
Trajectory-dependent threshold effects of proton stopping power in LiF nanosheets†
Ya-Ting
Sun
a,
Feng
Wang
 *a and
Cong-Zhang
Gao
*b
*a and
Cong-Zhang
Gao
*b
aSchool of Physics, Beijing Institute of Technology, Beijing 100081, China. E-mail: wangfeng01@tsinghua.org.cn
bInstitute of Applied Physics and Computational Mathematics, Beijing 100088, China. E-mail: czgao88@hotmail.com
First published on 30th May 2024
Abstract
We conducted a study on the trajectory-dependent threshold effects of proton stopping power in LiF nanosheets using time-dependent density functional theory non-adiabatically coupled to the molecular dynamics. This study covered protons with initial velocities in the range of 0.1–1.0 a.u., offering a vast amount of detailed information on the electronic structure during the stopping process with superior spatial and temporal resolution. Our results show that the impact parameters of incident protons play a crucial role in determining the threshold behavior of proton stopping power in LiF nanosheets. Most importantly, we found that close collisions do not exhibit a discernible threshold. In addition, the research results also revealed the time dependence of the number of electrons occupying the atomic orbitals of F and Li as protons pass through the nanosheets.
1 Introduction
A thorough comprehension of the way in which particle radiation interacts with materials is of utmost importance in addressing various significant issues across a range of fields,1,2 including nuclear industry3–5 and space technology6 applications, high-energy density physics,7 radio-therapy in medicine,8–10 fundamental research laboratories,11 and the manufacturing of integrated circuits. Usually, the basic process of collision between incident particles and target materials includes electron transitions (excitation, charge transfer, ionization) and energy transfer of nuclear degrees of freedom.12 Therefore, incident particles mainly lose energy through two mechanisms: one the nuclear stopping power (Sn) at low projectile velocities, in which the projectile particle mainly transfers energy to nuclei without electronic excitations, and another the electron stopping power (Se) at high projectile velocities, in which the projectile particle mainly transfers energy to electrons. Comparatively speaking, predicting the energy loss of electrons is still difficult due to their quantum mechanical properties.13 Over the past few decades, both theoretical and experimental physicists have shown great interest in studying the Se(v) of materials.14,15When the projectile particles pass through the free electron gas, the Se(v) is approximately proportional to the particle velocity v (a kind of Ohms law). The proportionality law holds well for metals and plasma, but for insulators, having an ionization energy gap, Se(v) dependence must exhibit a threshold, as was predicted in ref. 16 and 17. Experimental confirmation of this threshold has been achieved for gaseous targets,18 but conducting similar experiments with solid targets poses additional challenges. This is because low-energy projectiles stop too quickly in solids, necessitating the use of very thin target materials. To enhance the visibility of the threshold effect and facilitate its observation, it is advantageous to choose a target material with a wider ionization energy gap. LiF is one of the materials with a large energy gap. Nevertheless, even for LiF, the experimental transmission of protons and antiprotons through thin films19,20 did not show any observable threshold. This could be attributed to the possibility that the energies used in these experiments were not sufficiently low, or that the averaging over projectile impact parameters masked the threshold. Eventually, the threshold was observed in LiF targets for grazing incidence (surface channeling)21 and backscattering.22 Surface channeling differs from transmission in that it lacks close encounters between the projectile and target atoms, suggesting that it is in close collisions where the threshold may be absent.
On the other hand, it is important to acknowledge that fully atomistic first-principles simulations offer a vast amount of detailed information regarding the stopping process that cannot be obtained from experimentally tabulated data. In fact, several modern first-principles simulations have provided a comprehensive description of the stopping processes with superior spatial and temporal resolution compared to current experimental capabilities. For instance, earlier density functional theory (DFT) computer simulations determined the gap in Se(v) for H, He, and Ne targets.23 Additionally, Pruneda et al.24 have identified the existence of a threshold in time-dependent density functional theory (TDDFT) simulations, although they did not thoroughly investigate it, seemingly limiting their study to protons passing through the center of the LiF cell, where the threshold effect is most pronounced.
In order to fully understand the fascinating threshold behaviors of the stopping process, it is important to consider the physics of the threshold effects themselves and take into account the projectile's impact parameters. To address these points, this study focuses on the example of a proton in a LiF nanosheet. It is worth noting that the stopping power of a proton in bulk LiF has been extensively studied both experimentally20–22 and theoretically,24–27 providing a basis for validating our results in the bulk limit. However, no model calculations have been conducted for LiF nanosheets covering the velocity range of protons from 0.1 a.u. to 1.0 a.u.
The remaining sections of this article are structured as follows. Section 2 provides an introduction to the calculation methods and model construction employed for studying protons in LiF nanosheets. Section 3 presents a comprehensive analysis of the simulation results and offers explanations for various observed phenomena. Finally, in Section 4, the conclusions drawn from this study are summarized. It is important to note that atomic units (ħ = me = e = 1) are consistently utilized unless explicitly mentioned.
2 Computational method and model
The Se(v) of protons in LiF nanosheets was investigated using a time-dependent density functional molecular dynamics (TDDFT-MD) model that combined Ehrenfest molecular dynamics (EMD) and real-time TDDFT (rt-TDDFT).28,29 In the collision system, all particles are divided into two categories: one is the ion core (atomic nucleus and inner electron), which is regarded as the classical charged point particle with coordinates Rj (j = 1…Nion); the other is the valence electrons, which is described by the single-particle wave function ψi (i = 1…Ne). It should be noticed that in the case of the Li atom, the electrons of the inner 1s orbitals are considered explicitly in the calculation. In TDDFT, all observables are represented by a function of the electronic density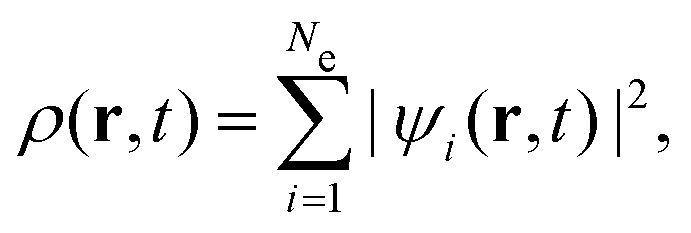 | (1) |
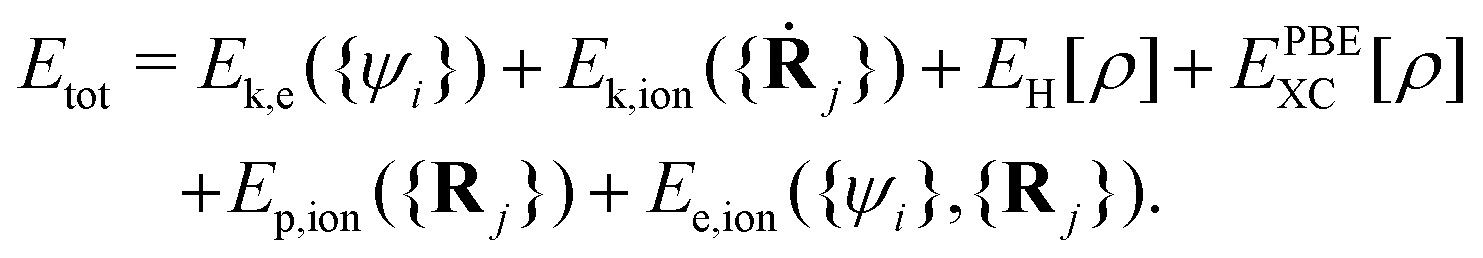 | (2) |
It includes the kinetic energy of electrons (Ek,e) and ion cores (Ek,ion), the coulomb energy between valence electrons (EH), the exchange–correlation (XC) energy of electrons (EPBEXC), and the interaction potential energy between ion cores (Ep,ion), as well as the interaction energy between electrons and ion cores (Ee,ion).
The time-dependent Kohn–Sham (KS) equation describes the evolution of the electronic structure:
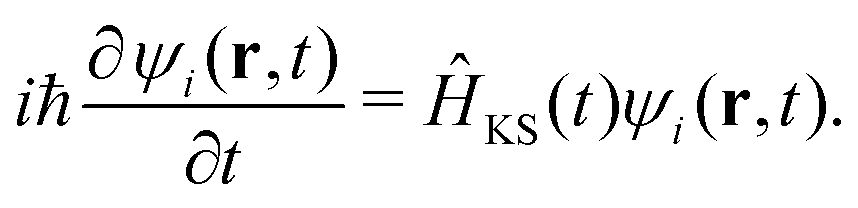 | (3) |
Ĥ KS represents the Hamiltonian of the system, specifically expressed as:
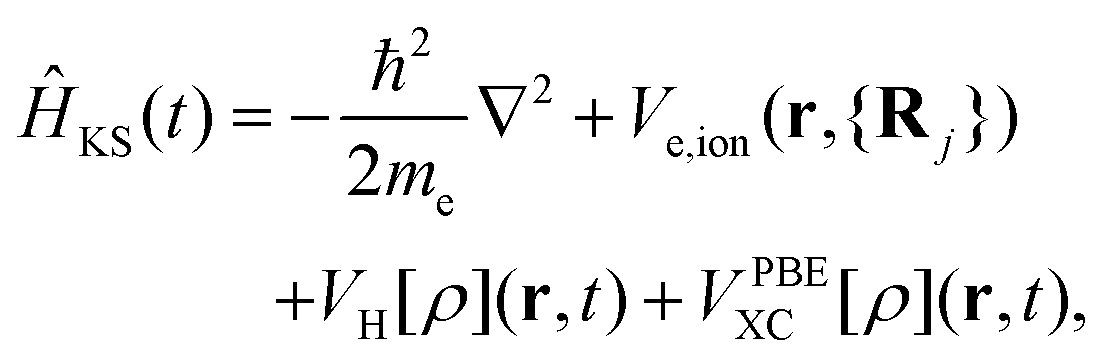 | (4) |
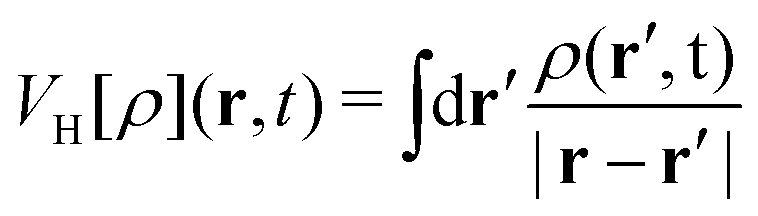 represents the Hartree potential, which represents the Coulomb potential between valence electrons. And VPBEXC represents the XC potential using the adiabatic generalized gradient approximation (GGA) based on the Perdew–Burke–Ernzerhof (PBE) functional.30
represents the Hartree potential, which represents the Coulomb potential between valence electrons. And VPBEXC represents the XC potential using the adiabatic generalized gradient approximation (GGA) based on the Perdew–Burke–Ernzerhof (PBE) functional.30
Here, the time-dependent wave function ψi expands into the adiabatic eigenstate ϕl,
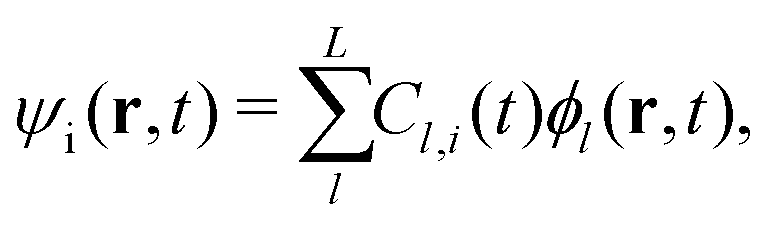 | (5) |
The evolution of the wave function ψi can be transformed into the evolution of the coefficient Cl,i = 〈ϕl|ψi〉 using eqn (5). And the evolution of the adiabatic state ϕl is given by
| ĤKS(t)ϕl(r,t) = εl(t)ϕl(r,t), | (6) |
The Newton's equation controls the motion of ion cores:
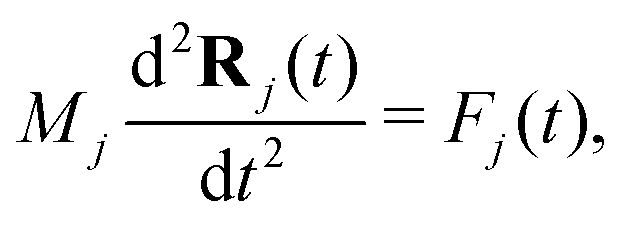 | (7) |
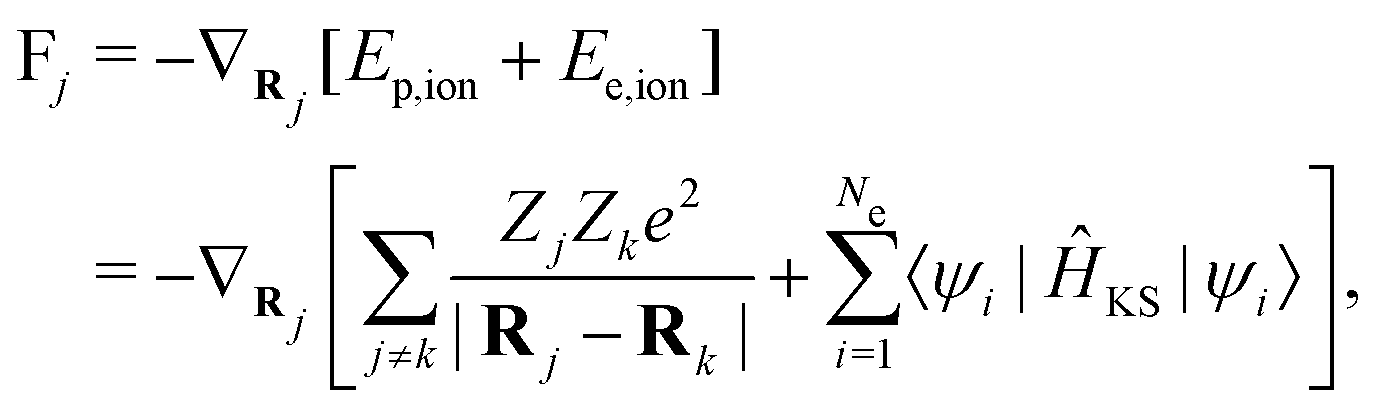 | (8) |
To perform TDDFT-MD simulations, we utilized PWmat31,32 software which uses the plane wave pseudopotential Hamiltonian and is optimized for graphics processing units (GPUs),31,32 introducing a modified Ehrenfest (ME) dynamics to correct a detailed balance problem in the original algorithm.33
In our simulations, we employed the Δt × ν ∼ 1.24 × 10−3 Å method to set the time step for protons of varying velocities,34,35 which is utilized to guarantee the convergence of the total energy. The computer time required to complete a single rt-TDDFT calculation is 48 hours with four GeForce GTX GPUs. The calculation was performed using a 2 × 2 × 1 k-point grid, and the optimized norm-conservinng vanderbilt pseudopotential36 was employed to describe interactions between valence electrons and ionic cores, including ion nuclei such as Li3+, F5+ (1s2) and H+. The plane wave expansion basis was used, with a cut-off energy of 116 Ry (ref. 24), corresponding to a real space grid size of 56 × 56 × 320. Our simulation involved a total of 640 electrons and 500 adiabatic states were calculated to set the simulation in motion. A cubic unit cell of the LiF crystal, taken from ref. 37, contains 8 atoms with a lattice parameter of 4.082 Å. This unit cell is expanded into a 2 × 2 × 4 supercell and placed in a simulation box with dimensions of 8.164 × 8.164 × 46.328 Å3. During the simulation process, periodic boundary conditions are used, and a vacuum layer with a thickness of 15 Å is taken in front of the front and rear surfaces of the LiF nanosheet to shield against the influence of other potential around it.
Before performing rt-TDDFT calculations, we performed DFT calculations on the LiF nanosheet to obtain an initial wave function, ensuring full convergence of calculations. In addition, the test calculations used to obtain the numerical parameters consistent with the calculated results are briefly described in the ESI† (SCI.A).38
The initial position of the proton is placed 11.38 Å away from the surface of the LiF nanosheet and moves in the positive direction along the z-axis. To demonstrate the variation trend of the Se(v) from channel to off-channel, three cases with different impact parameters (i.e., different closest distances to any target atom surrounding the projectile) were selected to represent channel, off-channel, and the transition between them. As depicted in the top view shown in Fig. 1(a), the center position between the F–F (Li–Li) atomic lines is defined as channel (channel-1), the position at 2/3 distance from the center point is defined as off-channel (channel-3), and the center position between two points is chosen as the transition point (channel-2). Due to the periodic arrangement of the crystal, the projectile will periodically pass through F atoms and Li atoms. It is worth noting here that due to the influence of Ehrenfest dynamics, the x and y coordinates of protons may slightly change (due to the symmetry of collision geometry, the displacement in the x-direction is consistent with the displacement in the y-direction).
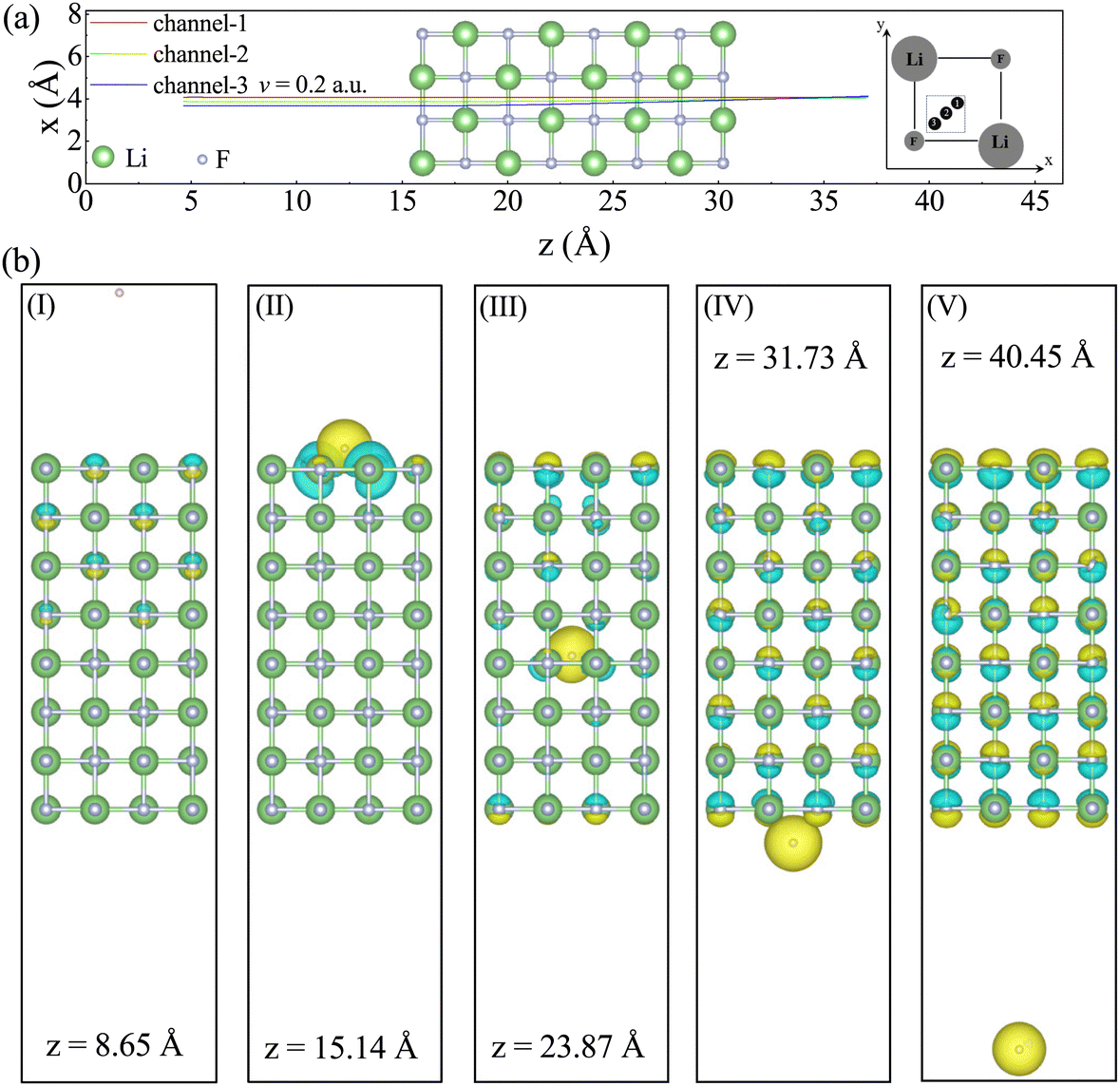 | ||
| Fig. 1 (a) The relationship between the displacement in the x-direction of the incident proton with an incidence velocity of 0.2 a.u. and the displacement in the z-direction during this simulation process. Among them, the background atomic structure shows the slices of LiF nanosheets along the y-direction. The inset provides a clear depiction of the model coordinate axis direction, alongside a top view of channel-1, channel-2, and channel-3. The green ball represents Li atoms, and the gray ball represents F atoms. (b) The representative ion positions and electron density differences in the five stages of (I)–(V), with the projectiles located at z = 8.65, 15.14, 23.87, 31.73, and 40.45 Å, respectively. | ||
For the convenience of further discussion, we divide the collision process into five stages as follows: (I) pre-entry stage, where the proton is far away from the front surface of the LiF nanosheet; (II) entry stage, where the proton enters the front surface of the LiF nanosheet; (III) internal stage, where the proton is inside the LiF nanosheet; (IV) exit stage, where the proton leaves the back surface of the LiF nanosheet; (V) post-exit stage, where the proton is far away from the back surface of the LiF nanosheet. In order to visually depict the characteristics of ion configuration and electron distribution in the five stages, Fig. 1(b) shows the representative ion positions and electron density differences in the five stages, with the projectiles located at z = 8.65, 15.14, 23.87, 31.73, and 40.45 Å, respectively.
3 Results and discussion
3.1 Kinetic energy loss of protons
As protons pass through the LiF nanosheet, they transfer energy to the system. We can quantify the transfer of energy by calculating the rate at which protons lose energy. In the simulation process, the proton moves in the positive direction of the z-axis. Since it has small displacements (the specific values can be found in Table S1 of the ESI†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38) in the x-direction and y-direction at low velocities, we can approximate its motion as a straight line along with the z-direction.
38) in the x-direction and y-direction at low velocities, we can approximate its motion as a straight line along with the z-direction.
Fig. 2 illustrates the kinetic energy loss of the proton as it moves along channel-1, plotted against its displacement. And the kinetic energy loss of the proton as a function of the displacement for channel-2 and channel-3 can be found in the ESI†38 (see Fig. S3). The instantaneous energy loss of a proton can be defined as the derivative of its energy with respect to its displacement, i.e., Se(v) = −dEk,proton/dz. In stage (I), proton kinetic energy remains relatively stable. In stage (II), there may be fluctuations in proton kinetic energy due to the exchange of charges between proton and target atoms. These fluctuations are more prominent at lower velocities. In stage (V), the proton kinetic energy gradually reaches a state of balance. To determine the equilibrium Se(v), we calculate the average of the instantaneous electronic stopping power between the two vertical dashed lines, as shown in Fig. 2. The periodic oscillation is caused by the lattice structure of the LiF nanosheet.
 | ||
| Fig. 2 The kinetic energy loss ΔEk,proton = Ek,proton(0) − Ek,proton(z) of protons as a function of displacement for different velocities along channel-1. | ||
In Fig. 3, the experimental values of Draxler et al.22 and Møller et al.,20 as well as the theoretically calculated TDDFT values of Pruneda et al.,24 are presented respectively. The Stopping and Range of Ions in Matter (SRIM)39 value is also given. It is important to highlight that the SRIM results are derived semi-empirically through the averaging of data from various incident directions with different impact parameters. Consequently, the model does not explicitly consider the specific channeling conditions that are the focus of our calculations. The numerical values obtained in this simulation align with the variation trend of the SRIM-2013 curve,40 as well as the experimental and theoretical reference values, proving the rationality and appropriateness of the model parameters used. The experimental value is determined by averaging all impact parameters. However, in the current simulation, only three specific incident trajectories were selected, resulting in the simulated values being lower than the experimental values. It is important to mention that the Se(v) value calculated by Pruneda et al. is not very consistent with the experiment because they only simulated the proton passing through the LiF center. When the velocity of the proton in channel-1 and channel-2 is 0.2 a.u., the threshold effect described in ref. 17 and 24 will appear, but in channel-3, this threshold effect will disappear. The appearance of velocity thresholds in LiF is attributed to the inability of incident ions to excite electrons at low velocities, resulting in threshold effects due to the existence of the electron excitation gap. This has been demonstrated by numerous studies.17,20
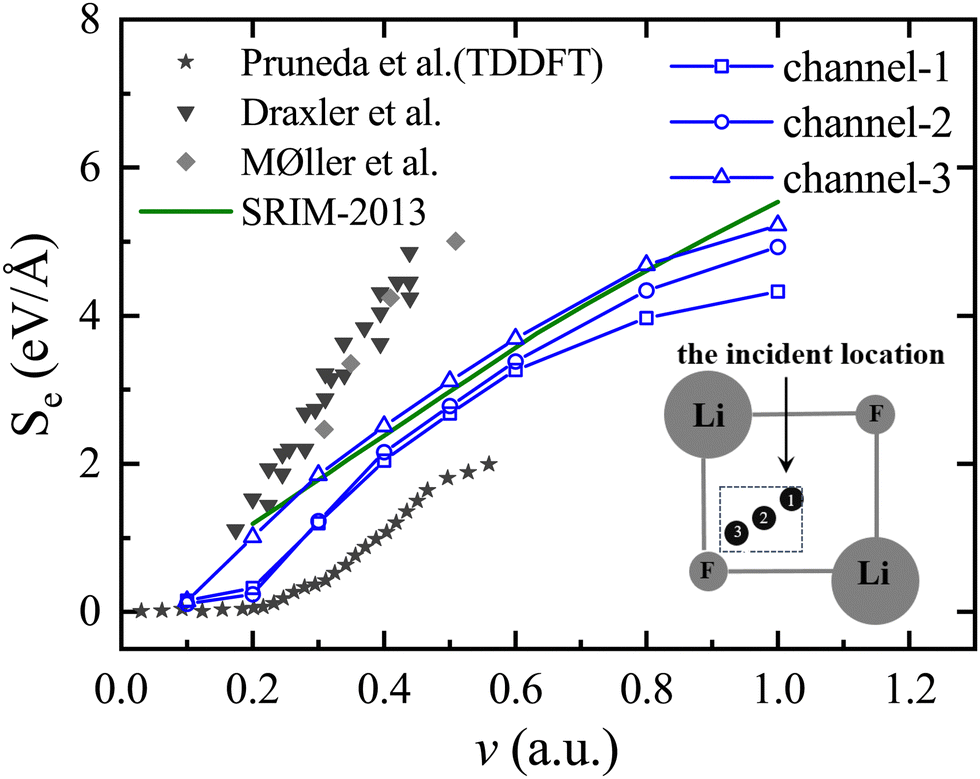 | ||
| Fig. 3 The electronic stopping power of the LiF nanosheet against protons moving along different trajectories is taken as a function of velocity, where blue curves with different shapes represent different incident trajectories, the ★ represents the theoretical values of TDDFT for Pruneda et al.,24 the ▼ represents the experimental values for Draxler et al.,22 the ✦ represents the experimental values for Møller et al.,20 and green curves represent the values for the SRIM-2013 curve.40 The inset shows the top view of channel-1, channel-2, and channel-3. | ||
Fig. 4 shows the variation of the total potential energy (Ep) and total kinetic energy (Ek) of the system with proton displacement as the proton passes through the LiF nanosheet along channel-1 with an initial velocity of 0.2 a.u., where
| Ek = Ek,ion({Ṙj}), | (9) |
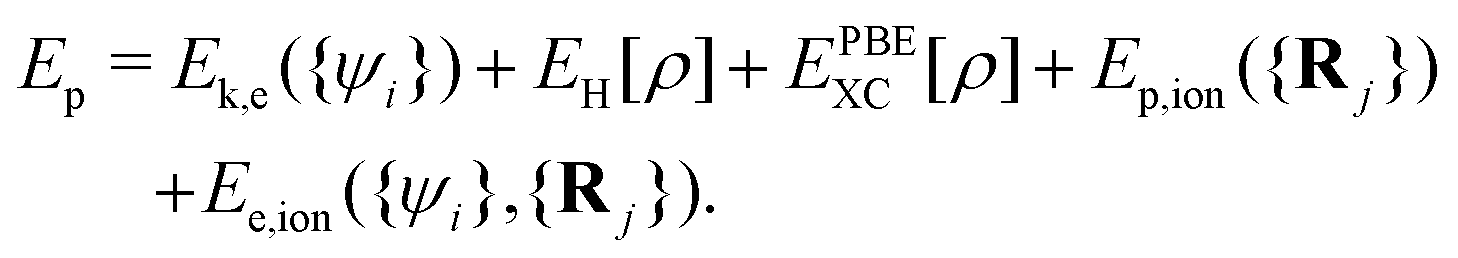 | (10) |
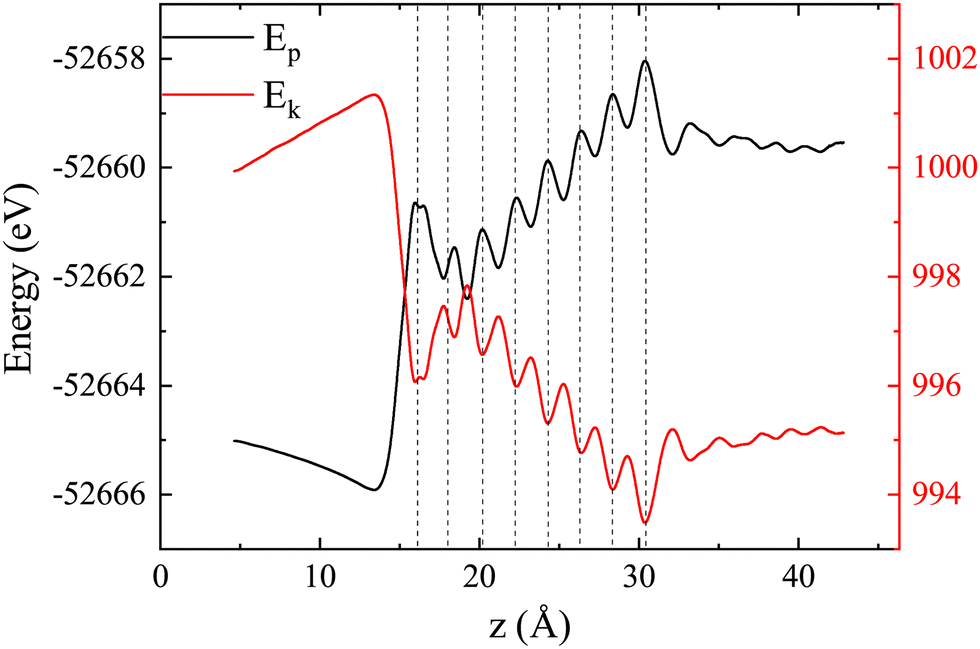 | ||
| Fig. 4 The total potential energy (Ep) and total kinetic energy (Ek) of the system when the proton is moving with an incidence velocity of 0.2 a.u. along channel-1. The vertical black dashed lines are indications of the positions of LiF atom layers. | ||
The sum of these two energies is the total energy of this system, which is a straight line (thus not shown). Overall, it is evident that the potential energy increases, while the kinetic energy decreases. Meanwhile, the total energy remains constant, indicating that kinetic energy is converted into potential energy.
3.2 Density of states and number of electrons
To gather detailed information about the electronic states involved in the stopping process, we calculated the adiabatic density of states (DOS) for different positions of protons along three distinct incident trajectories in the LiF nanosheet, as shown in Fig. 5 and Fig. S4, S538 (see the ESI†). Here total DOS of the system is defined as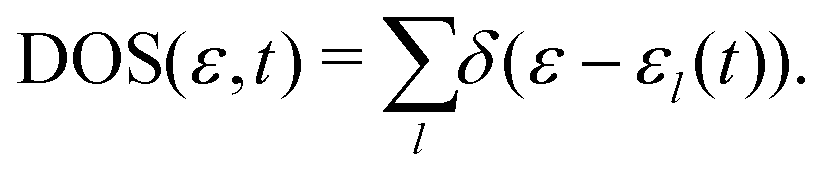 | (11) |
 | ||
| Fig. 5 Snapshots of DOS, ODOS, and PDOS show the projectiles with an incidence velocity of 0.2 a.u. located at z = 4.63, 8.65, 15.14, 23.87, 31.73, and 40.45 Å, respectively, under channel-1. The initial position of the Fermi level is indicated. The dashed ellipse represents the position of the adiabatic eigenstates that contribute to the H(1s) state, while the number of electrons occupying the H(1s) state within the elliptical area is shown above it. | ||
The delta function, denoted as δ(ε − εl(t)), has been approximated by a Gaussian function in our numerical calculations with a standard deviation of 0.05 a.u. In essence, DOS is a statistical distribution that represents the number of adiabatic eigenvalues per unit energy interval. Each adiabatic eigenvalue εl(t) corresponds to an adiabatic eigenstate ϕl(t), and thus the statistical distribution of adiabatic eigenvalues is a reflection of the statistical distribution of adiabatic eigenstates, which is also referred to as the DOS.
Furthermore, the occupied DOS (ODOS) can be expressed as:
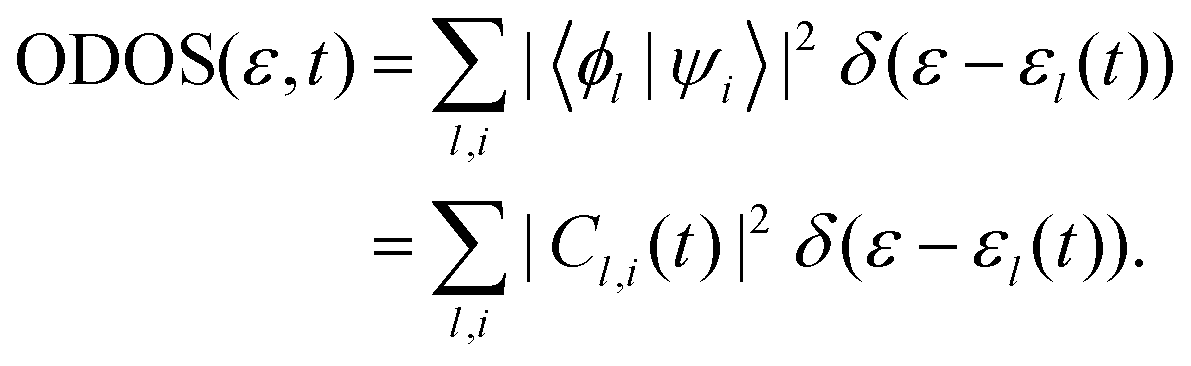 | (12) |
It is important to mention that the total DOS depicted in Fig. 5 and Fig. S4, S538 (see the ESI†) is unoccupied in the high-energy state. This suggests that the 500 adiabatic eigenstates utilized in this simulation are sufficient to fully describe the electron excitation.
In order to further describe the charge transfer process of the projectile inside the material, the projected DOS (PDOS) of the projectile's jth atomic orbital φj is expressed as:
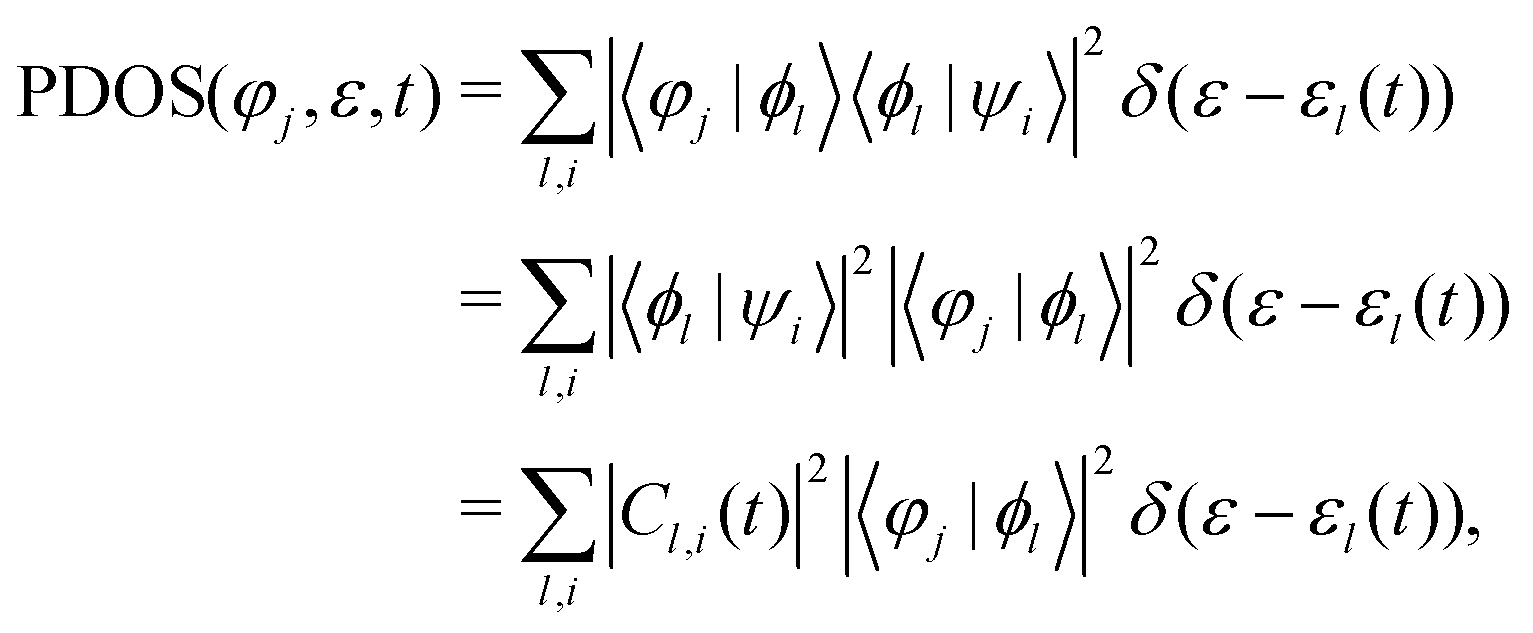 | (13) |
It is important to mention that the higher the excited state, the larger the volume occupied. In the case of the LiF nanosheet, there is limited space to accommodate electrons captured by projectiles. Therefore, only the 1s state electrons are most likely to survive in collision processes.
To facilitate further discussion, we classify the energy level of εl(t) into three distinct ranges based on its proximity to the Fermi level EF at initial time, as outlined below: (1) deep-level range (Dl), where εl(t) is significantly distant from EF; (2) shallow-level range (Sl), where εl(t) is in close proximity to EF; and (3) middle-level range (Ml), where εl(t) is positioned between Dl and Sl.
Fig. 5 and Fig. S4, S538 (see the ESI†) show the representative distribution of DOS, ODOS and PDOS in the five stages for an incident velocity of 0.2 a.u. under three channels with the projectiles located at z = 8.65, 15.14, 23.87, 31.73, and 40.45 Å, respectively. When analyzing Fig. 5 and Fig. S4, S538 (see the ESI†) combined with Table 1, we can observe several interesting findings regarding the same phenomenon but with different incident trajectories as follows:
| Stage | z (Å) | Channel-1 | Channel-2 | Channel-3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D l | M l | S l | C l | D l | M l | S l | C l | D l | M l | S l | C l | ||
| 4.63 | 128.0 | 128.0 | 384.0 | 0.0 | 128.0 | 128.0 | 384.0 | 0.0 | 128.0 | 128.0 | 384.0 | 0.0 | |
| (I) | 8.65 | 127.974 | 127.974 | 384.227 | 0.06 | 128.076 | 127.986 | 383.878 | 0.27 | 127.942 | 127.899 | 384.004 | 0.155 |
| (II) | 15.14 | 127.974 | 127.886 | 384.028 | 0.112 | 127.92 | 128.002 | 383.808 | 0.27 | 128.003 | 128.063 | 383.924 | 0.01 |
| (III) | 23.87 | 128.01 | 128.006 | 382.7068 | 0.856 | 127.923 | 128.013 | 382.685 | 0.973 | 127.936 | 127.996 | 382.719 | 1.135 |
| (IV) | 31.73 | 127.984 | 128.016 | 382.516 | 0.827 | 127.965 | 127.98 | 382.522 | 0.905 | 127.983 | 127.989 | 382.485 | 1.038 |
| (V) | 40.45 | 128.0 | 128.0 | 382.453 | 0.698 | 128.0 | 128.0 | 382.5432 | 0.6485 | 128.0 | 128.0 | 382.458 | 0.833 |
(1) In stage (I), only shallow-level electrons contribute to the H(1s) state, although this contribution is small, suggesting that protons only capture electrons from the shallow-level to the 1s orbital of H with a very small probability.
(2) In stage (II), as the contribution of shallow-level electrons to the H(1s) state increases significantly, the contribution of middle-level electrons also becomes apparent. Additionally, combined with Fig. 1b (II), it is important to note that surface holes have been created when the proton enters the front surface of the LiF nanosheet.
(3) In stage (III), as the holes vanish, electrons in the conduction band are produced. Simultaneously, the contribution of shallow-level electrons to the H(1s) state reduces, while the contribution of middle-level and deep-level electrons to the H(1s) state emerges.
(4) In stage (IV), the number of electrons being excited to the conduction band keeps growing. As the contribution of deep-level electrons to the H(1s) state disappears, the contribution of shallow-level electrons to the H(1s) state reappears.
(5) In stage (V), after the collision, when the projectile and target are far apart, it is observed that the deep and middle levels are completely filled with electrons, while the shallow-levels are not fully filled with electrons. The number of electrons reduced at the shallow-level is equal to the number of electrons that have been captured and excited by protons to the conduction band.
Interestingly enough, as shown in stages (I–III), the process of charge transfer occurs sequentially. At first, electrons are captured from shallow energy levels. As the collision continues, the phenomenon of capturing electrons from deeper levels starts to take place. In stages (I–V), compared to the case where the projectile is at z = 4.63 Å, the energy levels of the occupied adiabatic eigenstates shift towards lower energies. According to the explanation in ref. 25, this result is not surprising. When a proton approaches F−, its positive charge effectively increases the nuclear charge of F− by 1. This results in an isoelectronic configuration similar to a neutral Ne atom in the unified-atom limit. In other words, it is as if F− is replaced by a neutral Ne atom with all electrons tightly bound. Another reason for the decrease in energy levels is that LiF nanosheets cause the ionization of some electrons into the vacuum. As a consequence, the valence band is shifted to lower energies.
When analyzing Fig. 5 and Fig. S4, S538 (see the ESI†) combined with Tables 1 and 2, we can observe several interesting findings regarding the different phenomena with different incident trajectories, as follows: (1) more conduction band electrons are produced through channel-3 trajectories. (2) In stage (II), protons following channel-3 trajectories tend to generate fewer surface holes (the surface hole numbers corresponding to channel-1, chennel-2, and channel-3 are 0.77564, 0.65386, and 0.62884, respectively). (3) In stage (V), after the collision, when the projectile and target are far apart, protons following channel-3 trajectories tend to capture fewer electrons. Overall, through a comprehensive analysis of Fig. 5 and Fig. S4, S538 (see the ESI†) combined with Tables 1 and 2, we can obtain a large amount of detailed information about the electronic state of the stopping process, and be able to gain valuable insights into the interaction between protons and LiF nanosheets.
| Stage | z (Å) | Channel-1 | Channel-2 | Channel-2 |
|---|---|---|---|---|
| 4.63 | 0.0 | 0.0 | 0.0 | |
| (I) | 8.65 | 0.0419 | 0.04466 | 0.04244 |
| (II) | 15.14 | 1.62518 | 1.65972 | 1.57595 |
| (III) | 23.87 | 1.39064 | 1.36596 | 1.29722 |
| (IV) | 31.73 | 1.17126 | 1.12012 | 1.03637 |
| (V) | 40.45 | 0.84948 | 0.80829 | 0.70972 |
The average number of electrons ne captured by the jth atomic orbital of the atom is expressed as follows:
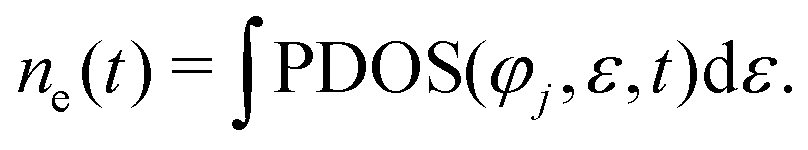 | (14) |
Fig. 6 shows the difference Δne = ne(t1) − ne(t0) of ne in different energy levels, respectively. Here, t1 represents the proton moving to a specific position, and t0 represents the proton at its initial moment. It is worth noting that a positive value of Δne indicates the gain of electrons, while a negative value of Δne indicates the loss of electrons. The designations “F-2s”, “F-2p”, “Li-1s”, and “Li-2s” refer to the respective atomic orbitals. Initially, there are 123.69 electrons in the F-2s orbital, 350.48 electrons in the F-2p orbital, 127.55 electrons in the Li-1s orbital and 38.34 electrons in the Li-2s orbital.
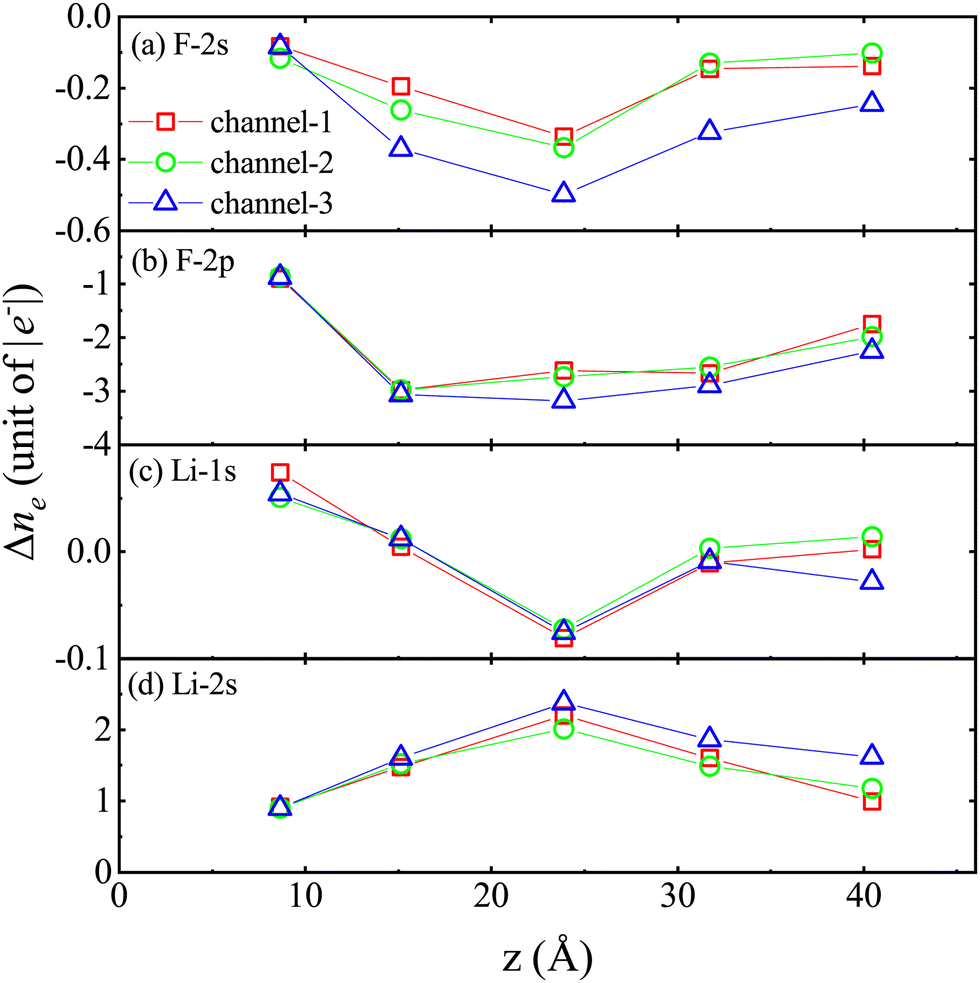 | ||
| Fig. 6 The differences Δne in the average electron numbers of F and Li atoms at different energy levels at z = 8.65, 15.14, 23.87, 31.73, and 40.45 Å when a proton is incident at a velocity of 0.2 a.u., where the energy levels are (a) F-2s, (b) F-2p, (c) Li-1s, and (d) Li-2s. | ||
The analysis of the Δne reveals the following observations:
(1) In stage II–V, compared to the trajectories of channel-1 and channel-2, the decrease in the number of inner valence electrons occupying the F-2s orbital is more significant under the trajectories of channel-3, indicating that when the incident trajectory of protons is very close to the F atom, the energy gap does not hinder the excitation of inner valence electrons (see Fig. 6a).
(2) In the II–V stage, compared to the orbitals of channel-1 and channel-2, there is a greater reduction in the number of valence electrons occupying the F-2s orbitals in the channel-3 orbitals (see Fig. 6b).
(3) The increase and decrease in the number of inner valence electrons occupying the Li-1s orbital undergo an alternating process (see Fig. 6c).
(4) In stages (II–V), compared to the trajectories of channel-1 and channel-2, the increase in the number of valence electrons occupying the Li-2s orbital is more significant under the trajectories of channel-3 (see Fig. 6d).
(5) During the stopping process, the excitation mechanism of electrons is mainly the transition from the F-2p orbital to the Li-2s orbital.
As is well known, the energy loss process is related to the velocity of incident ions. We also provided results in the ESI†38 (see Fig. S6–S9) at a proton velocity of 0.5 a.u., indicating the same electron excitation mechanism.
It is interesting to compare the present results with those of ref. 25, which initially computed the dynamics of the electronic DOS during the passage of protons through LiF clusters. The present calculations, which combine EMD and rt-TDDFT, incorporate dynamic effects that were not considered in the quasistatic treatment described in ref. 25. Both calculations show that the proton's location in LiF causes the system's valence band to shift towards lower energy, despite using different LiF models in the two calculations. Although there are many papers published on energy loss by slow ions in LiF, this article is focused on two complementary aspects: (1) we investigated how the existence of a velocity-dependent threshold for proton stopping power depends on proton impact parameters. (2) We explored the time dependence of electron capture by the proton as it passes through the nanosheet.
As is well known, the ability of the proton to form molecular orbitals with the target ions and to capture electrons constitutes an additional channel for energy loss for the stopping power. Fig. 7, along with Table 2, illustrates the time dependence of electron capture by the proton with a velocity of 0.2 a.u. as it passes through the nanosheet in channel-1, channel-2, and channel-3. Several interesting findings can be observed as follows: (1) the average amount of charge transferred between protons and LiF nanosheets depends on the position of the protons within the nanosheet. (2) Protons that follow the channel-3 trajectory tend to capture fewer electrons. (3) The average number of electrons captured by protons in channel-3 differs significantly from the other two channels. This is not unexpected, as the availability of electrons in matter may be influenced by the impact parameter. However, it is important to note that the proton, being a singly charged particle, can only accommodate a maximum of two electrons forming H and H−, respectively.
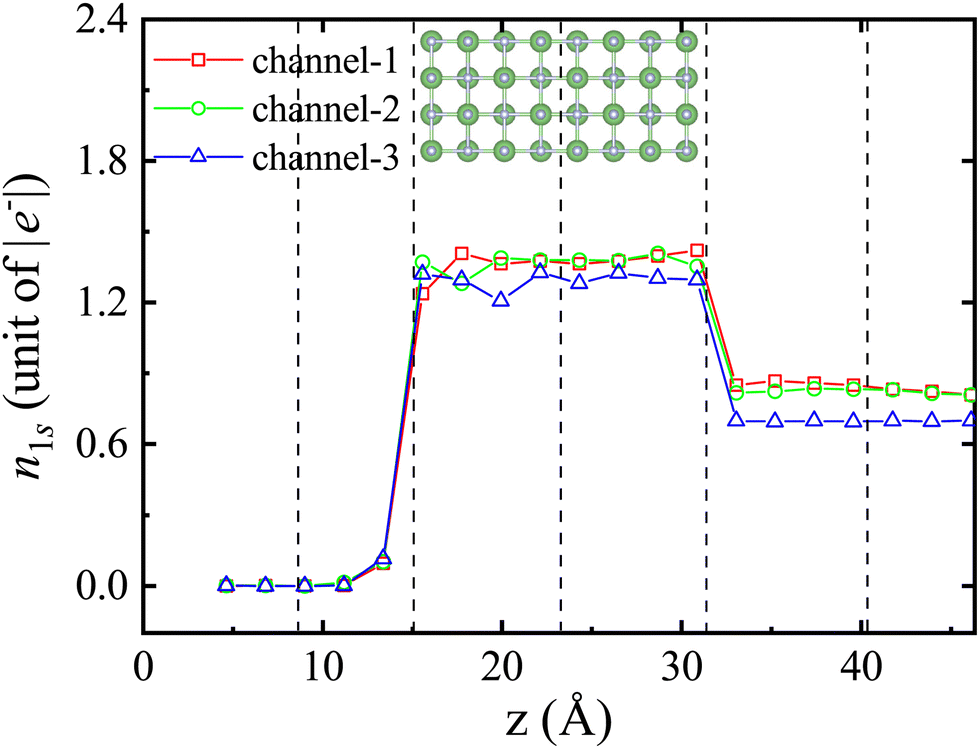 | ||
| Fig. 7 The average number of electrons captured n1s of a proton with a velocity of 0.2 a.u. on different incident trajectories as a function of displacement. The inset shows the position of the LiF nanosheet, where the black dotted lines represent three channels with the projectiles located at z = 8.65, 15.14, 23.87, 31.73, and 40.45 Å. | ||
In all, in terms of trajectory dependence, it is important to mention that electrons in LiF are primarily confined within the fluorine cores. If the proton projectile passes at a sufficient distance from the F nucleus, the energy gap will prevent electron excitation. However, if the proton passes near the F nucleus, it can directly collide with the electrons and excite them, regardless of the gap. This is particularly relevant in the case of surface channeling experiments,21 where protons do not closely approach the fluorine nuclei. This is similar to channel-1 and channel-2 situations in terms of dynamics. Therefore, the threshold effect will no longer exist when following the channel-3 trajectory.
It is important to recognize that, in general, an exact quantum mechanical treatment of the dynamics of many-electron systems is computationally impractical, except for a few special cases. As a result, various models41–43 have been developed to approximate the problem in a numerically feasible way. These models introduce uncertainties, which can be divided into two categories: model uncertainties, which depend on the specific model used and are often poorly understood, and numerical uncertainties, which arise from convergence and other numerical issues associated with the chosen grid or basis set. Similar to static DFT, TDDFT is theoretically exact but requires approximations for the accurate time-dependent XC potential, which incorporates nonlocality in both space and time, as well as the derivative discontinuity property. Currently, popular applications of TDDFT primarily rely on adiabatic approximation, which uses the instantaneous time-dependent density to calculate the XC potential from a known static XC potential. However, it remains unclear whether improving the XC potential in static DFT necessarily leads to a better description in TDDFT.
To address this issue, many benchmark studies44,45 have investigated the performance of using the static XC potential within the adiabatic approximation for TDDFT calculations. However, these studies have mostly focused on linear and perturbative responses, neglecting the regime of nonlinear and non-perturbative responses in strong-excitation processes where TDDFT is computationally advantageous. When applying TDDFT to describe the interaction between ions and materials, the discrepancy between different time-dependent XC potentials diminishes as the ion velocity increases. This is because the ion velocities become comparable to the static electric field experienced by an electron in its energy band,46 which is significantly larger than the XC potential associated with electron–electron interactions. Consequently, the importance of the XC potential decreases with higher velocities due to the dominant external field.47
It should be noted that quantitative agreement between the model and experimental results is not expected without using the correct time-dependent XC potential and atomic pseudopotentials that accurately represent bound and excited states. In this study, we perform benchmark calculations using the adiabatic GGA for the time-dependent XC potential. The chosen benchmark problem involves the charge transfer of charged ions in nanosheets, making it a rigorous test for our new approach. Although the complex mechanisms are not fully captured by the GGA level of TDDFT, we believe that it is crucial to determine the extent to which charge transfer can be quantitatively described within the current theory. This result provides a first-order approximation to the intricate ion-material collision process.
4 Conclusions
In summary, we conducted a study on the trajectory-dependent threshold effects of proton stopping power in LiF nanosheets using time-dependent density functional theory non-adiabatically coupled to molecular dynamics, offering a vast amount of detailed information on the electronic structure in the stopping process with superior spatial and temporal resolution that cannot be obtained from experimentally tabulated data, obtaining a combination of interesting findings: (1) the impact parameters of incident protons play a crucial role in determining the threshold behavior of proton stopping power in the LiF nanosheet. (2) Close collisions of channel-3 do not exhibit a discernible threshold. (3) During the stopping process, protons capture fewer electrons when colliding closely along channel-3. (4) During the stopping process, the excitation mechanism of electrons is mainly the transition from the F-2p orbital to the Li-2s orbital. (5) The disappearance of the threshold effect is due to the excitation of F inner valence electrons when protons collide tightly along channel-3. This work validates the ability of the present methodology in dealing with ion collisions in nanosheets, which may have implications on the study of stopping power in more complex systems in the future. It is still an active field and many new results are expected in the future.Author contributions
Ya-Ting Sun: conceptualization (lead); data curation (lead); formal analysis (lead); methodology (lead); software (lead); visualization (lead); writing – original draft (lead); writing – review & editing (equal). Feng Wang: data curation (supporting); formal analysis (supporting); methodology (supporting); project administration (lead); resources (lead); writing – review & editing (equal). Cong-Zhang Gao: conceptualization (supporting); data curation (supporting); formal analysis (supporting); methodology (supporting).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 11774030) and the Beijing Natural Science Foundation (Grant No. 1242026).Notes and references
- R. S. Averback and T. D. de la Rubia, in Solid State Physics, ed. H. Ehrenreich and F. Spaepen, Academic Press, 1998, vol. 51, pp. 281–402 Search PubMed.
- M. Toulemonde, C. Dufour and E. Paumier, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 14362–14369 CrossRef PubMed.
- S. J. Zinkle and L. L. Snead, Scr. Mater., 2018, 143, 154–160 CrossRef CAS.
- G. Odette and B. D. Wirth, Handbook of Materials Modeling, Springer, Cham, 2005, pp. 999–1037 Search PubMed.
- F. F. Komarov, A. F. Komarov, V. V. Pil’ko and V. V. Pil’ko, J. Eng. Phys. Thermophys., 2013, 86, 1481–1484 CrossRef CAS.
- M. Yamaguchi, Sol. Energy Mater. Sol. Cells, 2001, 68, 31–53 CrossRef CAS.
- P. K. Patel, A. J. Mackinnon, M. H. Key, T. E. Cowan, M. E. Foord, M. Allen, D. F. Price, H. Ruhl, P. T. Springer and R. Stephens, Phys. Rev. Lett., 2003, 91, 125004 CrossRef CAS PubMed.
- L. K. Mansur, J. Nucl. Mater., 1994, 216, 97–123 CrossRef CAS.
- D. Schardt, T. Elsässer and D. Schulz-Ertner, Rev. Mod. Phys., 2010, 82, 383–425 CrossRef.
- M. Durante and J. S. Loeffler, Nat. Rev. Clin. Oncol., 2010, 7, 37–43 CrossRef PubMed.
- S. J. Zinkle and B. N. Singh, J. Nucl. Mater., 1993, 199, 173–191 CrossRef CAS.
- O. Knospe, J. Jellinek, U. Saalmann and R. Schmidt, Phys. Rev. A: At., Mol., Opt. Phys., 2000, 61, 022715 CrossRef.
- G. Schiwietz and P. L. Grande, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 84, 052703 CrossRef.
- C. P. Race, D. R. Mason, M. W. Finnis, W. M. C. Foulkes, A. P. Horsfield and A. P. Sutton, Rep. Prog. Phys., 2010, 73, 116501 CrossRef.
- A. A. Correa, Comput. Mater. Sci., 2018, 150, 291–303 CrossRef CAS.
- E. Fermi and E. Teller, Phys. Rev., 1947, 72, 399–408 CrossRef CAS.
- D. Semrad, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 1646–1652 CrossRef CAS PubMed.
- R. Golser and D. Semrad, Phys. Rev. Lett., 1991, 66, 1831–1833 CrossRef CAS PubMed.
- A. Imamogglu, H. Schmidt, G. Woods and M. Deutsch, Phys. Rev. Lett., 1997, 79, 1467–1470 CrossRef.
- S. P. Møller, A. Csete, T. Ichioka, H. Knudsen, U. I. Uggerhøj and H. H. Andersen, Phys. Rev. Lett., 2004, 93, 042502 CrossRef PubMed.
- C. Auth, A. Mertens, H. Winter and A. Borisov, Phys. Rev. Lett., 1998, 81, 4831–4834 CrossRef CAS.
- M. Draxler, S. P. Chenakin, S. N. Markin and P. Bauer, Phys. Rev. Lett., 2005, 95, 113201 CrossRef CAS PubMed.
- R. Cabrera-Trujillo, J. R. Sabin, Y. Öhrn and E. Deumens, Phys. Rev. Lett., 2000, 84, 5300–5303 CrossRef CAS PubMed.
- J. M. Pruneda, D. Sánchez-Portal, A. Arnau, J. I. Juaristi and E. Artacho, Phys. Rev. Lett., 2007, 99, 235501 CrossRef CAS PubMed.
- B. Solleder, L. Wirtz and J. Burgdörfer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125107 CrossRef.
- X. Qi, F. Bruneval and I. Maliyov, Phys. Rev. Lett., 2022, 128, 043401 CrossRef CAS PubMed.
- L. N. Serkovic Loli, E. A. Sánchez, O. Grizzi and N. R. Arista, Phys. Rev. A: At., Mol., Opt. Phys., 2010, 81, 022902 CrossRef.
- J. L. Alonso, X. Andrade, P. Echenique, F. Falceto, D. Prada-Gracia and A. Rubio, Phys. Rev. Lett., 2008, 101, 096403 CrossRef CAS PubMed.
- A. Castro, M. Isla, J. I. Martínez and J. Alonso, Chem. Phys., 2012, 399, 130–134 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- W.-L. Jia, J.-Y. Fu, Z.-Y. Cao, L. Wang, X.-B. Chi, W.-G. Gao and L.-W. Wang, J. Comput. Phys., 2013, 251, 102–115 CrossRef.
- W.-L. Jia, Z.-Y. Cao, L. Wang, J.-Y. Fu, X.-B. Chi, W.-G. Gao and L.-W. Wang, Comput. Phys. Commun., 2013, 184, 9–18 CrossRef CAS.
- J. Ren, N. Vukmirovic and L. W. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 205117 CrossRef.
- S.-M. Li, F. Mao, X.-D. Zhao, W.-Q. Jin, W.-Q. Zuo, B.-S. Li, F. Wang and F.-S. Zhang, Phys. Rev. B, 2022, 106, 014103 CrossRef CAS.
- S.-M. Li, F. Mao, X.-D. Zhao, B.-S. Li, W.-Q. Jin, W.-Q. Zuo, F. Wang and F.-S. Zhang, Phys. Rev. B, 2021, 104, 214104 CrossRef CAS.
- D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 085117 CrossRef.
- Web site of “primitive cell structure”, https://aflowlib.org/search/.
- The detailed computational information and additional results, are available in the ESI†.
- J. F. Ziegler, J. P. Biersack and M. D. Ziegler, The Stopping and Range of Ions in Matter, Ion Implantation Press, 2008 Search PubMed.
- Web site of “SRIM-2013”, https://www.srim.org/.
- J. A. S. David and S. Sholl, Density Functional Theory: A Practical Introduction, John Wiley & Sons, Inc, 2009 Search PubMed.
- C. A. Ullrich and Z.-H. Yang, Braz. J. Phys., 2014, 44, 154–188 CrossRef.
- K. Burke, J. Chem. Phys., 2012, 136, 150901 CrossRef PubMed.
- M. A. L. Marques, A. Castro and A. Rubio, J. Chem. Phys., 2001, 115, 3006–3014 CrossRef CAS.
- S. S. Leang, F. Zahariev and M. S. Gordon, J. Chem. Phys., 2012, 136, 104101 CrossRef PubMed.
- F. Calvayrac, P.-G. Reinhard, E. Suraud and C. Ullrich, Phys. Rep., 2000, 337, 493–578 CrossRef CAS.
- M. Marques and E. Gross, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00504j |
| This journal is © the Owner Societies 2024 |