
Halogen bond catalysis of the [4+2] cycloaddition reaction of 2-alkenylindoles: catalytic modes and stereoselectivity†
Ying
Li
,
Chang
Zhao
,
Huaiyu
Zhang
* and
Yanli
Zeng
 *
*
College of Chemistry and Materials Science, Hebei Normal University, Shijiazhuang, 050024, China. E-mail: yanlizeng@hebtu.edu.cn; huaiyu.zhang@hetu.edu.cn
First published on 4th December 2023
Abstract
Halogen bond donor catalysts have been widely used in organic reactions because they are environmentally friendly, inexpensive and recyclable. The [4+2] cycloaddition reaction is a key reaction in organic synthesis because of its ease of use, fast speed, and wide range of applications. In this work, halogen bond catalysis in the [4+2] cycloaddition reaction between 2-alkenylindoles was investigated based on DFT calculations. There are two modes of I⋯π halogen bond catalysis: either on the ethenyl of 2-alkenylindole (mode A) or on the five-membered ring of 2-alkenylindole (mode B). Both modes involve two steps: the formation of carbon–carbon bonds and the formation of six-membered rings. Gibbs free energy barriers were determined to investigate the stereoselectivity of the endo pathway and exo pathway. For mode A, the exo products were more easily generated when the substituent R = H, and the N–H⋯π interaction promoted high endo selectivity in the case of the substituent R = Ph. For mode B, an increasing proportion of endo products can be obtained in the order of catalyst I2, IBr and ICl. The π⋯π interaction of the substituent R = Ph promotes the [4+2] cycloaddition reaction, which is consistent with the experimental observation that R = Ph has a higher yield than R = H. The study of different catalytic modes and stereoselectivity would provide new ideas for the further study of the [4+2] cycloaddition reaction.
1. Introduction
Halogen bonds have been continuously developed in most chemical fields, from molecular recognition or drug design to supramolecular chemistry, materials science and catalytic chemistry.1 Halogen bonds2 are important noncovalent interactions that play an important role in catalysis, not only by controlling the reactivity of the conversion process but also by controlling selectivity to a considerable extent.3Halogen bond donor catalysts, as environmentally friendly and recyclable reagents, have been widely used in organic reactions in recent years.4,5 In 2008, Bolm et al.6 reported the Hantzsch ester reduction of 2-phenylquinoline activated by halogen bonds, which is the application of halogen bonds in catalytic reactions. In the following years, halogen bond donors have been used as catalysts in different reactions by activating various functional groups, such as amides,7,8 carbonyl groups,9–11 halides,12–14 nitro groups,15 and imines.16,17 In these reactions, halogen bond donor catalysts are mainly used to activate substrates containing lone pair-possessing heteroatoms to complete the catalytic reaction. However, there are fewer studies on halogen bond catalysis by activating the π-electrons of reactants in the [4+2] cycloaddition reaction.
The [4+2] cycloaddition reaction is a key reaction in organic synthesis because of its simple reaction, fast speed, wide range of application and mild reaction conditions. In 2019, Arai et al. first studied C–I···π halogen bond catalysis in the [4+2] cycloaddition reaction of 2-alkenylindoles by a cationic halogen bond donor catalyst.18 Subsequently, they used hypervalent cyclic dibenzoiodolium salts as halogen bond donors to catalyse the [4+2] cycloaddition reaction and obtained a high yield.19 Fernández et al. provided a detailed exploration of the nature of C–I···π halogen bonds and their role in organic catalysis using density functional theory (DFT) calculations.20
Dihalide molecules (I2, IBr, ICl) as neutral halogen bond donor catalysts have been widely investigated in cyclization reactions. I2 has been used as a catalyst and has shown good catalytic activity in [4+2] cycloaddition reactions.21,22 IBr has excellent diastereoselectivity in the low-temperature electrophilic cyclization of homallyl carbonates.23 ICl is generally easier to handle as a commercially available solution (1 M of dichloromethane/acetic acid), promoting its safe usage.24 Chimni et al. summarized the use of ICl for different types of cyclization reactions.25 Therefore, neutral halogen bond donor catalysts (I2, IBr, ICl) are expected to be studied for C–I···π halogen bond catalysis in the [4+2] cycloaddition reaction.
The [4+2] cycloaddition reaction is usually a one-step cyclization synergistic reaction, and the synergistic mechanism is not absolute, so there are great challenges in the research and control of the stepwise [4+2] cycloaddition reaction. It has been proven experimentally that stepwise cyclization can complete the [4+2] cycloaddition reaction (Diels–Alder reaction),26 but whether the halogen bond-catalysed [4+2] cycloaddition reaction can achieve the stepwise reaction mechanism is worthy of further study.
In this work, neutral halogen bond donors (I2, IBr, ICl) were used as catalysts, and the [4+2] cycloaddition reaction between the 2-alkenylindoles was investigated (Scheme 1). We will focus on the following points: (1) to explore the I⋯π halogen bond catalytic mechanism of the [4+2] cycloaddition reaction; (2) to search the I⋯π halogen bond catalytic modes of the [4+2] cycloaddition reaction; (3) to compare the stereoselectivity of the endo pathway and exo pathway; and (4) to give some insight into the relationship of the catalytic activity, catalytic modes, and stereoselectivity. Our study provides the basis for the selection of the catalytic mode and the control of stereoselectivity.
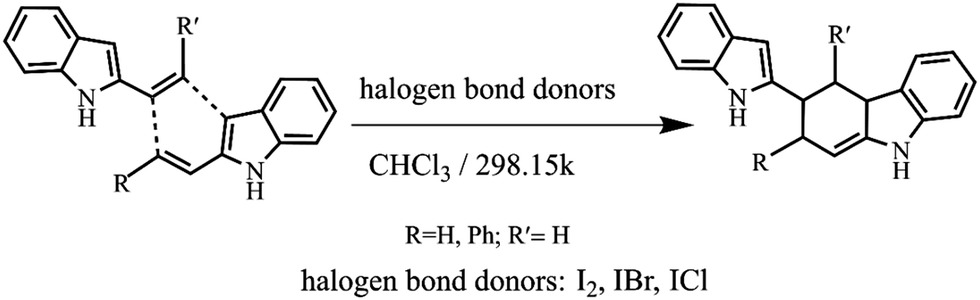 | ||
| Scheme 1 The [4+2] cycloaddition reaction between the 2-alkenylindoles catalysed by halogen bond donors (I2, IBr, ICl). | ||
2. Computational details
Density functional theory (DFT) calculations based on the M06-2X functional27 were performed using the Gaussian 16 package.28 The M06-2X functional was chosen because it provides a good explanation of halogen bonds29 and kinetics.30–32 The 6-311G** base set33 was used for C, H, N, and Cl atoms, and the aug-cc-pVTZ-PP basis set was used for Br and I atoms.34 The solvent effect in trichloromethane (CHCl3) was simulated by using the PCM implicit solvation model.35,36 The frequency of each optimized stationary point was calculated at the same theoretical level to verify the local minima point and transition state. The local minima points and transition states have zero and one imaginary frequency, respectively. In addition, it is confirmed by the intrinsic reaction coordinates (IRCs)37,38 that the transition state indeed connects the two related minima along the reaction path. To determine the effect of entropy on Gibbs free energy, the translation entropy correction of the solution was obtained by using Fang's THERMO program.39The electrostatic potential (ESP)40,41 was computed at the same level of theory and represented on the 0.001 a.u. electron density isosurface.42 The ESP and independent gradient model based on Hirshfeld partition (IGMH) were calculated with the Multiwfn program (version 3.8)43 and visualized by the VMD program.44 The 3D structure of the studied species was displayed by CYLview software.45
3. Results and discussion
3.1 The uncatalysed [4+2] cycloaddition reaction pathway between 2-vinylindoles
Fig. 1 shows the calculated Gibbs free energy curves of the uncatalysed [4+2] cycloaddition reaction of 2-vinylindoles, with the black and red lines denoting the endo and exo pathways, respectively. The total energies of the 2-vinylindoles are set to zero, and all the energies of the stationary points are relative to zero.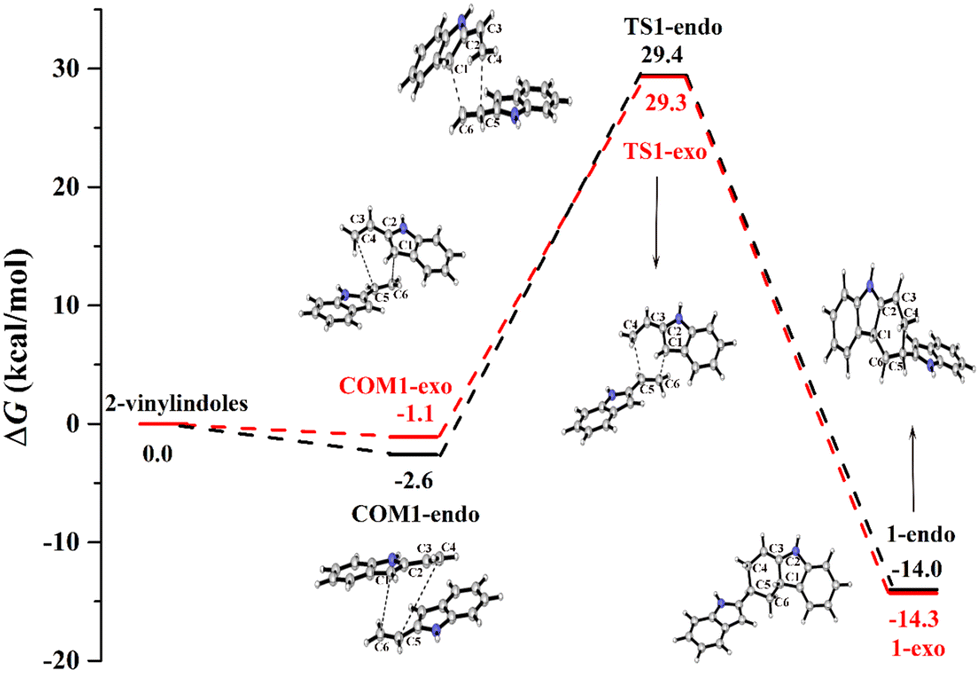 | ||
| Fig. 1 Gibbs free energy curves of the uncatalysed [4+2] cycloaddition reaction between 2-vinylindoles. | ||
For both the endo and exo pathways, when two 2-vinylindoles are close to each other, the complexes COM1-endo and COM1-exo are formed. Then, the C6 atom attacks the C1 atom, the C4 atom approaches the C5 atom, and products 1-endo and 1-exo are generated through TS1-endo and TS1-exo. The [4+2] cycloaddition reaction between the 2-vinylindoles involves a concerted mechanism, and the Gibbs free energy barriers (ΔG) of the endo and exo pathways were calculated to be 32.0 and 30.4 kcal mol−1, respectively. The Gibbs free energy barriers of TS1-endo and TS1-exo are high, which indicates that the [4+2] cycloaddition reaction is difficult to proceed without catalysts.
3.2 The mechanism of the I⋯π halogen bond catalytic [4+2] cycloaddition reaction between 2-vinylindoles
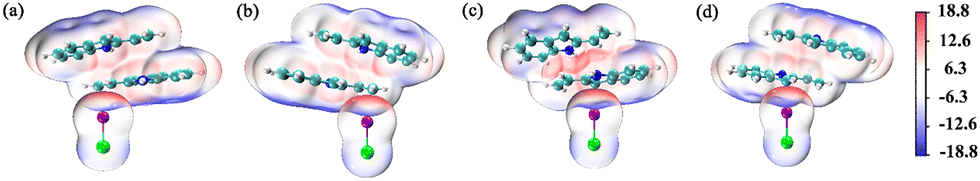
Halogen bond donors (I2, IBr, ICl) were used as catalysts to investigate the I⋯π halogen bond catalysis on the [4+2] cycloaddition reaction between 2-vinylindoles.Electrostatic potential (ESP) calculations for 2-vinylindole and halogen bond donors were performed, as shown in Fig. 2. A halogen atom X (X = Cl, Br, I) in a molecule is usually negatively charged. However, the anisotropic distribution of its electron density can lead to an electron-depleted σ-hole at the pole on the extension of the R–X bond and an electron-rich equatorial belt perpendicular to the bond direction.46 The halogen bond can be understood as the attraction between the σ-hole and the negative site of another molecule.47–49 For 2-vinylindole, the most negative values (Vs,min) exist outside the C5 and C6 atoms of ethenyl and the C7 atom of the five-membered ring, with Vs,min values of −16.0, −15.4 and −24.1 kcal mol−1, respectively. For I2, IBr, and ICl, the most positive values (Vs,max) outside the I atom along the I–X (X = I, Br, Cl) bond axis are 32.3, 43.0, and 50.8 kcal mol−1, respectively, increasing in the order of I2, IBr and ICl. Therefore, it could be predicted that I⋯π halogen bonds could be formed via either C5![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6 of ethenyl or via the C7 atom of the five-membered ring.
C6 of ethenyl or via the C7 atom of the five-membered ring.
 | ||
| Fig. 2 Electrostatic potential maps on the 0.001 a.u. (electrons per bohr3) contour of the electronic density (kcal mol−1): (a) 2-vinylindole; (b) I2; (c) IBr; (d) ICl. | ||
Fig. 3 depicts the ESP surfaces of COM1a-endo, COM1a-exo, COM1b-endo and COM1b-exo. The van der Waals surface penetration can be clearly observed between the ethenyl of 2-vinylindoles and catalyst ICl, as well as between the five-membered ring of 2-vinylindoles and catalyst ICl. Red and blue regions penetrate each other, reflecting the complementary characteristics of electrostatic potentials, verifying that the ethenyl and five-membered rings of 2-vinylindoles can form halogen bonds with the catalyst ICl, respectively.
 | ||
| Fig. 3 Electrostatic potential maps on the 0.001 a.u. (electrons per bohr3) contour of the electronic density (kcal mol−1): (a) COM1a-endo, (b) COM1a-exo, (c) COM1b-endo, (d) COM1b-exo. | ||
Fig. 4 shows the two catalytic modes of halogen bond activation to π electrons of the reactant. Mode A: The electrophilic catalysis of the I⋯π halogen bond between the catalyst and ethenyl of 2-vinylindoles; mode B: the electrophilic catalysis of the I⋯π halogen bond between the catalyst and the five-membered ring of 2-vinylindoles.
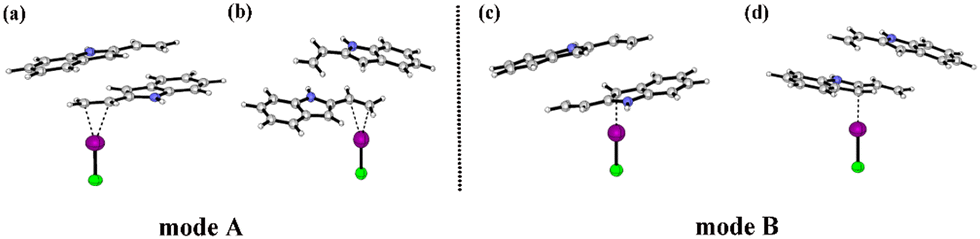 | ||
| Fig. 4 Two modes of halogen bond activation to the reactant. Mode A: I⋯π halogen bond catalysis on the ethenyl of 2-vinylindoles; Mode B: I⋯π halogen bond catalysis on the five-membered ring of 2-vinylindoles. (a) COM1a-endo, (b) COM1a-exo, (c) COM1b-endo, (d) COM1b-exo. | ||
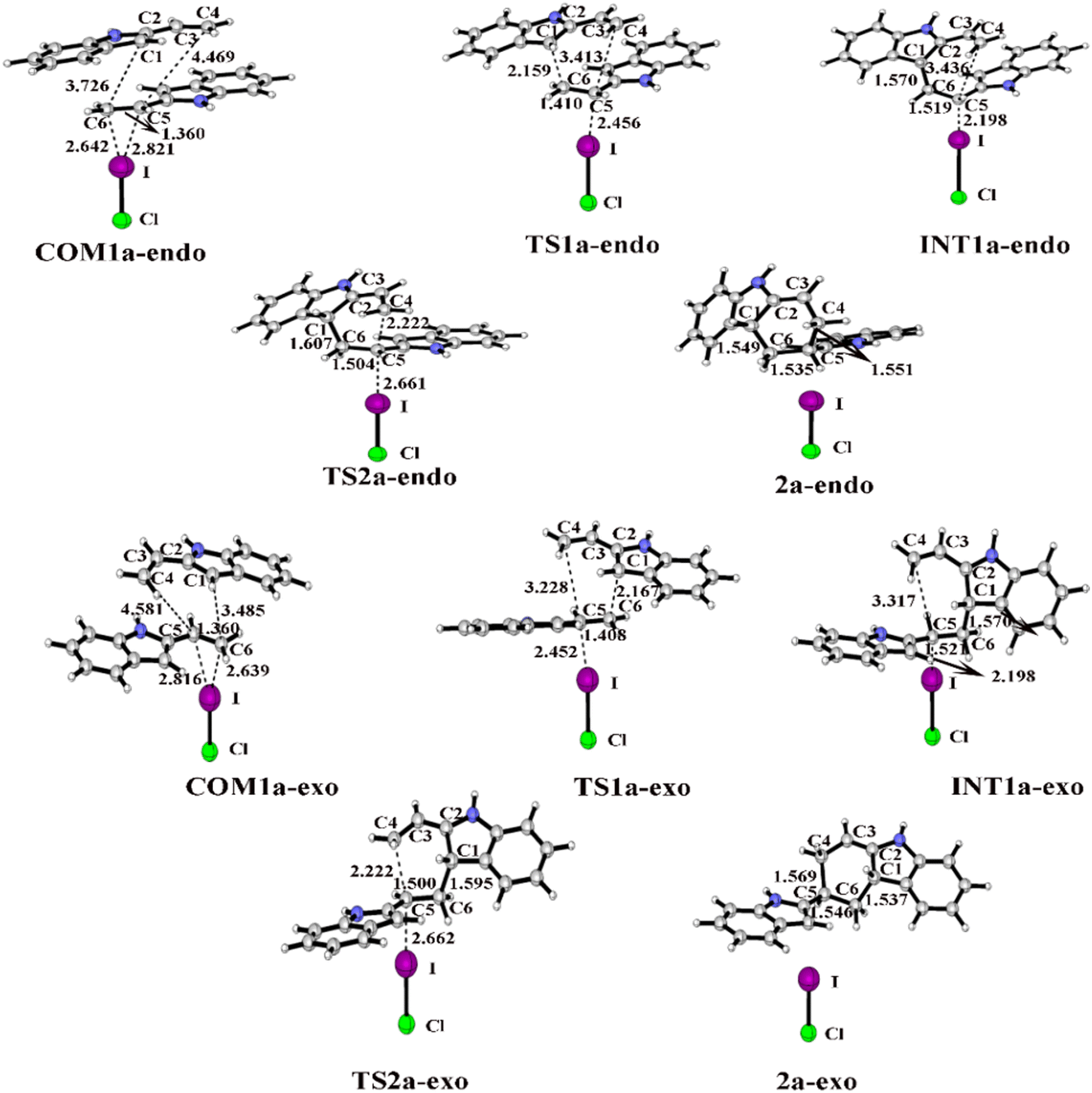 | ||
| Fig. 5 Optimized geometries and key bond lengths (in Å) along the IRC pathway of I⋯π halogen bond catalysis via ICl activation on the ethenyl of 2-vinylindole. | ||
When the catalyst attacks the ethenyl of 2-vinylindole, COM1a is formed. There are two stereo geometries in the endo and exo pathways, namely, COM1a-endo and COM1a-exo, respectively. From Tables S1 and S2 (ESI†), the distances of I⋯C5 and I⋯C6 in COM1a-endo and COM1a-exo are obviously shorter than the corresponding sum of van der Waals radii (3.68 Å) of I and C atoms, indicating that an I⋯π halogen bond is formed between the catalyst and ethenyl of 2-vinylindole. The I⋯C5 and I⋯C6 distances in COM1a-endo and COM1a-exo decrease along the sequence of I2, IBr, and ICl, which are in accordance with the Vs,max value of ESP. The larger the Vs,max value of the halogen bond donor catalyst is, the shorter the I⋯π halogen bond distances and the stronger the halogen bond interaction.
With I⋯π halogen bond catalysis, the [4+2] cycloaddition reaction of 2-vinylindoles undergoes a stepwise mechanism, which is different from the concerted mechanism of the uncatalysed reaction. Both the endo and exo pathways involve two steps: the step of C1–C6 bond formation and the step of six-membered ring formation. The stepwise pathway of the I⋯π halogen bond catalytic [4+2] cycloaddition reaction in this work agrees with that of the electric field–catalysed single-molecule Diels–Alder reaction, which has been observed and confirmed in the experiment.26
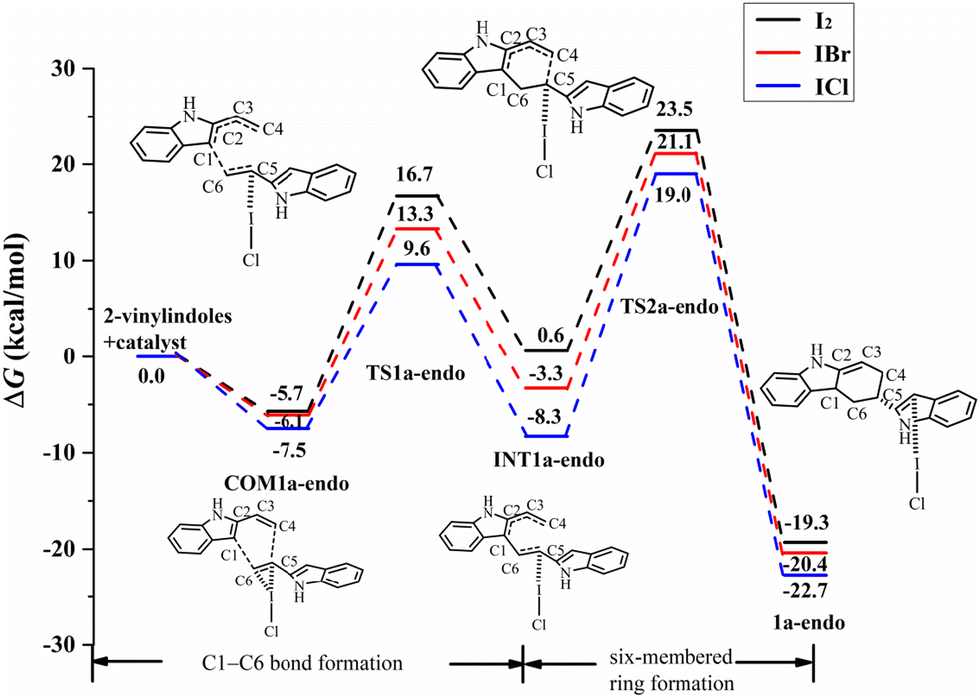
For the endo pathway of the [4+2] cycloaddition reaction, Fig. 6 shows the Gibbs free energy curves of the endo pathway via I⋯π halogen bond catalysis on the ethenyl of 2-vinylindoles. Taking the catalyst ICl as an example, the catalytic processes were investigated. The catalytic reaction starts from COM1a-endo with a Gibbs free energy of −7.5 kcal mol−1, and the distances of C1–C6, C4–C5, and C5–C6 are 3.726, 4.469 and 1.360 Å, respectively. After COM1a-endo, the C6 atom attacks the C1 atom, and the C5 atom approaches the C4 atom to form TS1a-endo. In TS1a-endo, the distances of C1–C6 and C4–C5 are 2.159 and 3.413 Å, respectively. After TS1a-endo, the C5–C6 distance is lengthened. When the C1–C6 distance reaches 1.570 Å, the C1–C6 bond is formed, and INT1a-endo is reached. The C1–C6 bond formation step occurs with a Gibbs free energy barrier (ΔG) of 17.1 kcal mol−1. After INT1a-endo, the C4–C5 distance continues to be shortened to form a six-membered ring product 2a-endo via TS2a-endo. When the distances of C1–C6 and C4–C5 become 1.549 and 1.551 Å, the product 2a-endo is formed. The six-membered ring formation step is the rate-determining step, with a ΔG of 27.3 kcal mol−1 relative to INT1a-endo.
 | ||
| Fig. 6 Gibbs free energy curves of the endo pathway via I⋯π halogen bond catalysis on the ethenyl of 2-vinylindole (mode A). | ||
For the exo pathway of the [4+2] cycloaddition reaction, Fig. 7 shows the Gibbs energy curves of the endo pathway via I⋯π halogen bond catalysis on the ethenyl of 2-vinylindole. The catalytic reaction starts from COM1a-exo with a Gibbs free energy of −6.4 kcal mol−1, and the distances of C4–C5, C1–C6 and C5C6 are 4.581, 3.485 and 1.360 Å, respectively. From COM1a-exo to INT1a-exo, the C1–C6 distance is shortened until the C1–C6 bond is formed. After INT1a-exo, the C4–C5 distance is constantly shortened, from 3.317 Å in INT1a-exo to 1.569 Å in 2a-exo. The ΔG values of the first step of C1–C6 bond formation and the second step of six-membered ring formation are 15.9 and 26.0 kcal mol−1, respectively.
 | ||
| Fig. 7 Gibbs free energy curves of the exo pathway via I⋯π halogen bond catalysis on the ethenyl of 2-vinylindole (mode A). | ||
The catalysts I2 and IBr also catalysed the [4+2] cycloaddition reaction by I⋯π halogen bonds. For I2, IBr, and ICl, the Gibbs free energies of COM1a-endo are −5.7, −6.1, and −7.5 kcal mol−1, respectively, and those of COM1a-exo are −4.6, −5.4, and −6.4 kcal mol−1, respectively, as shown in Fig. 6 and 7. According to the order of I2, IBr and ICl, the exothermic heat of COM1a increases with the gradual strengthening of the halogen bond. For the endo pathway catalysed by I2, IBr and ICl, the ΔG values needed for the formation of the C1–C6 bond are 22.4, 19.4, and 17.1 kcal mol−1, and those needed for the formation of the six-membered ring are 29.2, 27.2, and 27.3 kcal mol−1, respectively. For the exo pathway catalysed by I2, IBr and ICl, the ΔG values of C1–C6 bond formation are 21.2, 18.2, and 15.9 kcal mol−1, respectively, and those of six-membered ring formation are 27.0, 25.4, and 26.0 kcal mol−1, respectively.
Comparing the ΔG values of the two steps, the formation of the six-membered ring is the rate-determining step for mode A. For the process of six-membered ring formation, the ΔG values of the exo pathway are somewhat lower than those of the endo pathway, indicating that the [4+2] cycloaddition reaction via I⋯π halogen bond catalysis on the ethenyl of 2-vinylindole can proceed smoothly, the exo pathway is preferred, and the product of the exo pathway is the main product. In the products of 2a-endo and 2a-exo, the interaction between the catalysts (I2, IBr, ICl) and the substrate is a relatively weaker halogen bond than that of COM1a-exo and COM1a-endo, indicating that the catalysts (I2, IBr, ICl) can be separated easily and then carry out the catalytic cycle.
For the endo pathway of the [4+2] cycloaddition reaction, the optimized geometries and key bond lengths along the IRC pathway of the [4+2] cycloaddition reaction are shown in Fig. 8. Taking the ICl catalyst as an example, the distances of C1⋯C6 and C4⋯C5 in COM1b-endo are 3.748 and 4.524 Å, respectively, and the C1⋯C6 distance is shorter than that of C4⋯C5, indicating that C1⋯C6 forms a bond before C4⋯C5. The C1⋯C6 distance gradually shortened from 3.748 Å in COM1b-endo to 1.595 Å in INT1b-endo, and a C1–C6 bond was formed in INT1b-endo.
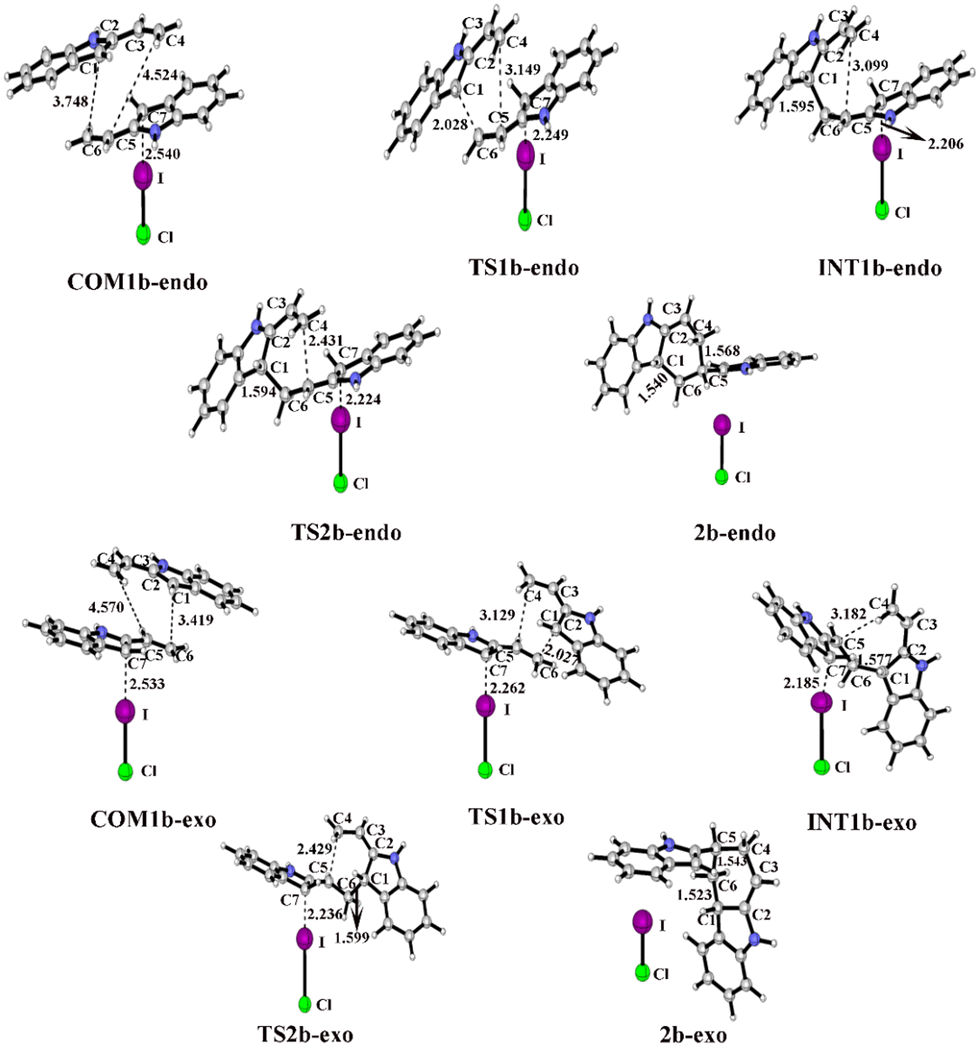 | ||
| Fig. 8 Optimized geometries and key bond lengths (in Å) along the IRC pathway of I⋯π halogen bond catalysis via ICl activation on the five-membered ring of 2-vinylindole. | ||
For the endo pathway of the [4+2] cycloaddition reaction, Fig. 9 shows the Gibbs free energy curves via I⋯π halogen bond catalysis on the five-membered ring of 2-vinylindole. COM1b-endo undergoes C1–C6 bond formation via TS1b-endo to INT1b-endo with a ΔG value of 18.0 kcal mol−1 relative to COM1b-endo, and C1–C6 bond formation is the rate-determining step for the endo pathway. After INT1b-endo, the endo pathway is accompanied by a shortening of the C4–C5 distance from 3.099 Å in INT1b-endo to 1.568 Å in 2c-endo. As shown in Fig. 9, the ΔG value of six-membered ring formation is 15.5 kcal mol−1 relative to COM1b-endo.
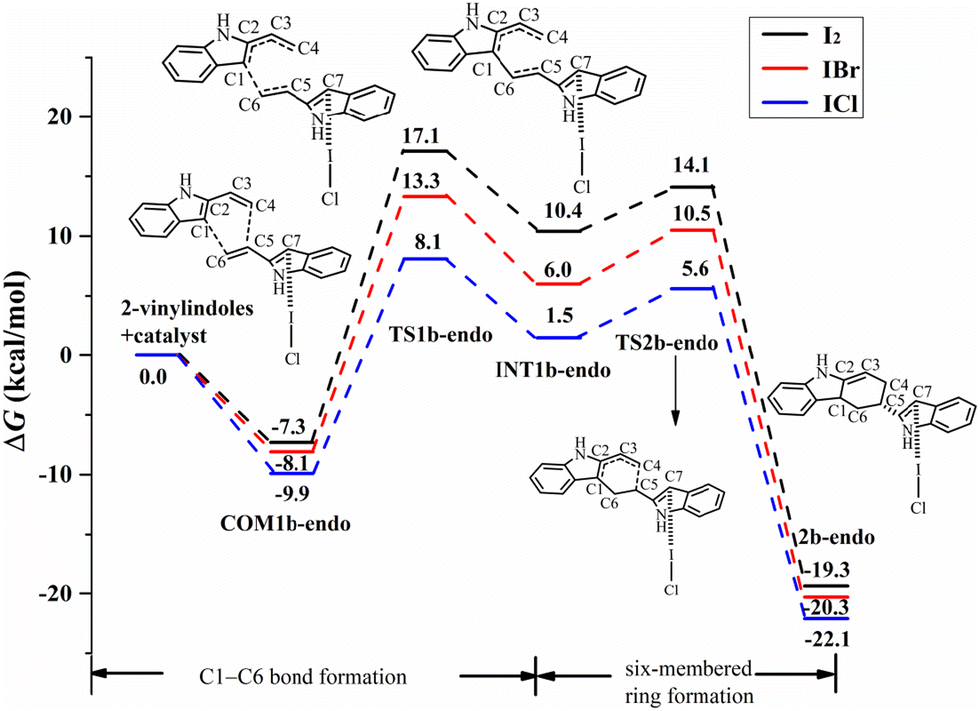 | ||
| Fig. 9 Gibbs free energy curves of the endo pathway via I⋯π halogen bond catalysis on the five-membered ring of 2-vinylindole (mode B). | ||
For the exo pathway of the [4+2] cycloaddition reaction, Fig. 10 shows the Gibbs free energy curves via I⋯π halogen bond catalysis on the five-membered ring of 2-vinylindole. A stable complex is formed via 2-vinylindoles with halogen bond donor ICl, namely, COM1b-exo, and the Gibbs free energy is −9.9 kcal mol−1. Then, INT1b-exo was formed by shortening the C1–C6 distance from 3.419 Å in COM1b-exo to 1.577 Å in INT1b-exo via TS1b-exo, which requires an energy barrier of 19.8 kcal mol−1, and C1–C6 bond formation is the rate-determining step for the exo pathway. After INT1b-exo, the C4–C5 distance (3.182 Å) in INT1b-exo was noticeably shortened compared to that in 2b-exo (1.543 Å). The calculated ΔG value for this step is 17.4 kcal mol−1 relative to COM1b-exo.
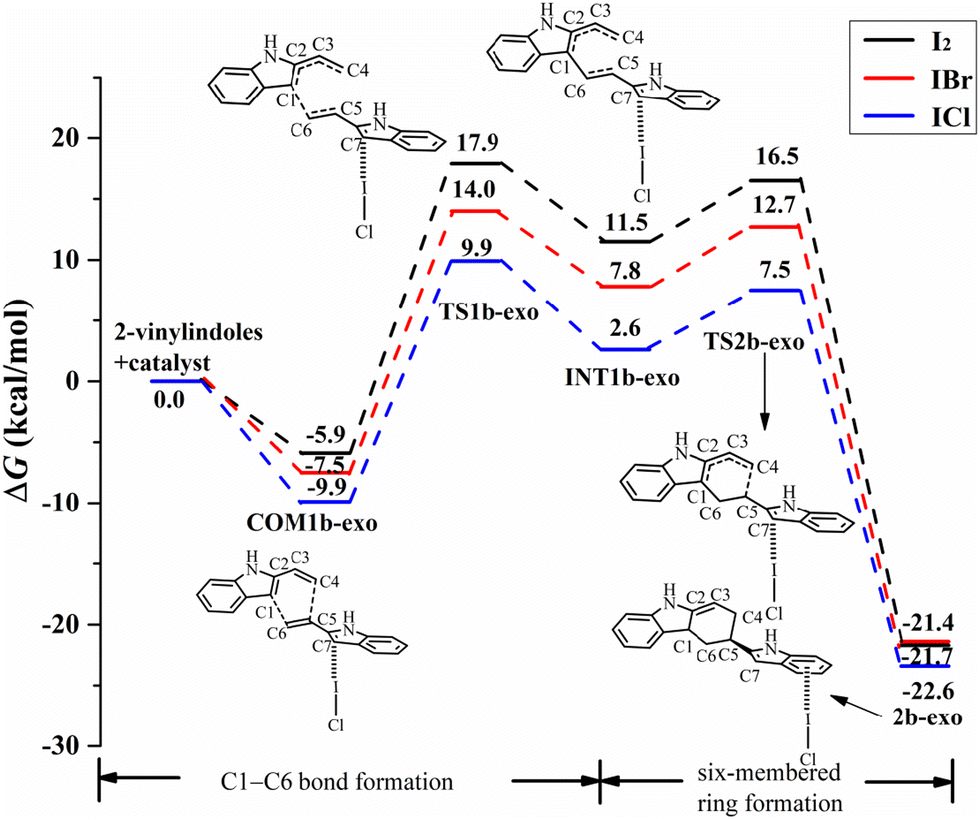 | ||
| Fig. 10 Gibbs free energy curves of the exo pathway via I⋯π halogen bond catalysis on the five-membered ring of 2-vinylindole (mode B). | ||
The catalysts I2 and IBr also catalysed the [4+2] cycloaddition reaction by I⋯π halogen bonds with the five-membered ring of 2-vinylindole, as shown in Fig. 9 and 10. The reaction mechanism is the same as that of ICl catalysis. The order of ΔG values of the halogen bond catalyst was I2 > IBr > ICl, and the catalytic activity of the halogen bond donor catalyst increased in the order of I2, IBr and ICl, which was consistent with the law of Vs,max value of ESP.
Comparing the Gibbs free energy barriers of the two steps of mode B, the C1–C6 bond formation step has a higher Gibbs free energy barrier than the six-membered ring formation step, so the C1–C6 bond formation step is the rate-determining step. For the rate-determining step, the ΔG value of the exo pathway catalysed by I2 is somewhat lower than that of the endo pathway, and the endo product can be obtained by I2 catalysis. For IBr and ICl, the ΔG values of the endo pathway are lower than those of the exo pathway, and the endo products are easier to obtain. Therefore, an increasing proportion of endo products can be obtained in the order of catalyst I2, IBr and ICl. It is further indicated that the catalytic activity of the catalyst has a certain effect on the stereoselectivity of the reaction. Different reaction products can be obtained by controlling the catalyst and different reaction modes.
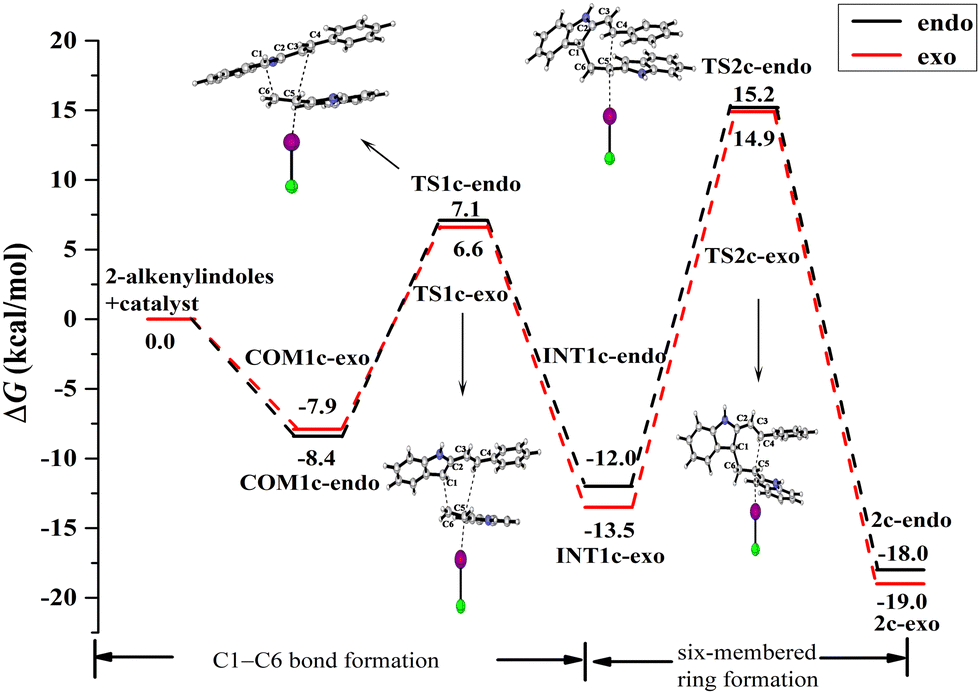 | ||
| Fig. 11 ICl-catalysed Gibbs free energy curves of the endo- and exo-pathways via mode A for the reactant substituent R = Ph. | ||
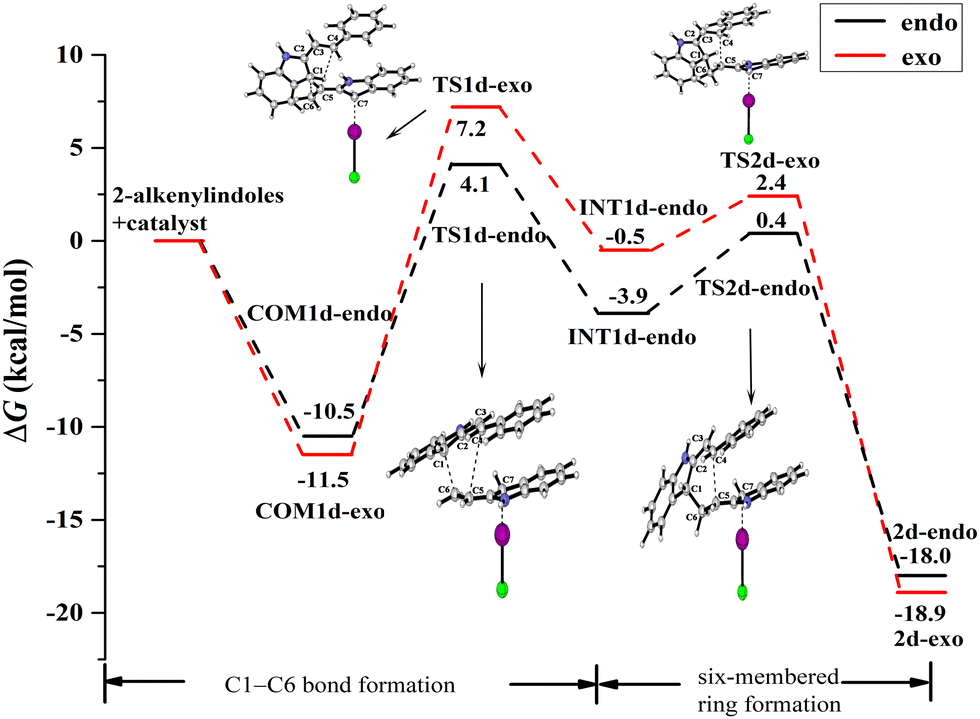 | ||
| Fig. 12 Gibbs free energy curves of endo and exo pathways via mode B when the reactant substituent R = Ph. | ||
For the endo pathway of mode A, the formation of the C1–C6 bond and the six-membered ring needs to overcome the ΔG of 15.5 kcal mol−1 and 27.2 kcal mol−1, respectively. For the exo pathway of mode A, the formation of the C1–C6 bond and the six-membered ring needs to overcome the ΔG of 14.5 kcal mol−1 and 28.4 kcal mol−1, respectively. For mode A, as shown in Fig. 13a and b, the N–H⋯π interaction exists between the two reaction substrates in COM1c-endo and INT1c-endo along the endo pathway, while the N–H⋯π interaction does not exist in COM1c-exo and INT1c-exo along the exo pathway. When the substituent R = H of mode A, the exo pathway is somewhat easier than the endo pathway. While for the substituent R = Ph of mode A, the N–H⋯π interaction makes the ΔG value of the endo pathway (27.2 kcal mol−1) lower than that of the exo pathway (28.4 kcal mol−1) at the rate-determining step. Therefore, the N–H⋯π interaction promotes high endo selectivity.
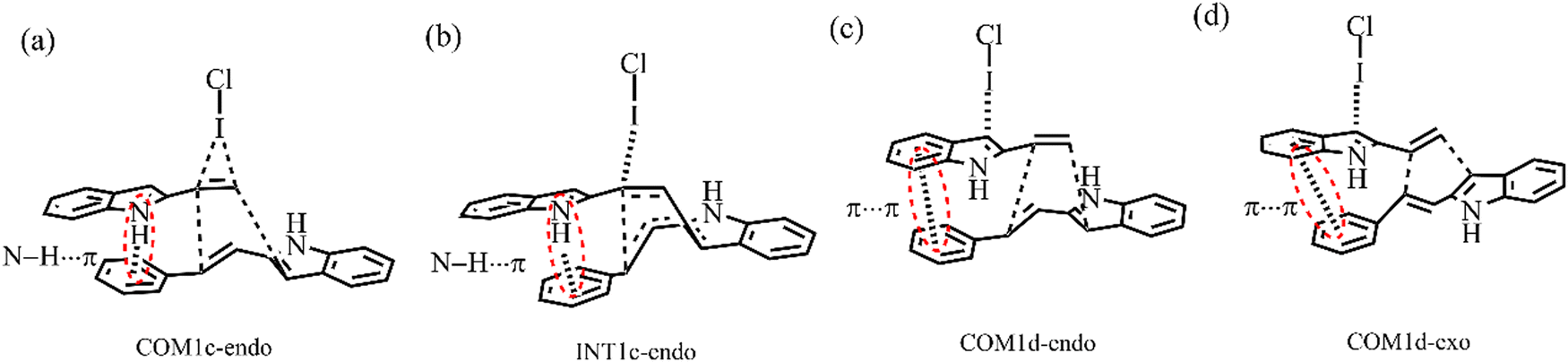 | ||
| Fig. 13 Structures of COM1c-endo, INT1c-endo, COM1d-endo, COM1d-exo. | ||
For the endo pathway of mode B, the formation of the C1–C6 bond and the six-membered ring formation needs to overcome the ΔG of 14.6 kcal mol−1 and 10.9 kcal mol−1, respectively. For the exo pathway of mode B, the calculated ΔG values for C1–C6 bond formation and six-membered ring formation are 18.7 and 13.9 kcal mol−1, respectively. The ΔG values of the endo pathway are lower than those of the exo pathway, the endo product is more easily generated for mode B, not only for the substituent is H but also Ph. When the substituent R = Ph, due to the π⋯π interaction between the two substrates in COM1d-endo and COM1d-exo (Fig. 13c and d), the ICl-catalysed ΔG values are significantly lower than R = H, which is consistent with the experimental observation that R = Ph has the higher yield than R = H.18 For the substituent R = Ph, the π⋯π interaction promotes the [4+2] cycloaddition reaction.
3.3 IGMH analysis
IGMH can intuitively understand the interaction of chemical systems by analysing molecular and periodic systems, weak interactions and chemical bonds.50,51Fig. 14 shows the IGMH plot of the COM1a-endo, COM1a-exo, COM1b-endo and COM1b-exo. Blue isosurfaces exist between the ethenyl of 2-vinylindoles and ICl in COM1a-endo and COM1a-exo, also exist between the five-membered ring of 2-vinylindoles and ICl in COM1b-endo and COM1b-exo. The IGMH plot analysis proves the weak attractive property of the interaction. Therefore, halogen-bonded interactions exist between the ethenyl of 2-vinylindoles and ICl, also exist between the five-membered ring of 2-vinylindoles and ICl.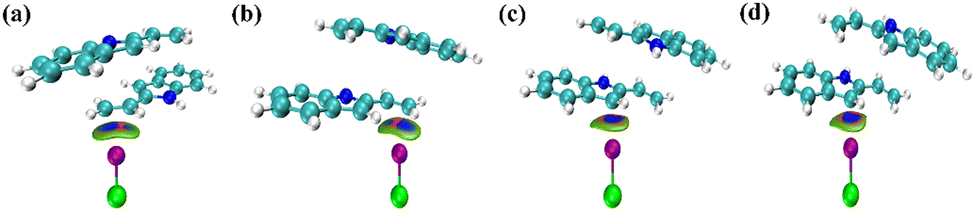 | ||
| Fig. 14 IGMH coloring isosurface maps for the COM1a-endo (a), COM1a-exo (b), COM1b-endo (c) and COM1b-exo (d). | ||
4. Conclusions
The halogen bond donor (I2, IBr, ICl)-catalysed [4+2] cycloaddition reaction between the 2-alkenylindoles was investigated based on DFT calculations. The catalytic modes, mechanism, and stereoselectivity have been systematically discussed. For the uncatalysed reaction, both the endo pathway and exo pathway are carried out in a concerted mechanism, and it is difficult to proceed at room temperature because of the high energy barriers.For the [4+2] cycloaddition reaction between 2-alkenylindoles, the catalytic process facilitated by halogen bond donors (I2, IBr, ICl) includes two modes: mode A refers to the electrophilic catalysis of the I⋯π halogen bond formed between the catalyst and ethenyl of 2-alkenylindole; mode B refers to the catalyst attacking the five-membered ring of 2-alkenylindole. Both mode A and mode B are stepwise and involve two steps: the formation of carbon–carbon bonds and the formation of six-membered rings. The formation of the six-membered ring is the rate-determining step for mode A, and the formation of the carbon–carbon bond is the rate-determining step for mode B.
Both catalytic modes include the exo pathway and endo pathway. For mode A, when the substituent R = H, the exo-pathway is somewhat easier than the endo-pathway. For the substituent R = Ph, the N–H⋯π interaction makes the reaction barrier of the endo-pathway lower than that of the exo-pathway at the rate-determining step. The N–H⋯π interaction promotes the high endo selectivity of mode A. For mode B, an increasing proportion of endo products can be obtained in the order of catalyst I2, IBr and ICl. For the substituent R = Ph, the π⋯π interaction results in lower reaction energies in both the endo and exo pathways compared with R = H at the rate-determining step, which is consistent with the experimental observation that R = Ph has a higher yield than R = H. Overall, different catalytic modes and stereoselectivities can expand the study of [4+2] cycloaddition reactions in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Natural Science Foundation of Hebei Province (B2022205022), the Science Research Project of Hebei Education Department (ZD2022103), the Innovation Capability Improvement Plan Project of Hebei Province (22567604H) and Graduate Innovation Ability Cultivation Funding Project of Hebei Province (Grants No. CXZZBS2023089).References
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Noncovalent Interactions in Catalysis, Royal Society of Chemistry, London, 2019 Search PubMed.
- A. E. Settle, L. Berstis, S. Zhang, N. A. Rorrer, H. Hu, R. M. Richards, G. T. Beckham, M. F. Crowley and D. R. Vardon, ChemSusChem, 2018, 11, 1768–1780 CrossRef CAS PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- C. Bolm, A. Bruckmann and M. Pena, Synlett, 2008, 900–902 CrossRef.
- A. Matsuzawa, S. Takeuchi and K. Sugita, Chem. – Asian J., 2016, 11, 2863–2866 CrossRef CAS PubMed.
- V. Mamane, P. Peluso, E. Aubert, R. Weiss, E. Wenger, S. Cossu and P. Pale, Organometallics, 2020, 39, 3936–3950 CrossRef CAS.
- S. Portela, J. J. Cabrera-Trujillo and I. Fernandez, J. Org. Chem., 2021, 86, 5317–5326 CrossRef CAS PubMed.
- H. Zhang and P. H. Toy, Adv. Synth. Catal., 2020, 363, 215–221 CrossRef.
- X. Liu, S. Ma and P. H. Toy, Org. Lett., 2019, 21, 9212–9216 CrossRef CAS PubMed.
- Y. Wang and P. Su, ACS Omega, 2020, 5, 21862–21872 CrossRef CAS PubMed.
- A. Dreger, E. Engelage, B. Mallick, P. D. Beer and S. M. Huber, Chem. Commun., 2018, 54, 4013–4016 RSC.
- K. Takagi, K. Yamauchi and H. Murakata, Chem. – Eur. J., 2017, 23, 9495–9500 CrossRef CAS PubMed.
- N. Tarannam, M. H. H. Voelkel, S. M. Huber and S. Kozuch, J. Org. Chem., 2022, 87, 1661–1668 CrossRef CAS PubMed.
- C. Zhao, C. Sun, X. Li and Y. Zeng, ChemistrySelect, 2021, 6, 12843–12851 CrossRef CAS.
- W. He, Y. C. Ge and C. H. Tan, Org. Lett., 2014, 16, 3244–3247 CrossRef CAS PubMed.
- S. Kuwano, T. Suzuki, M. Yamanaka, R. Tsutsumi and T. Arai, Angew. Chem., Int. Ed., 2019, 58, 10220–10224 CrossRef CAS PubMed.
- Y. Nishida, T. Suzuki, Y. Takagi, E. Amma, R. Tajima, S. Kuwano and T. Arai, ChemPlusChem, 2021, 86, 741–744 CrossRef CAS PubMed.
- S. Portela and I. Fernández, Eur. J. Org. Chem., 2021, 6102–6110 CrossRef CAS.
- P. Vermeeren, T. A. Hamlin, I. Fernandez and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2020, 59, 6201–6206 CrossRef CAS.
- S. P. Chavan, P. Sharma, G. Rama Krishna and M. Thakkar, Tetrahedron Lett., 2003, 44, 3001–3003 CrossRef CAS.
- J. J. Duan and A. B. Smith III, J. Org. Chem., 1993, 58, 3703–3711 CrossRef CAS.
- D. P. Day, N. I. Alsenani and A. A. Alsimaree, Eur. J. Org. Chem., 2021, 4299–4307 CrossRef CAS.
- S. Chimni and S. Singh, Synthesis, 2015, 1961–1989 CrossRef.
- C. Yang, Z. Liu, Y. Li, S. Zhou, C. Lu, Y. Guo, M. Ramirez, Q. Zhang, Y. Li, Z. Liu, K. N. Houk, D. Zhang and X. Guo, Sci. Adv., 2021, 7, eabf0689 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- S. Kozuch and J. M. Martin, J. Chem. Theory Comput., 2013, 9, 1918–1931 CrossRef CAS PubMed.
- J. Y. See, H. Yang, Y. Zhao, M. W. Wong, Z. Ke and Y.-Y. Yeung, ACS Catal., 2018, 8, 850–858 CrossRef CAS.
- H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808–5818 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- E. Cancès and B. Mennucci, J. Chem. Phys., 2001, 114, 4744–4745 CrossRef.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- K. Ishida, K. Morokuma and A. Komornicki, J. Chem. Phys., 1977, 66, 2153–2156 CrossRef CAS.
- D. C. Fang, THERMO, Beijing Normal University, Beijing, China, 2013 Search PubMed.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
- T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- J. S. Murray, P. Lane and P. Politzer, Int. J. Quantum Chem., 2007, 107, 2286–2292 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Model., 1996, 14, 33–38 CAS.
- C. Y. Legault, CYLview 20, Université de Sherbrooke, 2020, https://www.cylview.org Search PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- A. V. Rozhkov, E. A. Katlenok, M. V. Zhmykhova, A. Y. Ivanov, M. L. Kuznetsov, N. A. Bokach and V. Y. Kukushkin, J. Am. Chem. Soc., 2021, 143, 15701–15710 CrossRef CAS PubMed.
- T. Lu and Q. Chen, Comprehensive Computational Chemistry, 2024, vol. 2, pp. 240–264 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The I⋯C5 and I⋯C6 distances (in Å) in COM1a-endo catalysed by I2, IBr, and ICl; the I⋯C5 and I⋯C6 distances (in Å) in COM1a-exo catalysed by I2, IBr, and ICl; calculated Gibbs free energy and XYZ coordinates. See DOI: https://doi.org/10.1039/d3cp05479a |
| This journal is © the Owner Societies 2024 |