
Degradation of NTO induced by superoxide and hydroperoxyl radicals: a comprehensive DFT study†
Liudmyla K.
Sviatenko
 a,
Leonid
Gorb
bc and
Jerzy
Leszczynski
*a
a,
Leonid
Gorb
bc and
Jerzy
Leszczynski
*a
aInterdisciplinary Center for Nanotoxicity, Department of Chemistry, Physics & Atmospheric Sciences, Jackson State University, Jackson, Mississippi 39217, USA. E-mail: jerzy@icnanotox.org
bInstitute of Molecular Biology and Genetics, NAS of Ukraine, 150 Zabolotny Str., Kyiv, 03143, Ukraine
cQSAR Lab Sp. z o.o. Trzy Lipy 3, B, Gdansk, 80-172, Poland
First published on 29th November 2023
Abstract
Reactive oxygen species, produced in the aquatic environment under sunlight irradiation, actively take part in degradation of environmental pollutants. NTO (5-nitro-1,2,4-triazol-3-one), being a primary ingredient in a suite of insensitive munitions formulations, may be released into training range soils after incomplete detonations and dissolved in surface water and groundwater due to good water solubility. A detailed investigation of a possible mechanism for NTO decomposition in water induced by superoxide and hydroperoxyl radicals as one of the pathways for NTO environmental degradation was performed with a computational study at the PCM(Pauling)/M06-2X/6-311++G(d,p) level. Superoxide causes rapid deprotonation of NTO. Decomposition of NTO induced by hydroperoxyl radicals was found to be a multistep process leading to mineralization of the nitrocompound. The reaction process may begin with hydroperoxyl radical attachment to carbon atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond of NTO, then proceeds through rupture of C–N bonds and addition of water molecules leading to the formation of nitrous acid, ammonia, nitrogen gas, hydrazine, and carbon(IV) oxide. The obtained results indicate that the anionic form of NTO shows a higher reactivity towards hydroperoxyl radicals than its neutral form. Excitation of NTO by sunlight enables complete mineralization of NTO induced by superoxide. The calculated activation energies and exergonicity of the studied processes support the contribution of hydroperoxyl radicals and superoxide to the degradation of NTO in the environment into low-weight inorganic compounds.
N double bond of NTO, then proceeds through rupture of C–N bonds and addition of water molecules leading to the formation of nitrous acid, ammonia, nitrogen gas, hydrazine, and carbon(IV) oxide. The obtained results indicate that the anionic form of NTO shows a higher reactivity towards hydroperoxyl radicals than its neutral form. Excitation of NTO by sunlight enables complete mineralization of NTO induced by superoxide. The calculated activation energies and exergonicity of the studied processes support the contribution of hydroperoxyl radicals and superoxide to the degradation of NTO in the environment into low-weight inorganic compounds.
Introduction
Highly energetic substances, used as explosive materials, cause pollution when they enter the environment, creating a toxic hazard for the biota. All over the world, soils and natural waters are contaminated with such pollutants due to munitions dumping, military activities, industrial operations, etc. NTO (5-nitro-1,2,4-triazol-3-one) is a powerful insensitive explosive, present in industrial waste waters as well as in training fields.1 A variety of possible pathways for NTO decomposition in the environment make it of considerable interest for investigation into both experimental and computational chemistry. NTO dissolves easily in surface water and groundwater due to good water solubility2 and undergoes photo-transformation, which is a complex process that involves direct and indirect photolysis.3–8 Direct photolysis is considered as a hydrolytic decomposition of NTO in the triplet state. Indirect photolysis of NTO involves reactions with reactive oxygen species (singlet oxygen, superoxide, hydroperoxyl radical, hydroxyl radical, etc.), generated in water solution under UV-irradiation. The identified aqueous products of NTO photo-transformation are nitrite, nitrate, ammonium and 5-hydroxy-1,2,4-triazol-3-one or 1,2,4-triazolidine-3,5-dione – its isomer.3–8 Understanding of the mechanism of direct and indirect photochemical degradation of NTO is necessary in order to predict and control its environmental fate and persistence. Reactions of direct photolysis of NTO and its decomposition induced by singlet oxygen were modeled recently.9,10 In the present study, we examine the ability of superoxide and hydroperoxyl radicals to take part in degradation of NTO.Superoxide (O2−˙), as one of the reactive species, may be produced as a result of donation of an electron to oxygen by a photosensitizer after absorption of light.11 The conjugate acid of the superoxide is a hydroperoxyl radical (HO2˙), which is produced upon sunlight irradiation of components such as dissolved organic matter (DOM), nitrate, nitrite, and Fe(III) present in natural waters. It plays an important role in decomposition of organic pollutants.12–14 A hydroperoxyl radical is a stronger oxidant than a superoxide, which is evidenced from the experimental reduction potentials of 1.06 and 0.94 eV for HO2˙ and O2−˙, respectively.14 An experimental pKa of 4.88 indicates that in neutral pH, the major form of the superoxide reactant is the anionic form.15
The investigation of reactivity of several classes of organic compounds in nucleophilic substitution reactions with superoxide showed that the nucleophilic attack of O2−˙ at the O-alkyl carbon is a common mechanism of decomposition of organic carbonates, aliphatic carboxylic esters, lactones. However, N-alkyl substituted amides, lactams, and ethers (glymes) are stable against nucleophilic substitution by a superoxide.16 Nucleophilic reactions of O2−˙ with phenol esters of carboxylic acids proceed via attack at the carbonyl carbon.16 The one-electron transfer from O2−˙ was found as the major reaction channel for reaction of superoxide with 2,4,6-trinitrotoluene, while the proton abstraction reaction was negligible, which interestingly is in contrast to the observed deprotonation of 2,4-dinitrotoluene, 2-nitrotoluene, and quercetin.17,18 Reaction of superoxide with 5,5-dimethyl-1-pyrroline N-oxide occurs with the addition of O2−˙ to carbon atom of the double CN bond.19 The addition of HO2˙ to 5,5-dimethyl-1-pyrroline N-oxide is predicted to be more exoergic with a lower energy barrier than the addition of O2−˙. This is experimentally confirmed. The hydrogen atom abstraction and radical adduct formation were suggested as possible initial reaction routes between HOO˙ and coumarin derivatives.13
The current study is aimed to model NTO degradation induced by superoxide and hydroperoxyl radicals in aqueous solution to evaluate the possibility of NTO mineralization. Based on the structure of NTO and available literature data on mechanisms of reactions of superoxide and hydroperoxyl radicals with different classes of organic compounds13,14,16–19 the three directions for the reaction between superoxide and NTO will be considered: the nucleophilic attack of O2−˙ onto two carbon atoms of NTO, the deprotonation of NTO, and the one-electron transfer from O2−˙. The three pathways will also be discussed for the reaction of hydroperoxyl radicals with NTO: the attachment of HO2˙ to carbon atoms of NTO, the hydrogen atom transfer from NTO to HO2˙, and the one-electron transfer from HO2˙. The studied reactions will be modeled for NTO and its anion (NTOa). NTO deprotonated at the N4 position was chosen for calculation due to its increased stability as compared with NTO deprotonated at the N2 position.20
Methods
The geometries of all structures were optimized within the Gaussian 09 program21 using the M06-2X functional of the density functional theory (DFT) and 6-311++G(d,p) basis set.22–25 The M06-2X functional was recommended for applications involving thermochemistry, kinetics, and noncovalent interactions on the base of assessment of its performance over a broad amount of data.22 The influence of water bulk was simulated within the PCM model in combination with Pauling radii.26 Stationary points for modeled reactions were characterized as either local minima (reactant, intermediate, and product) having all real frequencies, or as transition states possessing only one imaginary frequency. This was carried out by calculation of the analytic harmonic vibrational frequencies at the PCM(Pauling)/M06-2X/6-311++G(d,p) theory level. The activation energy for electron transfer was calculated from Marcus theory using the internal reorganization energies and the reaction Gibbs free energies.27 The Cartesian coordinates for optimized local minima and transition states species are provided in the ESI.†Results and discussion
Superoxide induced degradation of NTO and NTOa
The reactions of NTO with superoxide in water in this study along with the corresponding Gibbs free energy diagram are shown in Fig. 1. Structures of the corresponding transition states are shown in Fig. S1 (ESI†). There are several possible directions for the initial attack of superoxide onto NTO. They are superoxide attachment to carbon atom of NTO, proton detachment from NTO, and electron transfer from superoxide to NTO. The possibility of superoxide addition to the C3 carbon atom can be excluded, because this would lead to intermediate INT1 lying 41.39 kcal mol−1 above the reactants. The obtained high activation energy for attachment of superoxide to carbon atom of the carbonyl group corresponds to the values reported in the study of mechanism of reaction between the superoxide and organic carbonates, aliphatic carboxylic esters, lactones.15 The addition of superoxide to carbon atom C5 requires an activation energy of 25.03 kcal mol−1, which might not be easily accessible at room temperature. Once this transition state is reached, unstable INT2 undergoes nearly barrierless exergonic C–N bond rupture leading to nitrite anion release.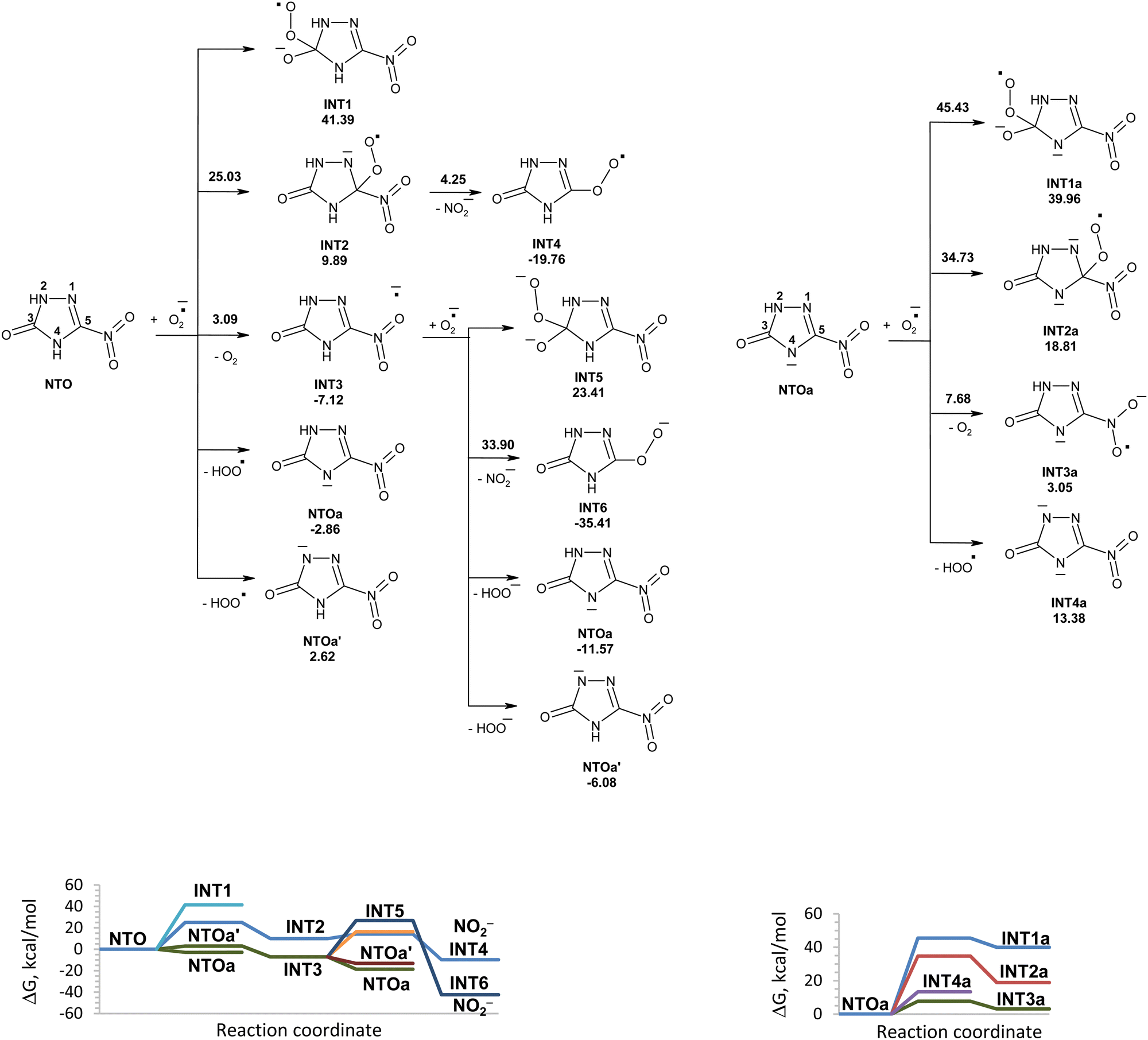 | ||
| Fig. 1 PCM(Pauling)/M06-2X/6-311++G(d,p) modeled pathways for NTO and NTOa decomposition in water induced by superoxide along with the corresponding Gibbs free energy diagram. In the top figure, numbers along with each of the arrow indicate the corresponding reaction barrier height, while numbers below structures indicate the corresponding reaction Gibbs free energy in kcal mol−1. | ||
The electron transfer from superoxide to NTO is easy to proceed with the formation of INT3, which is an anion radical of NTO. Unimolecular and hydrolytic decomposition of INT3 will not be realized due to high energy barriers that was studied earlier.28 Hydrogen atom transfer from INT3 to superoxide is a barrierless process. Hydrogen atom removal from atom 4 of INT3 is more favorable than that from position 2 and leads to NTOa. The addition of superoxide to carbon atom C3 of INT3 will not be realized due to high instability of formed intermediate INT5. An attack of superoxide onto carbon atom C5 of INT3 requires high activation energy of 33.90 kcal mol−1.
Proton transfer from nitrogen atom N4 of NTO to superoxide is more favorable than proton transfer from N2 atom of NTO and leads to more stable NTOa and hydroperoxyl radical. Based on the obtained results, deprotonation of NTO in position 4 is the most energy favorable process for the reaction of NTO with superoxide. Further possible degradation of NTOa induced by superoxide and hydroperoxyl radicals will be discussed below.
Various reactions of superoxide with anions of NTO were studied. They are attachment of superoxide to carbon atom of NTOa, proton detachment from NTOa, and electron transfer from superoxide to NTOa. The Gibbs free energy profile for the reaction of superoxide with anions of NTO is shown in Fig. 1. Nucleophilic addition to the carbon atoms of NTOa is unlikely to occur since the activation energy for this process is prohibitively high at room temperature (35–45 kcal mol−1). The one-electron reduction of NTOa by superoxide is endergonic (ΔGr = 3.05 kcal mol−1) and requires an activation energy of 7.68 kcal mol−1. Proton transfer from NTOa to superoxide gives an adduct that lies 13.38 kcal mol−1 above the reactants. The reactivity of NTOa is lower than reactivity of NTO in reaction with superoxide due to the requirement of an additional activation energy to overcome the Coulomb repulsion between the two anionic species.
The reactivity of NTO in the excited triplet state toward superoxide may differ from its reactivity in the ground singlet state. Excitation of NTO in the environment that results in its singlet–triplet transformation may occur after absorption of sunlight. Modeled reactions of NTO* (in triplet state) with superoxide in water along with the corresponding Gibbs free energy diagram are shown in Fig. 2. Comparative analysis of energetic parameters for interaction of NTO and NTO* with superoxide showed that excitation significantly facilitates the studied processes (Fig. 1 and 2). The most favorable direction is barrierless attachment of superoxide to carbon atom C5 of NTO* that leads to INT2, which easy losses nitrite anions. Formed INT4 isomerizes into slightly more stable INT7. Cleavage of O–O bonds in INT7 leads to cycle-opened intermediate INT9 via intermediate INT8. Hydroxyl radical, that was eliminated from INT7, may attack INT9. Hydrogen atom transfer from the nitrogen atom of INT9 to the hydroxyl radical has lower energy barrier than the addition of OH˙ to carbon atoms of double bond CO. Removal of hydrogen atoms from INT9 is accompanied by cleavage of C–N bonds and release of nitrogen gas and formation of INT11. Transformations of INT11 into intermediates INT14 and INT15 have similar energy barriers. The addition of molecules of water to INT11 leading to INT14 is more favorable as the reaction is exergonic, while the rupture of C–N bonds in INT11 with elimination of carbon(II) oxide is endergonic. The transfer of hydrogen atoms from INT14 to superoxide occurs without activation energy and is accompanied by rupture of C–N bonds with release of carbon(IV) oxide and isocyanic acid (INT16). Hydrolysis of isocyanic acid leads to intermediate INT17, which decomposes into ammonia and carbon(IV) oxide. Modeled pathways and Gibbs free energy calculations allow suggesting that degradation of NTO* in the triplet state induced by superoxide in aqueous solution may lead to its mineralization with formation of nitrite anion, ammonia, nitrogen gas, and carbon(IV) oxide (Fig. 2).
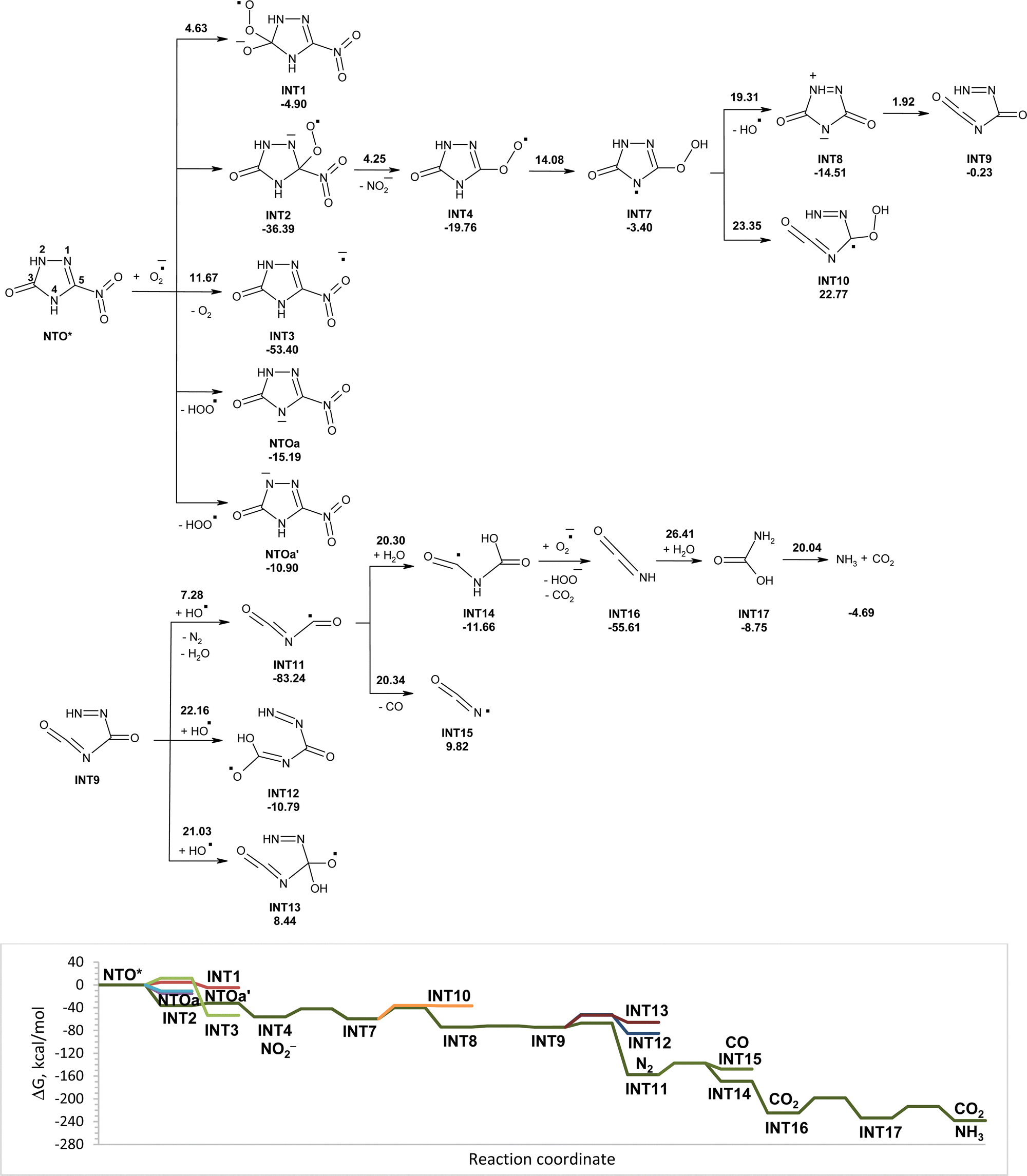 | ||
| Fig. 2 PCM(Pauling)/M06-2X/6-311++G(d,p) modeled pathways for NTO* (triplet state) decomposition in water induced by superoxide along with the corresponding Gibbs free energy diagram. In the top figure, numbers along with each of the arrow indicate the corresponding reaction barrier height, while numbers below structures indicate the corresponding reaction Gibbs free energy in kcal mol−1. | ||
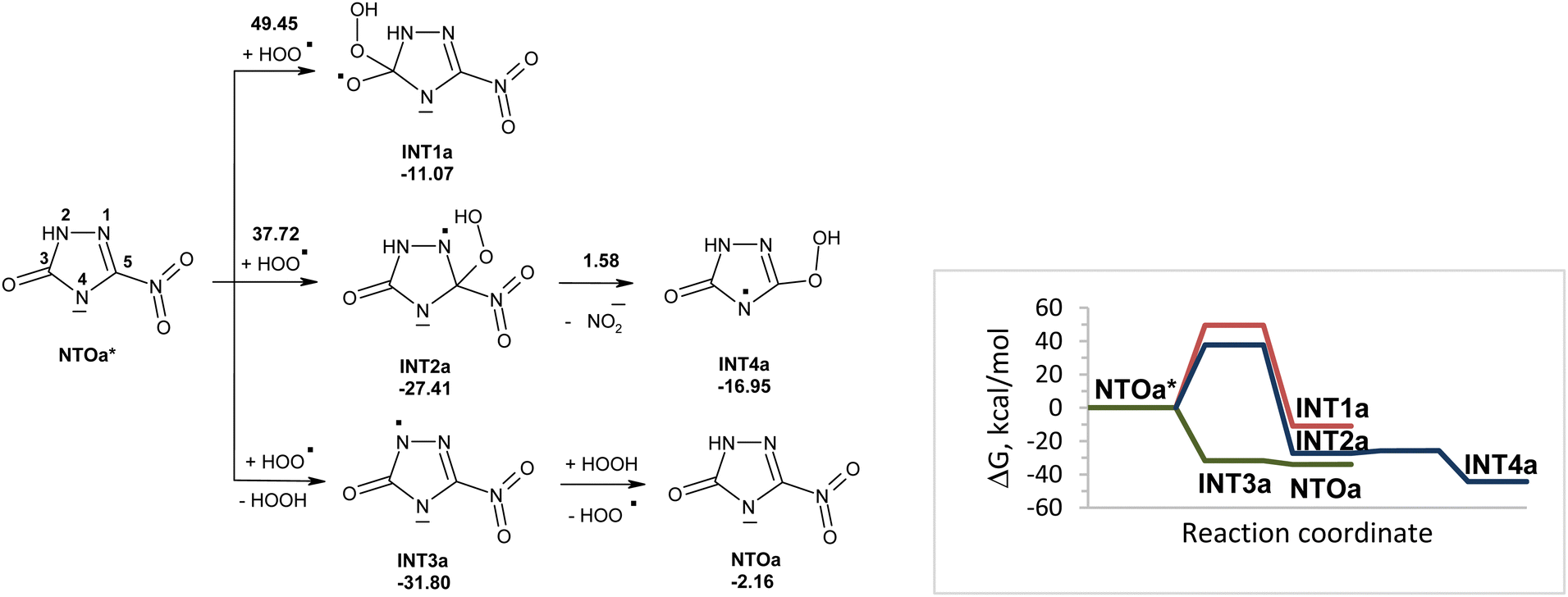
Reactions of NTOa* (in triplet state) with superoxide in water were also modeled (Fig. 3). The reactivity of NTOa in the excited triplet state toward superoxide increases significantly as compared to one in the ground singlet state (Fig. 1 and 3). The fastest processes are the electron transfer from superoxide to NTOa* and the proton transfer from NTOa* to superoxide. These processes lead to intermediates INT3a and INT4a, respectively. The attachment of superoxide to INT3a and INT4a is hard to occur due to high energy barriers of 33–45 kcal mol−1. Instead, these intermediates may be easy transformed into NTOa (in singlet state) by electron and proton transfer, respectively. Thus, superoxide is unlikely to contribute to the degradation of NTOa (Fig. 1 and 3).
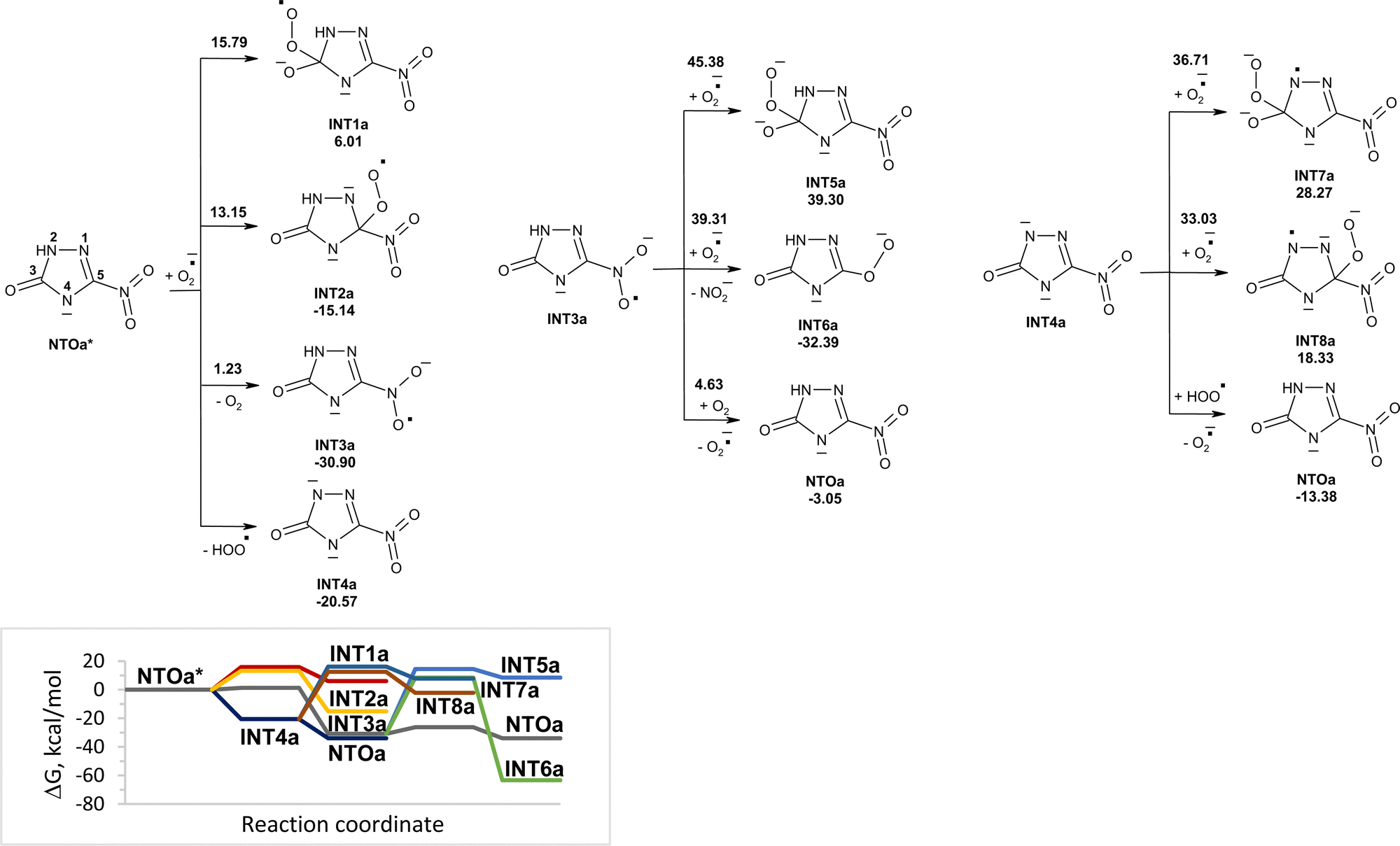 | ||
| Fig. 3 PCM(Pauling)/M06-2X/6-311++G(d,p) modeled pathways for NTOa* (triplet state) decomposition in water induced by superoxide along with the corresponding Gibbs free energy diagram. In the top figure, numbers along with each of the arrow indicate the corresponding reaction barrier height, while numbers below structures indicate the corresponding reaction Gibbs free energy in kcal mol−1. | ||
Hydroperoxyl radical induced degradation of NTO and NTOa
Modeled pathways for NTO decomposition in water induced by hydroperoxyl radicals along with the corresponding Gibbs free energy diagram are shown in Fig. 4. Structures of the corresponding transition states are shown in Fig. S2 (ESI†). The hydrogen atom transfer from NTO to HO2˙, the hydroperoxyl radical attachment to carbon atoms of NTO, and the one-electron reduction of NTO by HO2˙ are endergonic processes. The addition of HO2˙ to carbon atom C5 was found to be the most energy favorable pathway for the initial attack of HO2˙ onto NTO with an activation energy of 23.21 kcal mol−1. The formed adduct INT2 undergoes fast transformation into INT4 by C–N bond rupture and release of nitrite radicals. Interaction of nitrite radicals with HO2˙ results in formation of nitrous acid and oxygen gas. The reaction is exergonic and has an activation energy of 17.91 kcal mol−1. Elimination of a water molecule from INT4 leads to zwitterionic intermediate INT5 that undergoes nearly barrierless C–N bond rupture leading to cycle-opened intermediate INT6. Proton transfer between nitrogen atoms leads to decomposition of INT6 into isocyanic acid (INT10), carbon(II) oxide and nitrogen gas. The reaction is highly exothermic; however, activation barrier for the transfer is high to be realized at ambient temperature. An addition of water molecule to double bond CO of INT6 may result in the formation of intermediates INT7 and INT8. Hydrolytic rupture of C–N bonds may lead to INT9 and N2H2. Transformation of INT6 into INT7 is the most kinetically favorable pathway with the lowest activation energy of 24.34 kcal mol−1.
 | ||
| Fig. 4 PCM(Pauling)/M06-2X/6-311++G(d,p) modeled pathways for NTO decomposition in water induced by hydroperoxyl radicals along with the corresponding Gibbs free energy diagram. In the top figure, numbers along with each of the arrow indicate the corresponding reaction barrier height, while numbers below structures indicate the corresponding reaction Gibbs free energy in kcal mol−1. | ||
There are several pathways for unimolecular decomposition of INT7. Proton transfer from the oxygen atom to nitrogen atom results in formation of INT11 and INT12 and elimination of carbon(IV) oxide. A proton transfer from nitrogen to oxygen atom leads to rupture of two C–N bonds and formation of INT13, nitrogen gas, and carbon(II) oxide. A proton transfer between two nitrogen atoms yields INT14, nitrogen gas, and carbon(II) oxide. Decomposition of INT7 into INT11 and CO2 has the lowest activation barrier (20.60 kcal mol−1) among the studied pathways. Despite the reaction being endergonic the intermediate INT11 undergoes nearly barrierless C–N bond rupture, which leads to the release of isocyanic acid (INT10) and N2H2. Hydrolysis of isocyanic acid proceeds via two steps and results in formation of ammonia and carbon(IV) oxide. The disproportionation reaction of N2H2 leads to hydrazine (N2H4) and nitrogen gas.
Modeled pathways and Gibbs free energy calculations suggest that decomposition of NTO induced by hydroperoxyl radicals in aqueous solution may lead to its mineralization (Fig. 4). The predicted products are nitrous acid, ammonia, nitrogen gas, hydrazine and carbon(IV) oxide. There are a few steps with an activation energy more than 20 kcal mol−1 along the most energy favorable reaction channel. We suggest that use of additional water molecules for calculation may stabilize transition states and reduce activation energy. The slowest step in NTO decomposition is the hydrolysis of isocyanic acid with an activation barrier of more than 26 kcal mol−1. In recent work, we showed that acidic catalysis may considerably decrease the activation energy for the hydrolysis of isocyanic acid to 9.51 kcal mol−1.10
The attachment of hydroperoxyl radicals appears to be more energy favorable than the attachment of superoxide to carbon atom C5 of NTO (Fig. 1 and 4). Relatively higher rates of hydroperoxyl radical trapping as compared to those of superoxide were earlier demonstrated for nitrone 5,5-dimethyl-1-pyrroline N-oxide by comparative analysis of Gibbs free activation energy of O2˙− or HO2˙ addition to CN double bonds of nitrone in an aqueous system.19
Modeled pathways for decomposition of anions of NTO in water induced by hydroperoxyl radicals along with the corresponding Gibbs free energy diagram are shown in Fig. 5. Reaction of hydroperoxyl radicals with NTOa starts from the addition of HO2˙ to carbon atom C5 of the C5N1 double bond, that was found to be a more energy favorable pathway than the initial attack of HO2˙ onto the C3 atom of NTOa. The formed INT2a is less stable than reactants; however the intermediate fast transforms into more stable INT4a by release of nitrite anions. The activation energy for the interaction of hydroperoxyl radicals with NTOa is lower than that for the reaction of HO2˙ with NTO, which suggests higher reactivity of the deprotonated anionic form of NTO toward a hydroperoxyl radical. That is confirmed by a smaller value of energy gap between the frontier molecular orbitals of NTOa and HO2˙ (7.92 eV), than between ones of NTO and HO2˙ (8.65 eV). An alternative reaction pathway of initial transformation of NTO anions involving a hydrogen atom transfer from NTOa to HO2˙ is slightly endergonic and further decomposition of INT3a requires high activation energy, which suggests that this direction of NTOa transformation is hard to occur.
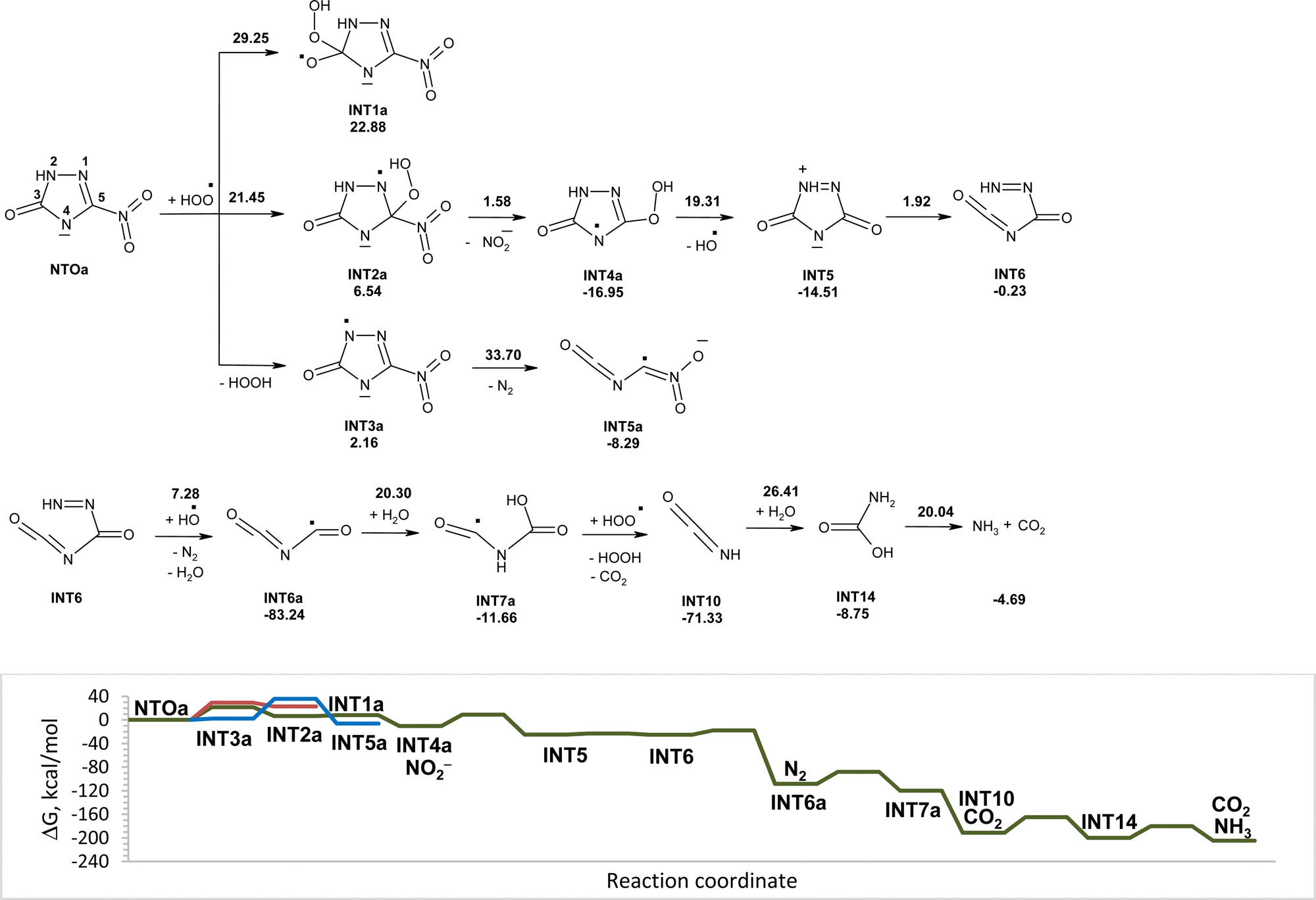 | ||
| Fig. 5 PCM(Pauling)/M06-2X/6-311++G(d,p) modeled pathways for NTOa decomposition in water induced by hydroperoxyl radicals along with the corresponding Gibbs free energy diagram. In the top figure, numbers along with each of the arrow indicate the corresponding reaction barrier height, while numbers below structures indicate the corresponding reaction Gibbs free energy in kcal mol−1. | ||
Cleavage of O–O bonds in INT4a leads to intermediate INT5 and hydroxyl radicals. The cycle opening in INT5 leads to intermediate INT6, which may be attacked by the hydroxyl radical. The transfer of the hydrogen atom from the nitrogen atom of INT6 to the hydroxyl radical is accompanied by cleavage of the C–N bond and release of nitrogen gas and formation of INT6a. The addition of molecules of water to INT6a results in the formation of INT7a. The hydroperoxyl radical captures the hydrogen atom from INT7a without an activation barrier that leads to rupture of C–N bonds with release of carbon(IV) oxide and isocyanic acid (INT10). Two-step hydrolysis of isocyanic acid yields ammonia and carbon(IV) oxide. Modeled pathways and Gibbs free energy calculations suggest that degradation of anions of NTO induced by hydroperoxyl radicals in aqueous solution may lead to its mineralization with formation of nitrite anions, ammonia, nitrogen gas, and carbon(IV) oxide (Fig. 5).
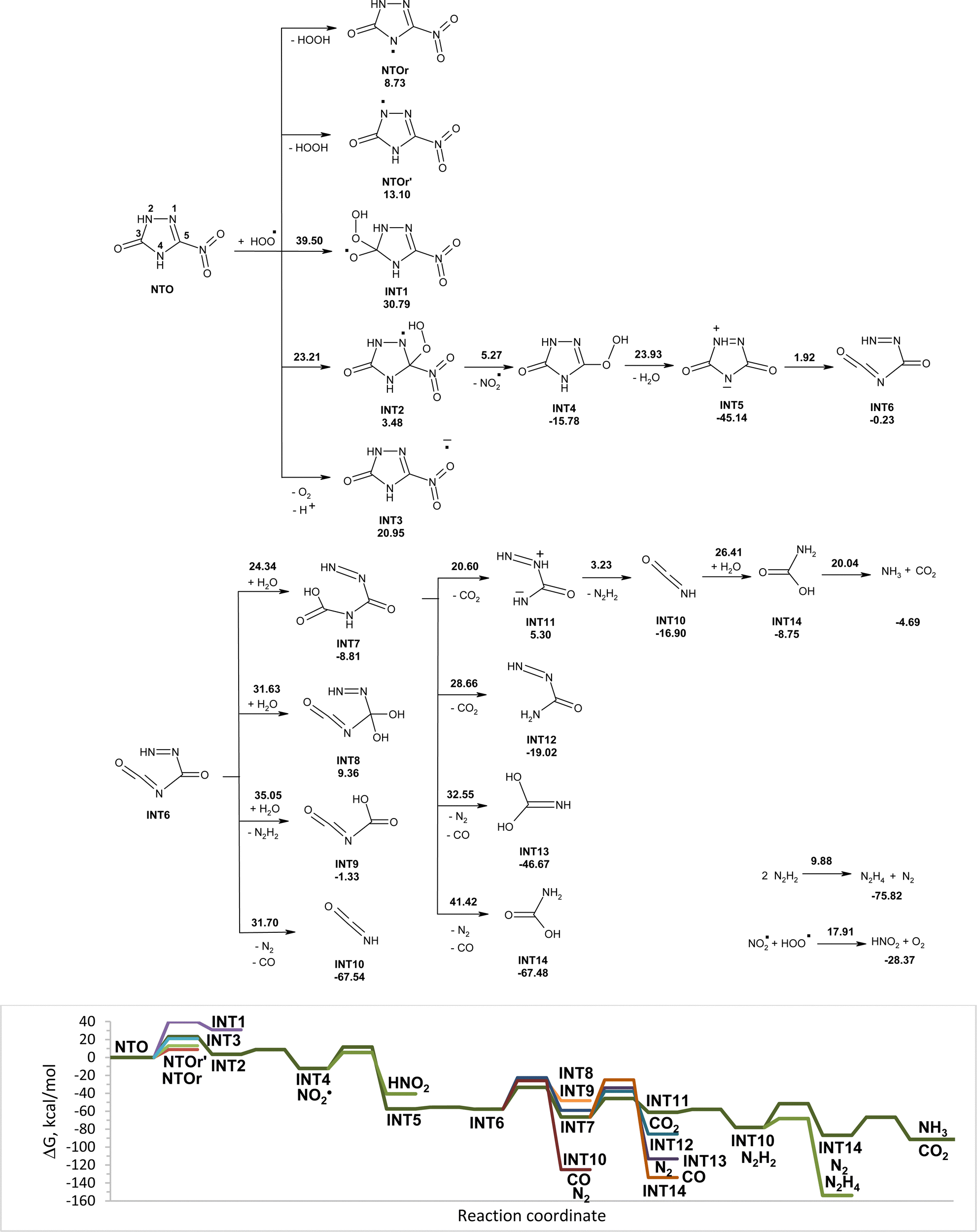
Modeled pathways for degradation of NTO* and NTOa* in the triplet state induced by hydroperoxyl radicals are shown in Fig. 6 and 7, respectively. Excitation of NTO and NTOa makes the addition of hydroperoxyl radicals hard to occur due to the high activation energy. Barrierless capture of hydrogen atoms from NTO* by HO2˙ leads to more stable intermediates NTOr and NTOr′ than the electron transfer from hydroperoxyl radicals to NTO*, leading to INT3 (Fig. 6). Radical intermediate NTOr is more stable than NTOr′ and may undergo an attachment of molecule of water with formation of INT18. The addition of hydroperoxyl radicals to NTOr requires higher activation energy than the addition of water molecules. The attack of water molecules onto carbon atom C5 is more favorable than the one onto atom C3. Intermediate INT18 easily loses nitrite radicals, which may transform into nitrous acid by fast capturing of hydrogen from INT21. Decomposition of INT22 into isocyanic acid (INT10) and INT28 as well as the addition of hydroperoxyl radicals to INT22 will not occur due to high activation energy and endergonicity. The hydrogen transfer from INT22 to hydroperoxyl radicals is a barrierless process leading to INT5, which undergoes rapid opening of five-membered cycles. The addition of water molecules to INT6 results in sequential release of carbon(IV) oxide, diimide and ammonia. Diimide can go through a disproportionation reaction to form nitrogen gas and hydrazine (Fig. 4).
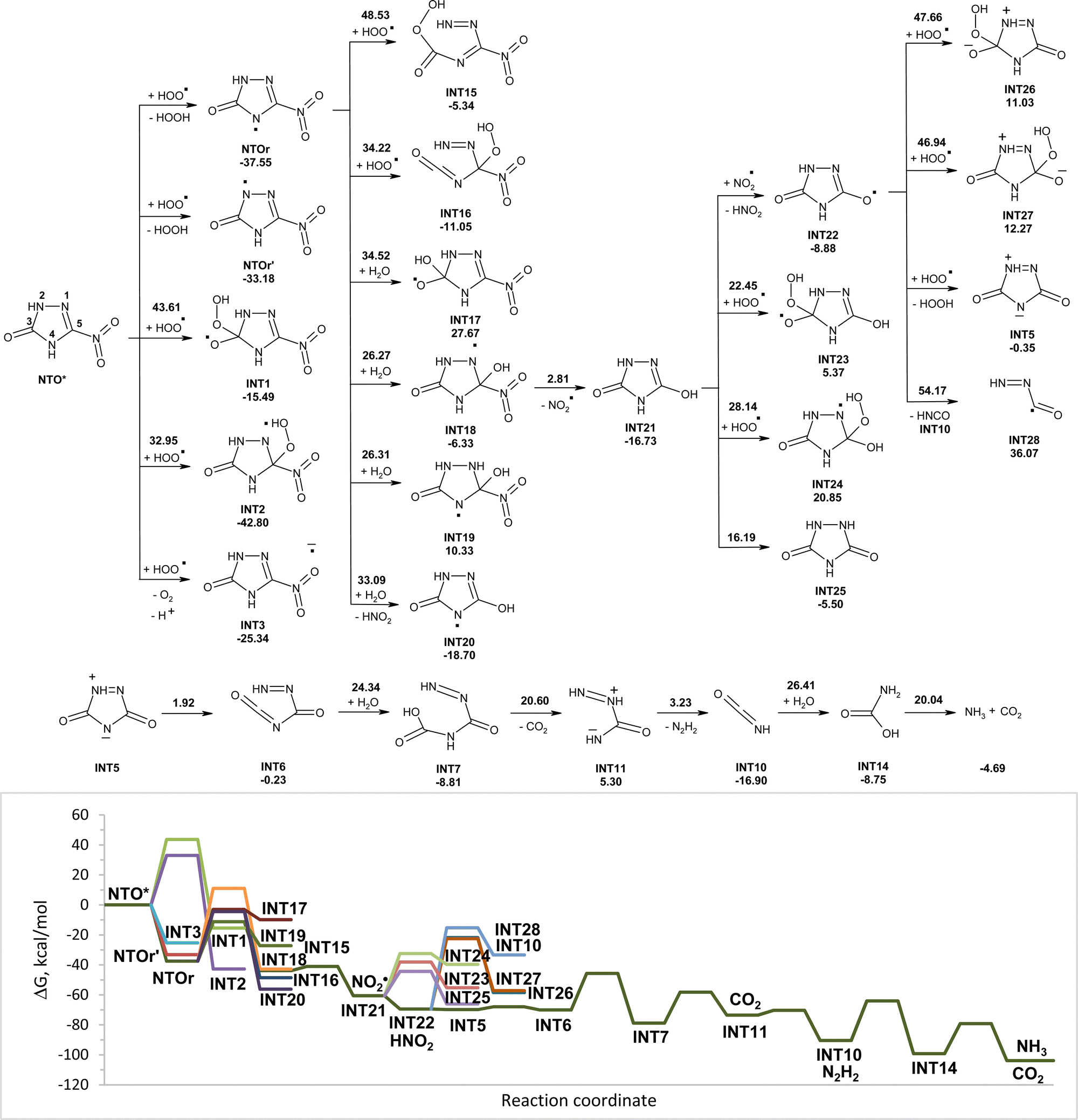 | ||
| Fig. 6 PCM(Pauling)/M06-2X/6-311++G(d,p) modeled pathways for NTO* (triplet state) decomposition in water induced by hydroperoxyl radicals along with the corresponding Gibbs free energy diagram. In the top figure, numbers along with each of the arrow indicate the corresponding reaction barrier height, while numbers below structures indicate the corresponding reaction Gibbs free energy in kcal mol−1. | ||
 | ||
| Fig. 7 PCM(Pauling)/M06-2X/6-311++G(d,p) modeled pathways for NTOa* (triplet state) decomposition in water induced by hydroperoxyl radicals along with the corresponding Gibbs free energy diagram. In the top figure, numbers along each of the arrow indicate the corresponding reaction barrier height, while numbers below structures indicate the corresponding reaction Gibbs free energy in kcal mol−1. | ||
Comparative analysis of NTO degradation in water induced by hydroperoxyl radicals for singlet and triplet states of NTO showed that in both cases NTO undergoes complete mineralization into nitrous acid, nitrogen gas, ammonia and carbon(IV) oxide (Fig. 4 and 6). The initial reaction of NTO with hydroperoxyl radicals is an addition of HO2˙ to carbon atom C5 of NTO. Excitation of NTO considerably increases the activation energy for the addition reaction, making hydrogen transfer from NTO* to hydroperoxyl radicals the most favorable initial reaction. Further transformations of INT1 (Fig. 4) and NTOr (Fig. 6) lead to INT5, and then the degradation proceeds in the same way for both cases.
Excitation of NTOa also facilitates hydrogen transfer from NTOa* to hydroperoxyl radicals (Fig. 7). However, formed INT3a may capture hydrogen from hydrogen peroxide that results in the formation of NTOa in the singlet state. Other possible decomposition pathways for INT3a, that is release of nitrogen gas and formation of INT5a (Fig. 5), was ruled out due to high activation barriers. Thus, hydroperoxyl radicals can contribute to degradation of NTOa in its singlet state (Fig. 5).
The current study established that superoxide and hydroperoxyl radicals may take an active part in the process of NTO degradation. Superoxide mainly participates in deprotonation of NTO in the singlet state. Excitation of NTO facilitates its degradation induced by superoxide, that may result in complete mineralization of NTO. The degradation pathway consists of an addition of superoxide to carbon atom of NTO, release of nitrite anion, cleavage of five-membered cycle, elimination of nitrogen gas, and hydrolysis leading to formation of carbon(IV) oxide and ammonia. Hydroperoxyl radicals appear to be more reactive than superoxide in reaction with NTO (in singlet state) on the basis of the calculated Gibbs free activation energies. Decomposition of NTO induced by hydroperoxyl radicals in water is a multistep process, which leads to mineralization of the studied nitrocompound. The initial nucleophilic addition of HO2˙ to NTO occurs at the carbon atom of double bond CN. Then sequential transformations occur involving release of nitrite radicals, cycle opening, water molecule addition, and breaking of C–N bonds. The predicted products are nitrous acid, ammonia, nitrogen gas, hydrazine, and carbon(IV) oxide. Degradation of NTO anions induced by hydroperoxyl radicals resulted in formation of nitrite anions, nitrogen gas, carbon(IV) oxide, and ammonia. Excitation of NTO causes a change of an initial step of reaction between NTO and hydroperoxyl radicals toward formation of NTO radicals. The addition of water molecules to the NTO radical leads to the release of nitrite radicals, which then transforms into nitrous acid, diimide, which may form nitrogen gas and hydrazine, carbon(IV) oxide and ammonia. Formation of nitrous acid (nitrite anion), ammonia, and carbon(IV) oxide is experimentally observed during photolytic decomposition of NTO in aqueous solutions.3–8
Conclusions
Superoxide and hydroperoxyl radicals are important reactive oxygen species produced in the aquatic environment under sunlight irradiation. They may cause decomposition of organic pollutants. The ability of superoxide and hydroperoxyl radicals to take part in degradation of NTO was studied at the PCM(Pauling)/M06-2X/6-311++G(d,p) theory level. Our results revealed that superoxide fast deprotonates NTO, while hydroperoxyl radicals may cause mineralization of NTO. Degradation of NTO induced by hydroperoxyl radicals proceeds through several steps, starting from hydroperoxyl radical attachment to carbon atoms of CN double bonds of NTO. Sequential cleavage of C–N bonds and addition of water molecules lead to such products of NTO degradation as nitrous acid, ammonia, nitrogen gas, hydrazine, and carbon(IV) oxide. The anionic form of NTO is more reactive toward interaction with hydroperoxyl radicals as compared with its neutral form. The absorption of UV-irradiation under environmentally relevant conditions causes singlet–triplet transformation of NTO that significantly affects its reactivity. In the reaction of triplet NTO with superoxide, the corresponding activation barriers are lowered, which allows starting a sequence of processes to achieve complete mineralization of NTO. Based on the energetics of the studied reaction pathways we suggest that indirect photolysis induced by hydroperoxyl radicals and superoxide may contribute to NTO degradation in the environment.
Author contributions
All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank ARO for financial support (grant number is W911NF-20-1-0116). The computation time was provided by the Mississippi Center for Supercomputer Research. The use of trade, product, or firm names in this report is for descriptive purposes only and does not imply endorsement by the U.S. Government. The findings of this report are not to be construed as an official Department of the Army position unless so designated by other authorized documents.Notes and references
- R. J. Spear, C. N. Louey and M. G. Wolfson, A preliminary assessment of 3-nitro-1,2,4-triazol-5-one (NTO) as an insensitive high explosive, DSTO Materials Research Laboratory, Maribyrnong, Australia, 1989.
- S. Taylor, E. Park, K. Bullion and K. Dontsova, Chemosphere, 2015, 119, 342–348 CrossRef CAS PubMed.
- K. Dontsova, S. Taylor, R. Pesce-Rodriguez, M. Brusseau, J. Arthur, N. Mark, M. Walsh, J. Lever and J. Simunek, Dissolution of NTO, DNAN, and insensitive munitions formulations and their fates in soils, ERDC/CRREL TR-14-23, Hanover, NH, 2014.
- A. Halasz, J. Hawari and N. N. Perreault, Environ. Sci. Technol., 2018, 52, 589–596 CrossRef CAS PubMed.
- J. B. Becher, S. A. Beal, S. Taylor, K. Dontsova and D. E. Wilcox, Chemosphere, 2019, 228, 418–426 CrossRef CAS PubMed.
- L. C. Moores, S. J. Jones, G. W. George, D. L. Henderson and T. C. Schutt, J. Photochem. Photobiol., A, 2020, 386, 112094 CrossRef CAS.
- L. C. Moores, A. J. Kennedy, L. May, S. M. Jordan, A. J. Bednar, S. J. Jones, D. L. Henderson, L. Gurtowski and K. A. Gust, Chemosphere, 2020, 240, 124958 CrossRef CAS PubMed.
- H. W. Schroer, E. Londono, X. Li, H. J. Lehmler, W. Arnold and C. L. Just, ACS ES&T Water, 2023, 3(3), 783–792 Search PubMed.
- L. K. Sviatenko, L. Gorb and J. Leszczynski, Struct. Chem., 2023, 34, 23–31 CrossRef CAS.
- L. K. Sviatenko, L. Gorb and J. Leszczynski, Phys. Chem. A, 2023, 127(12), 2688–2696 CrossRef CAS PubMed.
- R. M. Baxter and J. H. Carey, Nature, 1983, 306, 575–576 CrossRef CAS.
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS.
- D. S. Dimic, D. A. Milenkovic, E. H. Avdovic, D. J. Nakarada, J. M. Dimitric Markovic and Z. S. Markovic, Chem. Eng. J., 2021, 424, 130331 CrossRef CAS.
- W. H. Koppenol and J. Butler, Adv. Free Rad. Biol. Med., 1985, 1(1), 91–131 CrossRef CAS.
- D. Behar, G. Czapski, J. Rabani, L. M. Dorfman and H. A. Schwarz, J. Phys. Chem., 1970, 74(17), 3209–3213 CrossRef CAS.
- V. S. Bryantsev, V. Giordani, W. Walker, M. Blanco, S. Zecevic, K. Sasaki, J. Uddin, D. Addison and G. V. Chase, J. Phys. Chem. A, 2011, 115(44), 12399–12409 CrossRef CAS PubMed.
- D. T. Usmanov, S. Ninomiya, K. Hiraoka and S. Yamabe, Int. J. Mass Spectrom., 2020, 450, 116308 CrossRef CAS.
- Z. Dhaouadi, M. Nsangou, N. Garrab, E. H. Anouar, K. Marakchi and S. Lahmar, THEOCHEM, 2009, 904(1–3), 35–42 CrossRef CAS.
- F. A. Villamena, J. K. Merle, C. M. Hadad and J. L. Zweier, J. Phys. Chem. A, 2005, 109(27), 6083–6088 CrossRef CAS PubMed.
- L. K. Sviatenko, L. Gorb, D. Leszczynska, S. I. Okovytyy, M. K. Shukla and J. Leszczynski, J. Phys. Chem. A, 2019, 123, 7597–7608 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision A.01., Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- S. F. Sousa, P. A. Fernandes and M. J. Ramos, J. Phys. Chem. A, 2007, 111, 10439–10452 CrossRef CAS PubMed.
- W. J. Hehre, L. Radom, Pv. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, USA, 1986 Search PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- L. K. Sviatenko, L. Gorb, D. Leszczynska, S. I. Okovytyy, M. K. Shukla and J. Leszczynski, Struct. Chem., 2021, 32, 521–527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structures of transition states for NTO decomposition in water under action of superoxide and hydroperoxyl radicals. Cartesian coordinates for optimized local minima and transition state species. See DOI: https://doi.org/10.1039/d3cp05603a |
| This journal is © the Owner Societies 2024 |