
Substitutional control of non-statistical dynamics in the thermal deazetization of tetracyclic azo compounds†
Chandralekha
Hajra
and
Ayan
Datta
 *
*
School of Chemical Sciences, Indian Association for the Cultivation of Science, 2A and 2B Raja S. C. Mullick Road, Jadavpur – 700032, Kolkata, West Bengal, India. E-mail: spad@iacs.res.in; Tel: +91-33-24734971
First published on 23rd October 2024
Abstract
Dynamical control of reactivity for the deazetization of endo,endo-9,10-diazatetracyclo[3.3.2.02,4.06,8]dec-9-ene (3) is studied using on-the-fly quasi-classical trajectory (QCT) calculations at the density functional theory (DFT) level. Two degenerate homotropilidenes, 4 and 5, are formed simultaneously from a single transition state (TS). The ratio of the cyclohexadienyl substituted product, 4, and the dynamical product, i.e. bridgehead substituted product, 5, can be neatly controlled by tuning the topology of the potential energy surface (PES). A steep descent post-TS favors the cyclohexadienyl substituted product while a shallow descent increases the dynamical outcome. Chemical demonstration of the same is achieved by symmetrical and asymmetrical substitution of functional groups along the cleaving (C3–C4) bond. Asymmetric mono-functionalization makes the PES broader, thereby reducing the slope post-TS. This creates a favourable situation for the dynamical products, 5b–5d, to become the major ones. On the contrary, symmetric bi-functionalization makes the cyclohexadienyl substituted product, 4m–4o, overwhelmingly (>85%) predominating. As a corollary to this phenomenon, substitution of the C3–C4 bond by the heavier isotopologues of H/C restricts its motion along the IRC path by the Newtonian kinetic isotope effect. This facilitates bond-opening along the C10–C11 dynamical pathway. Hence, for isotopic substitution, the situation is reversed and the bifunctionalized 3 is more dynamically activated. Simultaneous substitution by the heavier isotopologue of C and H causes deviation from the geometric mean of individual isotopic substitution towards the dynamical product, 5. Therefore, the dynamic control in 3 becomes prominent either via functional group asymmetry or through a Newtonian kinetic isotope effect for symmetric bifunctionalization.
Introduction
Cyclic azo alkanes constitute a versatile class of organic compounds characterized by the presence of one or more azo functional groups. The diverse applications of these compounds arise from both the number of rings and their respective sizes. Their utility spans dyeing, pigment production, and the development of photochromic materials, among others.1–3 They have the ability to exist in different isomeric forms, dictated by the orientation of the rings relative to the azo group and other functional groups attached to the ring. This imparts significant diversity in their reactivity.4 The inherent reactivity of cyclic azo compounds depends critically on the strain within the rings. This enables them to undergo deazetization reactions wherein cleavage of the azo group occurs, further expanding their synthetic possibilities.5The smallest amongst the cyclic rings, cyclopropenes, show high reactivity in bioorthogonal reactions due to their highly strained structure. Recently, cyclopropene derivatives have gained huge attention as they have small sizes and hydrophobic properties. The rapid and selective reaction kinetics of tetrazine derivatives with cyclopropenes have been utilized for imaging purposes owing to the high contrast and minimal background signal.6,7 Inverse-electron demand Diels–Alder reactions between tetrazines and strained alkenes or alkynes have been reviewed by Boger.8 This results in the formation of dihydropyridazines or pyridazines with nitrogen gas being the sole byproduct. Prescher and co-workers have observed isomeric cyclopropenes displaying a variety of reactivities with tetrazines.9 Even though in general cyclopropenes do not react with tetrazine, Sauer and co-workers could form highly strained 3,4-diazanorcaradienes and 9,10-diazatetracyclo[3.3.2.02,4.06,8]dec-9-ene from the reaction of cyclopropene with tetrazines.10,11
Elimination of the azo-group from 9,10-diazatetracyclo[3.3.2.02,4.06,8]dec-9-ene (3) produces homotropilidenes. They show unique chemical reactivity because of their strained structure.12,13Scheme 1 shows the formation of 3 by the [4+2] cycloaddition of symmetric electron-deficient tetrazines (1) with two equivalents of cyclopropenes (2).14 Thermolysis of 3 produces homotropilidenes (4 and 5) through the elimination of a N2 molecule. The degeneracy between 4 and 5 can be broken to selectively prefer either of them based on the substituents on 3. The overall pathway along 1 → 4,5 is significantly thermodynamically downhill, which drives spontaneous product formation (see ESI,† Fig. S1).
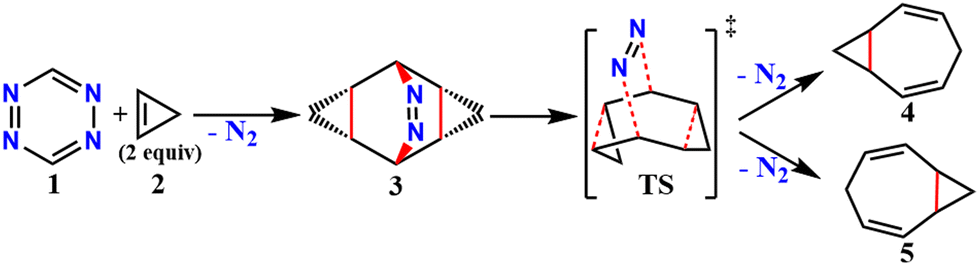 | ||
| Scheme 1 Formation of homotropilidenes through deazetization reaction from 3. | ||
One of the more popular frameworks to understand the selectivity of products is the transition state theory (TST) or variational transition state theory (VTST). Within this model, products are formed through competing independent transition-states and the free-energy difference amongst them can be utilized for a quantitative comprehension of the origin of product selectivity.15,16 However, an alternative product selectivity might arise when two products originate from a single transition state. In this scenario, the pathway of products shares a transition state, diverging after the valley-ridge inflection point when the potential energy surface bifurcates. Therefore, in such cases, post-TS effects like the vibrational motion and momenta of atoms along the non-intrinsic reaction co-ordinate (non-IRC) pathways become predominant.17–20 Tantillo et al. have studied post-TS bifurcations on several organic reactions.19–23 Houk and co-workers have studied non-statistical molecular dynamical effects in deazetization reaction of azoalkanes.24,25 Dynamical effects have been shown to be essential to describe the high selectivity in the [3,3] sigmatropic rearrangement for bispercyclic transition-state.26 Post-transition-state dynamics have been shown to be important for reactions in which the number of concerted processes is more than 3.27 Ess and co-workers have studied the nonstatistical intermediates and dynamically regulated routes for benzene reductive elimination from Mo/W-based organometallic complexes using direct-dynamics simulations.28 Mandal et al. have shown that deazetization reactions for semibullvalene formation, Au(I)-catalysed intramolecular Diels–Alder processes and the valence isomerism in benzene imine are richly controlled dynamical effects.29–31
The mechanism for the thermolysis of azo compounds is of importance32,33 and therefore, the thermolysis of 3 to form 4 and 5 is studied in the present article. The IRC and non-IRC preferences for 4 or 5 are investigated computationally by substituting functional groups (Fig. 1) on 3. Additionally, we carried out detailed quasiclassical trajectory simulations from the ambimodal transition state structure to examine dynamical behaviour. Newtonian kinetic isotope effects (KIEs)34 were computed to assess the kinetic selectivity via isotope labelling.
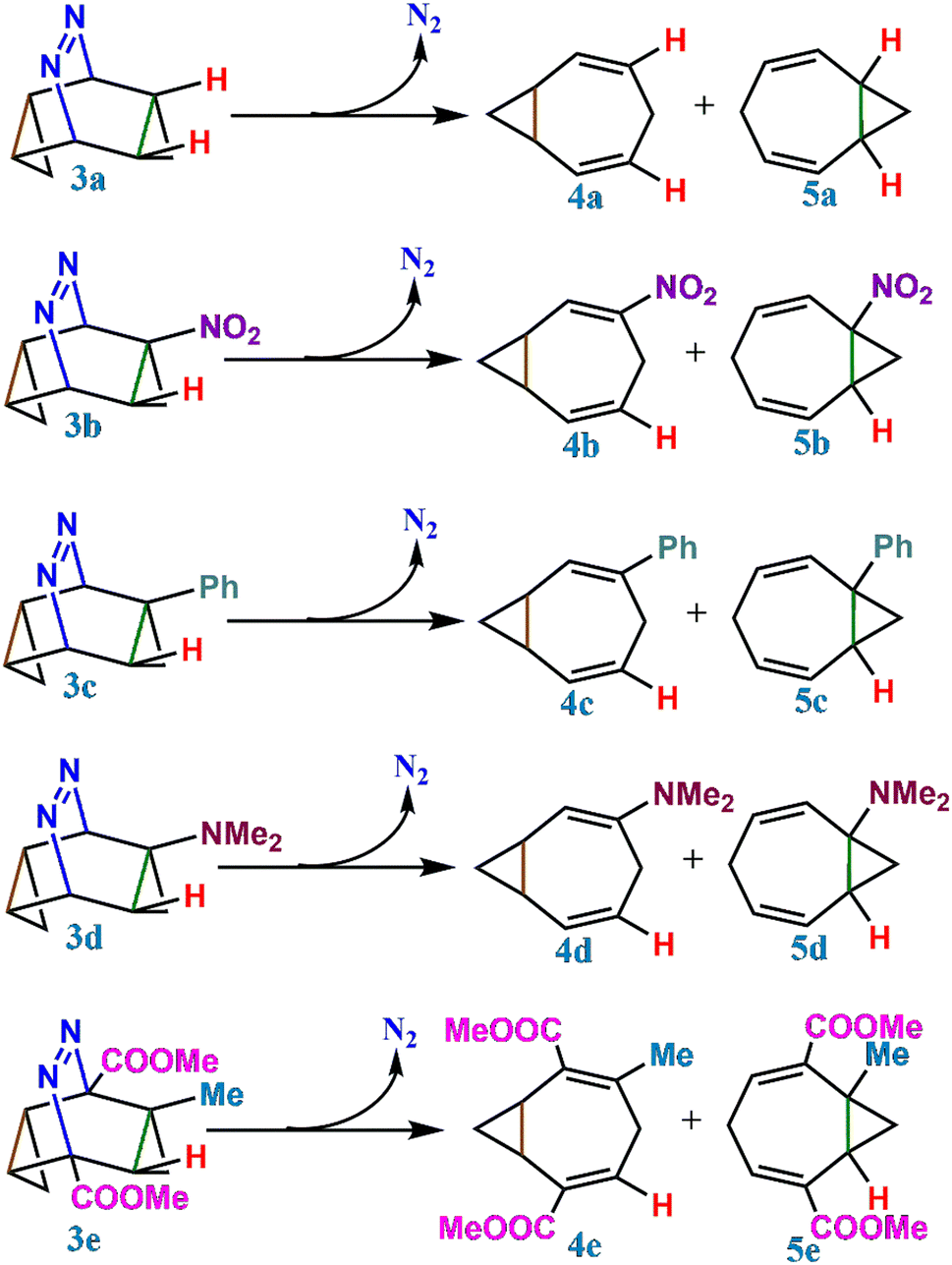 | ||
| Fig. 1 Formation of homotropilidenes (4 and 5) from endo,endo-9,10-diazatetracyclo[3.3.2.02,4.06,8]dec-9-ene (3) through deazetization reaction. | ||
Computational details
The structures for each molecule, along with the harmonic frequencies were computed using the Gaussian 16 program35 at the M06-2X/6-31G(d) level of theory.36,37 To ensure that transition states connect to the desirable stationary-points, intrinsic reaction coordinate (IRC) calculations were performed.38 Benchmarking of these calculations was performed at various levels of theory, including the B3LYP/6-31G(d), B3LYP/6-31+G(d,p) with dispersion corrections using Grimme's D3, and M06-2X/6-31+G(d,p) for a comparison of different theoretical methods.39–41 Additionally, single-point calculations were also performed at the DLPNO-CCSD(T)42,43/def2-TZVP level to further validate the DFT results (see ESI,† Table S1).44 Calculations at the DLPNO-CCSD(T)/def2-TZVP level are also in agreement with the DFT calculations. DLPNO-CCSD(T) calculations are done in ORCA 5.0.45 The 3D-view of the molecular structures was retrieved using Gauss View 6.1.1, a popular visualization tool that aids in the accurate depiction and analysis of complex molecular geometries.The quasiclassical trajectories (QCTs) were simulated via on-the-fly molecular dynamics (MD) using Singleton's ProgDyn code interfaced with Gaussian16, which calculates the force constant at each time-step along with trajectory formation at the M06-2X/6-31G(d) level of theory.46 ProgDyn generates the initial geometries from the precise frequency calculations based on the optimized transition state structures. All direct MD simulations were conducted at 298.15 K to mimic realistic reaction conditions. Normal mode sampling involved adding the zero-point energies for each real normal mode of the transition state, followed by Boltzmann sampling to generate a comprehensive set of coordinates for trajectory propagation. In these simulations, molecular configurations and velocities were propagated in both the forward and backward directions using the velocity Verlet algorithm with a timestep of 1 fs. This method ensured accurate propagation until either the starting material or the product was obtained. At each time step, the nuclei were propagated classically, while the electronic structures were calculated quantum mechanically using DFT (M06-2X/6-31G(d) level). Detailed thresholds for bond formations and trajectory terminations are provided in the ESI† for a reproducible workflow for these simulations.
Results and discussion
As shown in Table 1, the formation of both the homotropilidenes, 4 and 5, is thermodynamically favourable. For 3a, both 4a and 5a are degenerate but when a functional group is substituted on 3, the degeneracy of 4 and 5 is lifted making 4 relatively more stable than 5. The preferential stability of 4 can be understood by the electron-withdrawing/electron-donating properties of the substituents as well as steric effects. In the case of 4b and 5b, a H atom is substituted by the electron withdrawing –NO2 group which increases the s-character of the adjacent bond resulting in an enhanced bond-angle within the cyclopropane ring. This makes 5b relatively unstable (see, ESI,† Tables S2 and S3 for the % s-character from NBO analysis). For 4c, 5c and 4d, 5d, the –NMe2 and phenyl groups have +R effect. Both the groups are able to delocalize the electron-density on adjacent double-bonds. Therefore, 4c and 4d get stabilized.| ΔH | ΔG |
|
|
|
|
|
|---|---|---|---|---|---|---|
| 3a → 4a | −51.0 | −64.2 | 8.2 | 8.3 | 15.5 | 16.2 |
| 3a → 5a | −51.0 | −64.2 | ||||
| 3b → 4b | −52.7 | −66.4 | 8.0 | 7.7 | 14.8 | 16.1 |
| 3b → 5b | −52.5 | −65.6 | ||||
| 3c → 4c | −52.2 | −65.0 | 8.7 | 8.4 | 15.5 | 16.2 |
| 3c → 5c | −50.2 | −63.5 | ||||
| 3d → 4d | −50.9 | −64.0 | 9.2 | 9.1 | 16.6 | 19.1 |
| 3d → 5d | −50.3 | −63.3 |
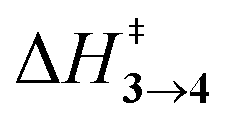
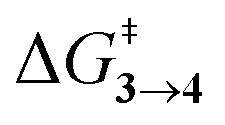
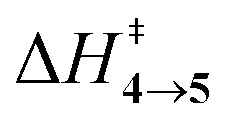
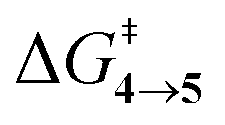
TS (3 → 4,5) involves the N2 elimination reaction. The free-energy barrier, 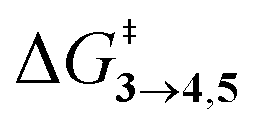 for 3b is the lowest in the series which again is correlated with the smaller % s – character of C–N making it weaker for easy dissociation. The barrier for the 4 → 5 interconversion is significantly higher than the initial product formation barrier. It is worthwhile to mention here that the endo conformers of 3(a–d) are considered here for the thermal deazetization reactions as the barriers are significantly higher for the same in the cases of the exo-conformers, 3# (see ESI,† Table S10). Houk and co-workers have found that the high thermal barrier in the case of the exo-conformers is due to the di-radical nature of the transition-states.24 In fact, deazetization reactions for the exo-conformers occur exclusively under photochemical conditions.10–13
for 3b is the lowest in the series which again is correlated with the smaller % s – character of C–N making it weaker for easy dissociation. The barrier for the 4 → 5 interconversion is significantly higher than the initial product formation barrier. It is worthwhile to mention here that the endo conformers of 3(a–d) are considered here for the thermal deazetization reactions as the barriers are significantly higher for the same in the cases of the exo-conformers, 3# (see ESI,† Table S10). Houk and co-workers have found that the high thermal barrier in the case of the exo-conformers is due to the di-radical nature of the transition-states.24 In fact, deazetization reactions for the exo-conformers occur exclusively under photochemical conditions.10–13
The steepest decent path from 3viaTS in mass-weighted coordinates with infinitesimal atomic velocity as represented by intrinsic reaction coordinate (IRC) calculations connects to 4. For reactant 3a, products 4a and 5a are the same. However for transition states TS(b–d), IRC connects 3(b–d) to the more stable 4(b–d). For 3(a–d), the valley-ridge inflection (VRI) point is found on the right-hand side of the transition-state that is towards the product. In all cases, only product 4 is obtained from IRC calculations except for degenerate 3a which connects TS with TS4→5. Nevertheless, previous experiments on similar reactions have shown that both the products, namely 4 and 5, are found. Additionally from Table 1, it is clear that the reaction barrier from 4 to 5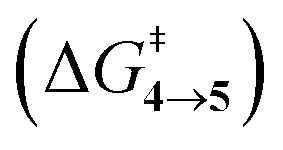 is much higher (∼15–19 kcal mol−1) than
is much higher (∼15–19 kcal mol−1) than 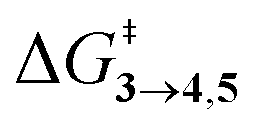 . Therefore, the thermal interconversion from the cyclohexadienyl substituted product (4) to the bridgehead substituted product (5) is unlikely at room temperature. This implies that there must be a post transition-state bifurcation on PES. As shown in Scheme 1, the TS bifurcates into 4 or 5. The bifurcation of PES is caused at the VRI point beyond which trajectories have an option to reach alternative products.
. Therefore, the thermal interconversion from the cyclohexadienyl substituted product (4) to the bridgehead substituted product (5) is unlikely at room temperature. This implies that there must be a post transition-state bifurcation on PES. As shown in Scheme 1, the TS bifurcates into 4 or 5. The bifurcation of PES is caused at the VRI point beyond which trajectories have an option to reach alternative products.
In this regard, we have conducted quasi-classical trajectory (QCT) analyses in the gas-phase. Since the species involved in the reaction are not overtly polar, consideration of implicit polar solvent models should not affect the path significantly. The results of the implicit solvation model of the barriers are reported in Tables S7–S9 (ESI†). The M06-2X/6-31G(d) level of theory is employed for all the trajectory propagations. Trajectories were initiated from transition-state ensembles of TS created using ProgDyn and propagated via classical equations of motion, with forces calculated quantum-mechanically in real-time. Transition-state ensembles were generated by performing Boltzmann sampling of geometries using normal-mode sampling, adding zero-point energy for each real normal mode, and incorporating thermal energy at 298 K to the structures. To verify the robustness of the sampling method, we have plotted an overlay of 30 sampled transition-state structures of TS and plotted the C3–C4 vs. C10–C11 bonds of 100 sampled transition state structures of TS in Fig. 2. We can see that the transition-state structure lies in a zone for which bond lengths are 1.59 ± 0.004 Å. 98% of transition-state bond lengths are in this region.
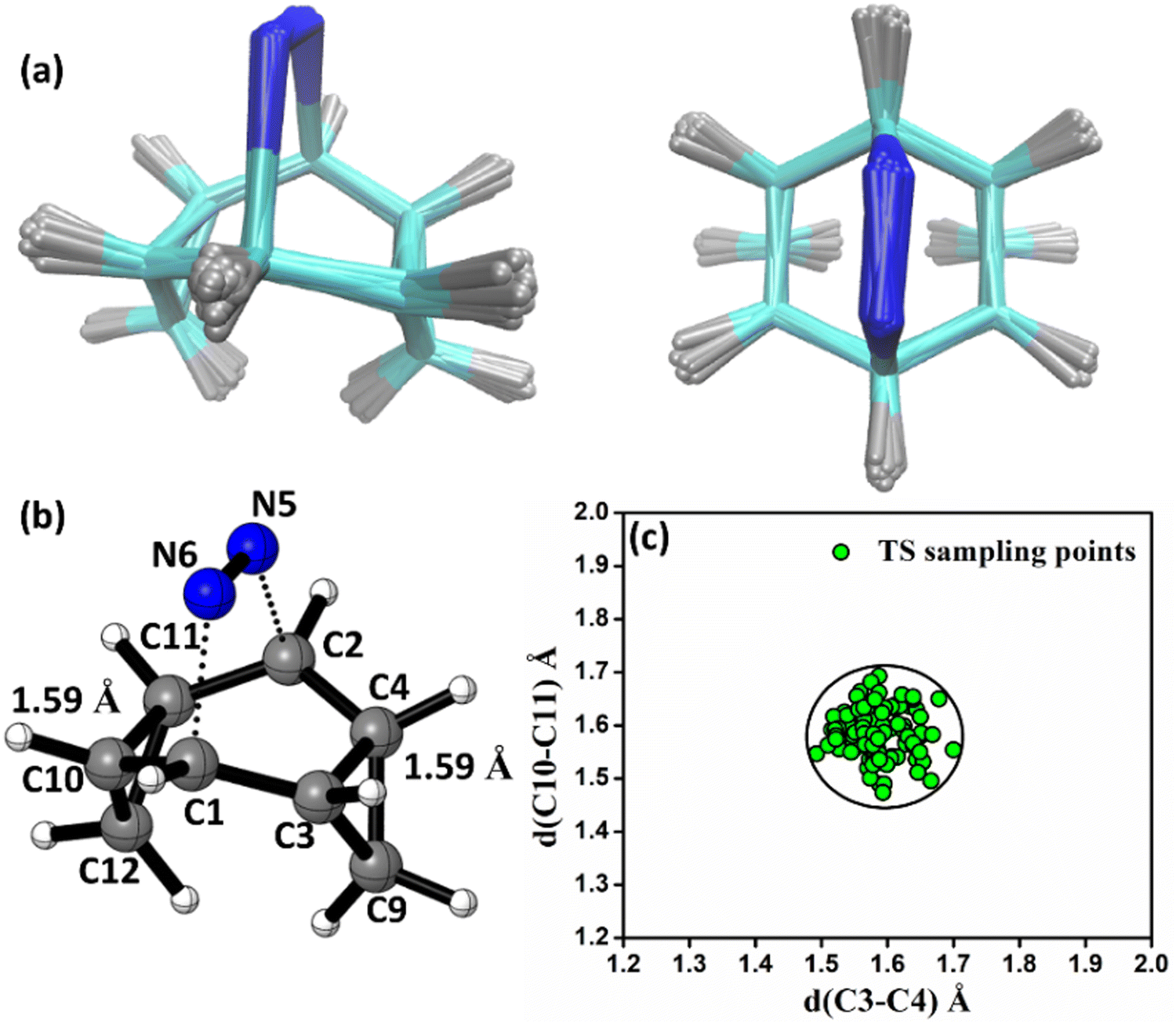 | ||
| Fig. 2 (a) Side and top views of the overlay of 30 sampled TS structures, TS-a. (b) Transition-state (TS-a) obtained along the IRC path between 3a and 4a at the M062X/6-31G(D) level of theory. (c) Scatter plot in C3–C4 and C10–C11 bond lengths across 100 sampled TS (TS-a) structures. | ||
To determine selectivity between products 4 and 5, MD reaction trajectory calculations have been performed. Amongst a total of 363 trajectories that were propagated, 336 (92%) were productive for 3a. Within the productive trajectories, 163 (44.9%) form product 4a while 173 (47.7%) reached product 5a and only 5 (1.5%) trajectories went through TS4a→5a to reach either 4a or 5a. This is in agreement with the intuitive expectation for degenerate product formation producing a numerical 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5a (1:1.06) ratio ∼1. From Table 2, we can see that for the reactant 3b where the substitutional degeneracy is broken, the product ratio between 4b and 5b gets altered from unity. In fact, the bridgehead substituted product, 5b, becomes the major product. Even larger population of the bridgehead substituted product is observed in 3c. 3d also shows that the bridgehead substituted product is major (Fig. 3).
:5a (1:1.06) ratio ∼1. From Table 2, we can see that for the reactant 3b where the substitutional degeneracy is broken, the product ratio between 4b and 5b gets altered from unity. In fact, the bridgehead substituted product, 5b, becomes the major product. Even larger population of the bridgehead substituted product is observed in 3c. 3d also shows that the bridgehead substituted product is major (Fig. 3).
| Reactant | Total trajectories | Productive trajectories | Trajectories reaching 4 (IRC) (cyclohexadienyl substituted product) | Trajectories reaching 5 (dynamical) (bridgehead substituted product) |
4:5 |
|---|---|---|---|---|---|
| 3a | 363 | 336 | 163 | 173 | 1:1.06 |
| 3b | 431 | 394 | 186 | 208 | 1:1.12 |
| 3c | 786 | 720 | 320 | 400 | 1:1.25 |
| 3d | 506 | 476 | 224 | 252 | 1:1.12 |
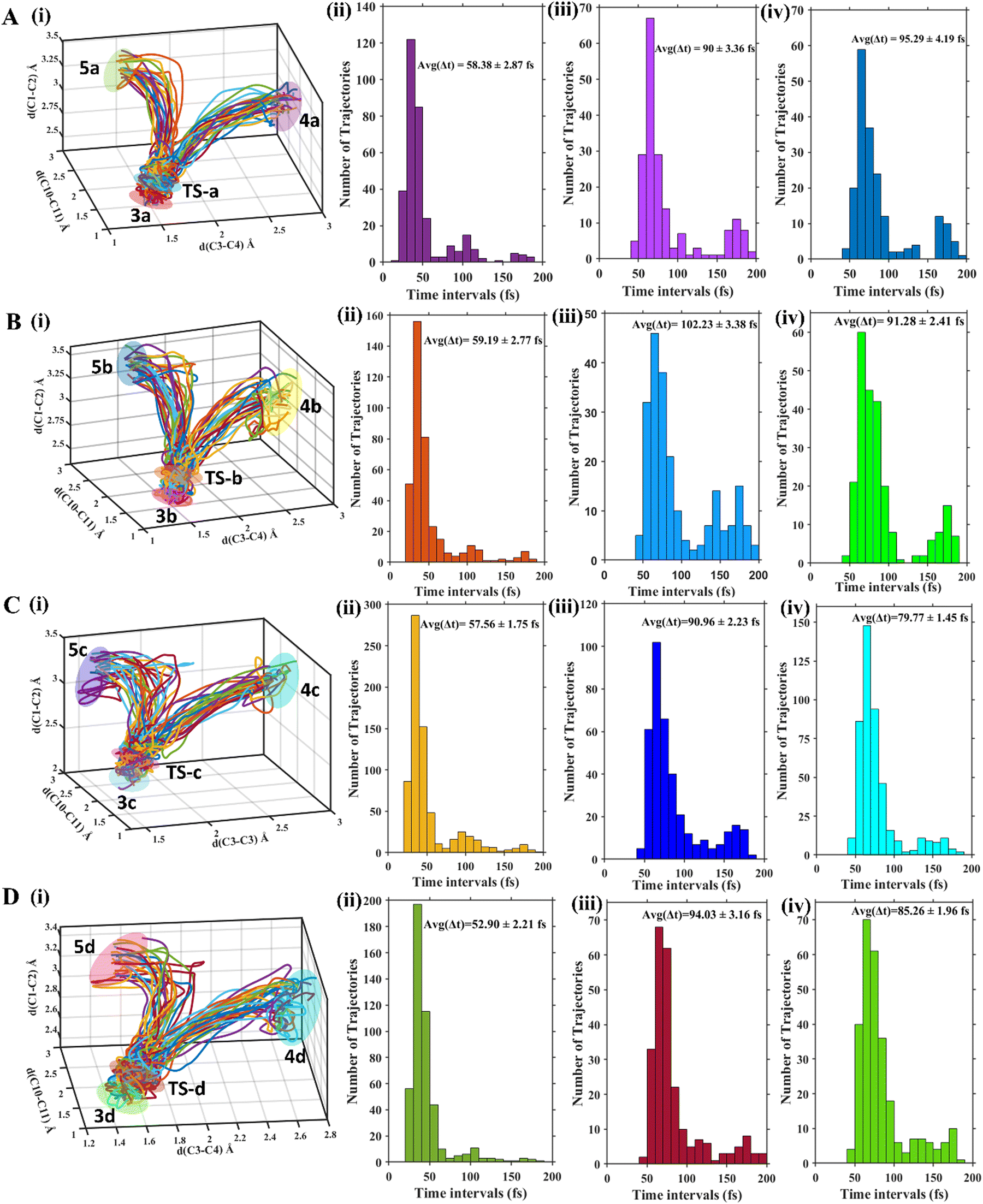 | ||
| Fig. 3 (i) 50 representative trajectories for the formation of 4 and 5 from 3 and average time required for (ii) 3 → TS, (iii) TS → 4, and (iv) TS → 5 for 3(a–d) in (A), (B), (C) and (D), respectively. | ||
In the case of 3a, the average time to reach TS (TS-a) from reactant (3a) is 58.38 ± 2.87 fs and the times taken to form product 4a from TS-a and 5a from TS-a are almost equal. For 4a, it is 90 ± 3.36 fs and for 5a it is 95.29 ± 4.19 fs. Therefore, for the degenerate products 4a and 5a the potential energy surfaces after bifurcation are identical making the product-ratio ∼1. For 3b, the time to reach TS-b from 3b is 59.19 ± 2.77 fs and the times to form product 4b and 5b are 102.23 ± 3.38 fs and 91.29 ± 2.41 fs, respectively. The faster formation of the bridgehead substituted product explains its higher population. 3c forms 5c (t = 79.77 ± 1.45 fs) faster than 4c (t = 90.96 ± 2.23 fs). 3d requires 94.03 ± 3.16 fs and 85.26 ± 1.96 fs to form the cyclohexadienyl substituted and bridgehead substituted products, respectively. For all the cases (3a–3d), one observes at-least 50% bridgehead substituted products. What accounts for such pronounced non-IRC behaviour in these reactions?
To address this, we have further studied the dynamical (bridgehead substituted product) and cyclohexadienyl substituted product distributions in a series of mono and bi-substitutionally modified endo,endo-9,10-diazatetracyclo[3.3.2.02,4.06,8]dec-9-ene (3e–3o) in Table 3. The presence of two functional groups on the same side makes the reactions more symmetrical. Formally, the monosubstituted 3 has only C1 symmetry (see Table 2) while the bi-substituted ones preponderantly have a Cs point-group.47 Comparing the mono and bi-fluorine substituted analogues in 3e and 3f, respectively, the percentage of the dynamical product sharply decreases from 47% to 29%. Analogously, for mono and bi-substituted –Cl groups in 3g and 3h, the contribution of the bridgehead substituted products are 41% and 25%, respectively. Similarly, one observes a strong correlation with asymmetrical functional group substitutions for –CH3, –CN, –NO2, –Ph and –NMe2. The percentages of dynamical products for the mono substituted analogues are 48%, 37%, 53%, 56% and 53% while the same for bi-substitutions are 36%, 15%, 15%, 12% and 13% only. Fig. 4 shows the sharp reduction in the dynamical product (bridgehead substituted product) formation as the substitution in the rings become more symmetric.
| Entry |
|
Total trajectories | Productive trajectories | Trajectories reached 4 | Trajectories reached 5 |
4:5 |
|
ΔG4 (kcal mol−1) | ΔG5 (kcal mol−1) |
|
|---|---|---|---|---|---|---|---|---|---|---|
| 3e | R1 = F, R2 = H | 269 | 244 | 129 | 115 | 1.12:1 |
7.6 | −68.4 | −65.2 | 19.2 |
| 3f | R1 = F, R2 = F | 262 | 240 | 171 | 69 | 2.48:1 |
6.6 | −77.3 | 67.3 | 24.3 |
| 3g | R1 = Cl, R2 = H | 392 | 368 | 218 | 150 | 1.45:1 |
7.7 | −66.7 | −64.3 | 18.2 |
| 3h | R1 = Cl, R2 = Cl | 422 | 389 | 291 | 98 | 2.97:1 |
7.7 | −70.5 | −62.7 | 20.1 |
| 3i | R1 = CH3, R2 = H | 488 | 446 | 234 | 212 | 1.10:1 |
8.0 | −64.4 | −63.3 | 17.2 |
| 3j | R1 = CH3, R2 = CH3 | 604 | 554 | 354 | 200 | 1.77:1 |
8.5 | −65.2 | −60.6 | 18.7 |
| 3k | R1 = CN, R2 = H | 408 | 369 | 231 | 138 | 1.67:1 |
7.8 | −68.2 | −65.0 | 16.5 |
| 3l | R1 = CN, R2 = CN | 324 | 298 | 251 | 47 | 5.34:1 |
7.5 | −74.1 | −64.9 | 18.5 |
| 3m | R1 = NO2, R2 = NO2 | 379 | 351 | 299 | 52 | 5.75:1 |
6.6 | −76.8 | −65.6 | 21.9 |
| 3n | R1 = Ph, R2 = Ph | 431 | 398 | 341 | 49 | 6.96:1 |
10.6 | −66.5 | −62.2 | 15.4 |
| 3o | R1 = NMe2, R2 = NMe2 | 332 | 313 | 272 | 41 | 6.63:1 |
10.0 | −67.4 | −54.7 | 16.5 |
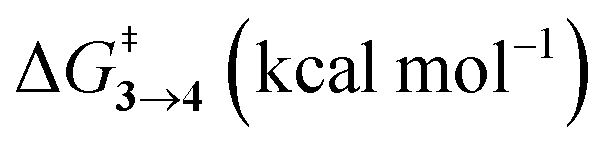
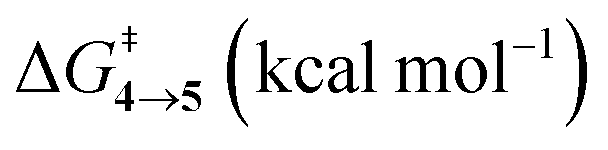
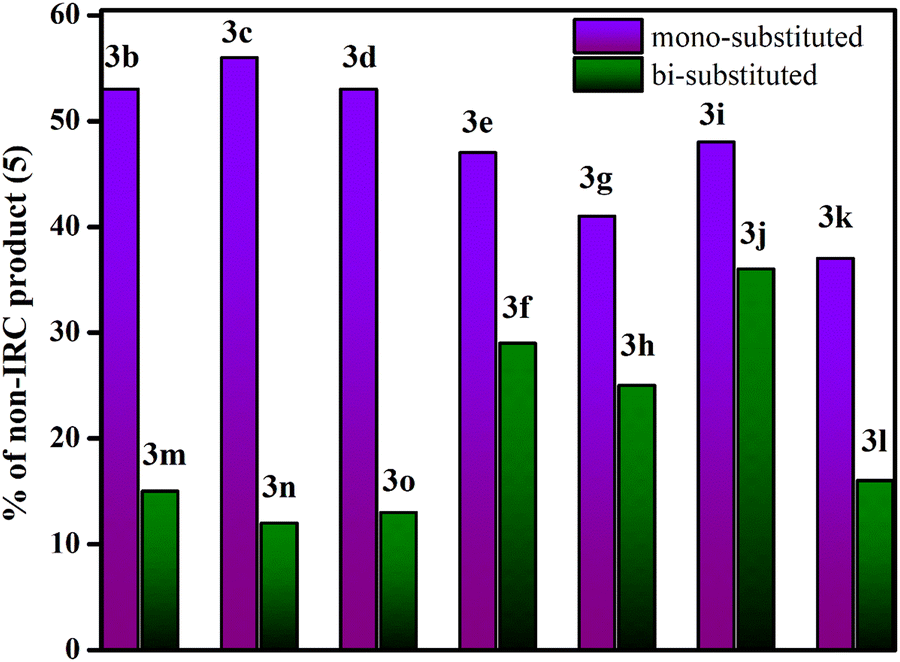 | ||
| Fig. 4 Comparative profile for the percentage of bridgehead substituted products (5) in mono-substituted and bi-substituted endo,endo-9,10-diazatetracyclo[3.3.2.02,4.06,8]dec-9-ene (3b–3o). | ||
To understand the preference of the mono-substituted 3 towards the dynamical products we have analysed the minimum energy pathway (MEP) along the intrinsic reaction coordinate (IRC). Comparing the MEP vs. IRC for the mono and bi-substituted analogues in Fig. 5, we observe that post the transition-state, the doubly substituted molecules descend steeply towards the product. On the other hand, the mono-substituted compounds exhibit a much flatter and plateaued potential energy surface beyond the TS. The greater slope in the case of the former ensures that from the transition-state the molecules have a greater tendency to fall in the product 4 zone (cyclohexadienyl substituted product) and the possibility for bifurcation post-TS is reduced. This suppresses the bridgehead substituted product, 5. This can also be understood from the bond-strengths of the C3–C4/C10–C11 bonds (see Fig. 2) in the TSs. For the mono-substituted cases, NBO analyses (see Table S4, ESI†) for the TS-structures show that the % s-character for the C3–C4 bond > C10–C11 bond, which implies that the C3–C4 bond is stronger than the C10–C11 bond. Therefore, cleavage of the C10–C11 bond should be relatively easier compared to C3–C4. As a result, the fraction of dynamical product 5 is relatively higher. But for the doubly substituted cases the situation gets reversed. The % s-character becomes smaller for the C3–C4 bond than the C10–C11 bond. Therefore, the cyclohexadienyl substituted product 4 gets preference. Additionally, comparing the product stability between the cyclohexadienyl substituted and bridgehead substituted products, the free energy difference ΔΔG(4–5) is much higher (5–13 kcal mol−1) for the doubly substituted cases with respect to the mono-substituted ones (1–3 kcal mol−1). This leads to a steeper MEP for the IRC path in the case of the doubly substituted molecules.
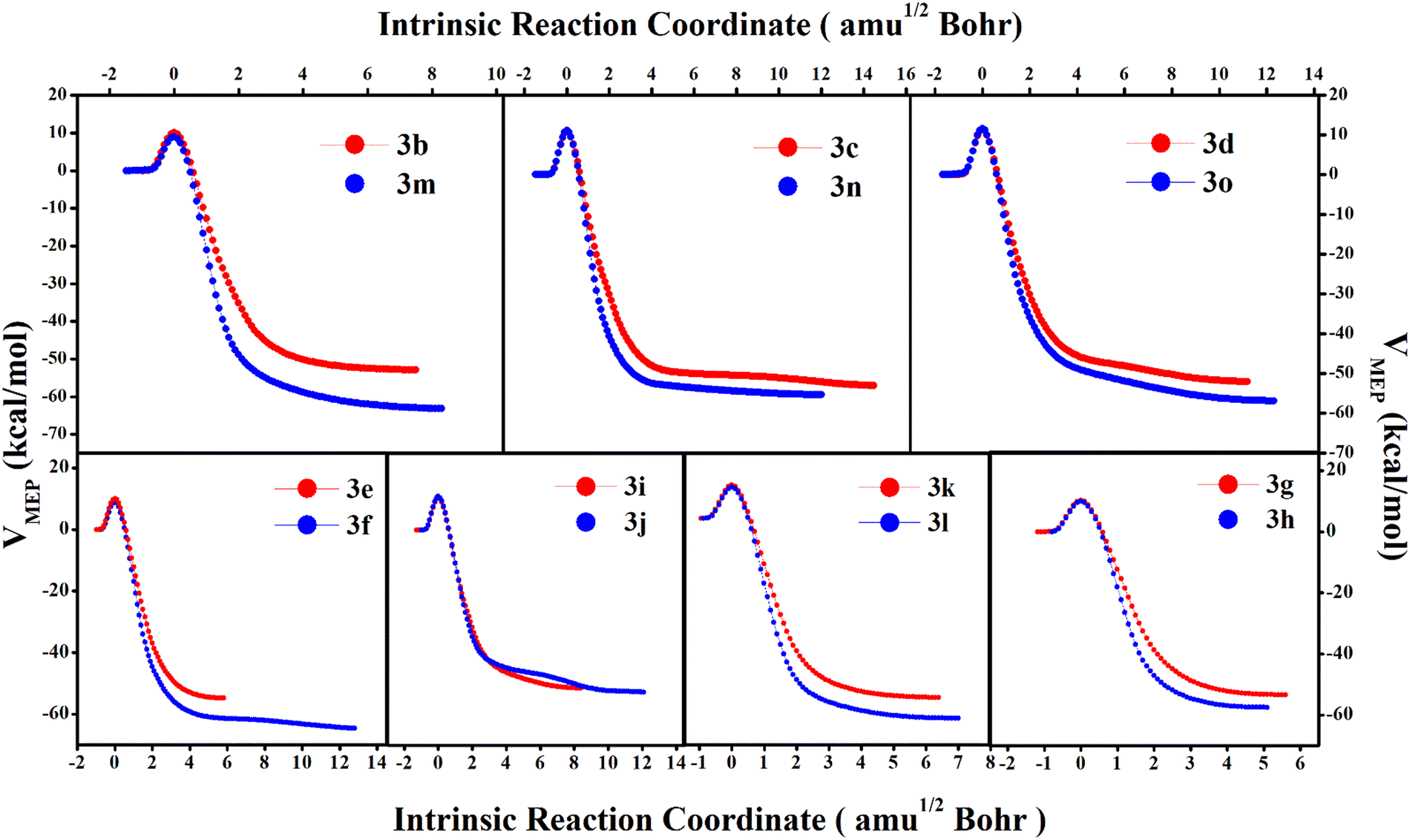 | ||
| Fig. 5 The minimum energy path along the intrinsic reaction coordinate for 3 → 4 for mono and bi-substituted 3. | ||
Experimental detection of post-transition state bifurcation gets facilitated by replacing the lighter isotopes by their heavier isotopologue, like protons by D, carbon by C13 and nitrogen by N15etc. The product selectivity gets controlled by their differences in masses and this being a purely classical (non-quantum) phenomenon is called the Newtonian kinetic isotope effect (NKIE).48 The difference in momenta impacts the zero-point energies (ZPE) as well as the vibrational frequencies of the molecules involved in the reaction. QCT trajectory calculations are performed on the H/D, 12C/13C and 14N/15N isotopologues of 3a (see Fig. 6) and reported in Table 4. In Fig. 6 (I), when a single H is substituted by D, the 4:5 ∼1 because only one hydrogen is substituted at the distal position by deuterium. However, for bi-substitution in II, 4:5 = 0.91:1 indicating the preference towards the bridgehead substituted product. The higher mass along the C3–C4 bond restricts its bond vibration as a result of which bond opening along C10–C11 gets facile. This favours product 5. 14N/15N substitution in III hardly alters the 4:5 ratio from ∼1 as nitrogen is placed remote to the bond-opening site. A significant effect of isotopic substitution occurs for 12C/13C in IV as it is the primary bond-cleavage site. Increasing the heaviness of the C3–C4 bond restricts motion along the IRC path and significantly alters the product selectivity towards the dynamical product with 4:5 = 0.96:1. The effect gets further augmented when D is substituted in the 13C-isotopologue (see V) leading to a 4:5 = 0.91:1. Replacing both the C3 and C4 carbons atoms by 13C augments the KIE to make 4:5 as 0.85 in VI. The dynamic effects get most pronounced in VII where both the carbon and hydrogens are replaced by their heavier isotopes to make 4:5 = 0.65.
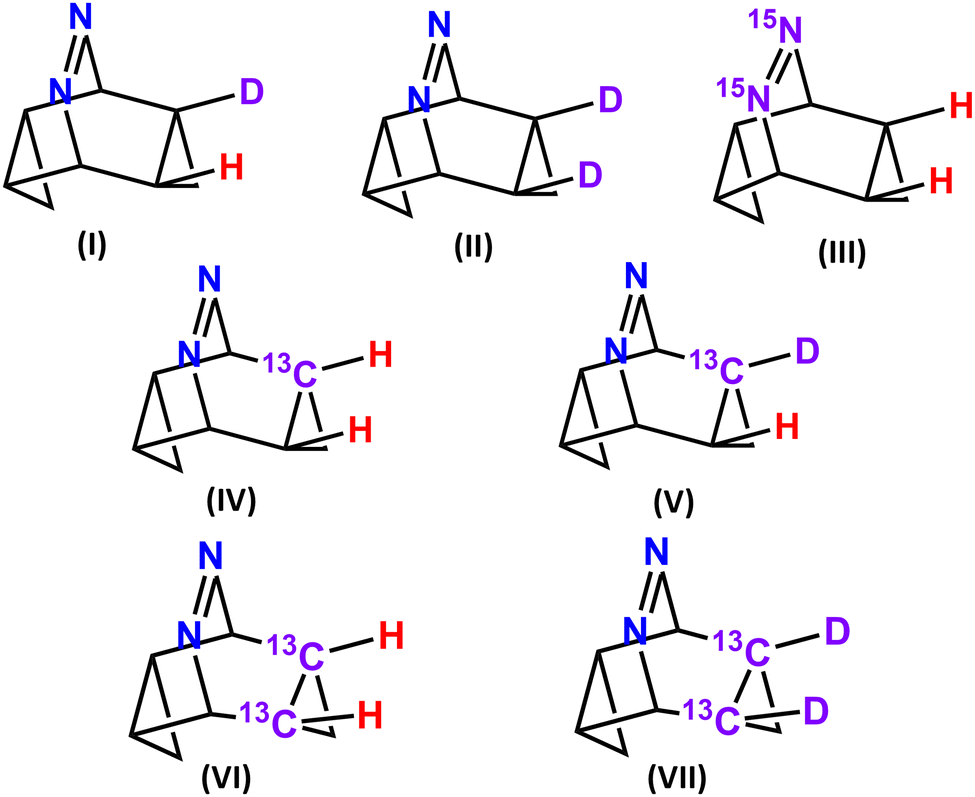 | ||
| Fig. 6 Seven isotopomers for 3a on which quasi classical trajectory calculations are performed. | ||
| Reactant | Total trajectory | Productive trajectory | Trajectory reached 5 | Trajectory reached 6 |
4:5 |
95% confidence |
|---|---|---|---|---|---|---|
| I | 417 | 382 | 191 | 191 | 1:1 |
1.22–0.81 |
| II | 538 | 511 | 243 | 268 | 0.91:1 |
1.08–0.76 |
| III | 1558 | 1436 | 727 | 709 | 1.02:1 |
1.14–0.92 |
| IV | 394 | 364 | 178 | 186 | 0.96:1 |
1.18–0.78 |
| V | 352 | 327 | 156 | 171 | 0.91:1 |
1.14–0.73 |
| VI | 366 | 331 | 152 | 179 | 0.85:1 |
1.06–0.68 |
| VII | 495 | 461 | 182 | 279 | 0.65:1 |
0.79–0.54 |
Dynamic effects get amplified cooperatively as the C3–C4 bond gets substituted by heavier isotopes. For mono-substitution, simultaneous replacement of one C by 13C and the H-atom connected to it in V by D, produces 4:5 = 0.91 which is lower than the product of the individual I and IV. Similarly, 4:5 for VII (= 0.65) < II × VI (= 0.77).
Conclusions
Post-transition state bifurcation on a potential energy surface produces multiple products. While the product derived from the intrinsic reaction coordinate (IRC) is rather straightforward to envision from the transition-state theory (TST) the same fails to provide a quantitative understanding of the dynamical products. The situation becomes further complicated in molecules with complex topography in the PES. By controlling the slope of the PES beyond the TST, the product selectivity can be dictated. Asymmetry in the functional group substitution makes the surface shallower, thereby increasing the population of the dynamical product. On the other hand, symmetric substitution makes the PES steeper resulting in the expected cyclohexadienyl substituted product. An exalted s-orbital character of the crucial C–C bond can be achieved through asymmetric substitution that favors dynamical products (bridgehead substituted product). Functionalization induced alteration of the PES, therefore, provides an experimentally amenable route for exact control of the dynamical effects in a reaction. In fact, the product ratio between the cyclohexadienyl substituted and bridgehead substituted products can be tuned either way just by the symmetry of the substitution in 3. Considering the marked difference between the fate of the reaction, the trade-off between the cyclohexadienyl substituted product and dynamical product bridgehead substituted alternative can be an excellent test to experimentally determine the non-statistical dynamical role of functional-group asymmetry. Additionally, isotopic substitution provides an alternate route towards magnifying the dynamical aspects in the symmetrically substituted reactants. Therefore, the extent to which non-statistical dynamics can affect reactivity, can be explicitly calibrated through both functional group and isotopic substitutions. We expect that at-least one of the two approaches would be experimentally substantiated for the thermal deazetization in 3.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
CH thanks CSIR for the senior research fellowship vide 09/0080(18469)/2024-EMR-I. AD thanks TRC-DST for partial funding.References
- G. Sudesh Kumar, Azo Functional Polymers: Functional Group Approach in Macromolec, CRC Press, 1992 Search PubMed.
- R. S. Aliabadi and N. O. Mahmoodi, Synthesis and Characterization of Polypyrrole, Polyaniline Nanoparticles and Their Nanocomposite for Removal of Azo Dyes; Sunset Yellow and Congo Red, J. Cleaner Prod., 2018, 179, 235–245 CrossRef.
- S. Aiken, R. J. L. Edgar, C. D. Gabbutt, B. M. Heron and P. A. Hobson, Negatively Photochromic Organic Compounds: Exploring the Dark Side, Dyes Pigm., 2018, 149, 92–121 CrossRef CAS.
- P. S. Engel, Mechanism of the Thermal and Photochemical Decomposition of Azoalkanes, Chem. Rev., 1980, 80(2), 99–150 CrossRef CAS.
- R. A. Bartsch, Y. M. Chae, S. Ham and D. M. Birney, Experimental and Theoretical Studies on the Thermal Decomposition of Heterocyclic Nitrosimines1, J. Am. Chem. Soc., 2001, 123(31), 7479–7486 CrossRef CAS PubMed.
- D. M. Patterson and J. A. Prescher, Orthogonal Bioorthogonal Chemistries, Curr. Opin. Chem. Biol., 2015, 28, 141–149 CrossRef CAS.
- K. Lang and J. W. Chin, Bioorthogonal Reactions for Labeling Proteins, ACS Chem. Biol., 2014, 9(1), 16–20 CrossRef CAS.
- D. L. Boger, Diels-Alder Reactions of Heterocyclic Aza Dienes. Scope and Applications, Chem. Rev., 1986, 86(5), 781–793 CrossRef CAS.
- D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H.-W. Shih, K. N. Houk and J. A. Prescher, Isomeric Cyclopropenes Exhibit Unique Bioorthogonal Reactivities, J. Am. Chem. Soc., 2013, 135(37), 13680–13683 CrossRef CAS PubMed.
- H. D. Fühlhuber, C. Gousetis, T. Troll and J. Sauer, Photolysen von tetracyclischen azoverbindungen (,-9,10-diazatetracyclo[3.3.2.02,4.06,8]dec-9-ene) ein leichter zugang zu substituierten homotropilidenen, Tetrahedron Lett., 1978, 19(41), 3903–3906 CrossRef.
- R. Dyllick-Brenzinger, J. F. M. Oth, H. D. Fühlhuber, C. Gousetis, T. Troll and J. Sauer, Einfluss von Substituenten Auf Die Gleichgewichtslage Unsymmetrisch Substituierter Und Die Geschwindigkeit Der Valenzisomerisierung Symmetrisch Substituierter Homotropilidene, Tetrahedron Lett., 1978, 19(41), 3907–3910 CrossRef.
- C. Gousetis and J. Sauer, Ein beitrag zum mechanismus der photolyse tetracyclischer azoverbindungenx) (exo,exo-9,10-diazatetracyclo[3.3.2.02,4,06,8]dec-9-ene), Tetrahedron Lett., 1979, 20(15), 1295–1298 CrossRef.
- J. Sauer, P. Bäuerlein, W. Ebenbeck, R. Dyllick-Brenzinger, C. Gousetis, H. Sichert, T. Troll and U. Wallfahrer, The Cycloaddition-Cycloelimination Pathway to Homotropilidenes − Synthesis and Properties of Homotropilidenes, Eur. J. Org. Chem., 2001,(14), 2639–2657 CrossRef.
- J. Sauer, P. Bäuerlein, W. Ebenbeck, C. Gousetis, H. Sichert, T. Troll, F. Utz and U. Wallfahrer, [4+2] Cycloadditions of 1,2,4,5-Tetrazines and Cyclopropenes − Synthesis of 3,4-Diazanorcaradienes and Tetracyclic Aliphatic Azo Compounds, Eur. J. Org. Chem., 2001,(14), 2629–2638 CrossRef.
- D. Truhlar and B. Garrett, Variational Transition State Theory, Annu. Rev. Phys. Chem., 1980, 35, 159–189 CrossRef.
- B. C. Garrett and D. G. Truhlar, Transition state theory, Encycl. Comput. Chem., 1998, 5, 3094, DOI:10.1002/0470845015.CTA014.
- D. H. Ess, S. E. Wheeler, R. G. Iafe, L. Xu, N. Celebi-Olçüm and K. N. Houk, Bifurcations on Potential Energy Surfaces of Organic Reactions, Angew. Chem., Int. Ed., 2008, 47(40), 7592–7601 CrossRef.
- P. Collins, B. K. Carpenter, G. S. Ezra and S. Wiggins, Nonstatistical Dynamics on Potentials Exhibiting Reaction Path Bifurcations and Valley-Ridge Inflection Points, J. Chem. Phys., 2013, 139(15), 154108 CrossRef.
- H.-H. Chuang, D. J. Tantillo and C.-P. Hsu, Construction of Two-Dimensional Potential Energy Surfaces of Reactions with Post-Transition-State Bifurcations, J. Chem. Theory Comput., 2020, 16(7), 4050–4060 CrossRef.
- S. R. Hare and D. J. Tantillo, Post-Transition State Bifurcations Gain Momentum – Current State of the Field, J. Macromol. Sci., Part A: Pure Appl. Chem., 2017, 89(6), 679–698 CAS.
- D. J. Tantillo, Beyond Transition State theory—Non-Statistical Dynamic Effects for Organic Reactions, in Advances in Physical Organic Chemistry, ed. I. H. Williams and N. H. Williams, Academic Press, 2021, ch. 1, vol. 55, pp. 1–16 Search PubMed.
- Y. J. Hong and D. J. Tantillo, A Potential Energy Surface Bifurcation in Terpene Biosynthesis, Nat. Chem., 2009, 1(5), 384–389 CrossRef CAS PubMed.
- S. R. Hare, A. Li and D. J. Tantillo, Post-Transition State Bifurcations Induce Dynamical Detours in Pummerer-like Reactions, Chem. Sci., 2018, 9(48), 8937–8945 RSC.
- K. S. Khuong and K. N. Houk, One-Bond, Two-Bond, and Three-Bond Mechanisms in Thermal Deazetizations of 2,3-diazabicyclo[2.2.2]oct-2-Enes, Trans-Azomethane, and 2,3-diazabicyclo[2.2.1]hept-2-Ene, J. Am. Chem. Soc., 2003, 125(48), 14867–14883 CrossRef CAS.
- L. Törk, G. Jiménez-Osés, C. Doubleday, F. Liu and K. N. Houk, Molecular Dynamics of the Diels–Alder Reactions of Tetrazines with Alkenes and N2 Extrusions from Adducts, J. Am. Chem. Soc., 2015, 137(14), 4749–4758 CrossRef.
- R. Villar López, O. N. Faza and C. Silva López, Dynamic Effects Responsible for High Selectivity in a [3,3] Sigmatropic Rearrangement Featuring a Bispericyclic Transition State, J. Org. Chem., 2017, 82(9), 4758–4765 CrossRef PubMed.
- M. Castiñeira Reis, C. S. López, O. Nieto Faza and D. J. Tantillo, Pushing the Limits of Concertedness. A Waltz of Wandering Carbocations, Chem. Sci., 2019, 10(7), 2159–2170 RSC.
- J. I. Wheeler, A. J. Schaefer and D. H. Ess, Trajectory-Based Time-Resolved Mechanism for Benzene Reductive Elimination from Cyclopentadienyl Mo/W Phenyl Hydride Complexes, J. Phys. Chem. A, 2024, 128(24), 4775–4786 CrossRef CAS.
- N. Mandal and A. Datta, Dynamical Effects along the Bifurcation Pathway Control Semibullvalene Formation in Deazetization Reactions, J. Phys. Chem. B, 2018, 122(3), 1239–1244 CrossRef CAS PubMed.
- N. Mandal, A. Das, C. Hajra and A. Datta, Stereoelectronic and Dynamical Effects Dictate Nitrogen Inversion during Valence Isomerism in Benzene Imine, Chem. Sci., 2022, 13(3), 704–712 RSC.
- N. Mandal and A. Datta, Gold(I)-Catalyzed Intramolecular Diels–Alder Reaction: Evolution of Trappable Intermediates via Asynchronous Transition States, J. Org. Chem., 2018, 83(18), 11167–11177 CrossRef CAS PubMed.
- E. L. Allred and J. C. Hinshaw, Thermolysis of Azo-Compounds: A Reactivity Factor of 10 17, J. Chem. Soc. D, 1969,(18), 1021–1022 RSC.
- M. B. Reyes and B. K. Carpenter, Mechanism of Thermal Deazetization of 2,3-Diazabicyclo[2.2.1]hept-2-Ene and Its Reaction Dynamics in Supercritical Fluids, J. Am. Chem. Soc., 2000, 122(41), 10163–10176 CrossRef.
- K. K. Kelly, J. S. Hirschi and D. A. Singleton, Newtonian Kinetic Isotope Effects. Observation, Prediction, and Origin of Heavy-Atom Dynamic Isotope Effects, J. Am. Chem. Soc., 2009, 131(24), 8382–8383 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120(1), 215–241 Search PubMed.
- P. C. Hariharan and J. A. Pople, The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies, Theor. Chim. Acta, 1973, 28(3), 213–222 CrossRef.
- K. Fukui, The Path of Chemical Reactions – the IRC Approach, Acc. Chem. Res., 1981, 14(12), 363–368 CrossRef.
- A. Becke, A New Mixing of Hartree-Fock and Local Density-Functional Theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef.
- V. A. Rassolov, M. A. Ratner and J. A. Pople, 6-31G* Basis Set for Third-row Atoms, J. Comput. Chem., 2001, 22(9), 976–984 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, Natural Triple Excitations in Local Coupled Cluster Calculations with Pair Natural Orbitals, J. Chem. Phys., 2013, 139(13), 134101 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, Natural Triple Excitations in Local Coupled Cluster Calculations with Pair Natural Orbitals, J. Chem. Phys., 2013, 139(13), 134101 CrossRef PubMed.
- E. Schiavo, K. Bhattacharyya, M. Mehring and A. A. Auer, Are Heavy Pnictogen-π Interactions Really “π Interactions”?, Chemistry, 2021, 27(58), 14520–14526 CrossRef PubMed.
- F. Neese, Software Update: The ORCA Program System—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12(5), e1606 Search PubMed.
- D. A. Singleton, C. Hang, M. J. Szymanski and E. E. Greenwald, A New Form of Kinetic Isotope Effect. Dynamic Effects on Isotopic Selectivity and Regioselectivity, J. Am. Chem. Soc., 2003, 125(5), 1176–1177 CrossRef PubMed.
- For 3m, the flexibility of rotations along the –NO2 groups causes symmetry-breaking from Cs to C1 point-group.
- Note that the Newtonian kinetic isotope effect is a purely classical momentum driven process and differs from kinetic isotope effects (both primary and secondary) where quantum mechanical tunnelling also plays a significant role. See, S. Karmakar and A. Datta, Heavy-atom Tunneling in Organic Transformations, J. Chem. Sci., 2020, 132, 127, DOI:10.1007/s12039-020-01809-x.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates, energies, and harmonic frequencies, free-energy barriers, calculations for NBO analyses and QCT input. See DOI: https://doi.org/10.1039/d4cp03447c |
| This journal is © the Owner Societies 2024 |