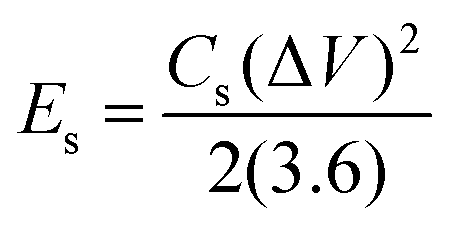Novel electrospun bead-like Ag2MoO4 nanofibers coated on Ni foam for visible light-driven heterogeneous photocatalysis and high-performance supercapacitor electrodes†
Received
29th September 2023
, Accepted 21st November 2023
First published on 11th December 2023
Abstract
Novel Ag2MoO4 nanocomposite fibers were designed to enhance the photocatalytic response and supercapacitor performance of MoO3 grown via the sol–gel electrospinning technique. The Ag2MoO4 nanocomposite fibers exhibit a high specific surface area of 49.3 m2 g−1 comprising nanobeads that aggregate in the fibrous structure. The photodegradation efficiency of Ag2MoO4 was evaluated as 62% under visible light irradiation which improved to 71% with heterogeneous photocatalysis. The Ag2MoO4@Ni foam exhibited a low Rct of 19.6 Ω, and an enhanced specific capacitance of 1445 F g−1 was obtained at 1 A g−1, with 93% of its initial capacitance remaining after 5000 cycles. In addition, the Ag2MoO4//activated carbon asymmetric supercapacitor possesses an excellent energy density of 76.6 W h kg−1 at 743.2 W kg−1 and a noteworthy cycling durability of 91% after 5000 cycles. Our findings demonstrate that the electrospun Ag2MoO4@Ni foam is an important and inexpensive electrode material for supercapacitor applications and visible light-driven heterogeneous photocatalysis, drawing on the synergic effects of Ag and Mo to exhibit much better performance.
Introduction
Nowadays, water pollution and energy crises worldwide force researchers to develop multi-functional and highly efficient nanomaterials.1–3 Supercapacitors are an attractive energy storage solution due to their ease of maintenance, environmental friendliness, safety, robustness, high power density, and high capacitance.4–6 The performance of supercapacitors is heavily influenced by the electrode material, which should exhibit superior conductivity, excellent stability, high specific surface area, and exceptional reversibility, and use low-cost and abundant materials.5,7 The photocatalytic activity of semiconductors is a promising clean energy technology and environmental remedy, with efficient photocatalysts characterized by wide-ranging photoabsorption and low recombination rates of electrons and holes.7–11 The design of heterojunctions for efficient electrochemical energy storage and environmental remediation is promising for future energy and environmental applications.5 Heterogeneous photocatalysis has become a popular method for dye removal, with nickel foam being a suitable catalyst carrier due to its thermal stability, flexibility, large surface area, porous structure, and high electrical conductivity.12 The shape of nanomaterials is crucial in optimizing their properties and potential applications, particularly in the field of environmental remediation. Morphology influences the surface area, crystal facets, interfaces, and active sites, ultimately affecting efficiency.13 Fibrous morphologies are particularly advantageous for photocatalytic reactions due to their high aspect ratio, which provides a greater surface area for catalysis.14 Additionally, their high surface-to-volume ratios facilitate electron transport and accelerate the decomposition of dye molecules. Recent research has shown that one-dimensional nanomaterials such as fibers, wires, tubes, and rods have great potential in photocatalyst activity, energy conversion, and energy storage devices.15,16 To develop specialized electrode materials for various applications, research in this field focuses on designing and synthesizing new electrode materials and investigating structure–function correlations of nanomaterials with particular morphologies.14,17
Electrospinning is a simple process for producing nanofibers (NFs) of multiple shapes and sizes which is scalable and continuous, offering numerous benefits including high surface-to-volume ratios, tunable porosities, and the ability to alter chemical composition for specific needs in energy devices.18,19 Calcination can convert polymeric nanofibers into extremely conductive metal oxides without losing flexibility. Hierarchical nanostructures formed of low-dimensional materials reduce internal resistance for electron transport and shorten ion diffusion paths, improving electrochemical performance.5,20–23 Metal molybdates like XMoO4 (X = Mg, Mn, Zn, etc.) are excellent options for photocatalytic activities and supercapacitor electrodes due to their synergistic effect between metal and molybdate species.24–29 Ag2MoO4 nanostructures have been shown to be useful for semiconductor photocatalyst and energy storage applications,30–32 but there has been no report on synthesis of electrospun Ag2MoO4 for these applications. In this study, we utilized a sol–gel electrospinning technique to prepare Ag/Mo-based polymeric nanofibers which were then converted to Ag2MoO4 nanostructures after calcination at high temperatures. Energy storage and photocatalytic applications of nanomaterials are strongly influenced by morphology, specific surface area, and band structure, so we studied these common features of the prepared nanofibers using analytical tools to better analyse the charge storage mechanism and optical response. Our results indicate that Ag2MoO4@Ni foam exhibits good photodegradation efficiency, superior specific capacitance and cycling stability. Furthermore, the Ag2MoO4//activated carbon asymmetric supercapacitor possesses a great energy density and excellent cycling stability, making it a significant inexpensive electrode active material for supercapacitor applications.
Experimental
Reagents and chemicals
All chemicals used in this study were reagent grade or higher and were utilized without further purification. Ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24·4H2O), silver nitrate hexahydrate (Ag(NO3)2·6H2O), ethanol (99.9% purity), carbon black, citric acid, polyvinylidene fluoride (PVDF) and C5H9NO (NMP) were purchased from Merck. Polyvinylpyrrolidone (PVP) (MW = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000) was obtained from Sigma-Aldrich.
300000) was obtained from Sigma-Aldrich.
Synthesis of MoO3 and Ag2MoO4 nanostructures
For the synthesis of electrospun MoO3, ammonium heptamolybdate tetrahydrate, 0.1 mmol, and a small amount of citric acid were dissolved in 5 mL of deionized water under magnetic stirring. Furthermore, the PVP solution was prepared by dissolving 1 g of PVP in 10 mL of ethanol. The precursor sol was ultimately made by combining the two solutions for 12 hours with magnetic stirring. Slightly altered from a comparable procedure, Ag2MoO4 NFs were produced using a similar method. Along with 0.1 mmol ammonium heptamolybdate, 1.4 mmol silver nitrate was dissolved in deionized water. The precursor solution was loaded into a plastic syringe, and 17 kV was applied between the collector and needle. The rate of feeding was 0.5 mL h−1. The needle was placed 12 cm away from the collector. The as-spun fibers were dried at 80 °C and calcined at 550 °C for two hours.
Manufacture of electrodes
Interconnecting porous nickel foams (Latech Scientific Supply Pte. Ltd, Singapore) were completely cleaned with ultrasonic devices using HCl, acetone, and deionized water before being dried in an oven for two hours at 80 °C. The MoO3@Ni foam and Ag2MoO4@Ni foam electrodes were prepared by combining the carbon black, binder (PVDF) and active materials in NMP in a weight ratio of 1:1:8. After that, the electrodes were put in the oven and heated to 80 °C. Asymmetric supercapacitors were built to investigate the potential use of the Ag2MoO4@Ni foam electrode in energy storage. Ag2MoO4@Ni foam was employed as the positive electrode and activated carbon (AC) was used as the negative electrode.
Characterization
Field emission scanning electron microscopy (FESEM, MIRA3, TESCAN-XMU) and energy-dispersive spectroscopy (EDS, MIRA3, TESCAN-XMU) were applied to examine the surface morphology and elemental composition of nanostructures. The crystallographic constitution and phase integrity of the specimens were investigated using an X-ray diffractometer (XRD, PANalytical X’Pert Pro) with Cu-Kα radiation. X-ray photoelectron spectroscopy (XPS, UHV analysis system, SPECS, Al Kα = 1486.6 eV) was utilized to examine the chemical state of the elements that make up the electrospun Ag2MoO4 nanostructures. Fourier transform infrared spectroscopy (FTIR, Bruker Alpha) was applied to detect the functional groups in the samples. The Raman spectra were recorded using Teksan Takram P50C0R10 upon excitation by a laser beam with a wavelength of 532 nm for excitation. The BELSORP-mini II equipment was used to conduct a BET study on the surface area of the synthesized materials.
Photocatalytic study
The photocatalytic activity of the samples was investigated by assessing the degradation of MB in water utilizing a 200 W tungsten lamp as the visible light source (the emission range is 400–700 nm). This allowed us to assess the ability of the samples to remove organic pollutants from water. We combined 100 mL of a 10 mg L−1 (ppm) MB dye solution with the catalyst and agitated in the dark for 40 minutes at room temperature to achieve the adsorption–desorption equilibrium of dye on the catalyst surface. The samples were then exposed to visible light for two hours, with samples collected every 30 minutes at equal intervals. The collected samples were analysed using a CARY 100 Scan UV-Vis spectrophotometer to determine the absorption spectra and evaluate the degradation of the MB dye by observing changes in the primary absorption peak intensity. For subsequent runs, the samples were separated from the solution and washed with water and ethanol. We then coated 20 mg of Ag2MoO4 on two pieces of nickel foams (1 cm × 1 cm) and dried them completely in an oven. The coated Ni foams and dye solution were agitated in the dark for 40 minutes at room temperature to achieve the adsorption–desorption equilibrium of dye on the foams.
Electrochemical measurements
The electrochemical properties of nanostructures were examined utilizing cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) on a three-electrode system (Zahner Zennium device) electrochemical workstation. The electrochemical characteristics of the designed samples were investigated using KOH solution (3 M) as an electrolyte. Platinum wire (Pt) served as the counter electrode, whereas Ag/AgCl was employed as the reference electrode. The samples were utilized as the working electrode.
Results and discussion
Morphological and elemental analyses
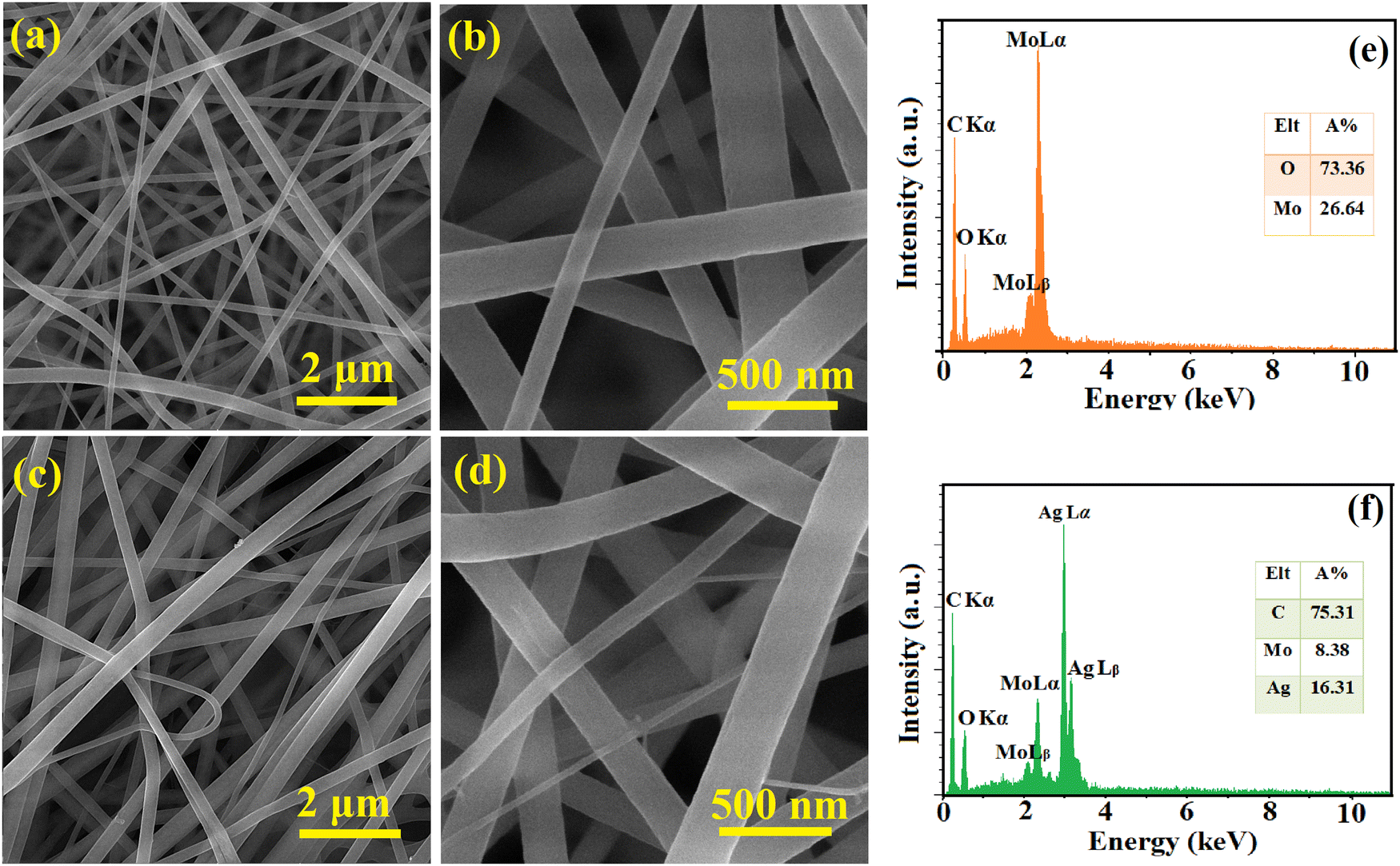
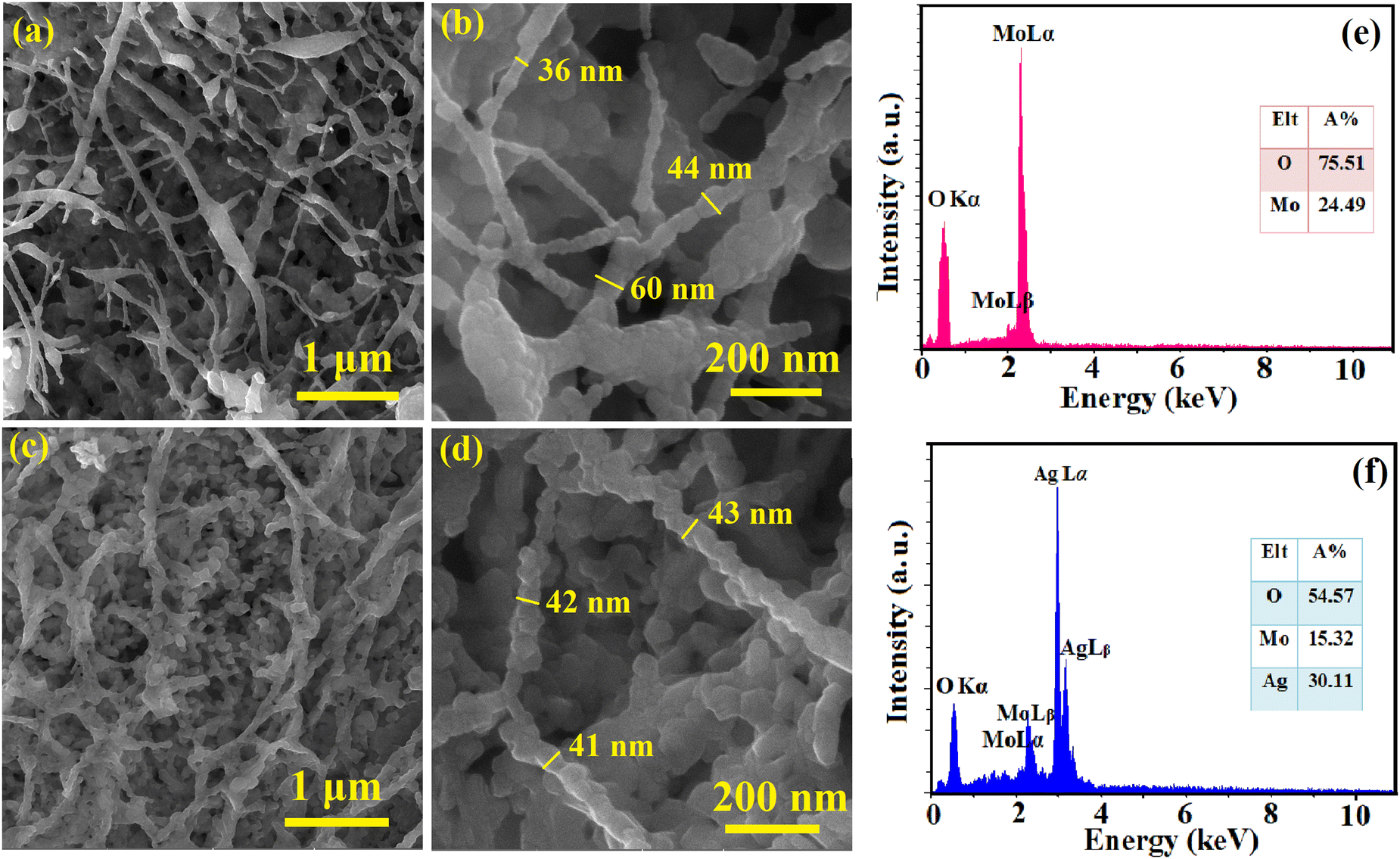
The surface morphology and elemental composition of the as-spun NFs and calcined samples were evaluated using FESEM and EDS, respectively, and the results are presented in Fig. 1 and 2. The as-spun NFs have a continuous, defect-free and randomly oriented structure with a nearly uniform diameter. Upon calcination, the surface morphology was altered, and the diameter of the NFs decreased due to the shrinkage and wrinkling of the composites. The samples were formed into packed particles due to the complete disintegration of polymers at high temperatures during calcination. The one-dimensional MoO3 and Ag2MoO4 nanostructures had a bead-like morphology. A dense matrix that is interconnected was formed by growing Ag2MoO4 nanoparticles on the nanofibers. The EDS results showed a carbon-related peak in the as-spun NFs due to the presence of PVP in their structure which disappeared after calcination. The MoO3 sample encompassed oxygen and molybdenum in correct proportions (inset of Fig. 2e). Additionally, the atomic proportion of the component elements in Ag2MoO4 was near stoichiometry (4:1:2 for O, Mo, and Ag, respectively), as illustrated in the inset of Fig. 2f.
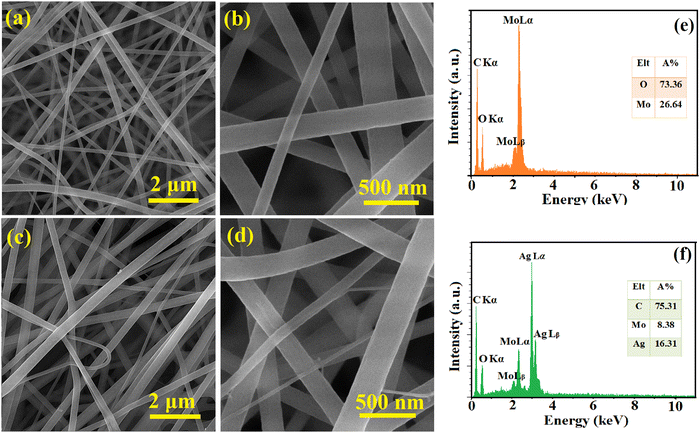 |
| | Fig. 1 FESEM pictures of (a) and (b) MoO3/PVP and (c) and (d) Ag2MoO4/PVP as-spun NFs at various magnifications. The EDS results of (e) MoO3/PVP and (f) Ag2MoO4/PVP as-spun NFs. | |
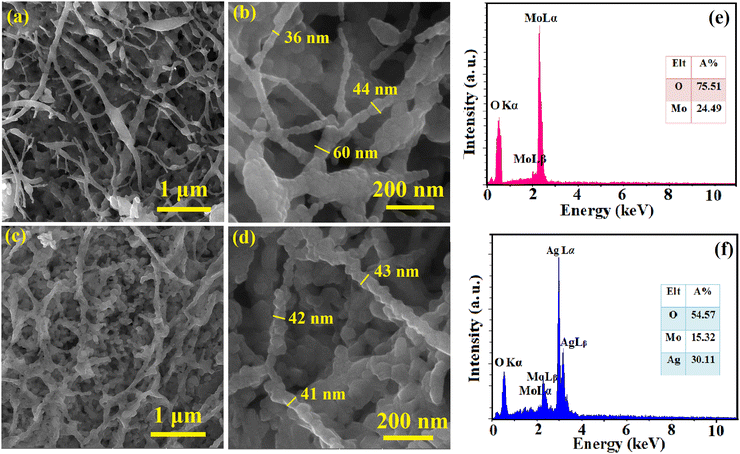 |
| | Fig. 2 FESEM pictures of (a) and (b) MoO3 and (c) and (d) Ag2MoO4 samples. The EDS results of (e) MoO3 and (f) Ag2MoO4 nanostructures. | |
XRD analysis
The crystal structure and phase purity of the produced metal oxides were analysed using X-ray diffraction (XRD), and the results are presented in Fig. 3. The XRD pattern of MoO3 comprises several peaks, occurring at 2θ of 23.3, 25.7, 27.4, and 38.9°, which correspond to the (110), (040), (021), and (060) planes, respectively. The orthorhombic crystal structure and the Pbnm space group of α-MoO3 are both well-referenced to the standard pattern (JCPDS no. 00-035-0609),33 which has an extremely anisotropic double-layer structure in the [010] direction constructed by corner-sharing MoO6 octahedra.33 The crystalline silver molybdate (β-Ag2MoO4) was demonstrated by the sharp and intense diffraction peaks corresponding to JCPDS no. 08-0473.32 The spinel-type cubic structure (β-Ag2MoO4) with the space group Fd3m and point group symmetry O7h could be precisely indexed to all XRD peaks of Ag2MoO4. The electrospun Ag2MoO4 sample possessed a high degree of crystallinity as indicated by the presence of pointed and concentrated diffraction peaks, with no signature of impurity phases in the XRD patterns of the samples.34 The Rietveld refinement was used to determine the average crystallite size (D) and lattice strain (ε) of the samples using HighScore Plus (Malvern Panalytical) software. The computed D values were 51.6 and 25.0 nm for MoO3 and Ag2MoO4 nanostructures, respectively, while the obtained lattice strains were 0.25 and 0.55 for MoO3 and Ag2MoO4 nanostructures, respectively.
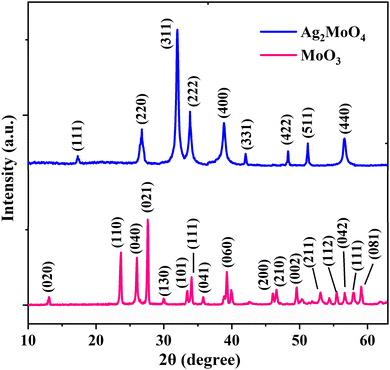 |
| | Fig. 3 XRD patterns of the MoO3 and Ag2MoO4 samples. | |
XPS studies
The electronic states and elemental composition of Ag2MoO4 NFs were studied utilizing XPS (Fig. 4). The Ag 3d, Mo 3d, and O 1s peaks were demonstrated in the survey spectrum of Ag2MoO4 NFs. Certain regions of the survey spectrum were further analysed to determine the precise electronic states and associated binding energies. The Mo 3d XPS results in Fig. 4b revealed Mo 3d5/2 and 3d3/2 spin–orbits at 232.3 and 235.7 eV, respectively.35,36 The Ag+ ionic spin–orbits 3d5/2 and 3d3/2 were located at 374.4 and 368.5 eV, respectively, and the lack of elemental silver was confirmed by the extended Ag 3d XPS spectra (Fig. 4c).37 These results clearly demonstrate the electronic states of two transition metal elements (Ag+ and Mo6+) in Ag2MoO4. The magnified O 1s spectrum (Fig. 4d) indicates the existence of the O2− state of lattice oxygen (Mo–O) at 530.5 and 532.3 and –OH (from adsorbed oxygen species) at 533.5 eV.38,39 Based on the XPS outcomes, the phase structure of Ag2MoO4 was composed of the +1, +6, and −2 valence states of Ag, Mo, and O, respectively.
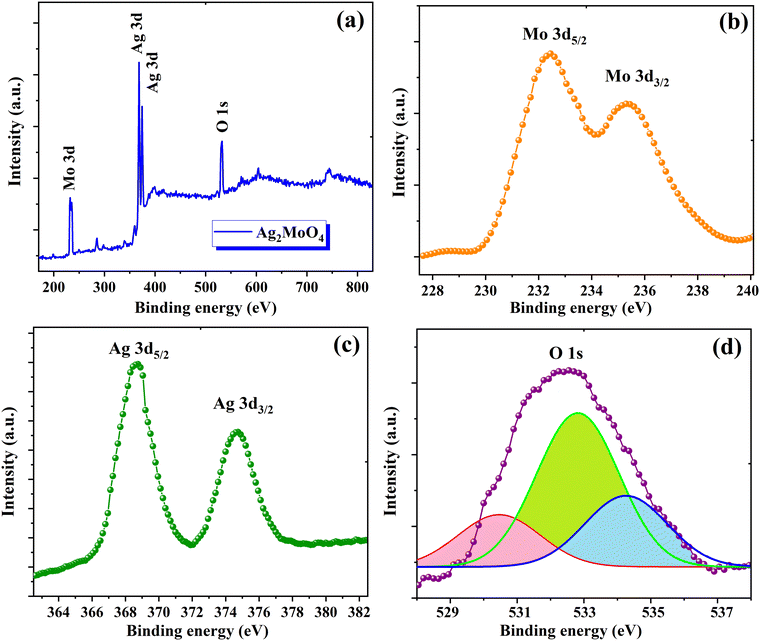 |
| | Fig. 4 XPS of Ag2MoO4: (a) survey spectra; high-resolution spectra of (b) Mo 3d, (c) Ag 3d and (d) O 1s. | |
Surface area and porosity measurement
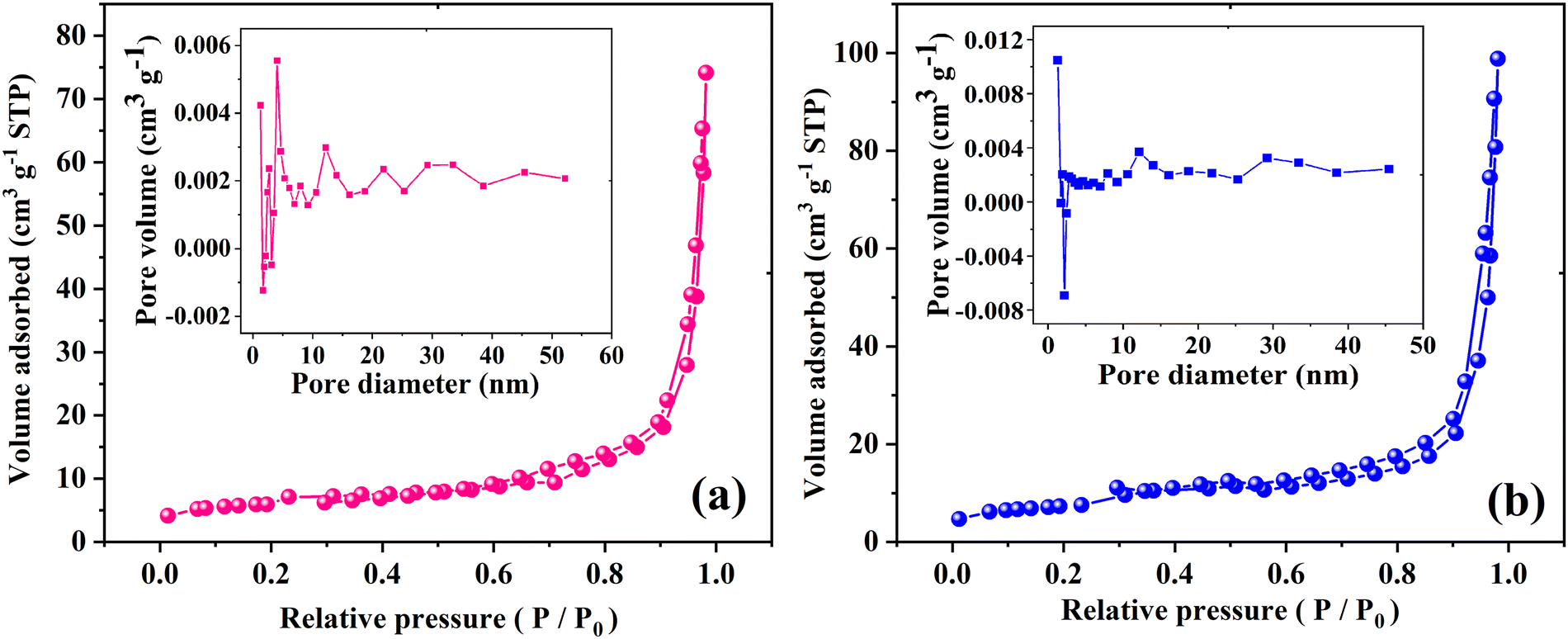
The surface area and pore size of the samples were studied using nitrogen adsorption/desorption isotherms as demonstrated in Fig. 5. The acquired isotherms for the MoO3 and Ag2MoO4 NFs are type IV, based on what is known as the Brunauer–Deming–Deming–Teller classification, which demonstrates the existence of mesopores that develop from the internal aggregation of nanoparticles. Type H3 is characterized by non-limiting adsorption at high P/P0, which indicates that the samples have slit-like pore geometries. According to the included curves, electrospun MoO3 and Ag2MoO4 have average pore diameters of 21.43 and 23.55 nm, respectively. The computed specific surface areas of MoO3 and Ag2MO4, which are 40.15 and 49.33 m2 g−1, respectively, are particularly noteworthy. The small average crystallite size of Ag2MO4 can be the reason for obtaining a large specific surface area. Previous research has reported the specific surface area of silver molybdate with different morphologies between 1.03 and 34.90 m2 g−1.30,40–44
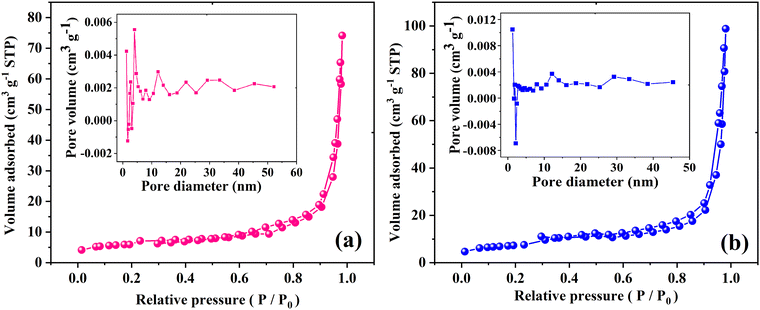 |
| | Fig. 5 N2 adsorption/desorption isotherms (the insets show the pore size distribution) of (a) MoO3 and (b) Ag2MoO4. | |
FTIR and Raman studies
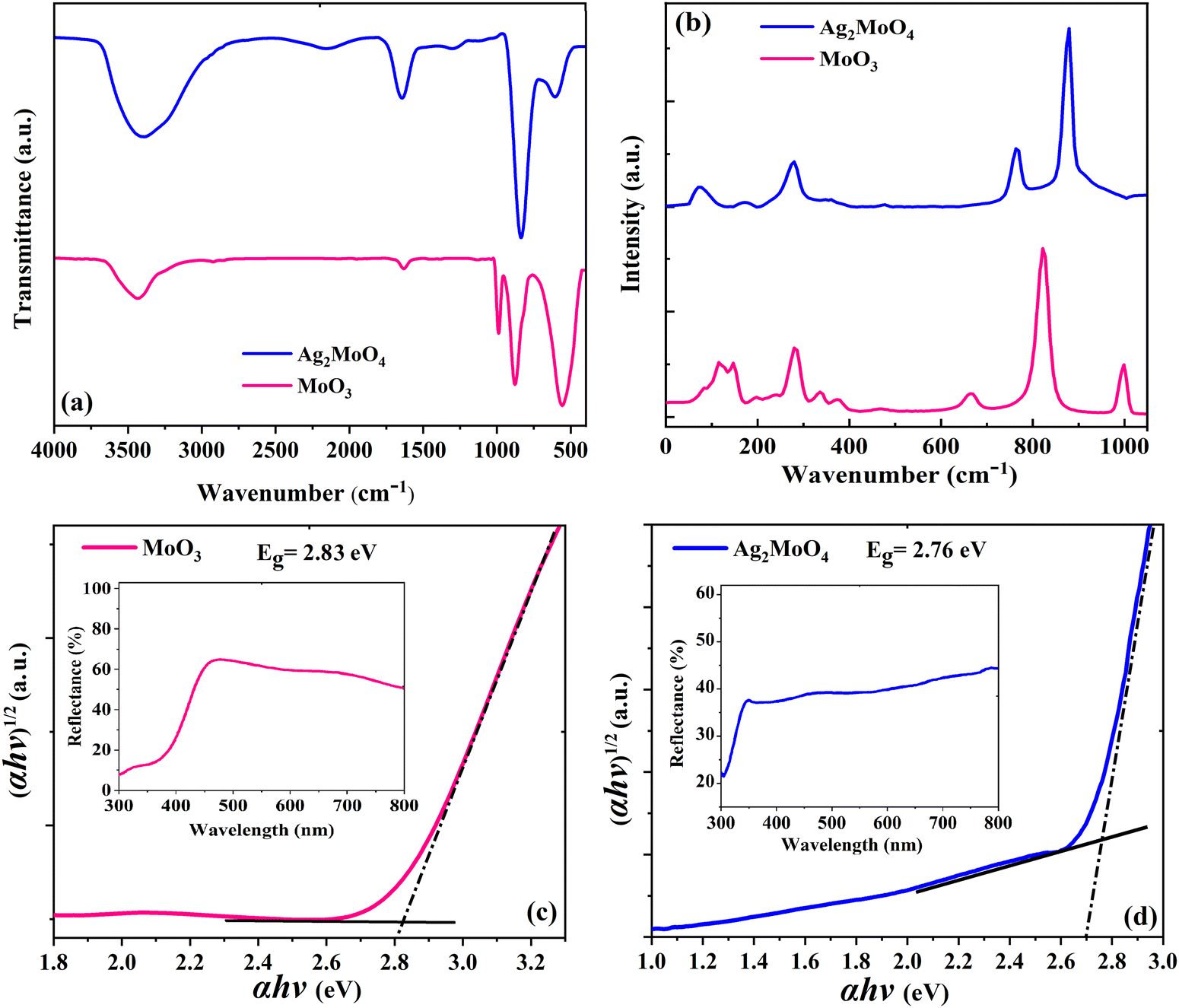
Elongation and O–H–O twisting vibrations of the water molecules are characterized by the peak around 1640 cm−1 and the broad band around 3600 cm−1, respectively, which are adsorbed onto the surface of the samples due to humidity.45 The pristine MoO3 sample in Fig. 6a displays three distinctive peaks around 560, 870, and 990 cm−1, which correspond to the Mo–O–Mo bending vibration mode, Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration mode, and asymmetric Mo–O bonds of molybdenum oxide, respectively.46 The Ag2MoO4 sample in Fig. 6a shows a peak around 600 cm−1, which is related to the Ag–O stretching vibration.45 The anti-symmetric Mo–O stretching of the tetrahedral MoO42− ion is responsible for the strong peak around 820 cm−1.37,45Fig. 6b shows the Raman spectra of electrospun MoO3 and Ag2MoO4 samples. The Raman spectra provide accurate data on molecular vibration and rotation. The sharp peaks indicate a highly ordered structure.47 For MoO3 in Fig. 6b, the peaks at 380, 340, and 289 cm−1 are related to O–Mo–O scissoring, O–Mo–O bending, and OMoO wagging, respectively. Furthermore, the peaks at 666 cm−1 are caused by O–Mo–O stretching, while the peaks at 822 cm−1 and 994 cm−1 are caused by asymmetric and symmetric stretching vibrations of the terminal MoO bonds, respectively.48,49 For the Ag2MoO4 in Fig. 6b, the peak at 278 cm−1 is attributed to the structural vibrations in the octahedral clusters [AgO6],50 while the torsional vibrations of molybdenum and oxygen atoms in the O–Mo–O bonds of tetrahedral clusters [MoO4] have caused modes at 90, 352, and 762 cm−1.51 The symmetric stretching vibrations of oxygen atoms in the [← O ← Mo → O →] bonds are responsible for the mode at 872 cm−1.50 These outcomes correspond to the data observed from the XRD structural characterization. Slight alterations in the positions of Raman modes can be attributed to various factors, including preparation procedures, average crystal size, interaction forces between ions, and the degree of structural order in the lattice.45
O stretching vibration mode, and asymmetric Mo–O bonds of molybdenum oxide, respectively.46 The Ag2MoO4 sample in Fig. 6a shows a peak around 600 cm−1, which is related to the Ag–O stretching vibration.45 The anti-symmetric Mo–O stretching of the tetrahedral MoO42− ion is responsible for the strong peak around 820 cm−1.37,45Fig. 6b shows the Raman spectra of electrospun MoO3 and Ag2MoO4 samples. The Raman spectra provide accurate data on molecular vibration and rotation. The sharp peaks indicate a highly ordered structure.47 For MoO3 in Fig. 6b, the peaks at 380, 340, and 289 cm−1 are related to O–Mo–O scissoring, O–Mo–O bending, and OMoO wagging, respectively. Furthermore, the peaks at 666 cm−1 are caused by O–Mo–O stretching, while the peaks at 822 cm−1 and 994 cm−1 are caused by asymmetric and symmetric stretching vibrations of the terminal MoO bonds, respectively.48,49 For the Ag2MoO4 in Fig. 6b, the peak at 278 cm−1 is attributed to the structural vibrations in the octahedral clusters [AgO6],50 while the torsional vibrations of molybdenum and oxygen atoms in the O–Mo–O bonds of tetrahedral clusters [MoO4] have caused modes at 90, 352, and 762 cm−1.51 The symmetric stretching vibrations of oxygen atoms in the [← O ← Mo → O →] bonds are responsible for the mode at 872 cm−1.50 These outcomes correspond to the data observed from the XRD structural characterization. Slight alterations in the positions of Raman modes can be attributed to various factors, including preparation procedures, average crystal size, interaction forces between ions, and the degree of structural order in the lattice.45
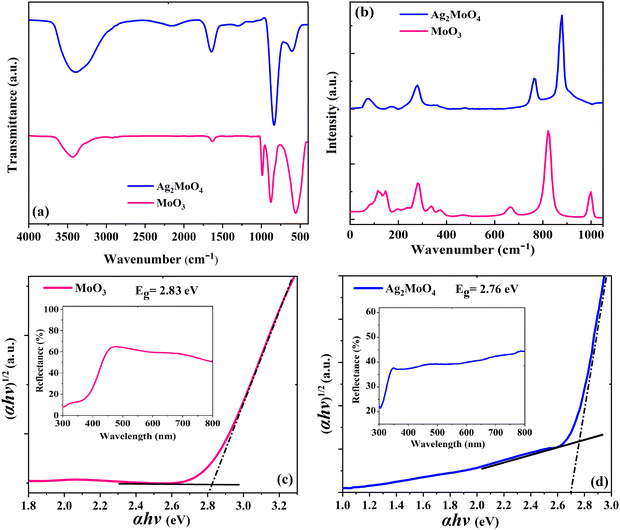 |
| | Fig. 6 (a) FTIR spectra of the samples. (b) Raman spectra of the samples. Band gap plot of (c) MoO3 and (d) Ag2MoO4. | |
DRS analysis
The optical characteristics of the samples as measured by UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS) are indicated in Fig. 6c and d. This analysis offers data on semiconductor band gaps and electronic transition states. To determine the optical band gap energy (Eg) of samples, a modified Tauc's equation was implemented:52where A represents the proportionality constant, hν represents the photon energy, Eg signifies the optical band gap, and n denotes a constant associated with various kinds of electronic transitions (n = 3 for an indirect forbidden transition, n = 2 for an indirect allowed transition, n = 2 for an indirect allowed transition, n = 1.5 for an immediate forbidden transition, and n = 0.5 for a direct allowed transition).33 The α depends on F(R), the Kubelka–Munk function,14 which is estimated using eqn (2):| | 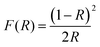 | (2) |
where R denotes the reflectance of the samples. The cross point between the baseline and the extrapolated lines in the plots of (αhυ)2versus hυ defines the band gap energy.53 The optically determined band gap energies for electrospun MoO3 and Ag2MoO4 were around 2.83 and 2.76 eV, respectively. The obtained bandgaps are in agreement with some previous reports (precipitation method prepared MoO3 (2.96 eV),54 green synthesis obtained MoO3 (2.57 eV),55in situ synthesized Ag2MoO4 (2.37 eV),39 thermal decomposition prepared Ag2MoO4 (2.81 eV)56). Phase defects, crystallite size, size distribution, and morphology can affect their band gap values.57 According to the XRD results, the lattice strain of Ag2MoO4 is smaller than that of MoO3 which can change the band gap values.52 On the other hand, the Burstein–Moss (B–M) shift can be another reason for the band gap increase. The Fermi level rise in the conduction band (CB) can raise the Eg due to the B–M shift.57,58 Also, indirect electronic transitions can affect the optical absorption spectra of MoO3 and Ag2MoO4. In this physical phenomenon, electrons in minimum energy in the CB states may return to maximum energy in the valence band (VB) states following the electronic absorption process, but only at distinct points of the Brillouin zone.59
Photocatalytic application
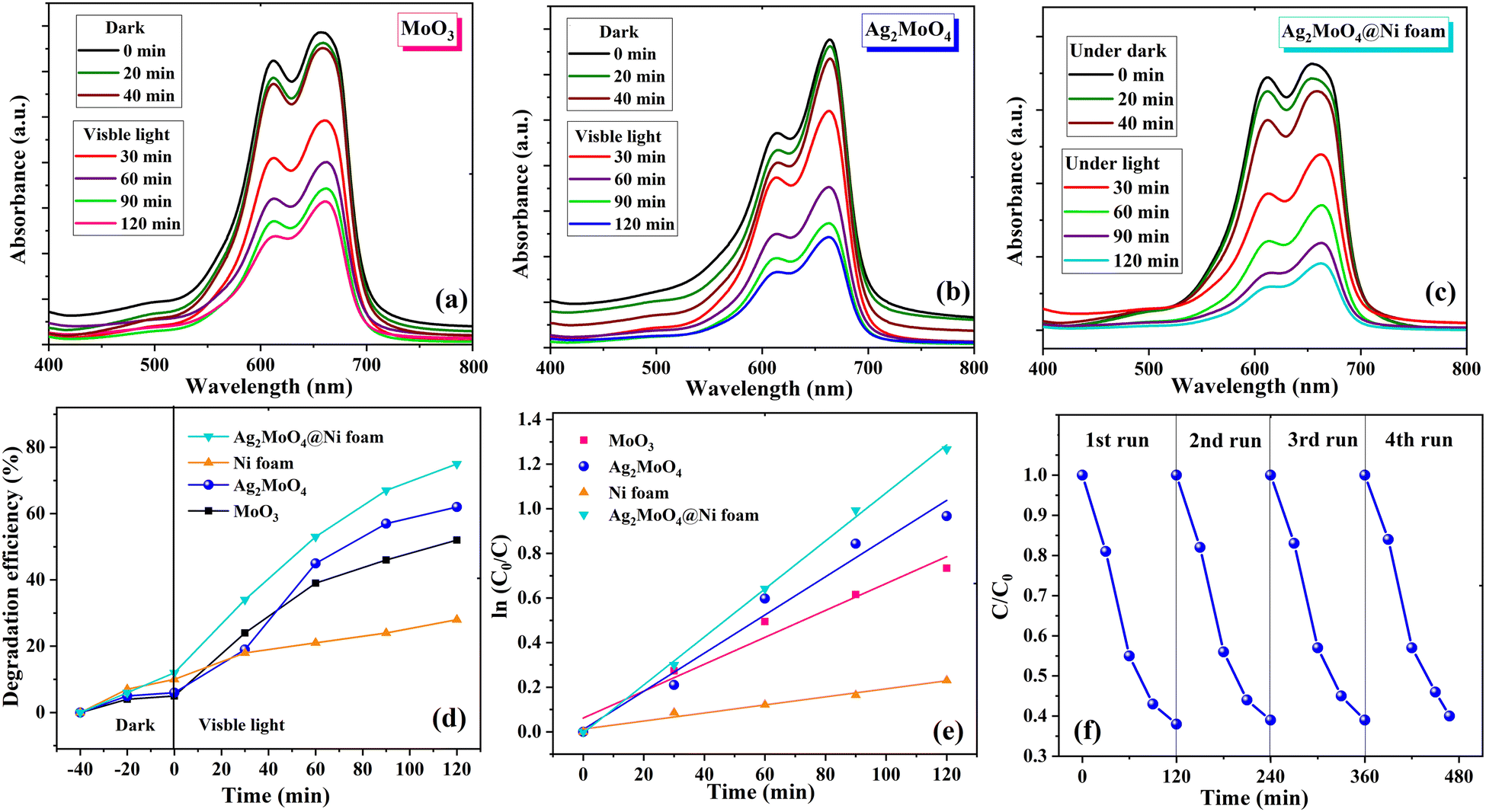
The photocatalytic activity of MoO3, Ag2MoO4, pure Ni foams and Ag2MoO4@Ni foams was investigated by the degradation of MB in solution under visible light irradiation. The experiment involved exposing 100 mL of 10 ppm MB solution with photocatalyst samples to visible light at different time intervals ranging from 0 to 120 min. The absorption spectra did not show any significant change in peak strength without light according to the UV-Vis spectra. As demonstrated in Fig. 7a–c, during spectrophotometry measurements, the solutions were exposed to visible light for various durations, and the photocatalytic degradation of MB dye was plotted against the modification in concentration. The results showed that, as the radiation exposure increased, the absorption peak of MB dye gradually dropped concerning its initial concentration. Notably, Ag2MoO4 NFs exhibited a higher potential for destroying the MB dye than MoO3. The interaction between dye molecules and ˙OH radicals or the formation of intermediate photoproducts is the primary cause of the deterioration of MB aqueous solution when illuminated with visible light.60,61 In order to investigate how well bead-like Ag2MoO4 NFs can keep their ability for photocatalytic activity, we performed a recyclability test. Fig. 7f shows that the photocatalytic activity does not change much for four consecutive cycles. Therefore, the fabricated bead-like Ag2MoO4 NFs are stable and can be utilized in industries. To evaluate the influence of the substrate on the photodegradation activity of Ag2MoO4@Ni foam electrodes under visible light irradiation, two pieces of pure Ni foams were placed into the dye solution for 120 min. Then, two pieces of Ag2MoO4@Ni foam electrodes and dye solution were agitated in the dark for 40 minutes at room temperature to achieve adsorption–desorption equilibrium on the foams. After that, we started the photocatalytic process of Ag2MoO4@Ni foam electrodes. Fig. 7d illustrates the MB degradation efficiency with irradiation time by MoO3, Ag2MoO4, pure Ni foams and Ag2MoO4@Ni foams, respectively. For MoO3 and Ag2MoO4, it was discovered that MB dye solution was destroyed by 57 and 62% during 120 minutes of irradiation, respectively. Table 1 shows the comparison of the photocatalytic degradation efficiency of the present work with some available literature.62–68 The fibrous morphology, high surface area, narrow bandgap energy and strong light absorption of silver molybdate synthesized in this work may contribute to its superior photocatalytic performance compared to previous reports. Additionally, the prepared Ag2MoO4 exhibits a larger specific surface area and pore diameter, providing more active sites for binding and decomposing MB dye molecules under visible light.60 The high surface-to-volume ratios also promote electron transport and speed up the decomposition of MB.61 Furthermore, surface defects can enhance the photocatalytic activity of nanoparticles by generation of reactive oxygen species (ROS: ˙OH and ˙O2−).9 The following reactions represent a possible reaction mechanism for the degradation of MB dye utilizing Ag2MoO4 nanostructures.69| | | Ag2MoO4 + hν → Ag2MoO4 (eCB− + hVB+) | (3) |
| | | e− + ˙O2− + H+ → OH− + ˙OH | (6) |
| | | ˙OH/˙O2− + MB → H2O + CO2 | (7) |
The better photodegradation properties of heterogeneous Ag2MoO4 grown on Ni foam compared to Ag2MoO4 nanofibers against MB can be attributed to several factors. Firstly, the heterogeneous Ag2MoO4 grown on Ni foam provides a synergistic effect due to the presence of both Ag2MoO4 and Ni foam. This combination enhances the separation of electron–hole pairs and provides a larger surface area for MB adsorption and degradation, leading to more efficient photocatalytic activity. Secondly, the Ag2MoO4 supported on Ni foam has enhanced light absorption due to the presence of Ni foam, which acts as a light reflector and increases the amount of light absorbed by the Ag2MoO4. This results in a more efficient photocatalytic activity.
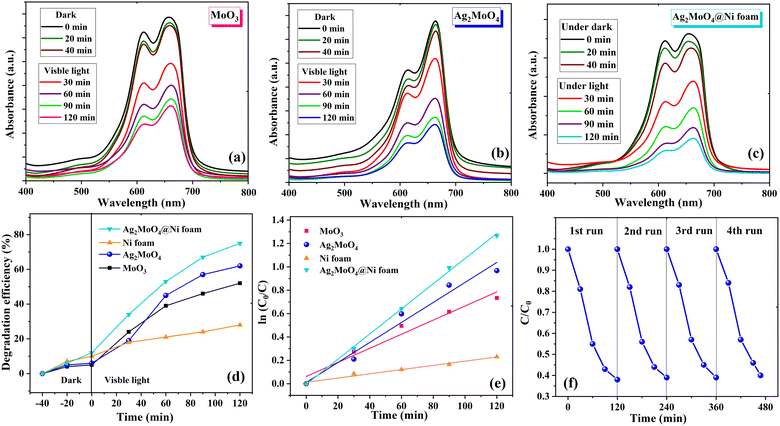 |
| | Fig. 7 UV-visible adsorption spectra of MB solution with (a) MoO3, (b) Ag2MoO4 and (c) Ag2MoO4@Ni foam in the dark and under visible light irradiation. (d) The photodegradation efficiency of samples. (e) Kinetic analysis of samples. (f) The recyclability test of the Ag2MoO4 sample for the degradation of MB solution. | |
Table 1 Comparison of degradation efficiency for different photocatalysts
| Catalyst |
Dye |
Cons. of dye (mg L−1) |
Amount of catalyst (mg mL−1) |
Degradation (%) |
Reaction time (min) |
Preparation method |
Ref. |
| Ag2MoO4 |
RhB |
10 |
0.6 |
15 |
90 |
Chemical deposition |
62
|
| Ag2MoO4 |
RhB |
9.5 |
4 |
22.2 |
6 |
Hydrothermal |
63
|
| Ag2MoO4 |
MB |
20 |
1 |
26 |
6 |
Anion-exchange |
8
|
| Ag2MoO4 |
4-CPh |
10 |
1 |
30 |
80 |
Anion-exchange |
64
|
| Ag2MoO4 |
MB |
5 |
— |
32 |
50 |
Electrospinning |
65
|
| Ag2MoO4 |
MB |
10 |
0.3 |
43.9 |
90 |
Precipitation |
66
|
| Ag2MoO4 |
MB |
10 |
— |
54.6 |
160 |
Sonochemical |
67
|
| Ag2MoO4 |
MB |
10 |
0.15 |
59.8 |
180 |
Co-precipitation |
68
|
| Ag2MoO4 |
MB |
10 |
0.15 |
62 |
250 |
Sonochemical |
40
|
| Ag2MoO4 |
MB |
10 |
0.2 |
62 |
120 |
Electrospinning |
This work |
| Ag2MoO4@Ni |
MB |
10 |
0.2 |
71 |
120 |
Electrospinning |
This work |
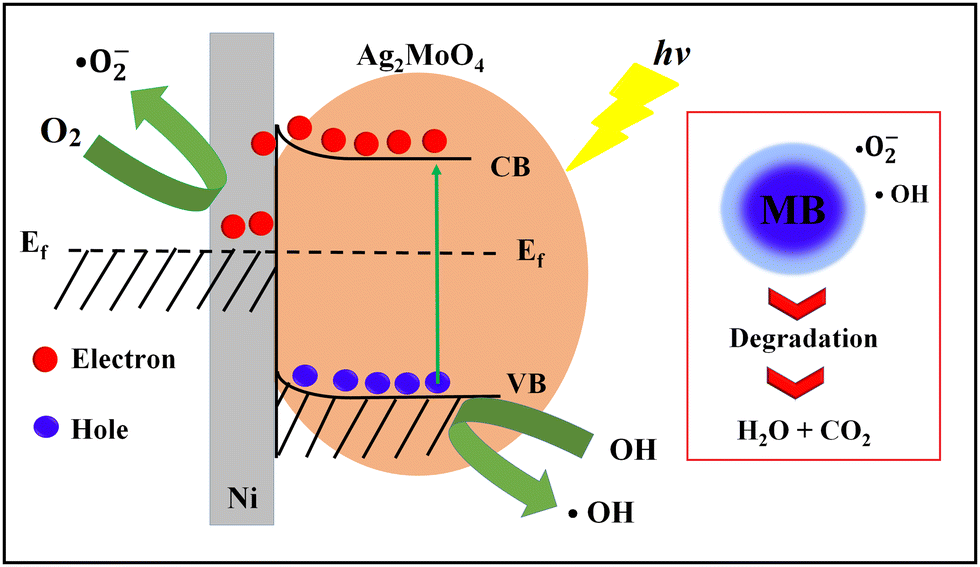
Finally, the heterogeneous Ag2MoO4 grown on Ni foam has improved electron transfer due to the presence of Ni foam, which acts as an electron mediator and facilitates the transfer of electrons from the valence band to the conduction band of Ag2MoO4, leading to the generation of ROS that can degrade MB.12,70–73 The formation of a Schottky barrier at the junction of Ni metal and the Ag2MoO4 semiconductor results in Fermi level alignment due to electron transfer from the material with the higher Fermi level to the one with the lower level.75 A diagram of this junction and a band bending diagram of Ag2MoO4/Ni are illustrated in Fig. 8. The Ni metal acts as an effective electron trap to receive photoelectrons from the Ag2MoO4 upon excitation, improving charge carrier separation and reducing recombination, as charges cannot flow in the opposite direction. The electrons are captured by dissolved O2 adsorbed on the surface of nickel foam to produce a superoxide anion (˙O2−), which is a powerful radical capable of decomposing organic pollutants. Meanwhile, the holes react with OH− and H2O to form ˙OH, which can oxidize and degrade almost all organic molecules.74,75 The linear relationship between lnC0/C and t in Fig. 7e demonstrates that the photodegradation mechanism is related to effectively pseudo-first-order kinetic equation:
| | 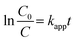 | (8) |
where
kapp is the degradation rate constant, and
C0 and
C are the initial and final concentrations of the dye at time
t.
54 The photodegradation rate constant (
kapp) is calculated to be 6.03 × 10
−3, 8.56 × 10
−3 and 10.84 × 10
−3 min
−1 for MoO
3, Ag
2MoO
4 and Ag
2MoO
4@Ni foam electrodes, respectively.
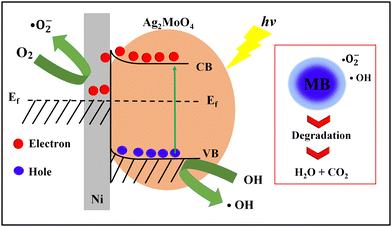 |
| | Fig. 8 Schematic photodegradation and band bending diagram of heterogeneous Ag2MoO4@Ni. | |
Electrochemical studies
In Fig. 9, the impact of Ag incorporation on the electrochemical performance of MoO3 was probed through the analysis of cyclic voltammetry (CV) data. The CV analysis highlights the distinct electrochemical behaviour between MoO3 and Ag2MoO4, with MoO3 exhibiting redox peaks due to reversible redox processes of Mo(VI) ↔ Mo(V),76 and Ag2MoO4 NFs displaying redox reactions of Ag ↔ Ag(II), with molybdenum atoms not participating in the reaction.28 Both electrodes exhibit typical pseudocapacitive behaviour and the synergistic effect of Ag and Mo enhances the electrical conductivity and electrochemical performance of the electrode material, as evidenced by the larger CV area and higher specific capacitance of Ag2MoO4 compared to MoO3. As the scan rate increased, there was a modest sloping of the redox peaks towards higher and lower voltages. In pseudocapacitive electrodes, these phenomena are generally attributable to the increased internal diffusion resistance and electrode polarization.25,77 The power-law relationship can be employed to differentiate between the diffusion and surface-controlled charge storage processes:78where i, ν, and a(b) stand for the current density, scan rate, and variable parameters. The slope of log(i) against log(ν) typically demonstrates the b-value. If the value of b is almost equal to 1, the charge storage process is surface-controlled, whereas a value of almost 0.5 for b implies a diffusion-controlled process.25 The b-values for MoO3 and Ag2MoO4 were calculated to be around 0.56 and 0.64 (Fig. S1, ESI†), specifying the mechanism of charge storage is mostly diffusion-controlled. The fraction of the diffusion- or capacitive-controlled process of electrodes at different scan rates was calculated using Dunn's relation:78where ν indicates the scan rate, k1 and k2 are constants, and i(ν) is the current response at a given voltage. The k1ν and k2v1/2 correspond to the current contributions from the surface-controlled effects and the diffusion-controlled intercalation process, respectively. The slope and intercept of the linear relationship between i(ν)/ν1/2 and ν1/2 define the values of k1 and k2 (see Fig. S1, ESI†). Voltammetric responses for MoO3@Ni and Ag2MoO4@Ni electrodes at a scan rate of 80 mV s−1 are shown in Fig. S2 (ESI†). Fig. 9d demonstrates the surface-controlled (capacitive) contributions of the electrodes at different scan rates. The maximum capacitive contributions were 15 and 40% for MoO3@Ni foam and Ag2MoO4@Ni foam, respectively. To investigate the specific capacitance (Cs) and rate capabilities of the electrodes, GCD tests were conducted at various current densities (Fig. 10a and b). The sloping fluctuation in potential in discharge curves displayed their pseudocapacitive character, which may be attributed to the faradaic reactions at the electrolyte–electrode interface.63,65 Using eqn (11), where Cs (F g−1), I/m (A g−1), Δt (s), and ΔV (V) are the specific capacitance, current density, discharge time, and potential window, respectively,77 MoO3@Ni foam had Cs values of 1220, 1040, 988, 855, 800 and 733 F g−1 at current densities of 1, 3, 5, 7, 10 and 15 A g−1, while Ag2MoO4@Ni foam had Cs values of 1695, 1580, 1255, 1057, 977 and 933 F g−1 at the same current densities.| | 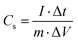 | (11) |
The calculated Cs values in this work were compared with previously reported values (see Table 2). The difference in the results may be related to the variety of morphology, specific surface area, preparation method, number of electroactive sites, phase purity and conductivity of the nanoparticles.25 An increase in charging/discharging current can decrease specific capacitance, possibly due to a boost in potential drop and a decrease in faradaic redox processes at high current densities.79 The superior electrochemical performance of Ag2MoO4@Ni foam can be attributed to the higher specific surface area (existence of more active sites) compared to MoO3 nanofibers, providing easier ion adsorption/desorption and improved capacitive performance. Additionally, Ag2MoO4 has higher electrical conductivity than MoO3 due to the presence of silver ions, which form a conductive network that facilitates electron transfer. Finally, the incorporation of Ag ions into the MoO3 crystal lattice improves the charge transfer kinetics of the material, leading to faster ion diffusion and better charge storage capacity.80–87 Coulombic efficiency 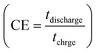 refers to the ratio of the amount of charge stored during discharge to the amount of charge supplied during charging.77 In other words, it measures how efficiently a supercapacitor can store and release electrical energy. MoO3@Ni foam had CE values of 79, 81, 83, 84, 85 and 88% at current densities of 1, 3, 5, 7, 10 and 15 A g−1, while Ag2MoO4@Ni foam had CE values of 66, 75, 89, 93, 97 and 98% at the same current densities. A substantial fraction of parasitic side-chain interactions between electrolyte contaminants and electrodes at low current densities such as the oxygen evolution reaction (OER) can reduce Coulombic efficiency. However, at higher current densities, the impact of the OER decreases, leading to an increase in coulombic efficiency. Ag2MoO4 has a higher coulombic efficiency than MoO3 due to its ability to store and release electrical energy more effectively. This suggests that Ag2MoO4 has a greater capacity for charge storage and experiences less energy loss during charge and discharge cycles, making it a more efficient material for use in supercapacitors. The unique properties of Ag2MoO4, such as its porous network, which provides better electrochemical accessibility of OH− ions to the surface due to high surface area, good conductivity, and stability under cycling conditions, could be responsible for this advantage.88–90 At a current density of 15 A g−1, the cycling performance of electrodes was additionally studied (Fig. 10c). MoO3@Ni foam and Ag2MoO4@Ni foam maintained 89 and 93% of Cs values, respectively, after 5000 cycles, indicating outstanding electrode stability. The cycling stability of Ag2MoO4 is higher than that of MoO3 due to the presence of silver ions, which not only enhance the electrical conductivity but also stabilize the crystal structure during repeated charge–discharge cycles.25
refers to the ratio of the amount of charge stored during discharge to the amount of charge supplied during charging.77 In other words, it measures how efficiently a supercapacitor can store and release electrical energy. MoO3@Ni foam had CE values of 79, 81, 83, 84, 85 and 88% at current densities of 1, 3, 5, 7, 10 and 15 A g−1, while Ag2MoO4@Ni foam had CE values of 66, 75, 89, 93, 97 and 98% at the same current densities. A substantial fraction of parasitic side-chain interactions between electrolyte contaminants and electrodes at low current densities such as the oxygen evolution reaction (OER) can reduce Coulombic efficiency. However, at higher current densities, the impact of the OER decreases, leading to an increase in coulombic efficiency. Ag2MoO4 has a higher coulombic efficiency than MoO3 due to its ability to store and release electrical energy more effectively. This suggests that Ag2MoO4 has a greater capacity for charge storage and experiences less energy loss during charge and discharge cycles, making it a more efficient material for use in supercapacitors. The unique properties of Ag2MoO4, such as its porous network, which provides better electrochemical accessibility of OH− ions to the surface due to high surface area, good conductivity, and stability under cycling conditions, could be responsible for this advantage.88–90 At a current density of 15 A g−1, the cycling performance of electrodes was additionally studied (Fig. 10c). MoO3@Ni foam and Ag2MoO4@Ni foam maintained 89 and 93% of Cs values, respectively, after 5000 cycles, indicating outstanding electrode stability. The cycling stability of Ag2MoO4 is higher than that of MoO3 due to the presence of silver ions, which not only enhance the electrical conductivity but also stabilize the crystal structure during repeated charge–discharge cycles.25
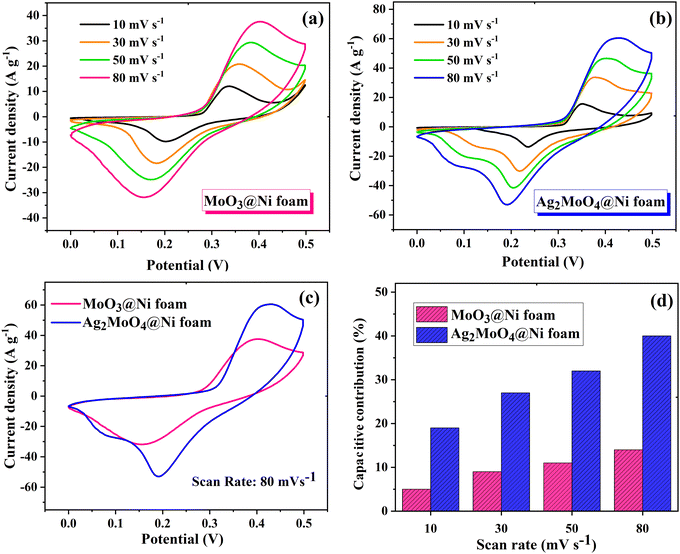 |
| | Fig. 9 The CV curves of (a) MoO3@Ni foam and (b) Ag2MoO4@Ni foam electrodes at various scan rates. (c) Comparison of the CV curves of electrodes at a scan rate of 80 mV s−1. (d) Capacitive-controlled contributions of the electrodes at different scan rates. | |
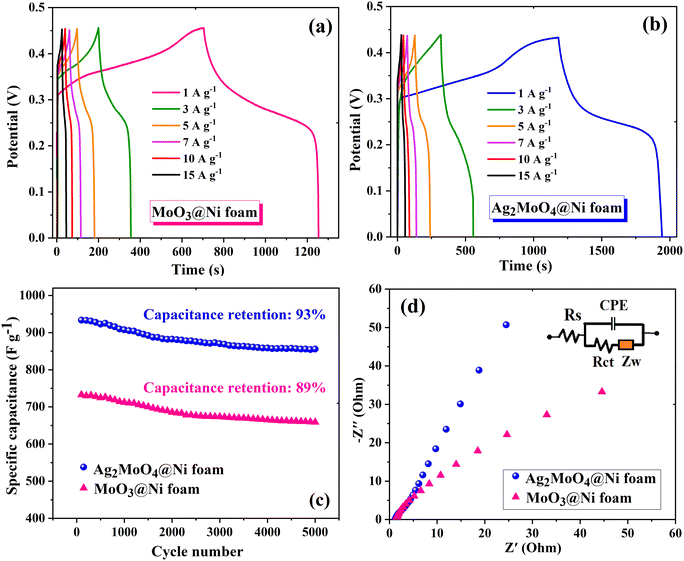 |
| | Fig. 10 GCD curves of (a) MoO3@Ni foam and (b) Ag2MoO4@Ni foam electrodes at various current densities. (c) Cycling stability of electrodes at a current density of 15 A g−1 after 5000 cycles. (d) The Nyquist plot of the electrodes (the inset shows the equivalent circuit used to fit the experimental data). | |
Table 2 Comparison of the specific capacitance and cycling stability of electrodes in this work with other reports
| Electrode material |
Electrolyte |
Substrate |
Specific capacitance (F g−1) |
Current density (A g−1) |
Preparation method |
% retention (cycles) |
Ref. |
| MoO3 |
2 M Li2SO4 |
Carbon cloth |
835 |
1 |
Electrodeposition |
98 (8000) |
80
|
| MoO3 |
1 M H2SO4 |
Titanium meshes |
527 |
1 |
Hydrothermal |
100 (10000) |
81
|
| MoO3 |
1 M H2SO4 |
Graphite sheet |
331.0 |
1 |
In situ preparation |
87.9 (10000) |
82
|
| MoO3 |
1 M H2SO4 |
Platinum foil |
267.0 |
1 |
Hydrothermal |
66.9 (3000) |
83
|
| MoO3 |
3 M KOH |
Ni foam |
1220 |
1 |
Electrospinning |
89 (5000) |
This work |
| ZnMoO4 |
2 M KOH |
Ni foam |
704 |
1 |
Hydrothermal |
93 (10000) |
84
|
| MnMoO4 |
2 M KOH |
Ni foam |
654 |
1 |
Solvothermal |
55 (1000) |
85
|
| NiMoO4 |
1 M KOH |
Ni foam |
1221 |
1 |
Electrodeposition |
82 (1000) |
86
|
| NiMoO4 |
1 M KOH |
Ni foam |
1694 |
1 |
Hydrothermal |
92 (9000) |
87
|
| Ag6Mo7O24 |
1 M Na2SO4 |
Glassy carbon |
971 |
1 |
Grinding method |
98 (5000) |
44
|
| Ag2MoO4 |
3 M KOH |
Ni foam |
1468 |
1 |
MOF-derived |
90 (5000) |
25
|
| Ag2MoO4 |
2 M KOH |
Ni foam |
2880 |
1 |
Conventional heating |
90 (5000) |
32
|
| Ag2MoO4 |
3 M KOH |
Ni foam |
1695 |
1 |
Electrospinning |
93 (5000) |
This work |
EIS measurement was utilized to investigate the electrochemical and ion transport behaviour of MoO3@Ni foam and Ag2MoO4@Ni foam electrodes. The Nyquist plots in Fig. 10d illustrate the impedance characteristics of the electrodes at frequencies ranging from 100 kHz to 10 mHz, with an inset depicting the appropriate circuit fitting. The slope of the curve indicates the diffusion resistance of the corresponding ionic charge, with Ag2MoO4@Ni foam exhibiting a lower diffusion resistance than MoO3@Ni foam. The Nyquist plot was simulated with ZView software and the corresponding equivalent circuit to explain the electrochemical behavior of electrodes which consisted of equivalent series resistance (sum of intrinsic resistance, electrode-to-current collector contact resistance and electrolyte ionic resistance, Rs), a charge transfer resistance (Rct), constant phase elements (CPE), and a Warburg element (ZW). The Rs values of MoO3@Ni foam and Ag2MoO4@Ni foam are estimated to be approximately 1.28 and 1.14 Ω, respectively. The increased conductivity of Ag2MoO4 causes a drop in the Rs value.91 A charge transfer discontinuity occurs at the electrolyte/electrode interface due to distinct conductivities of the electrode material and electrolyte, resulting in a charge transfer resistance (Rct) in the high-frequency zone with a small arc diameter. The Ag2MoO4@Ni foam exhibits a smaller Rct value (19.6 Ω) than MoO3@Ni foam (56.3 Ω), ensuring adequate electrolyte ion permeation and exceptional electrochemical performance.92
The route of ion diffusion is provided by the Warburg element (ZW), which can be deconvoluted to the diffusion resistance of the electroactive species in the host material (RW) and diffusion time constant (τ = L2/Dchem, where L is the maximum distance the electroactive species can diffuse into the host material and Dchem is the diffusivity).93,94 The Warburg diffusion impedance (ZW) was calculated by eqn (12):
| | 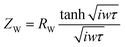 | (12) |
where
ω is the angular frequency. The
RW and
τ are 29.07 (25.20) and 0.43 (0.65) for MoO
3@Ni foam (Ag
2MoO
4@Ni foam), respectively.
93,94 The low
Rct and
RW values of Ag
2MoO
4@Ni foam indicated good capacitive performances with a short ion diffusion path.
95 The results of EIS can also be studied by the Bode plot (see Fig. S3, ESI
†). The Bode plots of the MoO
3@Ni foam and Ag
2MoO
4@Ni foam electrodes present a phase angle value of 57 and 70°, respectively, in the low frequency region. This indicates that the charge storage mechanism can be due to both the accumulation of ions at the electrode/electrolyte interface and a small contribution from faradaic processes. This confirms the pseudocapacitive behaviour of these active materials.
95 Fig. S4 (ESI
†) demonstrates the Nyquist plots for MoO
3@Ni foam and Ag
2MoO
4@Ni foam after 5000 cycles. The charge transfer resistance was slightly increased for MoO
3@Ni foam and Ag
2MoO
4@Ni foam, respectively, which confirms the stability of the electrodes. The increase of resistance is attributed to obstruction of the ion diffusion pathway during the charging–discharging process due to the partial deterioration of nanostructures.
96
To assess the real-world performance of the produced Ag2MoO4@Ni foam electrode in an aqueous asymmetric supercapacitor (ASC), a two-electrode Ag2MoO4//AC configuration was developed, where the negative and positive electrodes consisted of active carbon (AC@Ni foam) and electrospun Ag2MoO4@Ni foam, respectively. The mass loading ratio between the positive and negative electrodes was calculated using eqn (13), with a value of 0.317.
| | 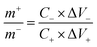 | (13) |
where Δ
V− (Δ
V+) and
C− (
C+) refer to the potential and specific capacitance of the negative (positive) electrode, respectively.
25Eqn (14) can be used to determine the specific capacitance of the electrode from CV curves:
65| | 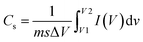 | (14) |
where
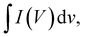 m
m (g), Δ
V (V), and
s (V s
−1) are the integrated area in the CV curve, active material mass, working potential window, and scan rate, respectively. To assess the operating voltage of the ASC device, a conventional three-electrode setup was employed to record the CV curve of the AC and Ag
2MoO
4@Ni foam electrodes at an 80 mV s
−1 scan rate (
Fig. 11a). Since the voltage window for Ag
2MoO
4@Ni foam equals 0 to +0.5 V and that for the AC@Ni foam equals −1 to 0 V, the ASC device possesses an operating voltage of 1.5 V. The AC CV curve exhibits a capacitive behaviour devoid of discernible redox peaks that would influence the electrochemical capabilities of the device.
Fig. 11b depicts the CV curves of the Ag
2MoO
4//AC electrode from 0 to 1.5 V using a variety of scan rates.
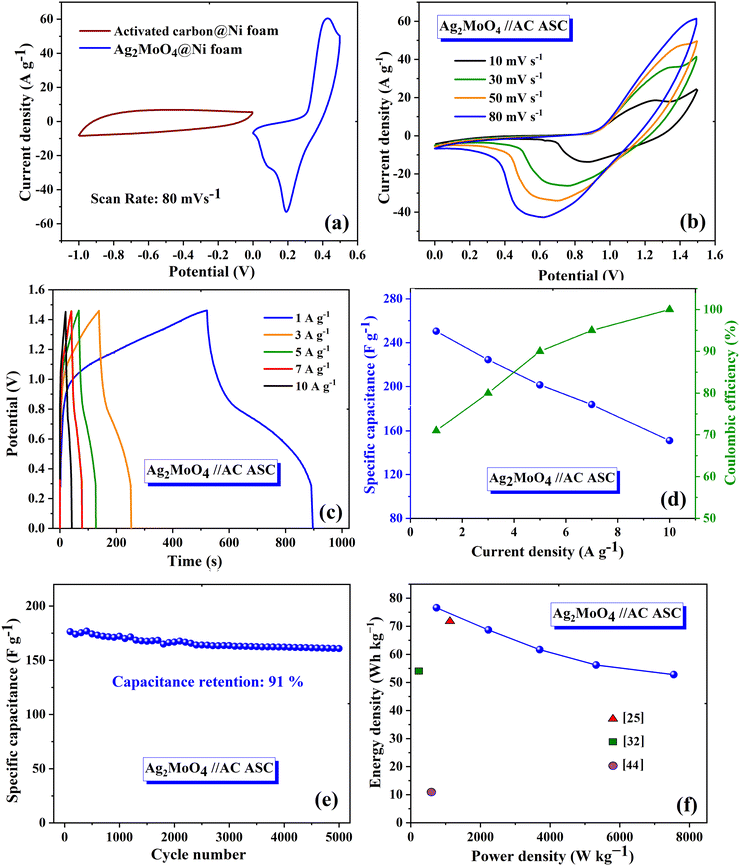 |
| | Fig. 11 (a) CV curves of AC@Ni foam and Ag2MoO4@Ni foam at 80 mV s−1 in the three-electrode configuration. (b) CV and (c) GCD curves of the Ag2MoO4//AC ASC device in the two-electrode system. (d) Specific capacitance and coulombic efficiency of the Ag2MoO4//AC ASC at different current densities. (e) Cycling stability performance of the Ag2MoO4//AC ASC at 15 A g−1. (f) Ragone plot. | |
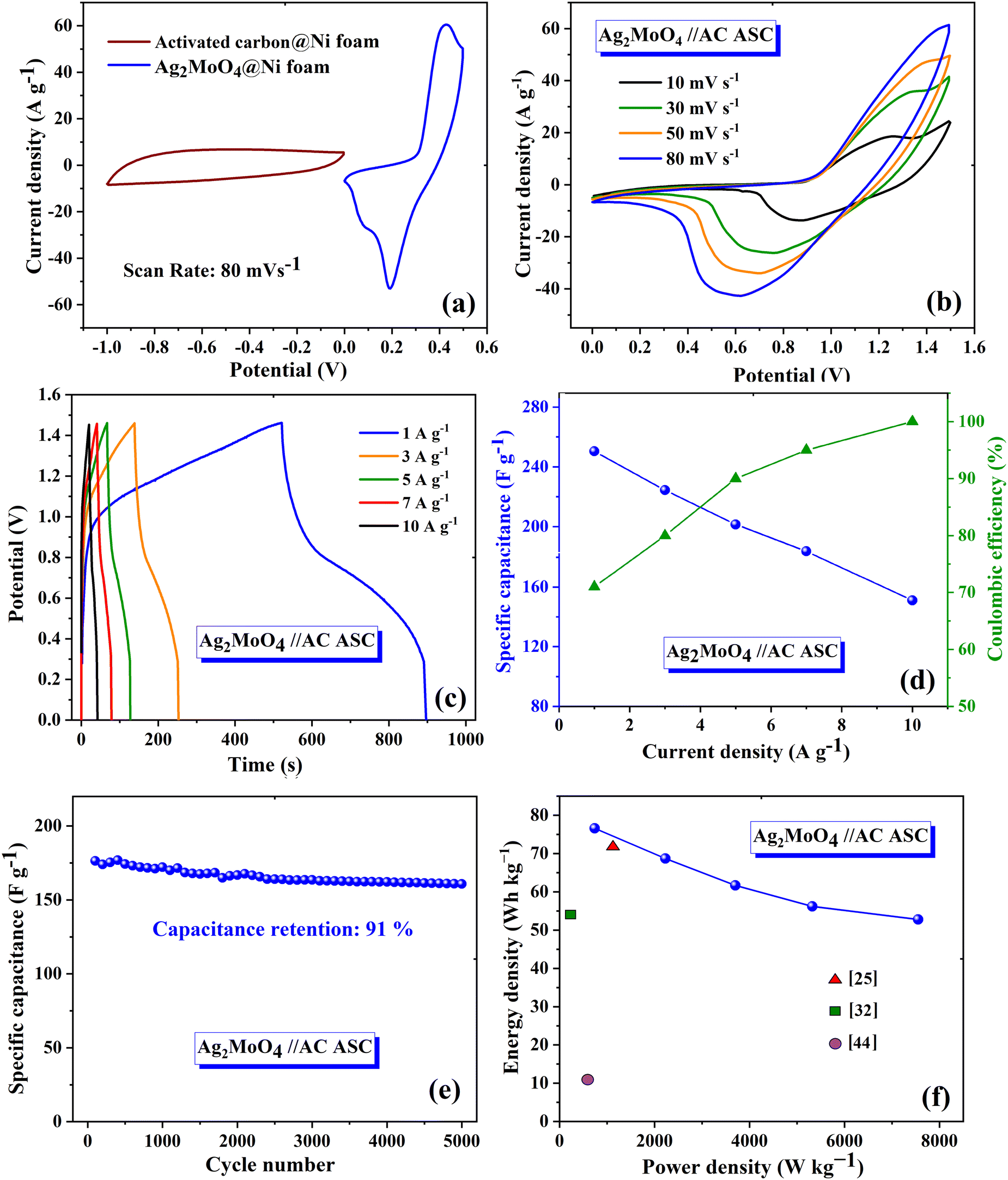
The ASC device exhibited excellent rate capability and charge/discharge reversibility, as evidenced by the broadening of the areas of the CV curves with increasing scan rate. The GCD curves of Ag2MoO4//AC are shown in Fig. 11c. The ASC exhibits a high specific capacitance of 250 F g−1 at 1 A g−1. The relationship between specific capacitance and current density is also displayed in Fig. 11d. Additionally, the CE results are shown in the same figure, with an increase in CE at higher current densities.72 According to eqn (15) and (16),65 the measured energy density (E) and power density (P) of the Ag2MoO4//AC device demonstrate exceptional findings of 76.6 W h kg−1 and 743.2 W kg−1 at 1 A g−1, proving the excellent functioning of the ASC device in energy storage applications.
| | 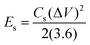 | (15) |
| | 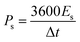 | (16) |
The capacitance retention of the Ag
2MoO
4//AC device during 5000 charge/discharge cycles is illustrated in
Fig. 11e, revealing an outstanding 91% retention rate and great CE. XRD analysis was conducted before and after the cycling stability test to examine the structural stability of the Ag
2MoO
4@Ni electrode (Fig. S5, ESI
†). The findings show that the peak intensity decreases slightly after the cycling stability measurements, which may be due to the slight lattice distortion caused by the expansion/contraction during electrochemical absorption/desorption.
97Fig. 11f depicts the Ragone plot, which validates the effectiveness of the electrode at different current densities and provides a comparison between the obtained results and previous reports.
Conclusions
In this work, we synthesized orthorhombic MoO3 and cubic Ag2MoO4 nanofibers by the electrospinning technique and studied their physical and electrochemical properties. The optical band gaps for electrospun MoO3 and Ag2MoO4 were found to be approximately 2.83 and 2.76 eV, respectively. The Ag2MoO4 NFs exhibited a high specific surface area of 49.33 m2 g−1 and delivered a photodegradation efficiency of 62% under visible light, which was higher than that of MoO3. We further improved the photodegradation efficiency to 71% using heterogeneous photocatalysis that included Ag2MoO4 NFs grown on nickel foam. The electrospun Ag2MoO4 nanocomposite electrode showed an excellent specific capacitance of 1445 F g−1 at 1 A g−1 and retained 93% of its initial capacitance after 5000 cycles at 15 A g−1. The Ag2MoO4@Ni foam electrode delivered a low charge transfer resistance of 19.6 Ω which was lower than that of MoO3. Moreover, the Ag2MoO4//AC asymmetric supercapacitor possesses an excellent energy density of 76.6 W h kg−1 at 743.2 W kg−1 and a noteworthy cycling durability of 91% after 5000 cycles at 15 A g−1. Our findings suggest that the electrospun Ag2MoO4@Ni foam is an important inexpensive electrode material for visible light-driven heterogeneous photocatalysis and supercapacitor applications, drawing on the synergic effects of Ag and Mo to exhibit much better performance.
Author contributions
Amirreza Safartoobi: conceptualization, investigation, writing – original draft. Jamal Mazloom: supervision, conceptualization, methodology, validation, writing – review & editing. Farhad Esmaeili Ghodsi: methodology, review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the University of Guilan Research Council for its financial support.
Notes and references
- T. Munawar, S. Sardar, F. Mukhtar, M. S. Nadeem, S. Manzoor, M. N. Ashiq, S. A. Khan, M. Koc and F. Iqbal, Phys. Chem. Chem. Phys., 2023, 25, 7010–7027 RSC.
- J. Wang, G. Yang, W. Lyu and W. Yan, J. Alloys Compd., 2016, 659, 138–145 CrossRef CAS.
- J. Wang, G. Wang, S. Wang, T. Haoc and J. Hao, Phys. Chem. Chem. Phys., 2023, 25, 26748–26766 RSC.
- M. A. A. Shanmuganathan, A. Raghavanab and S. Ghosh, Phys. Chem. Chem. Phys., 2023, 25, 7611–7628 RSC.
- S. A. Ansari and M. H. Cho, Sci. Rep., 2017, 7, 430055 Search PubMed.
- M. Safari, J. Mazloom, K. Boustani and A. Monemdjou, Sci. Rep., 2022, 12, 14919 CrossRef CAS PubMed.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 11, 4889–4899 CrossRef.
- Z. Wang, J. Zhang, J. Lv, K. Dai and C. Liang, Appl. Surf. Sci., 2017, 396, 791–798 CrossRef CAS.
- J. Mazloom and H. Zamani, J. Alloys Compd., 2018, 754, 238–246 CrossRef CAS.
- J. Wang, Y. Asakura and S. Yin, Nanoscale, 2019, 11, 20151–20160 RSC.
- Y. Asakura, Y. Inaguma, K. Ueda, Y. Masubuchi and S. Yin, Nanoscale, 2018, 10, 1837–1844 RSC.
- L. Hu, G. Zhang, M. Liu, Q. Wang, S. Dong and P. Wang, Sci. Total Environ., 2019, 647, 352–361 CrossRef CAS PubMed.
- A. K. Ganguli, G. B. Kunde, W. Raza, S. Kumar and P. Yadav, Molecules, 2022, 27, 7778 CrossRef CAS PubMed.
- R. Sidaraviciute, D. Buivydiene, E. Krugly, E. Valatka and D. Martuzevicius, J. Photochem. Photobiol., A, 2019, 368, 7–14 CrossRef CAS.
- A. Safartoobi, J. Mazloom and F. E. Ghodsi, Appl. Phys. A, 2022, 128, 13 CrossRef.
- L. Li, X. Sun, T. Xian, H. Gao, S. Wang, Z. Yi, X. Wue and H. Yang, Phys. Chem. Chem. Phys., 2022, 24, 8279–8295 RSC.
- J. Wang, Y. Asakura, T. Hasegawa and S. Yin, J. Chem. Eng., 2021, 423, 130220 CrossRef.
- M. M. Ghaziani, J. Mazloom and F. E. Ghodsi, J. Phys. Chem. Solids, 2021, 152, 109981 CrossRef.
- M. S. Choubari, S. Rahmani and J. Mazloom, Sci. Rep., 2023, 13, 7822 CrossRef PubMed.
- E. Samuel, A. Aldalbahi, M. E. Newehy, H. E. Hamshary and S. S. Yoon, Ceram. Int., 2022, 48, 18374–18383 CrossRef CAS.
- Y. Ma, J. Liu, Y. Lin and Y. Jia, Phys. Chem. Chem. Phys., 2023, 25, 8263–8828 RSC.
- J. Liu, K. Huang, H. L. Tang and M. Lei, Int. J. Hydrogen Energy, 2016, 41, 996–1001 CrossRef CAS.
- Y. Huang, F. Cui, Y. Zhao, J. Lian, J. Bao and H. Li, J. Alloys Compd., 2018, 753, 176–185 CrossRef CAS.
- W. Liu, Y. Cheng, S. Jin, K. Wang, J. Ma, B. Guan, Z. Ren, T. Tan and J. Wang, Phys. Chem. Chem. Phys., 2023, 25, 17583–17595 RSC.
- A. Safartoobi, J. Mazloom and F. E. Ghodsi, J. Energy Storage, 2023, 68, 107818 CrossRef.
- S. K. Ray and J. Hur, J. Ind. Eng. Chem., 2021, 101, 28–50 CrossRef CAS.
- L. Kamarasu, E. Sathiyamoorthi, S. S. Nannapaneni, S. Arunachalam, M. Arunpandian, J. Lee, P. P. Arumugam and N. K. Katari, Phys. B, 2023, 650, 414544 CrossRef CAS.
- Z. Zhang, Y. Liu, Z. Huang, L. Ren, X. Qi, X. Wei and J. Zhong, Phys. Chem. Chem. Phys., 2015, 17, 20795–20804 RSC.
- Q. Li, Y. Li, J. Zhao, S. Zhao, J. Zhou, C. Chen, K. Tao, R. Liu and L. Han, J. Power Sources, 2019, 430, 51–59 CrossRef CAS.
- C. A. Oliveira, D. P. Volanti, A. E. Nogueira, C. A. Zamperini, C. E. Vergani and E. Longo, Mater. Des., 2017, 115, 73–81 CrossRef CAS.
- D. G. D. Rocca, R. M. Peralta, R. A. Peralta and R. F. Peralta Muniz Moreira, React. Kinet., Mech. Catal., 2021, 132, 1–35 CrossRef.
- J. J. William, S. Balakrishnan, M. Murugesan, M. Gopalan, A. J. Brittenc and M. Mkandawire, Mater. Adv., 2022, 3, 8288 RSC.
- M. B. Rammal and S. Omanovic, Mater. Chem. Phys., 2020, 255, 123570 CrossRef CAS.
- A. Z. Darmian, H. Farsi, A. Farrokhi, R. Sarhaddic and Z. Li, Phys. Chem. Chem. Phys., 2021, 23, 9539–9552 RSC.
- P. Srinivasan and J. B. B. Rayappan, Mikrochim. Acta, 2019, 186, 797 CrossRef CAS.
- D. Wang, G. Du, D. Han, Q. Su, M. Zhang, S. Ding and B. Xu, J. Alloys Compd., 2021, 859, 157792 CrossRef CAS.
- J. V. Kumar, R. Karthik, S. M. Chen, V. Muthuraj and C. Karuppiah, Sci. Rep., 2016, 6, 34149 CrossRef CAS.
- Y. Song, W. Xie, Ch Yang, D. Wei, X. Su, L. Li, L. Wang and J. Wang, J. Mater. Res. Technol., 2020, 9, 5774–5783 CrossRef CAS.
- M. Abinaya, R. Rajakumaran, Sh. M. Chen, R. Karthik and V. Muthuraj, ACS Appl. Mater. Interfaces, 2019, 11, 38321–38335 CrossRef CAS.
- A. Syed, A. M. Elgorban and A. H. Bahkali, Colloids Surf., A, 2021, 629, 12737 Search PubMed.
- L. Li, D. Yin, L. Deng, S. Xiao, Y. Ouyan, K. K. Khaing, X. Guo, J. Wang and Z. Luo, New J. Chem., 2021, 45, 223–234 RSC.
- S. Kokilavani, A. A. Al-Kheraif, A. M. Thomas, A. Syed, A. M. Elgorban, L. L. Raju, A. Das and S. S. Khan, Phys. E, 2021, 133, 114767 CrossRef CAS.
- S. Kokilavani, A. Syed, B. H. Kumar, A. M. Elgorban, A. H. Bahkali, B. Ahmed, A. Das and S. S. Khan, Colloids Surf., A, 2021, 627, 127097 CrossRef CAS.
- X. Zhao, L. Gong, C. Wang, C. Wang, K. Yu and B. Zhou, Chem. – Eur. J., 2020, 26, 4613–4619 CrossRef CAS.
- E. A. C. Ferreira, N. F. Andrade Neto, A. A. G. Santiago, C. A. Paskocimas, M. R. D. Bomio and F. V. Motta, J. Mater. Sci.: Mater. Electron., 2020, 31, 4271–4278 CrossRef CAS.
- X. Zhang, X. Zeng, M. Yang and Y. Qi, ACS Appl. Mater. Interfaces, 2014, 6, 1125–1130 CrossRef CAS PubMed.
- V. Jadkar, A. Pawbake, R. Waykar, A. Jadhavar, A. Mayabadi, A. Date, D. Late, H. Pathan, S. Gosavi and S. Jadkar, J. Mater. Sci.: Mater. Electron., 2017, 28, 15790–15796 CrossRef.
- X. Guan, Y. Ren, S. Chen, J. Yan, G. Wang, H. Zhao, W. Zhao, Z. Zhang, Z. Deng, Y. Zhang, Y. Dai, L. Zou, R. Chen and C. Liu, J. Mater. Sci., 2020, 55, 5808–5822 CrossRef.
- S. Bai, C. Chen, R. Luo, A. Chen and D. Li, Sens. Actuators, B, 2015, 216, 113–120 CrossRef.
- G. S. Sousa, F. X. Nobre, E. A. A. Júnior, R. D. S. Bezerra, M. L. de Sá, J. M. E. de Matos and M. R. M. C. Santos, Environ. Nanotechnol. Monit. Manage., 2020, 14, 100379 Search PubMed.
- M. D. P. Silva, R. F. Gonçalves, I. C. Nogueira, V. M. Longo, L. Mondoni, M. G. Moron, Y. V. Santana and E. Longo, Spectrochim. Acta, Part A, 2016, 153, 428–435 CrossRef PubMed.
-
J. C. Tauc, Optical Properties of Solids, North-Holland, Amsterdam, 1972 Search PubMed.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9(23), 6814–6817 CrossRef PubMed.
- R. Rathnasamy and V. Alagan, Phys. E, 2018, 102, 146–152 CrossRef CAS.
- M. Abinaya, K. Saravanakumar, E. Jeyabharathi and V. Muthuraj, J. Inorg. Organomet. Polym. Mater., 2019, 22, 101–110 CrossRef.
- F. M. Sanakousar, C. C. Vidyasagar, D. B. Shikandar, V. M. Jiménez-Pérez, D. Mounesh, C. C. Viswanath and K. Prakash, J. Environ. Chem. Eng., 2023, 11, 109371 CrossRef.
- S. K. Sen, M. A. Jalil, M. Hossain, M. S. Manir, K. Hoque, M. A. Islam and M. N. Hossain, Mater. Today Commun., 2021, 27, 102404 CrossRef CAS.
- N. Manjula, A. R. Balu, K. Usharani, N. Raja and V. S. Nagarethinam, Optik, 2016, 127, 6400–6406 CrossRef CAS.
- F. S. Cunha, J. C. Sczancoski, I. C. Nogueira, V. G. de Oliveira, S. M. C. Lustosa, E. Longo and L. S. Cavalcante, CrystEngComm, 2015, 17, 8207–8211 RSC.
- A. L. A. Alotaibi, N. Altamimi, E. Howsawi, K. A. Elsayed, I. Massoudi and A. E. Ramadan, J. Inorg. Organomet. Polym. Mater., 2021, 31, 2017–2029 CrossRef.
- Y. Chen, C. Lu, L. Xu, Y. Ma, W. Hou and J. J. Zhu, CrystEngComm, 2010, 12, 3740–3747 RSC.
- J. Zhang and Z. Ma, J. Taiwan Inst. Chem. Eng., 2017, 71, 156–164 CrossRef CAS.
- Y. Y. Bai, Y. Lu and J. K. Liu, J. Hazard. Mater., 2016, 307, 26–35 CrossRef CAS PubMed.
- A. Abulizi, K. Kadeer, L. Zhou, Y. Tursun and T. Dilinuer, J. Taiwan Inst. Chem. Eng., 2018, 88, 243–251 CrossRef CAS.
- Y. Zhao, H. Yang, H. Hao, F. Zhu, G. Zhang, J. Bi, S. Yan and H. Hou, Langmuir, 2022, 38, 13437–13447 CrossRef CAS PubMed.
- Z. Jiao, J. Zhang, Z. Liu and Z. Ma, J. Photochem. Photobiol., A, 2019, 371, 67–75 CrossRef CAS.
- S. Balasurya, A. Das, A. A. Alyousef, A. Alqasim, N. Almutairi and S. S. Khan, J. Mol. Liq., 2021, 337, 116350 CrossRef CAS.
- S. Balasurya, S. Alfarraj, L. L. Raju, A. Chinnathambi, S. A. Alharbi, A. M. Thomas and S. S. Khan, Surf. Interfaces, 2021, 25, 101237 CrossRef CAS.
- S. Damdinova, W. Liu, S. Wang, B. Dugarov, Z. Su and Z. Cheng, Mater. Chem. Phys., 2019, 235, 121765 CrossRef CAS.
- D. Chatterjee and S. Dasgupta, Visible light induced photocatalytic degradation of organic pollutants, J. Photochem. Photobiol., C, 2005, 6, 186–205 CrossRef.
- J. Hao, X. Wang, F. Liu, S. Han, J. Lian and Q. Jiang, Sci. Rep., 2017, 7, 3021 CrossRef.
- X. Chen, L. Liu, Y. Feng, L. Wang, Z. Bian, H. Li and Z. L. Wang, Mater. Today, 2017, 20, 501–506 CrossRef.
- Y. Wang, Y. Liu, H. Wang, W. Liu, Y. Li, J. Zhang, H. Hou and J. Yang, ACS Appl. Energy Mater., 2019, 2, 2063–2071 CrossRef.
- Y. Xue, R. Su, G. Zhang, Q. Wang, P. Wang, W. Zhang and Z. Wang, RSC Adv., 2016, 6, 93370–93373 RSC.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Zh. X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- I. Shaheen and K. S. Ahmad, J. Sol-Gel Sci. Technol., 2021, 97, 178–190 CrossRef.
- M. Safari and J. Mazloom, J. Energy Storage, 2023, 58, 106390 CrossRef.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef.
- P. Siwatch, K. Sharma, N. Manyani, J. Kang and S. K. Tripathi, J. Alloys Compd., 2021, 872, 159409 CrossRef.
- N. Zhao, H. Fan, M. Zhang, J. Ma, Z. Du, B. Yan, H. Li and X. Jiang, Chem. Eng. J., 2020, 390, 124477 CrossRef.
- K. Zhou, W. Zhou, X. Liu, Y. Sang, S. Ji, W. Li, J. Lu, L. Li, W. Niu, H. Liu and S. Chen, Nano Energy, 2015, 12, 510–520 CrossRef CAS.
- H. Ji, X. Liu, Z. Liu, B. Yan, L. Chen, Y. Xie, C. Liu, W. Hou and G. Yang, Adv. Funct. Mater., 2015, 25, 1886–1894 CrossRef CAS.
- F. Jiang, W. Li, R. Zou, Q. Liu, K. Xu, L. An and J. Hu, Nano Energy, 2014, 7, 72–79 CrossRef CAS.
- Y. P. Gao, K. J. Huang, C. X. Zhang, S. S. Song and X. Wu, J. Alloys Compd., 2018, 731, 1151–1158 CrossRef CAS.
- B. Saravanakumar, S. P. Ramachandran, G. Ravi, V. Ganesh, A. Sakunthala and R. Yuvakkumar, Appl. Phys. A: Mater. Sci. Process., 2019, 125, 6 CrossRef.
- S. Peng, L. Li, H. B. Wu, S. Madhavi and X. W. Lou, Adv. Energy Mater., 2014, 5, 1401172 CrossRef.
- K. Xiao, L. Xia, G. Liu, S. Wang, L. X. Ding and H. Wang, J. Mater. Chem. A, 2015, 3, 6128 RSC.
- D. Gandl, X. Wu, F. Zhang, C. Wu and D. Q. Tan, ACS Omega, 2021, 6, 7615–7625 CrossRef.
- L. V. Quispe-Garrido, I. E. Monje, E. O. López, J. M. Gonçalves, C. S. Martins, G. Á. Planes, J. G. Ruiz-Montoya and A. M. Baena-Moncada, ACS Omega, 2022, 7(48), 43522–43530 CrossRef CAS PubMed.
- K. Wang, X. Wang, D. Zhang, H. Wang, Z. Wang, M. Zhao, R. Xi, H. Wu and M. Zheng, CrystEngComm, 2018, 20, 6940 RSC.
- X. Ji, D. Sun, W. Zou, Z. Wang and D. Sun, J. Alloys Compd., 2021, 876, 160112 CrossRef CAS.
- C. Bathula, I. Rabani, S. Ramesh, S. H. Lee, R. R. Palem, A. T. A. Ahmed, H. S. Kim, Y. S. Seo and H. S. Kim, J. Alloys Compd., 2021, 887, 161307 CrossRef CAS.
- C. A. Hall, A. Ignjatovic, Y. Jiang, P. A. Burr and A. Lennon, J. Electroanal. Chem., 2019, 850, 113379 CrossRef CAS.
- S. E. Chun, S. Pyun and G. J. Lee, Electrochim. Acta, 2006, 51, 6479–6486 CrossRef.
- R. H. ıquez, A. S. Mestra-Acosta, P. Grez, E. Muñoz, G. Sessarego, E. Navarrete-Astorga and E. A. Dalchielec, RSC Adv., 2023, 13, 10068 RSC.
- M. Zhang, H. Fan, N. Zhao, H. Peng, X. Ren, W. Wang, H. Li, G. Chen, Y. Zhu, X. Jiang and P. Wu, Chem. Eng. J., 2018, 347, 291–300 CrossRef.
- K. Lota, P. Swoboda, I. Acznik, A. Sierczyńska, R. Mańczak, Ł. Kolanowski and G. Lota, Curr. Appl. Phys., 2020, 20, 106–113 CrossRef.
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Farhad Esmaeili
Ghodsi
* and
Farhad Esmaeili
Ghodsi


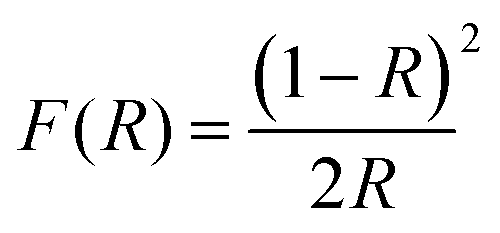
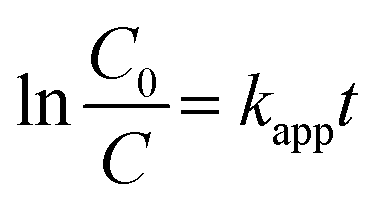
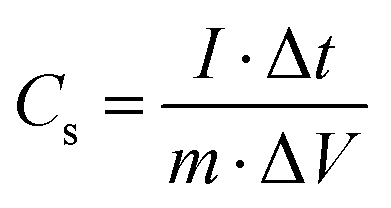
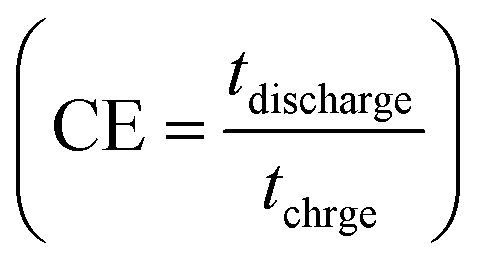 refers to the ratio of the amount of charge stored during discharge to the amount of charge supplied during charging.77 In other words, it measures how efficiently a supercapacitor can store and release electrical energy. MoO3@Ni foam had CE values of 79, 81, 83, 84, 85 and 88% at current densities of 1, 3, 5, 7, 10 and 15 A g−1, while Ag2MoO4@Ni foam had CE values of 66, 75, 89, 93, 97 and 98% at the same current densities. A substantial fraction of parasitic side-chain interactions between electrolyte contaminants and electrodes at low current densities such as the oxygen evolution reaction (OER) can reduce Coulombic efficiency. However, at higher current densities, the impact of the OER decreases, leading to an increase in coulombic efficiency. Ag2MoO4 has a higher coulombic efficiency than MoO3 due to its ability to store and release electrical energy more effectively. This suggests that Ag2MoO4 has a greater capacity for charge storage and experiences less energy loss during charge and discharge cycles, making it a more efficient material for use in supercapacitors. The unique properties of Ag2MoO4, such as its porous network, which provides better electrochemical accessibility of OH− ions to the surface due to high surface area, good conductivity, and stability under cycling conditions, could be responsible for this advantage.88–90 At a current density of 15 A g−1, the cycling performance of electrodes was additionally studied (Fig. 10c). MoO3@Ni foam and Ag2MoO4@Ni foam maintained 89 and 93% of Cs values, respectively, after 5000 cycles, indicating outstanding electrode stability. The cycling stability of Ag2MoO4 is higher than that of MoO3 due to the presence of silver ions, which not only enhance the electrical conductivity but also stabilize the crystal structure during repeated charge–discharge cycles.25
refers to the ratio of the amount of charge stored during discharge to the amount of charge supplied during charging.77 In other words, it measures how efficiently a supercapacitor can store and release electrical energy. MoO3@Ni foam had CE values of 79, 81, 83, 84, 85 and 88% at current densities of 1, 3, 5, 7, 10 and 15 A g−1, while Ag2MoO4@Ni foam had CE values of 66, 75, 89, 93, 97 and 98% at the same current densities. A substantial fraction of parasitic side-chain interactions between electrolyte contaminants and electrodes at low current densities such as the oxygen evolution reaction (OER) can reduce Coulombic efficiency. However, at higher current densities, the impact of the OER decreases, leading to an increase in coulombic efficiency. Ag2MoO4 has a higher coulombic efficiency than MoO3 due to its ability to store and release electrical energy more effectively. This suggests that Ag2MoO4 has a greater capacity for charge storage and experiences less energy loss during charge and discharge cycles, making it a more efficient material for use in supercapacitors. The unique properties of Ag2MoO4, such as its porous network, which provides better electrochemical accessibility of OH− ions to the surface due to high surface area, good conductivity, and stability under cycling conditions, could be responsible for this advantage.88–90 At a current density of 15 A g−1, the cycling performance of electrodes was additionally studied (Fig. 10c). MoO3@Ni foam and Ag2MoO4@Ni foam maintained 89 and 93% of Cs values, respectively, after 5000 cycles, indicating outstanding electrode stability. The cycling stability of Ag2MoO4 is higher than that of MoO3 due to the presence of silver ions, which not only enhance the electrical conductivity but also stabilize the crystal structure during repeated charge–discharge cycles.25


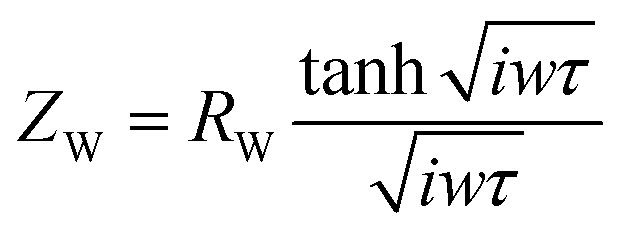
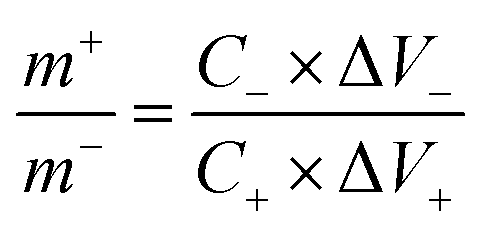

 m (g), ΔV (V), and s (V s−1) are the integrated area in the CV curve, active material mass, working potential window, and scan rate, respectively. To assess the operating voltage of the ASC device, a conventional three-electrode setup was employed to record the CV curve of the AC and Ag2MoO4@Ni foam electrodes at an 80 mV s−1 scan rate (Fig. 11a). Since the voltage window for Ag2MoO4@Ni foam equals 0 to +0.5 V and that for the AC@Ni foam equals −1 to 0 V, the ASC device possesses an operating voltage of 1.5 V. The AC CV curve exhibits a capacitive behaviour devoid of discernible redox peaks that would influence the electrochemical capabilities of the device. Fig. 11b depicts the CV curves of the Ag2MoO4//AC electrode from 0 to 1.5 V using a variety of scan rates.
m (g), ΔV (V), and s (V s−1) are the integrated area in the CV curve, active material mass, working potential window, and scan rate, respectively. To assess the operating voltage of the ASC device, a conventional three-electrode setup was employed to record the CV curve of the AC and Ag2MoO4@Ni foam electrodes at an 80 mV s−1 scan rate (Fig. 11a). Since the voltage window for Ag2MoO4@Ni foam equals 0 to +0.5 V and that for the AC@Ni foam equals −1 to 0 V, the ASC device possesses an operating voltage of 1.5 V. The AC CV curve exhibits a capacitive behaviour devoid of discernible redox peaks that would influence the electrochemical capabilities of the device. Fig. 11b depicts the CV curves of the Ag2MoO4//AC electrode from 0 to 1.5 V using a variety of scan rates.