
An α-benzithiazolyl 3-pyrrolyl BODIPY probe for ratiometric selective sensing of cyanide ions and bioimaging studies†
Kanhu Charan
Behera
a,
Roshnara
Mohanty
b and
Mangalampalli
Ravikanth
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: ravikanth@chem.iitb.ac.in; Web: https://www.ravikanthlab.com/
bCSIR – National Environmental Engineering Research Institute, Chennai Zonal Laboratory, Chennai 600113, India
First published on 26th January 2024
Abstract
A simple chromo-fluorogenic chemodosimeter probe, α-benzithiazolyl 3-pyrrolyl BODIPY, was synthesized by reacting α-formyl 3-pyrrolyl BODIPY with 2-aminothiophenol in DMF at reflux under basic conditions. The probe was structurally characterized by X-ray, HR-MS, and 1D & 2D NMR techniques. The X-ray structure revealed that the appended pyrrole was almost in the plane with a small deviation of 12.15° from the 12-atom mean plane of the BF2-dipyrrin core and the benzithiazolyl moiety was also deviated by 18.74° from the BF2-dipyrrin core. The α-benzithiazolyl 3-pyrrolyl BODIPY exhibits one intense absorption band at 608 nm and a less intense band at 412 nm corresponding to the 3-pyrrolyl BODIPY and benzithiazolyl moiety, respectively. The strongly fluorescent probe shows one intense emission band at 637 nm with a quantum yield of 0.48. The probe acted as an exclusive colorimetric and chemodosimetric sensor for CN− ions over other anions with high sensitivity (LOD = 13 nM) and quick response time (10 s) in an aqueous CH3CN medium. The CN− ion attacks the imine group of the benzithiazolyl moiety of 3via a nucleophilic addition reaction and converts the sp2 to sp3 carbon which disrupts the conjugation between the 3-pyrrolyl BODIPY and benzithiazolyl moieties, which is reflected in the clear colour change from red fluorescence to blue fluorescence as well as significant changes in the spectral and electrochemical properties. The detection of cyanide with the probe for biological applications was also performed with plant tissue. DFT/TD-DFT studies were in agreement with the experimental observations.
Introduction
The cyanide ion (CN−) is considered one of the very important anions with a wide range of applications in the context of chemical and industrial processes, including gold extraction, electroplating, and polymer processing.1–6 Cyanide gas is also used as a chemical warfare agent to exterminate pests and vermin. However, despite its wide use in the chemical industry, we all know that the CN− ion is highly toxic and one of the major pollutants, and harmful to humans and the environment. It disrupts cellular respiration by inhibiting cytochrome c oxidase, which prevents cells from utilizing oxygen.7–13 Thus, the recognition and detection of cyanide anions is of considerable interest at all times. The use of optical sensors for CN− ions, in which a change in colour and/or change in fluorescent wavelength and intensity are monitored, is a simple, inexpensive, and rapid method for the detection of CN− ions.14–16 Being highly nucleophilic and due to the strong binding nature of the CN− ion, the chemodosimetric approach in which the CN− ion attacks the fluorophore and causes an irreversible chemical reaction that is reflected in a change in colour or fluorescence of the fluorophore is one of the most accessible approaches for the detection of CN−.A pursual of the literature revealed that various methods13,17–19 are accessible for the detection of CN−, but many of them involve complex, costly procedures, intricate devices, and substantial sample quantities, and exhibit drawbacks such as high detection limits, low sensitivity, and lengthy measurement times.20 In contrast, fluorescent sensors provide a solution to the challenges mentioned by offering dual functionality through colorimetric and fluorometric signaling modes.21 While single emission measurements are influenced by concentration, device resolution, and environmental factors, ratiometric emission measurements remain unaffected by these factors as they are based on two distinct emission intensities. Fluorescence sensors employ various photophysical processes to detect CN− through different binding mechanisms, with intramolecular charge transfer (ICT) on/off being a commonly utilized mechanism. Many BODIPY-based sensors following this principle have been reported for CN− detection.22–24 However, these sensors face challenges such as the decomplexation of the –BF2 unit from the BODIPY moieties. To address the single wavelength measurement, evaluation of CN− in the aqueous medium, and its application to biological samples, we have developed a 3-pyrrolyl BODIPY-based fluorescent sensor capable of ratiometric CN− detection.
3-Pyrrolyl BODIPY 1, in which the BF2-dipyrrin (BODIPY) unit (Scheme 1) has an additional pyrrole ring at the 3-position of BODIPY, is a highly useful fluorescent dye and absorbs and emits at higher wavelength with good quantum yield compared to BODIPY.25 We25–28 and others29–31 have developed a much simpler and more rapid method to synthesize 3-pyrrolyl BODIPYs, which are now easily accessible. The appended pyrrole ring of 3-pyrrolyl BODIPY is highly susceptible to functionalization and several functional groups such as –Br, –CHO, –NO2, –CCTMS, etc. have been introduced at the α-position of 3-pyrrolyl BODIPY and the α-functionalized 3-pyrrolyl BODIPYs have been used further to prepare interesting 3-pyrrolyl BODIPY-based complex systems.26 In continuation of our work on 3-pyrrolyl BODIPY, we have functionalized α-formyl 3-pyrrolyl BODIPY 2 with 2-aminothiophenol in DMF at reflux under basic conditions to afford α-benzithiazolyl 3-pyrrolyl BODIPY 3 (Scheme 1). Compound 3 was designed using 3-pyrrolyl BODIPY as a signaling unit that is coupled to the benzithiazolyl moiety, causing electron delocalization across the entire molecule. Consequently, any receptor disruption induced by any anion may impair conjugation and produce significant colour changes and spectral shifts. Based on this concept, this modular chemical system, α-benzithiazolyl 3-pyrrolyl BODIPY 3 was synthesized, which acts as a specific chemodosimetric optical sensor for CN− ions by dual chromogenic and fluorogenic modes as described here. As far as we are aware, there aren’t many reports of CN− bioimaging using fluorescent probes in plant tissues due to cyanide's high toxicity, strong reactivity, high background fluorescence signal, intracellular localization, and thick plant tissue.32 Despite these difficulties, we have successfully demonstrated an α-benzithiazolyl 3-pyrrolyl BODIPY probe owing to its excellent deeper penetrating power into the cell, to visualize and investigate the spatial distribution and dynamics of cyanide ions in plant tissues.
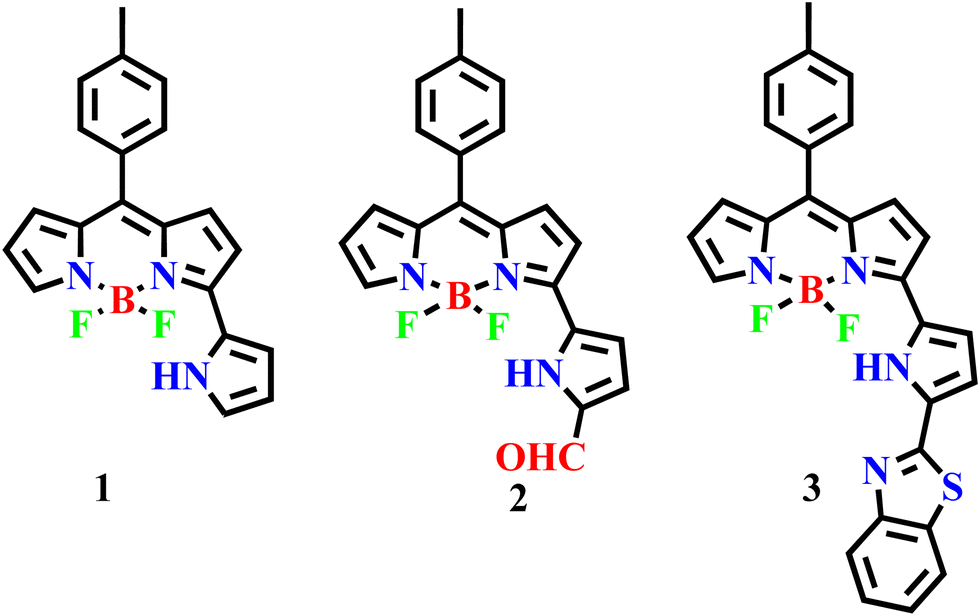 | ||
| Scheme 1 3-Pyrrolyl BODIPY and its further functionalization including α-benzithiazolyl 3-pyrrolyl BODIPY. | ||
Results and discussion
The required α-formyl 3-pyrrolyl BODIPY 2 was prepared in a series of steps by following our reported strategy.16 Compound 2 was treated with 2-aminothiophenol in the presence of Na2S2O5 in DMF at reflux for 3 h (Scheme 2). TLC analysis was used to track the reaction's progress. It was noticed that the disappearance of a more polar fluorescent red spot was associated with the starting compound 2 and the appearance of a less polar and less fluorescent blue spot corresponding to the desired product 3. After standard work-up, the crude compound was subjected to silica gel column chromatographic purification and afforded a pure blue solid of 3 in 30% yield. The HR-MS at 481.1484 confirmed the formation of compound 3.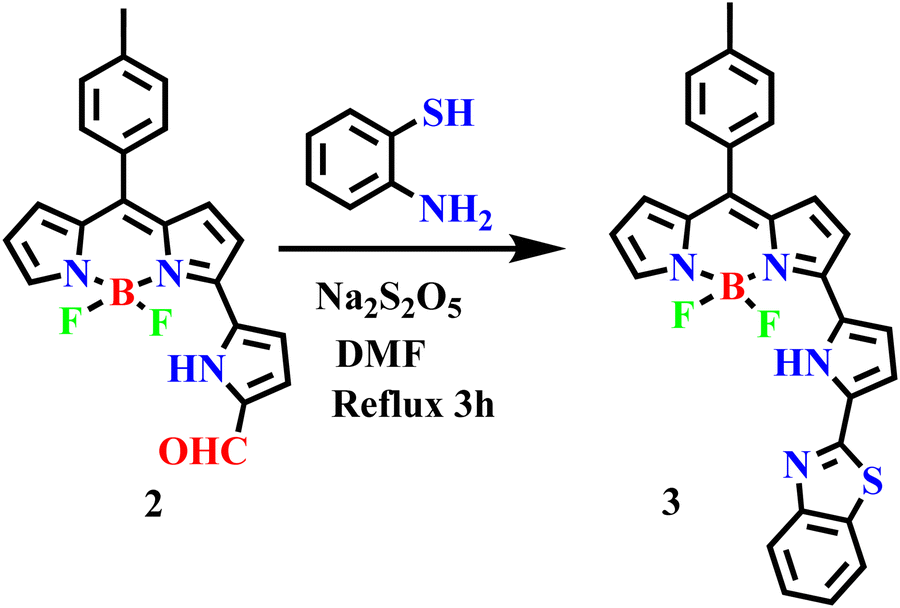 | ||
| Scheme 2 Synthetic procedure for 3. | ||
The compound 3 was freely soluble in all common organic solvents and was thoroughly characterized by 1D & 2D NMR and X-ray crystallography. The partial 1H–1H COSY, 19F, and 11B NMR of compound 3 are presented in Fig. 1. The 1H NMR spectrum of compound 3 showed well-separated resonances, which were identified based on their location and cross-peak correlations observed in 2D NMR (Fig. 1 and Fig. S4, ESI†). The NH proton signal of the appended pyrrole was shifted downfield at δ 11.36 ppm due to its involvement in hydrogen bonding with F− ions of the BF2 unit. In 1H NMR, the seven sets of resonances for seven pyrrolyl protons including dipyrrin and the appended pyrrole ring were observed in the region of 6.52–7.88 ppm whereas the aryl protons including a benzithiazolyl unit appeared in the region of 7.33–8.11 ppm. Furthermore, compound 3 showed a triplet resonance at δ 1.46 ppm in 11B NMR and a quartet resonance at δ −140.83 ppm in 19F NMR.
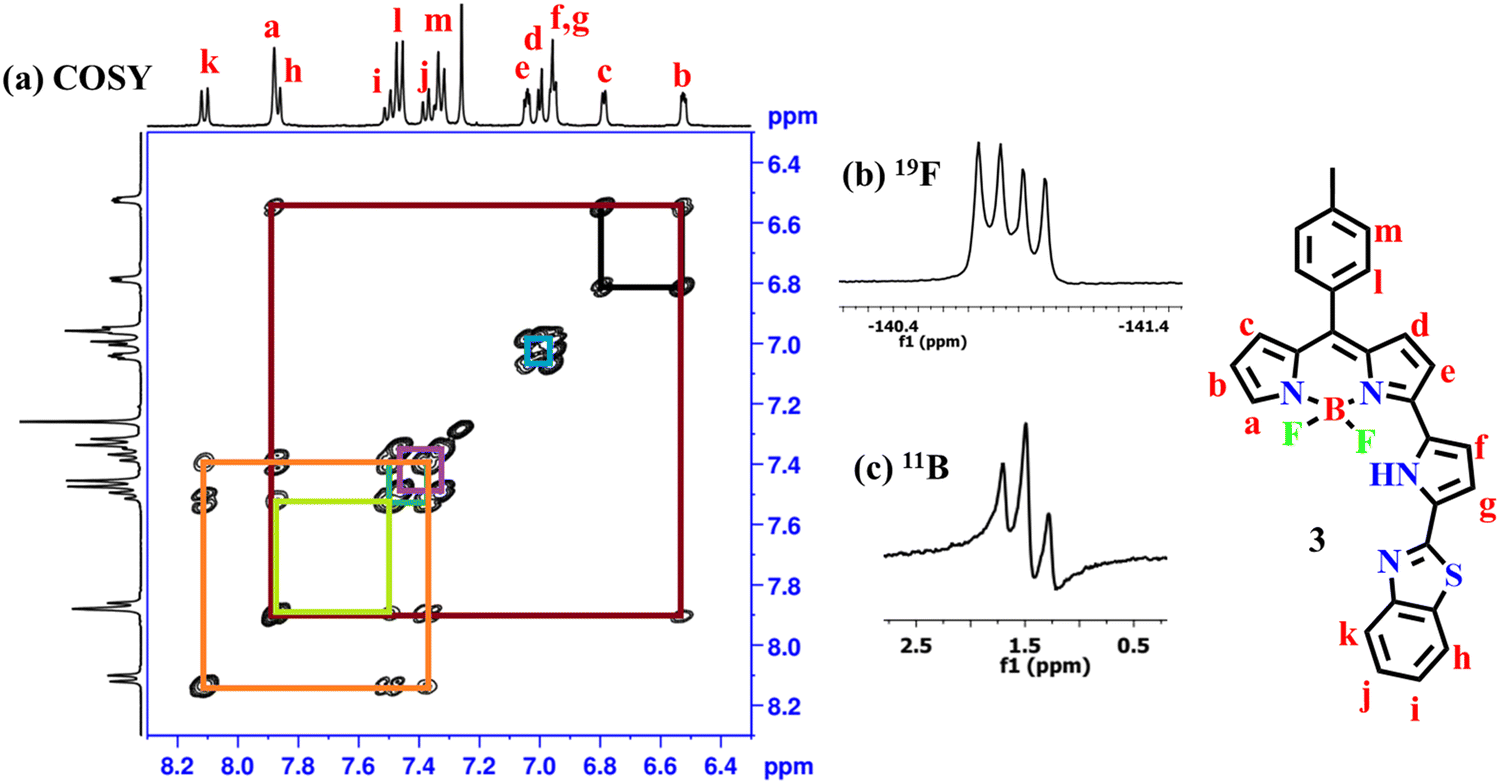 | ||
| Fig. 1 (a) 1H–1H NMR COSY, (b) 19F, and (c) 11B NMR spectra of compound 3 recorded in CDCl3 at RT. | ||
X-ray crystallography
The structure of 3 was elucidated by single-crystal X-ray crystallography (Fig. 2). The crystal was obtained by slow evaporation of CHCl3/n-hexane solution of 3 and the compound crystallized in the monoclinic space group C2/c. In the crystal structure of 3, the BF2-dipyrrin core was planar and the B(III) ion was coordinated with two pyrrole nitrogens of the dipyrrin unit and two axial fluoride ions in a tetrahedral geometry with bond angles of 107.51° (N2–B1–N1), 108.01° (F2–B1–F1), 109.64° (F2–B1–N1), 110.05° (F1–B1–N2) and 110.03° (F2–B1–N2). The meso-tolyl group was oriented at 56.95° relative to the planar BF2-dipyrrin core. The appended pyrrole was nearly in the plane with a small deviation of 12.15° from the 12-atom mean plane of the BF2-dipyrrin plane and the benzithiazolyl moiety was also deviated from the BF2-dipyrrin plane by 18.74°. The deviation of the appended pyrrole ring was reflected in their NH–F bond distances, which were 2.50 Å (NH–F1) and 2.16 Å (NH–F2). The bond lengths of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–S in the benzithiazolyl group in compound 3 were 1.305 Å, and 1.748 Å, respectively.
N and C–S in the benzithiazolyl group in compound 3 were 1.305 Å, and 1.748 Å, respectively.
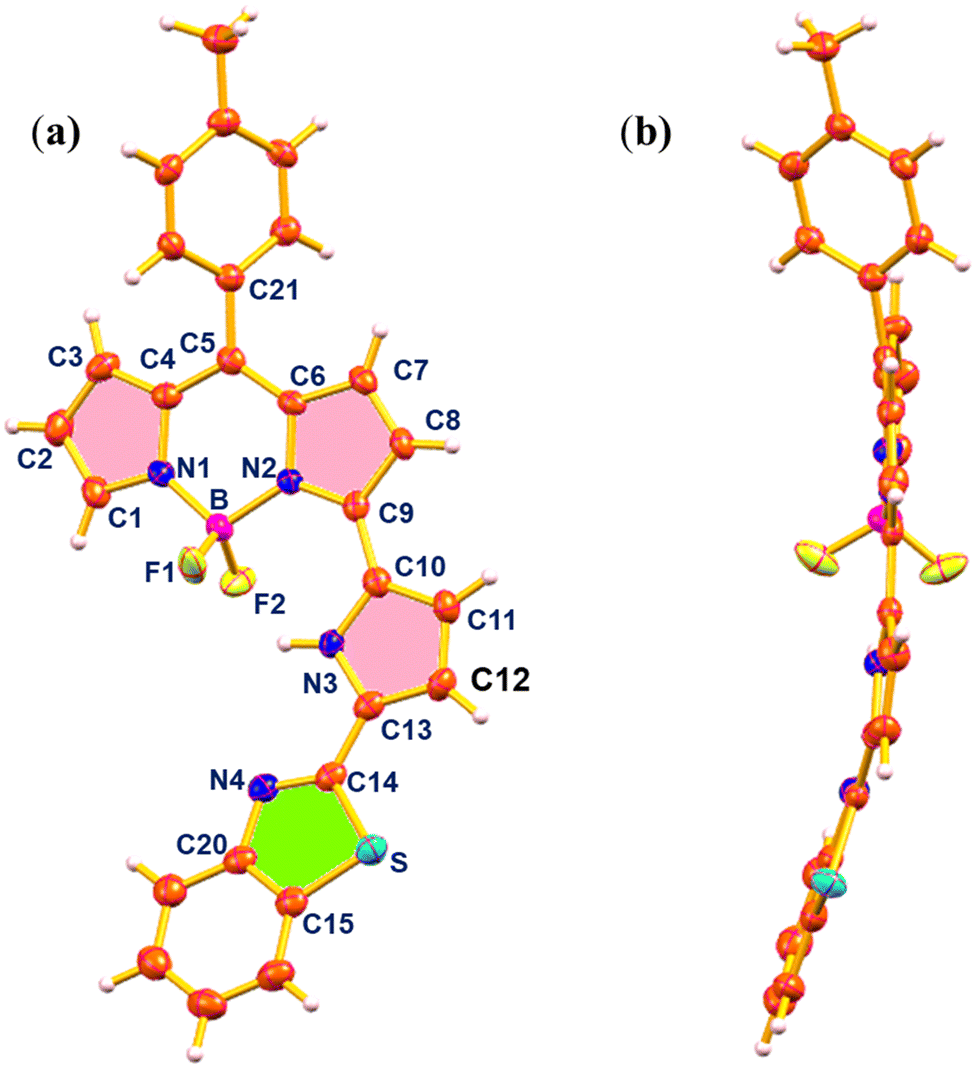 | ||
| Fig. 2 X-ray crystal structure of compound 3: (a) top view (thermal ellipsoids are drawn at 50% probability), and (b) side view (CCDC no. 2294221). | ||
Spectral and electrochemical properties
The absorption, fluorescence, and electrochemical properties of compound 3 were measured and the relevant figures are presented in Fig. 3. Compound 3 displayed one strong band at 608 nm accompanied by a shoulder band at 568 nm corresponding to the pyrrolyl BODIPY unit25 and a less intense broad absorption band at 412 nm due to the benzithiazolyl unit.33 Compound 3 upon excitation at 520 nm showed a strong fluorescence band at 637 nm with a quantum yield (ΦF) of 0.48. Thus, the strong fluorescence of compound 3 at 637 nm was tentatively attributed to the presence of ICT from the benzithiazolyl unit to the 3-pyrrolyl BODIPY unit within the molecule. The redox potentials were measured by cyclic voltammetry in CH2Cl2 using tetrabutylammonium perchlorate as a supporting electrolyte (Fig. 3(b)). Compound 3 showed two quasi-reversible reductions at ∼−0.91 and −1.33 V and one irreversible reduction at −1.78 V and didn’t show any oxidation, indicated that compound 3 was electron deficient in nature. | ||
| Fig. 3 (a) Absorption and fluorescence spectral profile of 3 in CH2Cl2, (b) cyclic voltammograms of the compounds 3 recorded in CH2Cl2 using a saturated calomel electrode as a reference electrode and 0.1 M TBAP as the supporting electrolyte at the scan rate of 50 mV s1; conditions: [3] = 10 μM (absorption) and 1 μM (emission), λex. = 520 nm, em./ex. b. p = 5 nm, RT. | ||
Sensing studies
The compound 3 was investigated for chemodosimetric sensing of anions by addition of 10 eq. of tetrabutyl ammonium salts of various anions such as ClO4−, NO3−, CN−, F−, C2O42−, Cl−, CH3CO−, Br−, PO43−, and SO42− to the solution of 3 in CH3CN–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 0.1 M PBS, pH 7.2). We did not observe any significant colour change or spectral profile change of compound 3 by the addition of any other anions except CN− ions. Upon addition of CN− ions to 3 in CH3CN–H2O, the absorption band at 608 nm decreased in intensity with the simultaneous appearance of a broad absorption band at 646 nm and color change of the solution from light violet to blue (Fig. 4(a)). Similarly, under excitation at 400 nm of compound 3, the intensity of the fluorescence band at 637 nm was significantly decreased with the simultaneous appearance of a fluorescence band at 456 nm exhibiting a large blue shift of ∼180 nm in the emission spectrum (Fig. 4(b)) with change in colour of the solution of 3 from red fluorescence to blue fluorescence (ΦF = 0.31) under UV light in the selective presence of CN− (Fig. 4(c)).
:1 v/v, 0.1 M PBS, pH 7.2). We did not observe any significant colour change or spectral profile change of compound 3 by the addition of any other anions except CN− ions. Upon addition of CN− ions to 3 in CH3CN–H2O, the absorption band at 608 nm decreased in intensity with the simultaneous appearance of a broad absorption band at 646 nm and color change of the solution from light violet to blue (Fig. 4(a)). Similarly, under excitation at 400 nm of compound 3, the intensity of the fluorescence band at 637 nm was significantly decreased with the simultaneous appearance of a fluorescence band at 456 nm exhibiting a large blue shift of ∼180 nm in the emission spectrum (Fig. 4(b)) with change in colour of the solution of 3 from red fluorescence to blue fluorescence (ΦF = 0.31) under UV light in the selective presence of CN− (Fig. 4(c)).
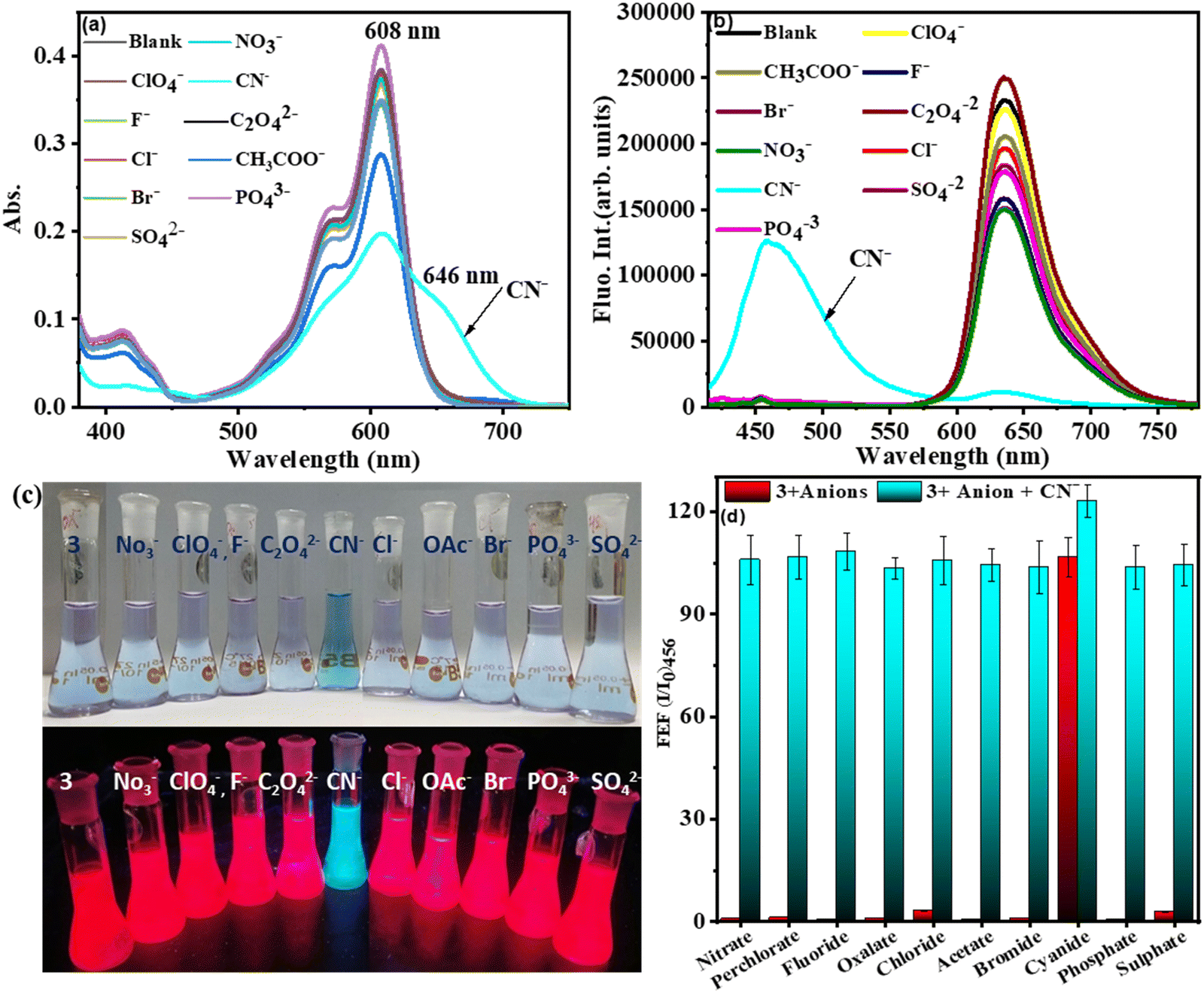 | ||
| Fig. 4 (a) Absorption and (b) and fluorescence spectral profile of 3 alone and in the presence of other anions in CH3CN–H2O (1:1, v/v, PBS, pH = 7.2), λex. = 400 nm, em./ex. b. p = 5 nm; (c) color change of solution of 3 in the presence of various anions under the naked eye and UV light, (d) the bar diagram to the corresponding FEF (at 456 nm) of 3 with anions used in this study (blue-colored bar); subsequent addition of CN− to the solution containing 3 and individual anions. (FEF (fluorescence enhancement factor) = I/I0, where I and I0 are the fluorescence intensity of compound 3 at specific emission wavelengths in the presence and absence of CN− respectively.) (Conditions: absorption: [3] = 5 μM; fluorescence: [3] = 1 μM.) | ||
However, no such fluorescence spectral changes were observed in the presence of other competitive anions. Thus, CN− induces selective dual colorimetric (light violet → blue (ε3/ε3+CN)608 = 1.9) and fluorometric (red fluorescent → blue fluorescent, (I3/I3+CN)637 = 19.78, (I3+CN/I3)456 = 22.90) signaling, identifying it as an excellent “turn-on” sensor for CN−. Furthermore, the appearance of a new peak in the lower wavelength region (blue shift) upon the addition of CN− to 3 was due to disruption in the π-conjugation over the entire molecule, resulting in the absence of intramolecular charge transfer.
The absorption spectral pattern of 3 was compared on the addition of CN− to the solution of 3 containing the other appropriate competitive anions to validate the cross-selectivity of 3 towards CN− over the other anions (Fig. 4(d)). In this test, the fluorescence emission of compound 3 (5 μM) was measured in the presence of 1 equiv. of appropriate anions followed by addition of 1 equiv. of cyanide. When cyanide was added to the mixture of 3 containing other anions, a new emission band at 456 nm appeared. As demonstrated in Fig. 3(d), there was no substantial variation in the emission of the 3 + CN− mixture in the presence of other competitive anions. These findings demonstrated that other anions do not interfere with the detection of CN− by compound 3 and compound 3 selectively senses the CN− ion.
We then performed systematic absorption and fluorescence titrations in PBS/CH3CN (1:1, v/v, pH = 7.2) medium by addition of increasing equivalents of CN− to compound 3. With the addition of increasing equivalents of CN− to solution 3, the intensity of the absorption band at 608 nm decreased with the simultaneous appearance and increase in the intensity of a new absorption band at 646 nm, resulting in an apparent change in the colour of compound 3 from light violet to blue. The fluorescence titration of 3 with CN− revealed that CN− induced fluorescent quenching at 637 nm with simultaneous significant enhancement in fluorescence intensity at 456 nm with one isosbestic point at 590 nm indicating the formation of a stable 3–CN product. The observed spectral changes in both absorption and fluorescence (blue shift) were attributed to the nucleophilic addition of cyanide ions to the imine bond of the benzithiazolyl group, which disrupts the π-conjugation of the receptor. Furthermore, the plot of fluorescence intensity ratio (I637/I456) as a function of increasing amounts of CN− (Fig. 5(d)) supported the ratiometric fluorescence sensing of CN−. The association constant for CN− using the Benesi–Hildebrand plot obtained34 from the emission spectrum (I456) was estimated to be 2.28 × 1010 M−1(Fig. 5(d) (inset)). The detection limit5 of 3 for CN− (Fig. S12, ESI†) was measured from the plot of fluorescence intensity ratio (I637/I456) versus concentration of CN− to be 13 nM, which is much less than the 1.9 μM of cyanide recommended by the WHO.35
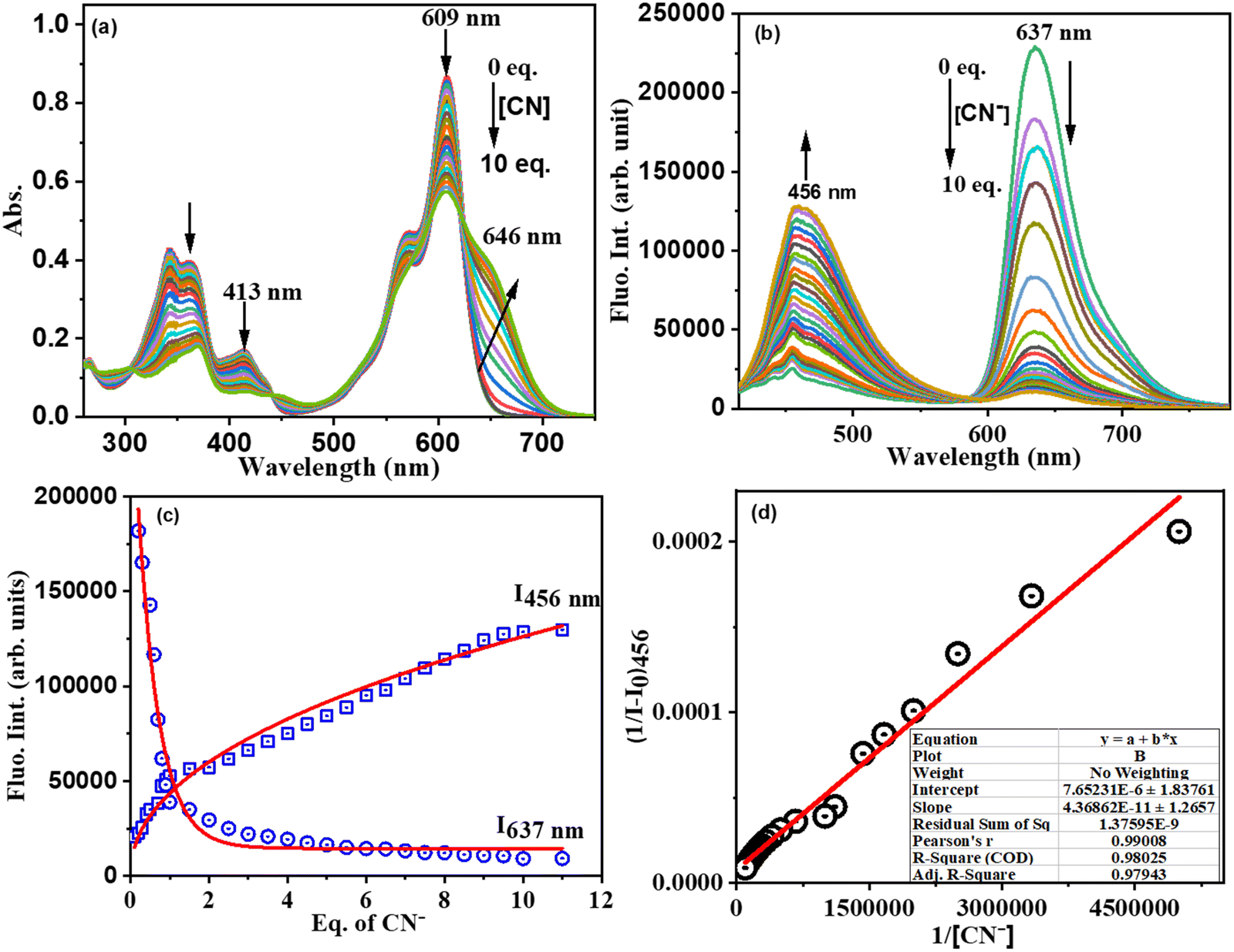 | ||
| Fig. 5 (a) Absorbance titration, (b) fluorescence titration profile of 3 as a function of equivalents of CN− added in CH3CN: H2O (1/1 v/v) medium, and (c) fluorescence spectral intensity at 456 nm and 637 nm versus equivalent of CN− and (d) linear regression to the plot (Benesi–Hildebrand plot) of 3 in the presence of CN− as a function of concentration of CN− for determination of the association constant (Ka) through a titration experiment. (Conditions: absorption: [3] = 10 μM; fluorescence: [3] = 1 μM.) | ||
The Job's plot indicates the 1:1 stoichiometry of 3 and CN− ions in the formation of the 3–CN complex (ESI), which was also supported by ESI-MS spectral analysis (ESI). The sensing response time of signaling probes is an important parameter in describing the sensing effectiveness. The rate of compound 3 interaction with CN− was calculated36 using their absorption spectral responses (abs = 456 nm) as a function of time, which followed first-order kinetics (A = A0e−kt). We have noted that the time driven CN− induced absorbance data were inferred to increase in absorbance intensity at 456 nm as a function of time up to 10 s and remained constant thereafter in a sigmoidal pattern after attaining the optimal intensity for this probe (ESI). The rate constant of CN− interaction with 3 was found to be 0.1169 s−1, implying a faster signaling response time.
Time-correlated single photon counting measurements for 3 and 3–CN in CH3CN–H2O (1:1 v/v, PBS, pH = 7.2) medium were investigated and the corresponding fluorescence decay profiles (excited at 400 nm and monitored at 456 nm and 637 nm) were fitted37 well to the mono or biexponential function (Fig. 6). The decay profile of 3 (λex.= 400 nm; λem.= 637 nm) was fitted to monoexponential with a lifetime of 4.43 ns (100%), whereas the decay profile for 3–CN fitted to bi-componential with the lifetime of 1.41 (22.65%, τ1) and 3.45 ns (77.35%, τ2), and the resulting τavg. was 1.53 ns. However, the decay profile for 3–CN at the emission wavelength of 456 nm exhibited bi-exponential fit with τ1 and τ2 being 0.98 ns (8.16%) and 2.05 ns (91.84%), respectively, and the average lifetime (τavg) was found to be 1.89 ns. Thus, the increase in the average lifetime at 456 nm and decrease in the average lifetime at 637 nm of 3–CN compared to 3 supports the ratiometric sensing of 3 towards CN− ions.
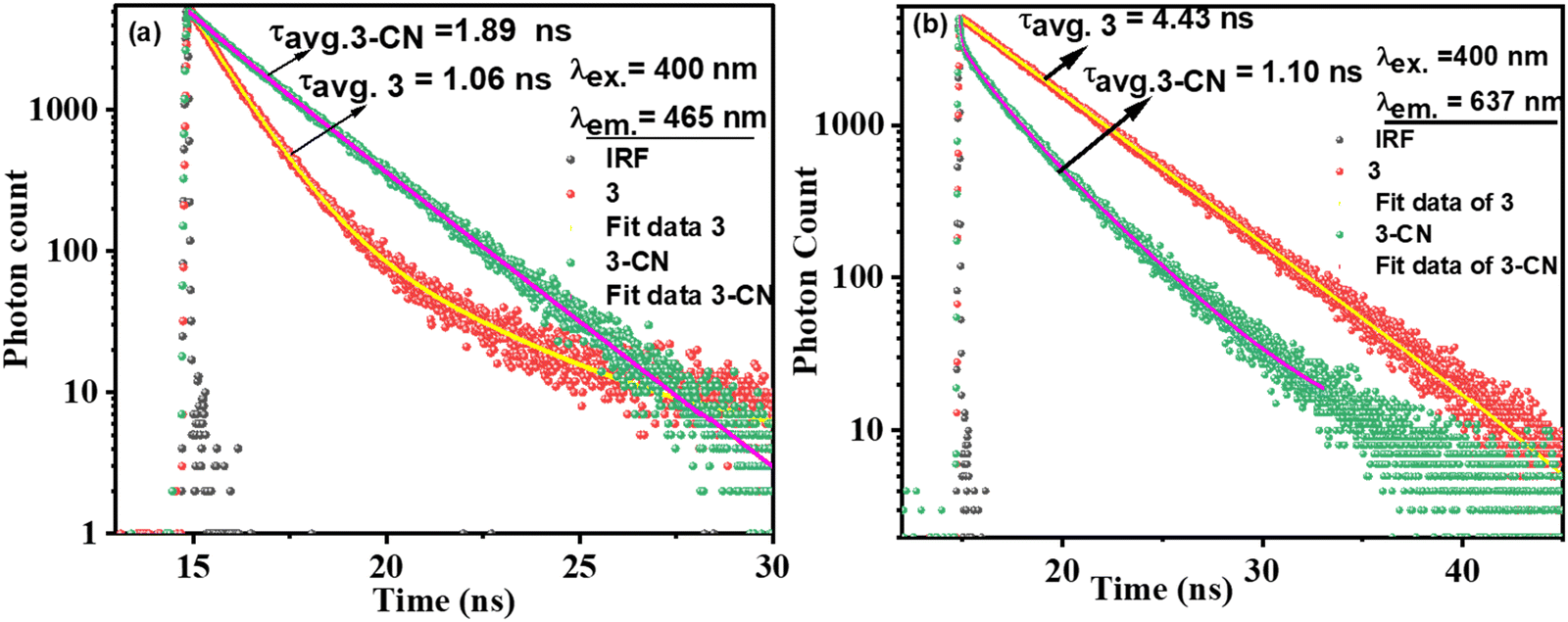 | ||
| Fig. 6 Fluorescence decay profile for 3 (1 μM) alone and in the presence of CN− upon excitation at λex. = 400 nm and emission at (a) 456 nm and (b) 637 nm. | ||
The interaction of CN− with compound 3 was also established by comparing 1H and 13C NMR spectra of 3 and 3–CN in CDCl3. In 1H NMR of 3–CN (Fig. 7), the new NH proton of the benzithiazole moiety (type n) was observed at 7.81 ppm, and the pyrrole protons of 3–CN showed either slight upfield or downfield shifts compared to 3. Interestingly, in 13C NMR (ESI), we noted the disappearance of the resonance at 151.12 ppm corresponding to imine C observed in 3 and the appearance of a peak at 63.40 ppm in 3–CN due to the quaternary carbon (C14) along with the peak at 129.32 ppm corresponding to the CN− in 3–CN. Additionally, the molecular ion peak (m/z)+ at 529.5601 [3–CN + Na]+ in the MS spectrum of 3–CN (ESI) implied their formation in a 1:1 stoichiometry. The comparison of the cyclic voltammograms of compounds 3 and 3–CN is presented in Fig. 8(b). Compound 3 showed two quasi-reversible reductions at ∼−0.91 and −1.33 V and one irreversible reduction at −1.78 V but no oxidation was noted. We tentatively attributed that the reductions at −0.91 V and −1.78 V were due the 3-pyrrolyl BODIPY moiety whereas the reduction at −1.33 V was due to the benzithiazole moiety. Interestingly, the compound 3–CN showed one quasireversible reduction at ∼−0.91 V and one irreversible reduction at −1.78 V, but the reduction at −1.33 V was absent, which clearly illustrates that the imine bond of benzithiazole was reduced by the nucleophilic addition of CN− (Fig. 8(a)). Thus, the spectral, electrochemical, and mass spectral analysis confirmed the site of attack of CN− at the carbon of CN of the benzithiazole moiety to form a 3–CN compound (Scheme 3).
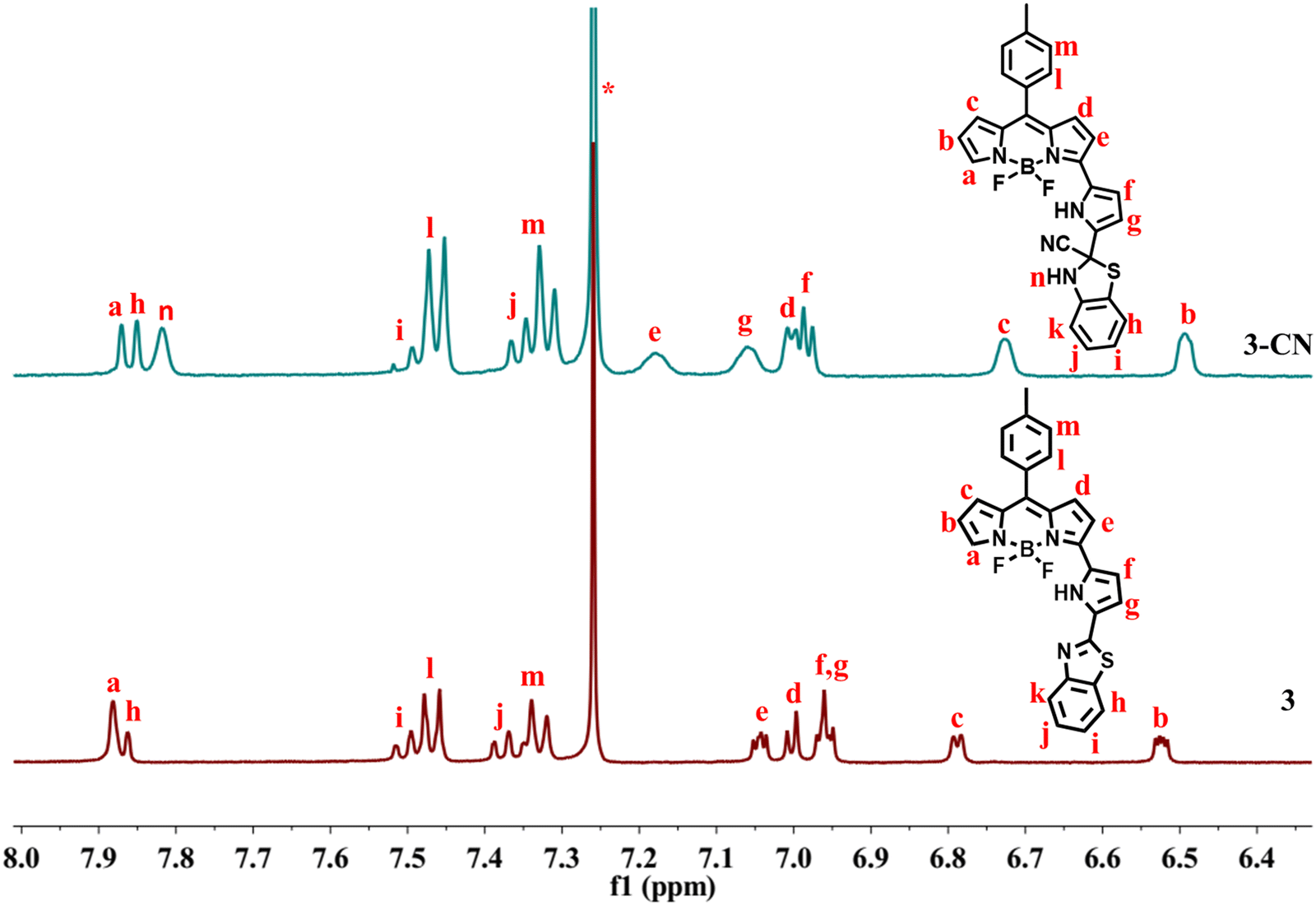 | ||
| Fig. 7 Partial 1H NMR of 3 and 3–CN in CDCl3 at RT. | ||
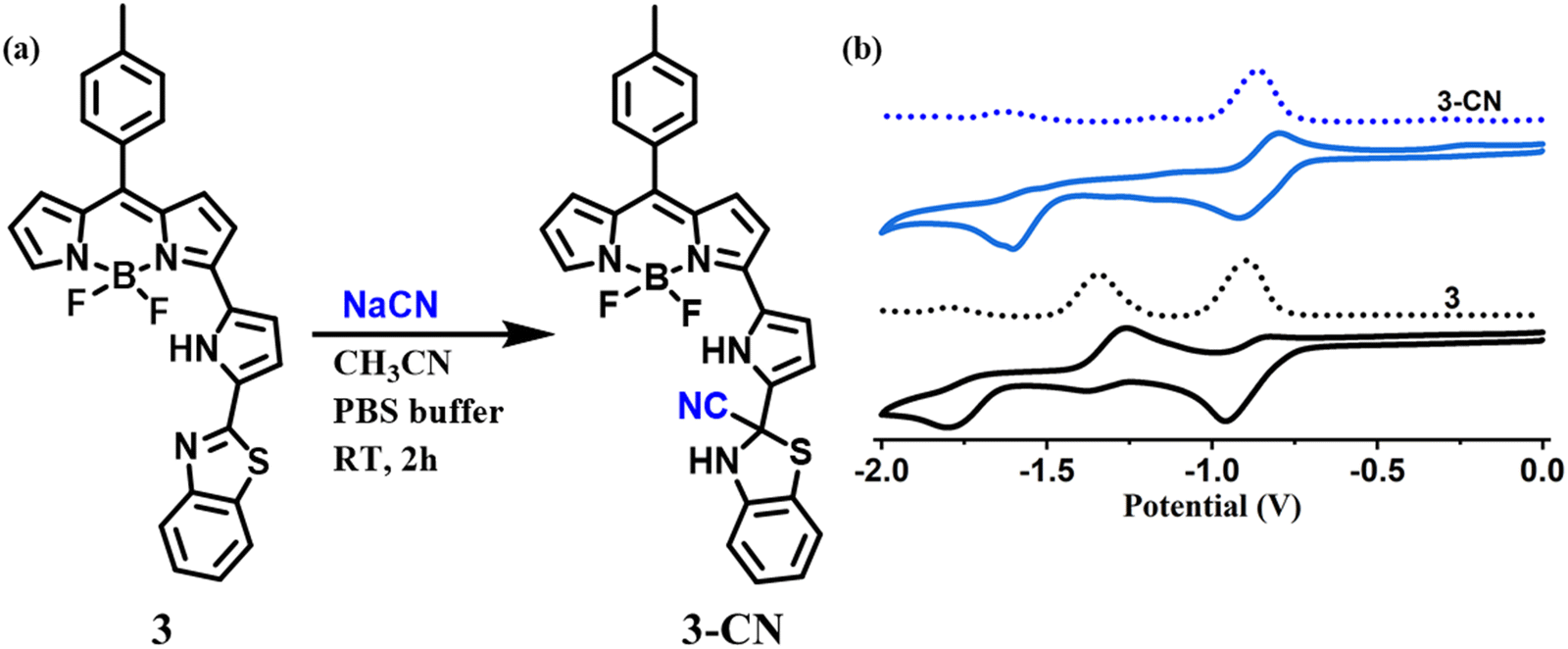 | ||
| Fig. 8 (a) Synthetic procedure of the 3–CN product, and (b) cyclic voltammograms of the compounds 3 and 3–CN recorded in CH2Cl2. | ||
DFT/TD-DFT studies
Density functional theory (DFT) calculations38,39 were carried out with the B3LYP functional using the 6-31g(d,p) basis set in the Gaussian 09 software package to comprehend the mechanism of interaction between 3 and CN−. The ground state optimized geometries of 3 and 3–CN are shown in Fig. S15 (ESI†). The geometry optimized structure at the ground state for compound 3 was in good agreement with the X-ray structure of 3.The optimized structures of 3 and 3–CN revealed that both the benzithiazolyl and 3-pyrrolyl BODIPY moieties in 3 were almost in a planar conformation and the benzithiazolyl moiety in 3–CN is deviated from the mean plane of BF2-dipyrrin by 82.29° compared to 18.74° in 3, which suggested that both moieties are not in a planar confirmation, resulting in disruption of the conjugation process in the entire molecule. The calculated frontier molecular orbital (HOMO−1, HOMO, LUMO, and LUMO+1) surface topologies of 3 and 3–CN are presented in Fig. 9. The electronic density of the HOMO of 3 was distributed on the α-pyrrolyl BODIPY and benzithiazolyl units and no distribution was noted on the meso-tolyl groups. However, the LUMO showed electron distribution only on the α-pyrrolyl BODIPY unit and some rare distribution on the meso-tolyl group, but no distribution was found on the benzithiazolyl unit, which suggests the ICT character from the benzithiazolyl unit to the α-pyrrolyl BODIPY in compound 3. However, the electron distribution of the HOMO and LUMO of 3–CN showed similar contributions only on the α-pyrrolyl BODIPY units, supporting blocking of the ICT character. Also, the electron distribution of the HOMO−1 and LUMO+1 of 3–CN showed only on the benzithiazolyl unit. Furthermore, the HOMO−1, LUMO, and LUMO+1 of 3 were stabilized compared to 3–CN but the HOMO of 3 was destabilized compared to 3–CN, resulting in a higher HOMO–LUMO energy gap (Eg = 2.56 eV) for 3–CN as compared to 3 (Eg = 2.36 eV).
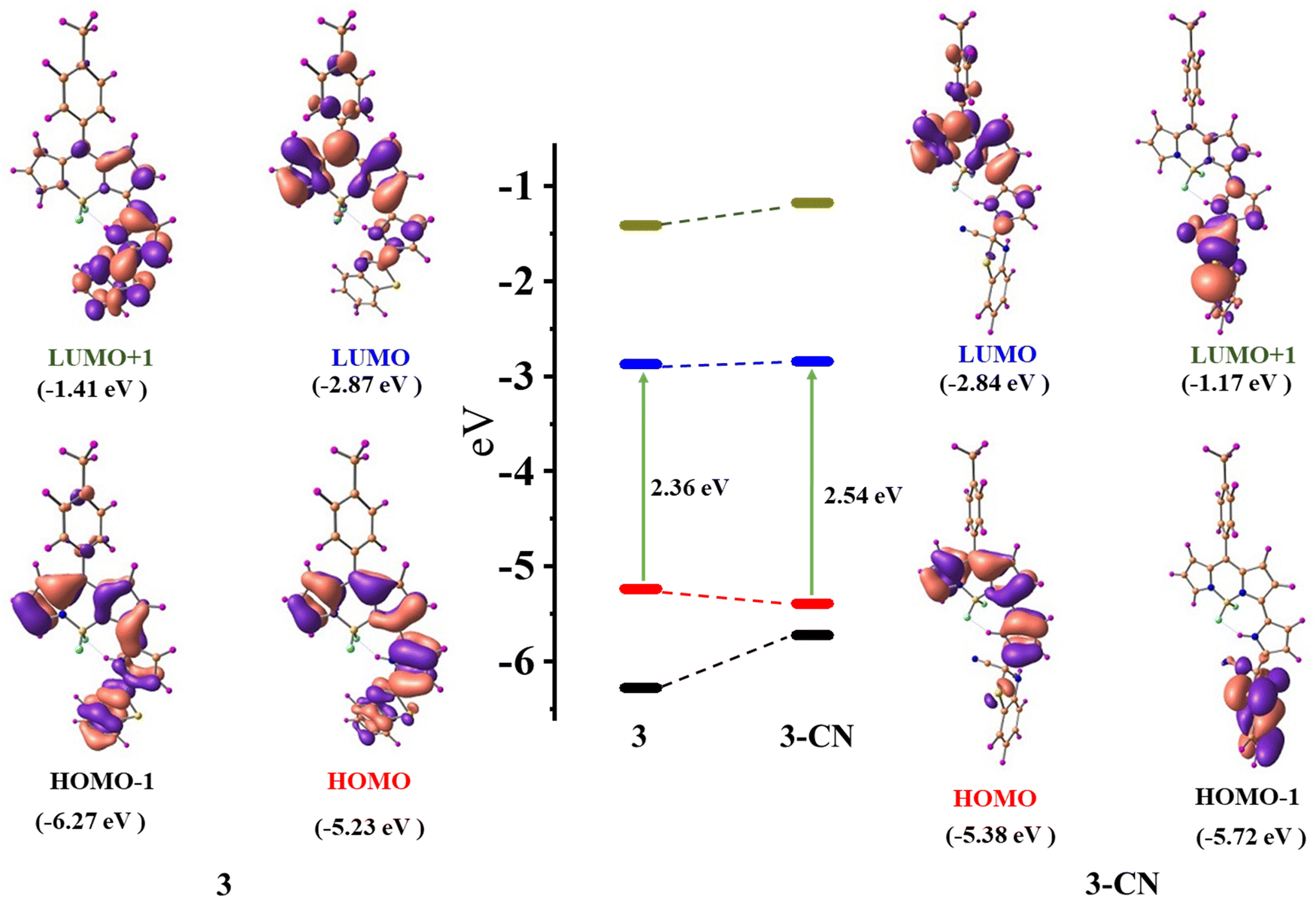 | ||
| Fig. 9 Selected frontier molecular orbital with energy level diagrams of compound 3 and 3–CN. | ||
TD-DFT studies40 were employed to assess the oscillator strength and excitation energies of the first S0 → Sn transitions, which were convincingly corroborated by the experimental absorption spectra of 3 and 3–CN, as shown in Fig. 10. The observed absorption bands at 608 nm, 412, and 341 nm of 3 in CH2Cl2 were mainly due to HOMO → LUMO, HOMO−1 →LUMO, and HOMO−1 →LUMO transitions, respectively. However, in 3–CN, the absorption bands at 608, and 371 nm were mainly due to the HOMO → LUMO and HOMO−2 → LUMO transitions, respectively. As a result, the structural and spectral properties obtained from DFT/TD-DFT calculations closely matched the results from the experiments.
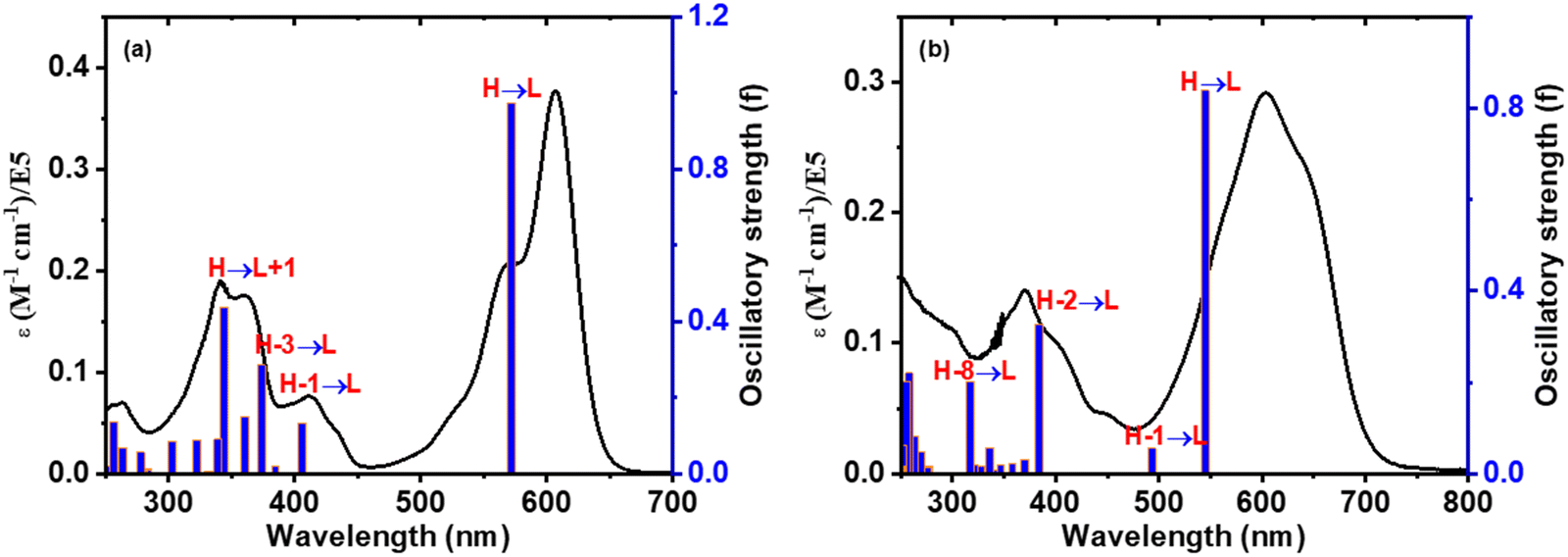 | ||
| Fig. 10 Comparison of the calculated excitation energies (blue vertical lines) and experimental absorption spectra (black colored line) of compounds (a) 3 and (b) 3–CN, respectively. | ||
Furthermore, the natural bond orbital (NBO) study of the title molecule 3 and 3–CN was computed at the B3LYP/6-31g level of the model.41 This analysis aimed to elucidate electron density delocalization and intra- and inter-molecular bonding, and explore charge transfer (CT) or the extent of conjugative interaction between the benzithiazolyl and 3-pyrrolyl BODIPY units in compound 3. Second-order perturbation theory, applied to the Fock matrix, was utilized to evaluate donor–acceptor interactions in the NBO investigation. The strength of interaction stabilization energy E(2), indicative of electron delocalization between the donor (i) and acceptor (j) (i → j), was assessed using the following equation.
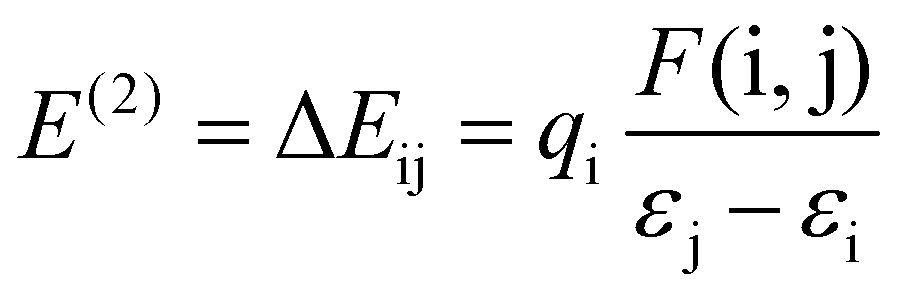
The greater stabilization energy E(2) value shows more intensive interaction amongst the electron acceptor and donor, i.e. the high electron donating ability and superior degree of conjugation of the entire system. The potential intensive interactions and electron dentistry transfer from lone pair/bond pair electrons to the anti-bonding orbitals along with the stabilization energy values were identified within numerous donor and acceptor interactions for 3. The results of the NBO analysis of compound 3 indicate that the intramolecular interactions occurred due to the overlap between the bonding lone pair of nitrogen (LP(1) N4) and antibonding C13–C14 orbitals with stabilization energy of 17.51 kJ mol−1, which results in the ICT initiated stabilization of compound 3. In addition, there are strong hyperconjugative interactions π (N4–C14) → π* (C13–C14) and π* (N4–C14) → π* (C13–C14) for compound 3, which are 43.98 and 370.49 kJ mol−1, respectively. However, these kinds of hyercojugative interactions were not found in 3–CN resulting in the weak nature of ICT. Also, hyper conjugative interactions σ (N4–H) → σ* (C14–S1), σ (C12–C13) → σ* (C≡N), and σ (C14–S1) → π* (C≡N) were found for compound 3–CN with stabilization energies of 16.23, 17.27, and 19.49 kJ mol−1, respectively, resulting in the failure of the ICT character from the benzithiazolyl to 3-pyrrolyl BODIPY unit in 3–CN. The charge analysis revealed that the charge on N and C in the imine bond of the benzothiazolyl unit of 3 was calculated to be 0.1778 and −0.0805 whereas that of 3–CN was found to be 0.3583 and −0.7601, respectively. We have also calculated the charge variation of the two main fragments, i.e., the benzithiazolyl and 3-pyrrolyl BODIPY units in 3 were found to be 0.2630 and −0.2630, whereas those in 3–CN were found to be 0.1818 and −0.1818, respectively. Hence, through the compelling experimental evidence and meticulous computational calculations, our investigation strongly supports the integration of photophysical processes into the ICT mechanism for cyanide sensing.
Bio-imaging studies
In vitro studies were performed with the tissue of Hybanthus enneaspermus, a medicinal plant with enormous pharmaceutical applications42 for the detection of cyanide ions accumulated by the plant with compound 3. The micropropagated plantlets of H. enneaspermus with healthy shoot and root systems were co-cultivated with tetrabutylammonium cyanide (TBACN) at varied concentrations (0 to 50 ppm) in the plant growth medium (Murashige and Skoog, 1/4th strength MS).43 Subsequently, another batch of in vitro plantlets maintained in the nutrient medium was labeled as control (i.e. without TBACN supply). The experiment was conducted over a period of 15 days in the plant culture.room provided with essential humidity (50–80%), temperature (25 ± 2 °C), and photoperiod (16 h, cool fluorescent light of 30 μmol m s−1), respectively (Fig. 11(a)). At the end of the culture period, cyanide toxicity was observed above 10 ppm (0.04 μM) of TBACN supply, and hence plants grown in 0.04 μM of TBACN were selected further for the bioimaging studies. TBACN treated plants as well as the control plants were cleansed and submerged in a solution of compound 3 at different time spans (15 minutes, 30 minutes, and 1 hour). Thereafter, the ability of compound 3 to sense the presence of cyanide ions in the plant cells, thin hand sections (around 15 μm) of roots and shoots were used at each time frame. These cross-sections were explored under an epifluorescence microscope (Nikon Eclipse E600, Japan) under an excitation wavelength range of 410–480 nm (40× magnification). However, the lack of bright blue colour in the control plant root (Fig. 11(c)), and the presence of shiny blue coloured emission in the xylem and cortex cells in the TBACN-treated plant root (Fig. 11(b)) supports the utilization of α-benzithiazolyl 3-pyrrolyl BODIPY in the sensitive detection of cyanide ions in the plant cells. Furthermore, the presence of cyanide in the root cells of the experimental medicinal plant as detected by the probe will facilitate its importance in traditional and herbal market safety testing procedures.
 | ||
| Fig. 11 Bioimaging studies. (a) The experimental plant H. enneaspermus treated in 10 ppm of TBACN, (b) cross section of H. enneaspermus root co-cultivated with 10 ppm of TBACN, followed by compound 3 treatment for 30 minutes, bright blue emissions in the xylem and cortex cells indicates cyanide deposition, and (c) cross section of H. enneaspermus root kept as a control, followed by probe treatment for 30 minutes. | ||
Conclusions
In conclusion, we developed a simple 3-pyrrolyl BODIPY based chromo-fluorogenic probe, α-benzithiazolyl 3-pyrrolyl BODIPY, that acts as an exclusive chemodosimetric sensor for CN− ions over other anions. The probe was synthesized by reacting α-formyl 3-pyrrolyl BODIPY with 2-aminothiophenol in DMF at reflux under basic conditions and the X-ray structure of the probe revealed that the appended pyrrole and benzithiazolyl units slightly deviated from the 12-atom mean plane of the BF2-dipyrrin plane. The probe absorbs strongly at 608 nm with a shoulder band at 568 nm due to the 3-pyrrolyl BODIPY unit with a less intense absorption band at 412 nm due to the benzithiazolyl unit. The probe is a strong red fluorescent dye with an intense emission band at 637 nm with a quantum yield (ΦF) of 0.48. Our studies revealed that the probe acts as an exclusive chemodosimetric CN− ion sensor by CN– attacking the imine bond of the benzithiazolyl unit via nucleophilic addition reaction, which is reflected in clear changes in the colour, absorption, and fluorescence properties. The CN− ion sensing behavior of the probe was also verified by NMR and mass spectral studies. DFT/TD-DFT studies were in agreement with the experimental observations. We also showed that the probe could detect intracellular CN− ions in plant tissue by fluorescence microscopy imaging studies. Thus, the results of this study would offer valuable information for developing future colorimetric optical sensors for toxic cyanide ions.Experimental
Synthesis of compounds
The 3-pyrrolyl BODIPY251 and α-formyl pyrrolyl BODIPY262 were synthesized following the method reported by our group.Synthesis of 3
A solution of α-formyl 3-pyrrolyl BODIPY (50 mg, 0.13 mmol) in anhydrous DMF (5 mL) was added to a solution of 2-aminothiophenol (15 mg, 0.12 mmol) and Na2S2O5 (28.5 mg, 0.15 mmol) and the reaction mixture was heated at reflux for 3 h with continuous stirring. The reaction mixture was brought to room temperature, water was added (30 mL), and it was extracted with CHCl3 (30 mL × 3). The solvent of the combined organic layers, after drying over Na2SO4, was removed under reduced pressure to afford the desired crude product 3 as a blue solid. The crude compound was subjected to silica gel (100–200 mesh) column chromatographic purification using petroleum ether/ethyl acetate (85:15% v/v) and afforded pure compound 3 in 30% yield.
Yield: 30% (15 mg); 1H NMR (400 MHz, CDCl3) δ 11.36 (1 H, s), 8.11 (1 H, d, J = 8.1 Hz), 7.87 (2 H, d, J = 7.7 Hz), 7.50 (1 H, d, J = 8.1 Hz), 7.46 (2 H, d, J = 8.0 Hz), 7.38 (1 H, d, J = 8.0 Hz), 7.33 (2 H, d, J = 7.9 Hz), 7.06–7.03 (1 H, m), 7.00 (1 H, d, J = 4.6 Hz), 6.95 (2 H, d, J = 4.5 Hz), 6.79 (1 H, d, J = 3.7 Hz), 6.52 (1 H, dd, J =3.9, 2.1 Hz), 2.48 (3H, s); 19F NMR (376 MHz, CDCl3) −140.87 (dd, J = 67.2, 33.0 Hz); 13C NMR (100 MHz, CDCl3) 158.0, 151.3, 150.1, 141.9, 140.6, 139.2, 137.5, 136.9, 134.9, 134.6, 133.1, 132.1, 131.8, 131.1, 129.6, 126.9, 124.6, 123.4, 123.3, 121.0, 120.8, 116.2, 112.6, 111.3, 21.47; 11B NMR (160 MHz, CDCl3) 1.46 (t, J = 33.7 Hz); HR-MS found for 3 (C27H19BF2N4S): 481.1483 (m/z)+.
Crystal data for 3 (CCDC no. 2294221): MF = C27H19BF2N4S; Mw = 480.33; block, olive crystals, monoclinic, space group C2/c, a = 19.5653(3) Å, b = 16.6631(2) Å, c = 14.1015(2) Å, α = 90, β = 107.245(2), γ = 90, U = 4390.68(12) Å3, T = 106 K, Z = 8, μ(Mo Kα) = 0.190 mm−1, F(000) = 1984.0, ρcalc = 1.453 mg m−3, 54489 reflection data with 317 parameters, 3848 [I ≥ 2σ(I)] unique reflections used in calculations. The final R1 = 0.0348, wR2 = 0.0962 (3845), S = 1.070.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
MR thanks CSIR (01(3036)/21/EMR-II), and J. C. Bose Fellowship (JCB/2021/000017) for financial support. KCB thanks IIT Bombay for the Institute Postdoctoral fellowship.References
- R. Gracia and G. Shepherd, Pharmacotherapy, 2004, 24, 1358–1365 CrossRef CAS PubMed.
- G. D. Shearer and K. C. Sellers, Vet. J., 1944, 100, 92–97 CAS.
- B. Vennesland et al. , Cyanide in Biology, Academic Press, San Diego, CA, 1981 Search PubMed.
- L. Nelson, J. Emerg. Nurs., 2006, 32, 8–11 CrossRef PubMed.
- C. Arivazhagan, R. Borthakur, R. Jagan and S. Ghosh, Dalton Trans., 2016, 45, 5014–5020 RSC.
- C. Gouda, D. Barik, C. Maitra, K.-C. Liang, F.-C. Ho, V. Srinivasadesikan, S. Chandran, S.-P. Wu, M.-C. Lin and H.-C. Lin, J. Mater. Chem. C, 2021, 9, 2321–2333 RSC.
- Q. Niu, L. Lan, T. Li, Z. Guo, T. Jiang, Z. Zhao, Z. Feng and J. Xi, Sens. Actuators, B, 2018, 276, 13–22 CrossRef CAS.
- G. Dorooshi, A. Dorostkar, A. Rahimi and S. Zoofaghari, Adv. Biomed. Res., 2020, 9, 42 CrossRef PubMed.
- J. O. Egekeze and F. W. Oehme, Tijdschr. Diergeneeskd., 1980, 105, 104–114 CAS.
- M. A. Holland and L. M. Kozlowski, Clin. Pharmacol., 1986, 5, 737–741 CAS.
- X. Yang, X. Chen, X. Lu, C. Yan, Y. Xu, X. Hang, J. Qu and R. Liu, J. Mater. Chem. C, 2016, 4, 383–390 RSC.
- K. Zuhra and C. Szabo, FEBS J., 2022, 289, 2481–2515 CrossRef CAS PubMed.
- Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC.
- P. Rathi, M. K. Chahal and M. Sankar, New J. Chem., 2017, 41, 11962–11968 RSC.
- L. A. Greenawald, G. R. Boss, J. L. Snyder, A. Reeder and S. Bell, ACS Sens., 2017, 2, 1458–1466 CrossRef CAS PubMed.
- S. Enbanathan, S. Munusamy, S. Ponnan, D. Jothi, S. Manoj Kumar and K. I. Sathiyanarayanan, Talanta, 2023, 264, 124726 CrossRef CAS PubMed.
- J. Ma and P. K. Dasgupta, Anal. Chim. Acta, 2010, 673, 117–125 CrossRef CAS PubMed.
- E. P. Randviir and C. E. Banks, TrAC, Trends Anal. Chem., 2015, 64, 75–85 CrossRef CAS.
- F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312–4324 RSC.
- Z. M. Dong, H. Ren, J. N. Wang, J. B. Chao and Y. Wang, Spectrochim. Acta, Part A, 2019, 217, 27–34 CrossRef CAS PubMed.
- G. Picci, R. Montis, A. M. Gilchrist, P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2024, 501, 215561 CrossRef CAS.
- W. Saiyasombat, U. Eiamprasert, T. Chantarojsiri, K. Chainok and S. Kiatisevi, Dyes Pigm., 2022, 206, 110643 CrossRef CAS.
- R. Guliyev, S. Ozturk, E. Sahin and E. U. Akkaya, Org. Lett., 2012, 14, 1528–1531 CrossRef CAS PubMed.
- L. Wang, H. Ding, X. Ran, H. Tang and D. Cao, Dyes Pigm., 2020, 172, 107857 CrossRef CAS.
- M. R. Rao, M. D. Tiwari, J. R. Bellare and M. Ravikanth, J. Org. Chem., 2011, 76, 7263–7268 CrossRef CAS PubMed; E. P. Randviir and C. E. Banks, TrAC, Trends Anal. Chem., 2015, 64, 75–85 CrossRef.
- T. Kaur, V. Lakshmi and M. Ravikanth, RSC Adv., 2013, 3, 2736–2745 RSC.
- S. Panchavarnam, P. Pushpanandan and M. Ravikanth, Inorg. Chem., 2022, 61, 1562–1570 CrossRef CAS PubMed.
- P. Chauhan, K. C. Behera and M. Ravikanth, Inorg. Chem., 2023, 62, 13919–13928 CrossRef CAS PubMed.
- J. Li, Q. Zhang, J. Yin, C. Yu, K. Cheng, Y. Wei, E. Hao and L. Jiao, Org. Lett., 2016, 18, 5696–5699 CrossRef CAS PubMed.
- M. Horetski, A. Gorlova, R. Płocińska, A. Brzostek, Y. Faletrov, J. Dziadek and V. Shkumatov, ChemistrySelect, 2022, 7, e202200506 CrossRef CAS.
- W. Miao, Z. Li, C. Yu, E. Hao and L. Jiao, J. Porphyrins Phthalocyanines, 2021, 25, 1119–1125 CrossRef CAS.
- M. R. S. A. Janjua, Open Chem., 2017, 15, 139–146 CAS.
- L. Wang, J. Zheng, S. Yang, C. Wu, C. Liu, Y. Xiao, Y. Li, Z. Qing and R. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 19509–19515 CrossRef CAS PubMed.
- I. D. Kuntz Jr, F. P. Gasparro, M. D. Johnston Jr and R. P. Taylor, J. Am. Chem. Soc., 1968, 90, 4778–4781 CrossRef.
- Guidelines for Drinking Water Quality, World Health Organization, Geneva, 1996 Search PubMed.
- K. C. Behera, B. N. Patra and B. Bag, Sens. Actuators, B, 2021, 338, 129861 CrossRef CAS.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- M. J. Gaussian, Gaussian 09, Revision B. 01, Gaussian, Inc, Wallingford, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
- T. R. K. Rana, A. Swain and G. Rajaraman, Dalton Trans., 2023, 52, 11826–11834 RSC.
- D. K. Patel, R. Kumar, K. Sairam and S. Hemalatha, Chin. J. Nat. Med., 2013, 11, 199–206 CAS.
- T. Murashige and F. Skoog, Physiol. Plant., 1962, 15, 473–497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data using HRMS, NMR spectra, and crystal data, detailed absorption, emission spectral pattern, DFT, TD-DFT, and TCSPC data of the compound are available. CCDC 2294221 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp05230c |
| This journal is © the Owner Societies 2024 |