented in the lower panel of Fig. 9. Both these parameters, i.e., γ and W, exhibit an inverse relationship with phonon lifetimes, providing additional insights. The large magnitude of γ and phase space W in the range of acoustic frequencies, which serve as the primary heat carriers, is evident from Fig. 9(a) and (d) and aligns with the lower κl of 2D GeS. On the other hand, a comparison of γ, W between 2D Ge2S and 2D GeS2 reveals that while the |γ|max of Ge2S (∼5) is greater than that of 2D GeS2 (∼3), the weighted phase space for Ge2S decreases significantly compared to GeS2 as the phonon frequency increases within the frequency range of 150 cm−1. Consequently, the weaker anharmonicity of 2D GeS2 is compensated by the larger three-phonon weighted phase space, resulting in a smaller κl compared to 2D Ge2S. The large W can be attributed to the higher phonon population in the lower-optical branches, as shown in Fig. 2(c), due to the greater number of atoms in the primitive unit cell of GeS2, increasing the probability of phonon–phonon scattering.53 Furthermore, the higher phase space [as observed in Fig. 9(e)] of GeS2 in the frequency range of 150 cm−1 supports our argument regarding its considerable anisotropic κl, attributed to both acoustic and low-frequency optical phonons within the 50–150 cm−1 frequency range.
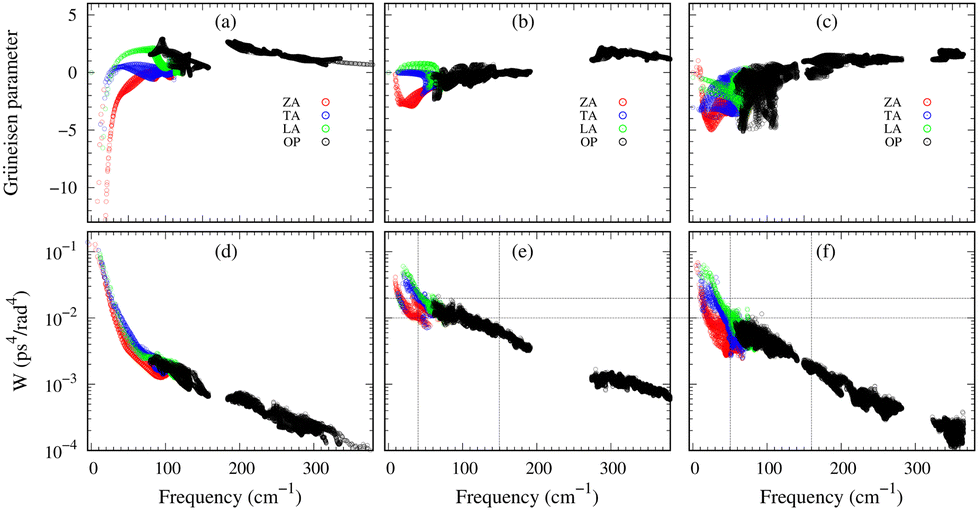 | ||
| Fig. 9 Phonon-mode-resolved Grüneisen parameter and the room temperature weighted phase space (W) for (a and d) 2D GeS, (b and e) 2D GeS2, and (c and f) 2D Ge2S. The gray dashed lines (in c and f) aid in visually comparing the W within the 50–150 cm−1 frequency range. | ||
4 Conclusion
In this manuscript, we present the discovery of novel energetically stable 2D configurations for Ge2S, GeS, and GeS2 structures, achieved through the utilization of the USPEX in conjunction with VASP. Their dynamic, elastic, and thermal stabilities are confirmed using state-of-the-art methods. Subsequently, we characterize their electronic structures, revealing the semiconducting nature with narrow, medium, and wide band gaps for Ge2S, GeS, and GeS2, respectively. Then, we focus on their lattice thermal conductivity (κl), which provides insights into their heat transport properties. All structures exhibit overall low κl values, with 2D GeS having the lowest value of 6.82 W m−1 K−1. We observe a significant decrease in κl of 2D Ge2S and GeS2 with decreasing grain size, suggesting that nanostructuring could further suppress their κl. Through comprehensive analysis of the underlying mechanisms, we uncover the factors contributing to the observed low κl in 2D GeS and the in-plane anisotropy in 2D GeS2. In 2D GeS, the combination of shorter phonon lifetimes, strong anharmonicity, higher weighted phase space, and smaller group velocities account for its low κl. The κl anisotropy of 2D GeS2 is attributed to the anisotropic group velocities of both acoustic and low-frequency optical phonons. The low-frequency optical phonons in 2D Ge2S and GeS2 exhibited lifetimes four times longer than that of 2D GeS, resulting in increased κl. These findings contribute to the fundamental understanding of heat transport in 2D GexSy compounds, which have potential in thermal management and related fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Ministry of Science and ICT (MSIT), government of South Korea (Grant No. 2021R1F1A1048555).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- J. Li, M. Chen, A. Samad, H. Dong, A. Ray, J. Zhang, X. Jiang, U. Schwingenschlögl, J. Domke, C. Chen, Y. Han, T. Fritz, R. S. Ruoff, B. Tian and X. Zhang, Nat. Mater., 2022, 21, 740–747 CrossRef CAS PubMed.
- Y. Anno, Y. Imakita, K. Takei, S. Akita and T. Arie, 2D Mater., 2017, 4, 025019 CrossRef.
- M. Zulfiqar, Y. Zhao, G. Li, Z. Li and J. Ni, Sci. Rep., 2019, 9, 4571 CrossRef PubMed.
- A. Shafique and Y.-H. Shin, Sci. Rep., 2017, 7, 506 CrossRef PubMed.
- X. Wang, W. Feng, C. Shen, Z. Sun, H. Qi, M. Yang, Y. Liu, Y. Wu and X. Wu, Front. Mater., 2021, 8, 709757 CrossRef.
- T. Jiang, Y.-J. Zhu, X.-J. Ye and C.-S. Liu, J. Appl. Phys., 2022, 132, 075001 CrossRef CAS.
- R. E. Abutbul, E. Segev, U. Argaman, G. Makov and Y. Golan, Adv. Mater., 2018, 30, 1706285 CrossRef PubMed.
- W. H. Zachariasen, Phys. Rev., 1932, 40, 917–922 CrossRef CAS.
- M. Shimada and F. Dachille, Inorg. Chem., 1977, 16, 2094–2097 CrossRef CAS.
- G. Dittmar and H. Schäfer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2060–2064 CrossRef.
- W. H. Zachariasen, J. Chem. Phys., 2004, 4, 618–619 CrossRef.
- Z. V. Popović, M. Holtz, K. Reimann and K. Syassen, Phys. Status Solidi B, 1996, 198, 533–537 CrossRef.
- Y. Li, G. Kuang, Z. Jiao, L. Yao and R. Duan, Mater. Res. Express, 2022, 9, 122001 CrossRef.
- A. J. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed.
- S. Lee, J.-E. Jung, H.-G. Kim, Y. Lee, J. M. Park, J. Jang, S. Yoon, A. Ghosh, M. Kim, J. Kim, W. Na, J. Kim, H. J. Choi, H. Cheong and K. Kim, Nano Lett., 2021, 21, 4305–4313 CrossRef CAS PubMed.
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Sahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS PubMed.
- X. Yu, L. Li, X.-W. Xu and C.-C. Tang, J. Phys. Chem. C, 2012, 116, 20075–20079 CrossRef CAS.
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef PubMed.
- A. R. Oganov, A. O. Lyakhov and M. Valle, Acc. Chem. Res., 2011, 44, 227–237 CrossRef CAS PubMed.
- A. Ali and Y.-H. Shin, Phys. Chem. Chem. Phys., 2022, 24, 29772–29780 RSC.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1998, 97, 2635 CrossRef.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- W. Li, J. Carrete, N. A. Katcho and N. Mingo, Comput. Phys. Commun., 2014, 185, 1747–1758 CrossRef CAS.
- W. Li, L. Lindsay, D. A. Broido, D. A. Stewart and N. Mingo, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 174307 CrossRef.
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355–10368 CrossRef CAS.
- B. D. Malone and E. Kaxiras, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 245312 CrossRef.
- B. Dong, Z. Wang, N. T. Hung, A. R. Oganov, T. Yang, R. Saito and Z. Zhang, Phys. Rev. Mater., 2019, 3, 013405 CrossRef CAS.
- A. K. Singh and R. G. Hennig, Appl. Phys. Lett., 2014, 105, 042103 CrossRef.
- M. Maździarz, 2D Mater., 2019, 6, 048001 CrossRef.
- X. Li, L. Tao, Z. Chen, H. Fang, X. Li, X. Wang, J.-B. Xu and H. Zhu, Appl. Phys. Rev., 2017, 4, 021306 Search PubMed.
- Z. Gao, X. Dong, N. Li and J. Ren, Nano Lett., 2017, 17, 772–777 CrossRef CAS PubMed.
- L. Zhu, G. Zhang and B. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 214302 CrossRef.
- G. Qin, X. Zhang, S.-Y. Yue, Z. Qin, H. Wang, Y. Han and M. Hu, Phys. Rev. B, 2016, 94, 165445 CrossRef.
- D. R. Clarke, Surf. Coat. Technol., 2003, 163–164, 67–74 CrossRef CAS.
- T. Feng, L. Lindsay and X. Ruan, Phys. Rev. B, 2017, 96, 161201 CrossRef.
- S. Kumar and U. Schwingenschlögl, Chem. Mater., 2015, 27, 1278–1284 CrossRef CAS.
- G. Qin, Q.-B. Yan, Z. Qin, S.-Y. Yue, M. Hu and G. Su, Phys. Chem. Chem. Phys., 2015, 17, 4854–4858 RSC.
- J. D. Wood, S. A. Wells, D. Jariwala, K.-S. Chen, E. Cho, V. K. Sangwan, X. Liu, L. J. Lauhon, T. J. Marks and M. C. Hersam, Nano Lett., 2014, 14, 6964–6970 CrossRef CAS PubMed.
- C. Kittel, Introduction to Solid State Physics, Wiley, 2004 Search PubMed.
- G. Qin, Z. Qin, W.-Z. Fang, L.-C. Zhang, S.-Y. Yue, Q.-B. Yan, M. Hu and G. Su, Nanoscale, 2016, 8, 11306–11319 RSC.
- J. Zhang, H. J. Liu, L. Cheng, J. Wei, J. H. Liang, D. D. Fan, P. H. Jiang and J. Shi, Sci. Rep., 2017, 7, 4623 CrossRef CAS PubMed.
- B. Peng, H. Zhang, H. Shao, Y. Xu, G. Ni, R. Zhang and H. Zhu, Phys. Rev. B, 2016, 94, 245420 CrossRef.
- B. Peng, H. Zhang, H. Shao, Y. Xu, X. Zhang and H. Zhu, Ann. Phys., 2016, 528, 504–511 CrossRef CAS.
- Q. Ren, Y. Li, Y. Lun, G. Tang and J. Hong, Phys. Rev. B, 2023, 107, 125206 CrossRef CAS.
- W. Li and N. Mingo, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 144304 CrossRef.
- C. Liu, C. Wu, T. Song, Y. Zhao, J. Yang, P. Lu, G. Zhang and Y. Chen, ACS Appl. Energy Mater., 2022, 5, 15356–15364 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04568d |
| This journal is © the Owner Societies 2024 |