
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of machine learning for predicting G9a inhibitors†‡
Mariya L.
Ivanova
 *,
Nicola
Russo
,
Nadia
Djaid
and
Konstantin
Nikolic
*,
Nicola
Russo
,
Nadia
Djaid
and
Konstantin
Nikolic
School of Computing and Engineering, University of West London, London W5 5RF, UK. E-mail: mariya.ivanova@uwl.ac.uk
First published on 2nd September 2024
Abstract
Object and significance: the G9a enzyme is an epigenomic regulator, making gene expression directly dependent on how various substances in the cell affect this enzyme. Therefore, it is crucial to consider this impact in any biochemical research involving the development of new compounds introduced into the body. While this can be examined experimentally, it would be highly advantageous to predict these effects using computer simulations. Purpose: the purpose of the model was to assist in answering the question of the potential effect that a compound under development could have on the G9a activity, and thus reduce the need for laboratory experiments and facilitate faster and more productive research and development. Solution: the paper proposes a cost-effective machine learning model that determines whether a compound is an active G9a inhibitor. The proposed approach utilises the already existing very extensive PubChem database. The starting point was the quantitative high-throughput screening assay for inhibitors of histone lysine methyltransferase G9a (also available on PubChem) which screened around 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds. For these compounds, datasets of 60 features were created. Then different ML algorithms were deployed to find the best performing one, which can then be used to predict if some untested compound would actively inhibit G9a. Results: six different ML classifiers have been implemented on five dataset variations. Different variants of the dataset were created by using two different data balancing approaches and including or not the influence of water solubility at a pH of 7.4. The most successful combination was a dataset with five features and a random forest classifier that reached 90% accuracy. The classifier was trained with 60244 and tested with 15062 compounds. Feature reduction was obtained by analysing three different feature importance algorithms, which resulted in not only feature reduction but also some insights for further biochemical research.
000 compounds. For these compounds, datasets of 60 features were created. Then different ML algorithms were deployed to find the best performing one, which can then be used to predict if some untested compound would actively inhibit G9a. Results: six different ML classifiers have been implemented on five dataset variations. Different variants of the dataset were created by using two different data balancing approaches and including or not the influence of water solubility at a pH of 7.4. The most successful combination was a dataset with five features and a random forest classifier that reached 90% accuracy. The classifier was trained with 60244 and tested with 15062 compounds. Feature reduction was obtained by analysing three different feature importance algorithms, which resulted in not only feature reduction but also some insights for further biochemical research.
1. Introduction
The euchromatic histone–lysine N-methyltransferase (G9a) was discovered around 30 years ago, and along with its epigenetic key role, it has been found that this enzyme is also a co-regulator of transcription factors and steroid receptors, as well as that it can suppress many types of cancer.1 The importance of this enzyme has prompted research into what machine learning (ML) can contribute to G9a studies.ML has become a very convenient and cost-effective way of conducting biochemical studies using only computer simulations, leading to many publications and review articles.2 Some of them investigate the use of ML and predictive modelling regarding the enzyme–substrate interaction,3,4 as well as the enzyme–chemical interaction to assess the effects on the enzyme activity.5 These molecular interactions can also be investigated using computational methods such as molecular dynamics, molecular docking, and Monte Carlo simulations. However, recently, ML techniques have been introduced, which significantly reduce the computing time and complexity of algorithms and allow for dealing with big datasets, covering large feature spaces. Furthermore, the ML approach offers other results beyond predictions. For example, feature importance analysis could offer important insights into the potential mechanisms of interaction.
Although ML is increasingly being used in the field of biochemical research,2 extensive searches of the available literature have not revealed any indications of the approach discussed in this article having previously been reported. Only a few G9a-related studies applying ML appeared to have been published. They are related to gene expression,6 investigations into hepatocellular carcinoma,7 or prediction of lysine methylation sites using CNNs.8 So, driven by the desire to develop an approach that can facilitate biochemical researchers, off-the-shelf ML algorithms and the world's largest collection of freely accessible chemistry information9 were utilised in the achievement of an ML model that can predict whether a newly obtained compound is an active G9a inhibitor.
The data source for the study was the open chemistry database PubChem.9 It contains diverse data, including over 1.6 million bioassays and a comprehensive description of the physical, chemical, structural and other properties of about 118 million compounds and 318 million substances.10 From this database, the PubChem AID 504332 bioassay11 (ESI, Fig. 1‡) related to G9a inhibition was initially selected and used for generation of targets. The bioassay is based on chemiluminescence AlphaScreen.12 Methylation has been measured through specific antibody-based detection, in conjunction with streptavidin-coated donor and anti-IgG antibody-coated acceptor beads. The method is particularly well suited for detection of inhibitors acting by the desired histone peptide competitive mechanism. Moreover, considering the importance of water to the physiology of the human body, a second bioassay (PubChem AID 1996;13 ESI, Fig. 2‡) related to the water solubility at a pH of 7.4 of the compounds was selected and used subsequently in the study.
Following the idea of performing the study at the lowest possible structural level of the compounds, the generation of datasets proceeded, utilising molecular data currently available on PubChem9 for the relevant compounds from the PubChem AID 504332 bioassay.11 The datasets were built incrementally, as described below in the Methodology section, 2.1. Dataset generation. Eventually, five datasets that differ from each other in size and content were created, which had no missing or negative values or categorical variables. These data sets were consequently used for training the machine learning classifiers.
The ML algorithms used in the study were taken from scikit-learn14 and PyTorch15 ML frameworks. The latter was used for building Artificial Neural Networks (ANNs), whose hyperparameters were tuned using the novel Define-by-Run style API (Application Programming Interface) Optuna.16 Since one of the datasets exceeded 600 thousand records, the API for Apache Spark–PySpark17 was used to handle the large dataset appropriately. Using these ML tools, the study was conducted, following the best ML practices18 because in this way the models developed through statistical learning are robust and the observed effects are reproducible. More details are provided below in the Methodology section, 2.2. Machine learning. The results of the cross-validations19 of these models were compared, and the best one was chosen for further investigation and fine tuning, to eventually achieve the desired predictive model.
In addition to the predictive aspects of deployed ML algorithms, feature importance analysis has been implemented, which could lead to some general insights useful for further research.
This study is focused on introducing a new methodology, which leverages readily available huge repositories of data such as PubChem to make a theoretical prediction about the effect of a compound on an enzyme and demonstrate it using the example of G9a inhibition as a classification problem. A separate study is underway which is investigating the efficacy of a compound or a substance on G9a activity.
2. Methodology
2.1 Dataset generation
737 rows of records, see ESI Fig. 1‡. Each row represents a different compound. The columns contain PubChem compounds and substance IDs, comments, outcomes, and 36 total columns with results, such as the type of activity observed; efficacy; potency; dose–response; variety of attributes related to a fit of the data to the Hill equation; activity at different concentrations in the range from 0.00366 μM to 186 μM11. For more details, see PubChem AID 504332 bioassay,11 result descriptions and ESI, Fig. 1‡.
Given the nature of the study, which was a binary classification, only the ‘phenotype’ and ‘PubChem activity outcome’ columns were taken into consideration. The ‘phenotype’ column contained values: inactive, inhibitor and activator, and the values in the other selected column were: inactive, inconclusive and active. The unique combinations between these two columns were ‘inactive–inactive’, ‘inhibitor–inconclusive’, ‘inhibitor–active’ and ‘activator–inactive’, so the ‘inhibitor–active’ combination was used for the “active-inhibitor” class (i.e. target 1), and the remaining combinations were used for the “other-than-active-inhibitor” class (i.e. target 0). Thus, the targets were created.
The expansion of the core datasets began with the addition of structural, chemical and physical properties and Quantitative Structure–Activity Relationship (QSAR) descriptors of the relevant compounds, all already computed in PubChem9 and/or Cactvs.20 These data were accessed through the PubChem portal.21
The features were: molecular weight; topological polar surface area;22 XLogP3;23 heavy atom count; hydrogen bond donor count; hydrogen bond acceptor count; formal charge; rotatable bond count; covalently bonded unit; the atomic coordinates;24 Simplified Molecular-Input Line-Entry (SMILES);25 molecular formulae. For more details, see ESI,‡ feature description, under Features imported from the PubChem database.
The complete list of all input features is given in ESI‡, feature description section.
Two balancing algorithms were used, where the Synthetic Oversample Technique (SMOTE)27 expanded the minority class and the Random Under Sampler (RUS)28 reduced the majority one. Thus, for dataset 1, implementing the RUS,28 the majority class was reduced to the number of minority ones, and the resulting dataset had 54 thousand rows (Fig. 1). On the other hand, SMOTE27 was used to create two datasets. For the first dataset, 40 thousand samples were randomly selected from the other-than-active-inhibitor class. This was done in order to explore a case where the data set was not highly imbalanced. After balancing it with SMOTE,27 the minority class increased from 27 thousand to 40 thousand and the dataset became 80 thousand rows. It was named dataset 2 (Fig. 1). For the second dataset balanced with SMOTE27 all samples from the other-than-active-inhibitor class were used. Thus, a big dataset of 613 thousand rows was created. It was named dataset 5 (Fig. 1).
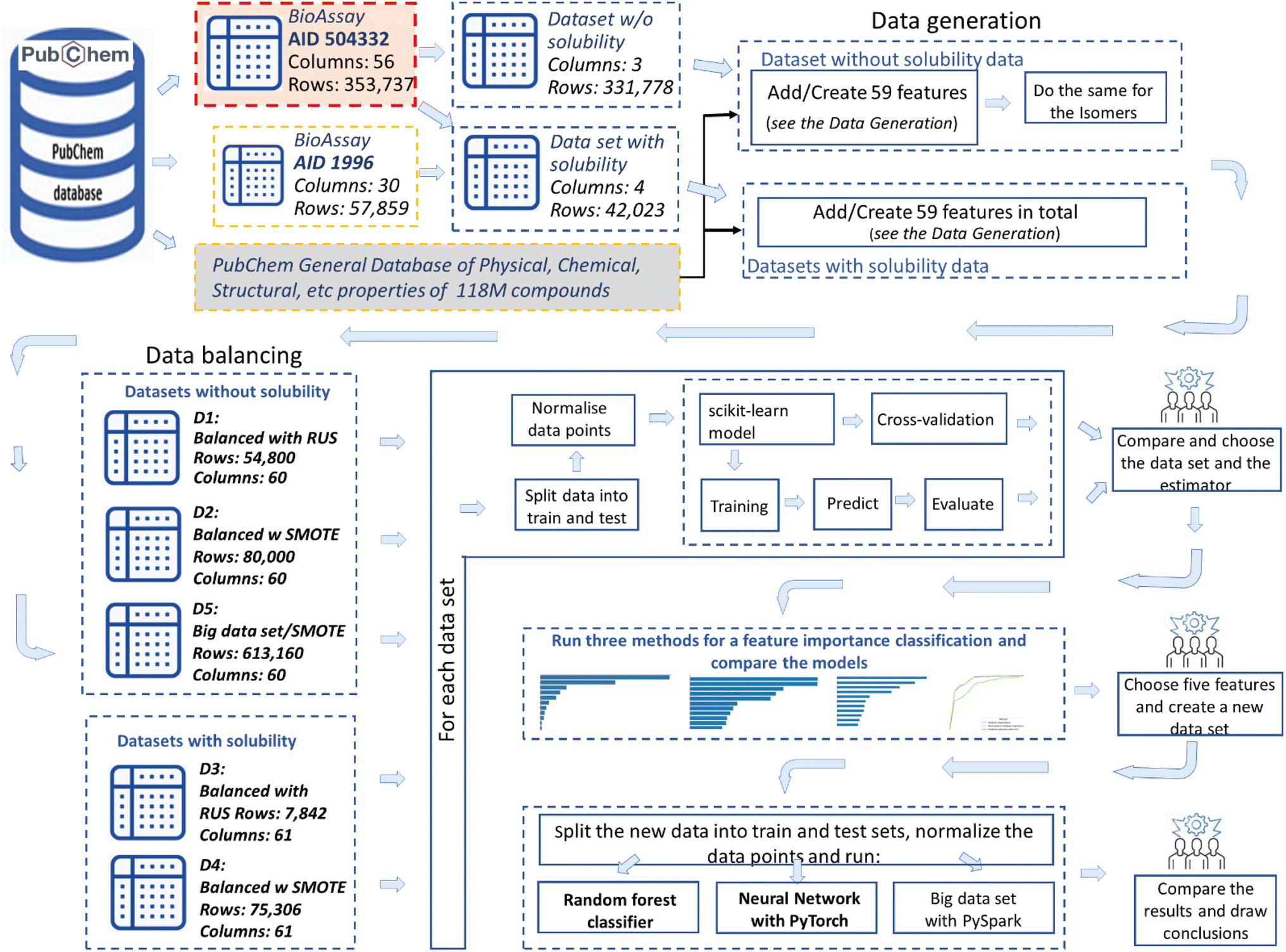 | ||
| Fig. 1 Methodology used in the study: (top part, from left to right) the input datasets were created by using the PubChem open-source database. The starting point is the bioassay AID 504332: Quantitative high-throughput screening assay for inhibitors of G9a which reports the experimental results for nominally 353737 compounds. Then a core dataset was created with 331778 rows (some duplicates of compounds were eliminated) and only three columns were kept: compound ID and the label (active-inhibitor and not-active-inhibitor). Then 58 features have been added to the dataset (data generation bloc), which were either directly taken from the PubChem general database, or calculated by us. Additionally, water solubility data from BioAssay AID 1996 was added. (bottom part) Five datasets (D1 to D5) were created, through implementation of two balancing algorithms (RUS and SMOTE) and excluding or including the water solubility feature. For each dataset five ML algorithms were deployed, and the predictive metrics were found and compared. Then the feature importance algorithm was run and most important features were identified. | ||
107 inhibitors (where only 4237 of them had water solubility results), 322625 (only 38890 with known water solubility results) non-inhibitors of G9a (all tested against G9a in the bioassay), and two different balancing algorithms:
- Dataset 1: w/o water solubility, RUS balancing, R = 54800, C = 60.
- Dataset 2: w/o water solubility, SMOTE balancing, R = 80000, C = 60.
- Dataset 3: with water solubility, RUS balancing, R = 7,842, C = 61.
- Dataset 4: with water solubility, SMOTE balancing, R = 75306, C = 61.
- Dataset 5: w/o water solub., Big Dataset, SMOTE, R = 613160, C = 60.
R – rows: number of compounds and C – columns: number of attributes
These datasets were subsequently used to train, predict and analyse the ML models (see Fig. 1, block: dataset creation).
2.2 Machine learning
In order to obtain ML training and validation datasets, the newly generated datasets were divided into data points and targets and then split into testing and training sets in a ratio of 80:20 (ESI, Fig. 3‡). Data normalisation was performed on the training data only. This was done after the train-test split to avoid getting unrealistically good results29 that could occur due to data leakage. The models were trained on the training sets (X_train and y_train). The predictions were obtained using test data points (X_test), and the model accuracy evaluation was based on the comparison between the predicted and actual value (y_test) (ESI, Fig. 3‡). The ML algorithms used at this stage of the study were: Decision Tree Classifier (DTC),30 Random Forest Classifier (RFC),31 Gradient Boost Classifier (GBC),32 XGBoost Classifier (XGBC)33 and Support Vector Classifier (SVC).34
For the best-model selection, the statistical cross-validation method19 was applied. Once the model was chosen, overfitting was checked. Since the indicator of overfitting is when there is a good performance of the training set and a poor generalisation performance,35 reaching a 5% difference between the training and testing results was tracked, and the hyperparameter of the model was chosen before the point.
For the feature reduction, three different feature importance methods were used. Each of them selected features in order of their importance according to the given method. Furthermore, each set of the first eleven features was used to explore the ML model behaviour. For this purpose, ML was performed by incrementally adding features one by one in the order of their importance. In this way, the feature importance algorithms not only reduced the features but also gave a hint as to which physical and chemical properties of compounds were most relevant for the inhibition of the G9a enzyme. When tracing of the ML models' behaviour was completed, the results were compared, and a set of features was set apart that achieved a satisfactory result the fastest. Thus, the final dataset was obtained.
With the intention of improving the performance of the selected classifier, the dataset with reduced features was included in the hyperparameter tuning36 of the selected classifier. The ML model was then used for new training, prediction and evaluation.
In addition to five ML algorithms, an ANN was built with PyTorch15 and trained with the final reduced dataset. To achieve an optimal performance, the hidden layers, the number of neurons, the learning rate and the optimiser of the ANN were tuned with the hyperparameter tuner Optuna.16
Regarding the big dataset, a PySpark17 session was established that implemented the Spark functionality, and then ML training, prediction and evaluation were done.
Both the data generation and ML were performed on the Jupyter Notebook because this computational environment allows the code and data to be supplemented with analysis, hypotheses, and conjecture in a research-friendly manner.37
3. Results and discussion
3.1 Performance metrics
Each of the ML algorithms (specified in the section Methodology, 2.2. Machine Learning) was deployed for each of the first four datasets listed in the section Methodology, 2.1.7. Final datasets for ML analysis. The results of the typical classifier metrics38 such as accuracy, precision, recall, F1 and ROC are presented in Table 1. It can be observed that the ML models deployed on expanded-balanced (SMOTE) datasets performed much better than the models applied on reduced-balanced (RUS) datasets. This result could be assigned to the volume of the data. However, this conclusion although correct in general, occasionally does not hold. For example, the big dataset (dataset 5) did not give the best results regardless of having the largest data volume (ESI, Table 6‡). Moreover, dataset 2 used for training, prediction, and evaluation in the same manner as dataset 4 did not give better results even though dataset 2 was bigger than dataset 4.| Classification estimators | |||||||
|---|---|---|---|---|---|---|---|
| 1.Algorithm | 2.Accuracy | 3.Precision | 4.Recall | 5.F1 | 6.ROC | ||
| Without Solubility data | |||||||
| RUS | 4 | XGBoost | 0.681 | 0.682 | 0.677 | 0.679 | 0.681 |
| 0 | SVM | 0.678 | 0.683 | 0.662 | 0.672 | 0.678 | |
| 2 | RandomForest | 0.673 | 0.676 | 0.663 | 0.669 | 0.673 | |
| 3 | GradientBoost | 0.651 | 0.659 | 0.625 | 0.641 | 0.651 | |
| 1 | Decision | 0.581 | 0.580 | 0.577 | 0.579 | 0.581 | |
| SMOTE | 2 | RandomForest | 0.746 | 0.764 | 0.719 | 0.741 | 0.746 |
| 4 | XGBoost | 0.742 | 0.771 | 0.698 | 0.732 | 0.742 | |
| 0 | SVM | 0.708 | 0.717 | 0.702 | 0.709 | 0.708 | |
| 3 | GradientBoost | 0.690 | 0.705 | 0.670 | 0.687 | 0.691 | |
| 1 | Decision | 0.636 | 0.640 | 0.643 | 0.641 | 0.636 | |
|
|||||||
| With Solubility data | |||||||
| RUS | 3 | GradientBoost | 0.637 | 0.643 | 0.616 | 0.629 | 0.637 |
| 0 | SVM | 0.634 | 0.646 | 0.593 | 0.618 | 0.634 | |
| 2 | RandomForest | 0.625 | 0.633 | 0.594 | 0.613 | 0.625 | |
| 4 | XGBoost | 0.619 | 0.621 | 0.608 | 0.615 | 0.619 | |
| 1 | Decision | 0.562 | 0.558 | 0.594 | 0.576 | 0.562 | |
| SMOTE | 2 | RandomForest | 0.948 | 0.959 | 0.939 | 0.949 | 0.948 |
| 4 | XGBoost | 0.945 | 0.981 | 0.910 | 0.944 | 0.946 | |
| 0 | SVM | 0.884 | 0.904 | 0.864 | 0.884 | 0.884 | |
| 3 | GradientBoost | 0.874 | 0.900 | 0.847 | 0.873 | 0.875 | |
| 1 | Decision | 0.868 | 0.863 | 0.882 | 0.872 | 0.868 | |
Focusing on the metric of accuracy, the cross-validation scores are shown in ESI, Table 1‡. The overall result was that the most successful classifier was the RFC,31 with the SMOTE balanced dataset which includes water solubility at a pH of 7.4 (dataset 4), reaching mean cross-validation score 95.1% with 0.21 standard deviation, followed by XGBoost33 with mean cross-validation score 94.6% with 0.07 standard deviation.
3.2 Feature importance
Three types of feature importance methods were used, namely feature importance of RFC,39 permutation feature importance40 and feature importance selected using the K highest score and chi-squared stats between each non-negative feature and class.41 These three methods provided three sets of features, with the features arranged in descending order (Fig. 2). The obtained lists of features were used to investigate how each feature affects the accuracy of the ML model. Fig. 3 demonstrates how inclusion of each feature one by one in order of importance affected the accuracy metric (also see the code in GitHub). The first two feature importance methods gave very similar results. Both reached their maximum accuracy at about the same time by including only their first five most important features. These features were: the hypothetical volume based on 2D atom coordinates (Volume_1); the relative proportion of sulphur (S_relative), nitrogen (N_relative) and carbon (C_relative) atoms to the total number of atoms of the compound; the mass proportion of sulphur (S) to the total mass of the atoms of the compound (ESI‡, data generation). The third feature importance algorithm, however, emphasised more on features that describe the physical size of the compounds. Overall, the feature importance algorithms pointed to chemical composition as a relevant factor for G9a inhibition, in particular, to the presence of sulphur in the compound. The biological significance of this insight remains to be investigated. | ||
| Fig. 2 Feature importance analysis using three different methods: (a) Random forest classifier, (b) Permutation feature importance, and (c) SelectKBest and Chi2 feature importance. | ||
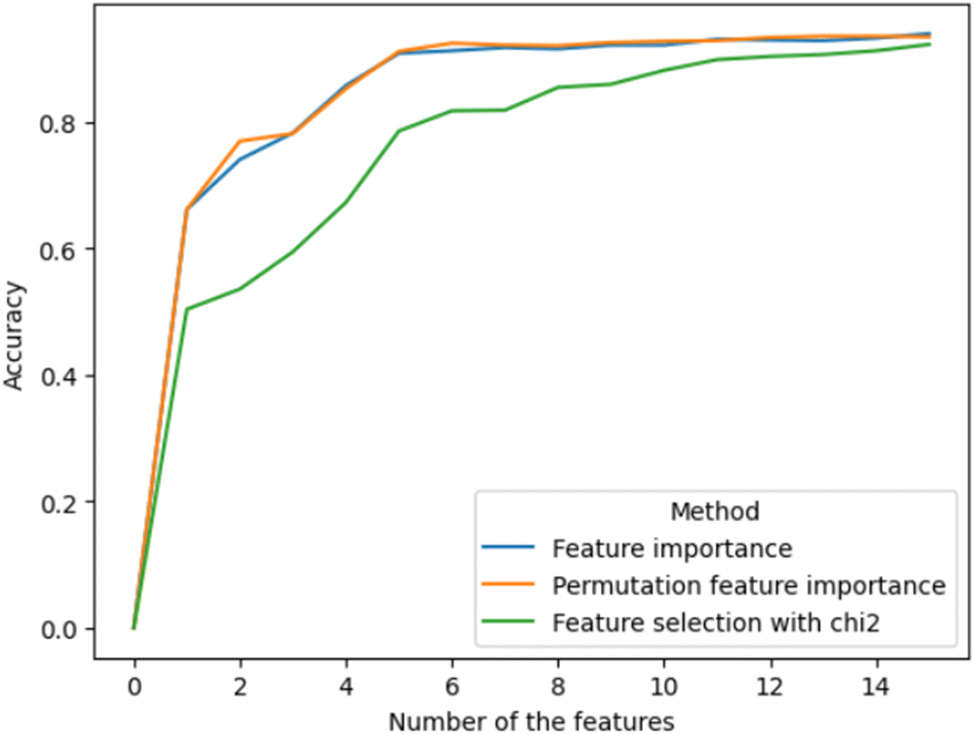 | ||
| Fig. 3 Tracing of accuracy when the features are added one by one in order of their importance. Different colours of the lines represent different feature importance methods. The results indicate that for the first two methods it is sufficient to take into account only the first 5 features, and the rest have a very small effect on the accuracy. | ||
3.3 Overfitting analysis
The presence of overfitting was carefully scrutinised initially for the best performing models for each dataset (ESI, Fig. 4‡) and later for the final model with five features (Fig. 4).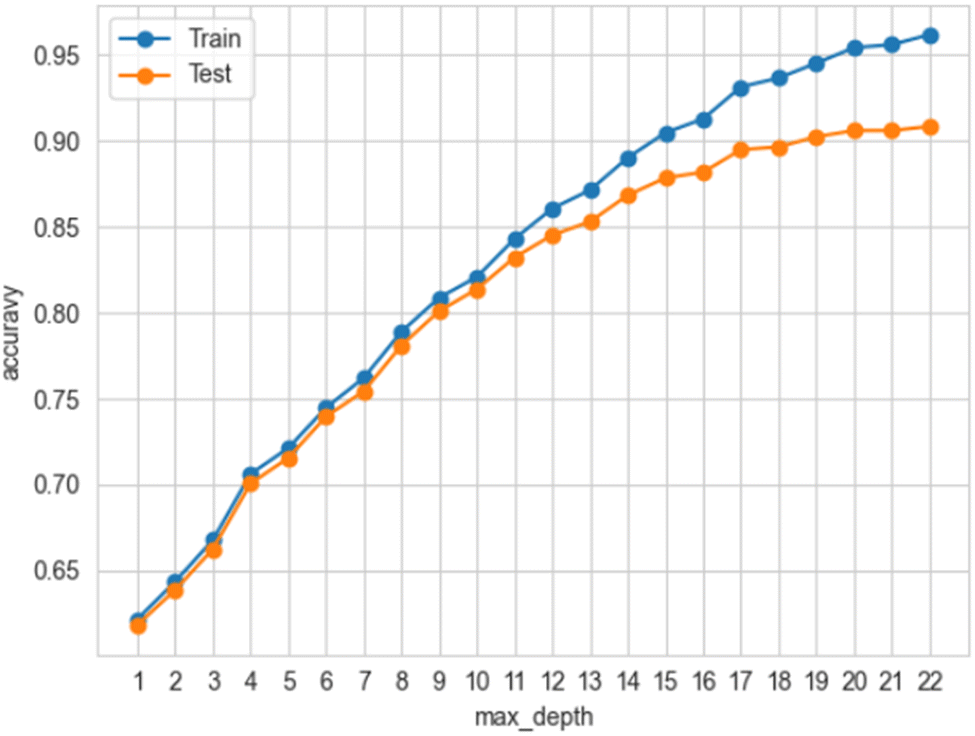 | ||
| Fig. 4 Accuracy vs. max_depth for the five-feature random forest classifier for Dataset 4, which indicates the onset of overfitting. Note how the divergence between training and testing accuracy is slower for a reduced number of features in comparison to when all features are included as in ESI, Fig. 4. | ||
As was mentioned in the section Methodology, 2.2. Machine learning, the point where the accuracy of the training and testing data deviated by more than 5% was taken as an indicator of where the overfitting begins to occur.35 So, bearing this indicator in mind, the panels in ESI, Fig. 4‡ were created, tracing the accuracy of the training and testing data of the best-performing algorithms during cross-validation.19 Cross-validation was performed for each estimator listed in the section Methodology, 2.2. Machine learning and for each one of the four datasets respectively. Amongst these combinations, the RFC with Dataset 4 balanced with SMOTE27 performed best. At max_depth (i.e., the maximum depth of the tree) 12 the deviation was 4.9% and at max_depth 13 it was 5.6% respectively, i.e., the model was overfitted, and thus the model with max_depth 12 that achieved test accuracy 85.23% was not overfitted. It turned out that the initially obtained result of 95% accuracy for this combination was overfitted. This happened because by default the hyperparameter max_depth is ‘none’, so since the number of levels/branches was not restricted, the RFC became complex, which in turn led to overfitting.
Similar tracing of overfitting35 was done for the final ML model with the RFC algorithm and Dataset 4 whose features were reduced to five. It turned out that the model reached 90% accuracy at max-depth 20 where the deviation was 4.8%, i.e. the model was not overfitted (ESI, Fig. 6‡). This increase in accuracy was expected because the reduction of the features decreased the complexity of the model, which in turn could lead to an increase in accuracy42
3.4 Optimal ML model and comparison with the ANN and PySpark results
Creating the optimal model for predicting G9a inhibition started with a retrain of the RFC by keeping only the aforementioned five features (which are the most important according to the first two feature importance algorithms above) of Dataset 4. It obtained 91.12% accuracy. Furthermore, with hyperparameter tuning,36 the model achieved a 91.29% best grid search score and 90.36% when it was run with the values recommended by the hyperparameter tuning. The hyperparameters and their values used for the tuning are presented in ESI, Table 2‡. The hyperparameter-tuned ML model was then explored for overfitting. Given that the deviation between the training and testing accuracy at max_depth 20 was 4.8% and at max_depth 21 was 5.0%, it was concluded that the model achieved an accuracy of 90% at max_depth 20 without being overfitted (Fig. 4). The prediction summary of the model is illustrated by the confusion matrix in ESI, Fig. 5‡, where true-positives (i.e., correctly predicted class 1 instances) are 6606 out of 7383 and true-negatives (i.e., correctly predicted class 0 instances) are 7011 out of 7679 per class. The classification report in ESI, Table 3‡ provides details about how the ML model has performed for each class.The Artificial Neural Network (ANN) used in the study was tuned using the hyperparameter optimization framework Optuna.16 The model was run six times. For each run, the hyperparameters, such as the number of layers, neurons, dropout regularization, optimiser and learning rate are presented in ESI, Table 4‡. The final result was calculated as the average of all six runs, so the ANN achieved an accuracy of 65% with 3.7% standard deviation (Fig. 5a and ESI, Table 5‡).
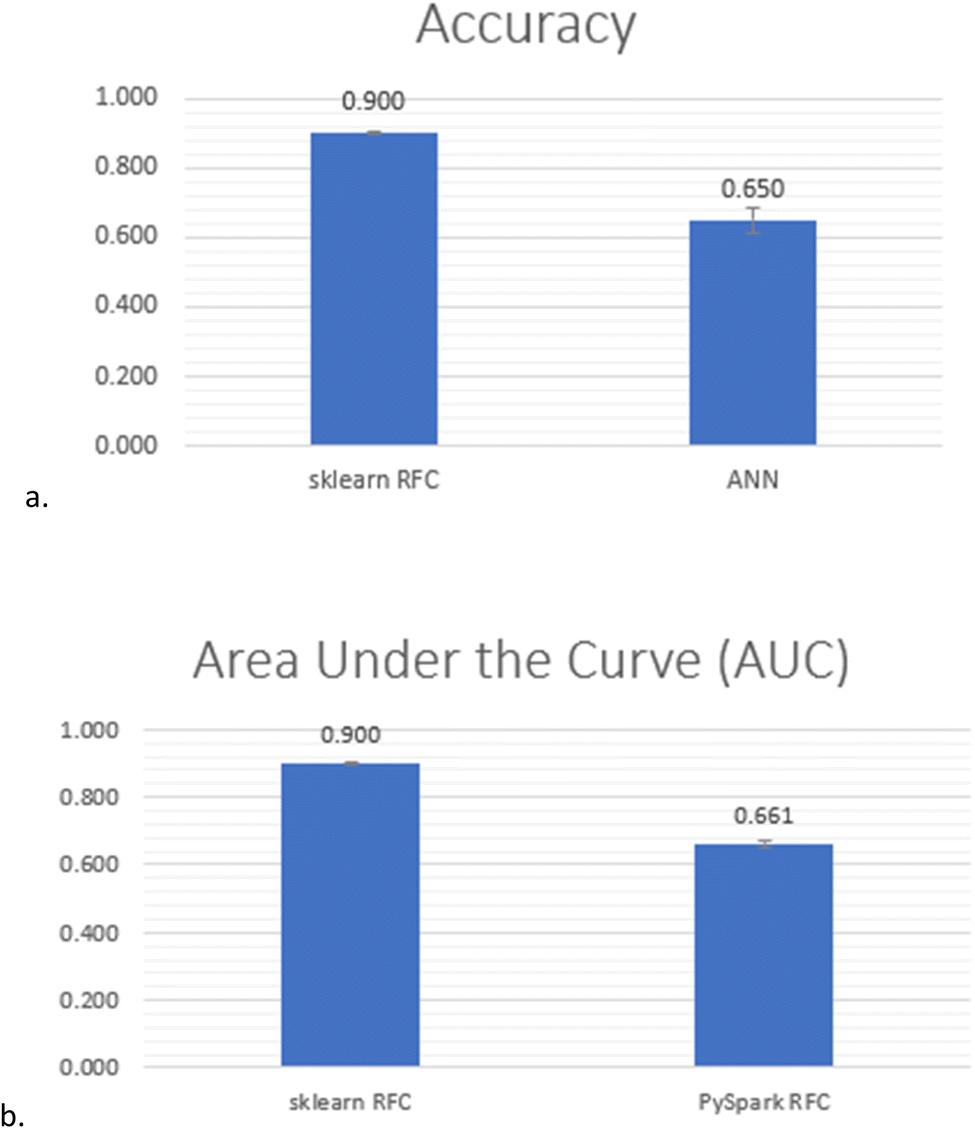 | ||
| Fig. 5 Final comparison between PySpark RFC, scikit RFC and ANN, (a) scikit RFC vs. ANN tuned with Optuna and (b) Scikit RFC vs. PySpark RFC. | ||
Finally, a modality of the RFC algorithm provided by PySpark17 was evaluated and compared with the results obtained for the same classifier provided by scikit-learn.14 The area under the receiver operating characteristic (ROC) curve (AUC) metric reached only 66.1% for the PySpark algorithm, in comparison to 90.0% obtained for scikit-learn (Fig. 5b and ESI, Table 6‡). So, the final comparison of the results (Fig. 5) nominated the random forest classifier of scikit-learn as the optimal algorithm for the study.
The code for this study was written in Python,43 and it is available on GitHub https://github.com/articlesmli/G9a_clsf.git.
4. Discussion and conclusions
Although the study was focused on the G9a enzyme, it also showed that data from bioassays, combined with the structural, chemical and physical properties and QSAR descriptors of the considered compounds available in databases (such as PubChem), along with off-the-shelf ML algorithms, can be utilised to predict the impact of new compounds on various biochemical processes, without the need for laboratory experiments.The datasets for the presented study were generated using qHTS and MLSMR bioassays along with aggregated data, all of which were provided by PubChem. Moreover, engineered features based on the PubChem database were added. The obtained five different datasets expectedly produced different results when used for ML, but it was unexpected that the bigger Dataset 2 did not perform better than the smaller Dataset 4 even though the ML approach for both of them was the same. The difference between these two datasets was only due to the crossover of the core bioassay with the bioassay containing water solubility data at a pH of 7.4. So, directions for further research emerged because, on the one hand, the water solubility data led to an increase in model accuracy, but on the other hand, the solubility feature did not exhibit significant importance during the calculation of the feature importance. Furthermore, the feature importance algorithms revealed that sulphur significantly influenced the ML model regarding G9a inhibition. So, the presented research not only developed an ML model but also raised questions whose answers would most likely contribute to studies related to G9a inhibition.
By using off-the-shelf ML libraries, the predictive model with the best performance was the random forest classifier from scikit-learn. The XGBoost classifier has also shown good results and even slightly outperformed the RFC on the precision metric (98.1% vs. 95.9%). However, the RFC was the classifier of choice because, first, the RFC performed the best on cross-validation (ESI, Table 1). Second, the deviation between the classification metrics, such as accuracy, precision, recall, F1 and ROC (Table 1) was the smallest for the RFC compared to the rest of the classifiers that was an indicator that amongst the selected classifiers, the RFC was the most reliable model for the given dataset.
The PubChem repository offers a huge number of variables that characterise various compounds and the possible number of derived features is theoretically unlimited, so the total number of features could easily run into thousands. However, to demonstrate the presented approach a more manageable number of features (which was 60) was used, and since the result (i.e. the predictive power of the algorithm) was quite high (90%), the number of features was not expanded, but it could be significantly increased in future studies, in order to investigate the impact of new features.
In the study, the charged compounds were intentionally removed from the dataset. However, given that histone proteins are positively charged and DNA is negatively charged, the study of how the charged compounds could improve the ML model remains open for further investigations. Also, the hyperparameter tuning of the final ML model did not improve accuracy significantly, but bearing in mind that the hyperparameter tuning combinations are unlimited, the option of more extensive hyper-parameter tuning of the final ML model that would improve it remains open for further research.
It is known that studies similar to the one presented can lead to some interesting biological implications. For example, QSAR studies could elucidate the importance of a specific class of descriptors in inducing anticancer activity against a particular type of cancer.44 The methodology and ML model presented in the paper could have some practical biological implications as well. For example, given the importance of G9a, any compound under development could easily be screened for its effect on this enzyme. Furthermore, feature importance analysis indicates which features of compounds and substances may be relevant for G9a inhibition and facilitate the design of new compounds accordingly. This also contributes to the desired explainable AI algorithms.45
In conclusion, the study not only developed a five-feature ML model that predicted with 90% accuracy whether a compound was a G9a inhibitor, but also raised questions for further research and paved the way towards the next study where, using the already existing datasets, the efficacy of the newly predicted G9a inhibitor will be forecasted.
Data availability
The code for this study can be found at https://github.com/articlesmli/G9a_clsf.git. The data that support the findings of this study are openly available in: PubChem; https://pubchem.ncbi.nlm.nih.gov/; https://pubchem.ncbi.nlm.nih.gov/bioassay/504332. https://pubchem.ncbi.nlm.nih.gov/bioassay/1996. GitHub; https://github.com/articlesmli/G9a_clsf.git.Author contributions
M. L. I. and K. N. conceptualized the project. M. L. I. and K. N. designed the methodology. M. L. I. and N. R. wrote the code and processed the data. K. N. and N. D. supervised the project. All authors were involved with the writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. L. I. thanks the UWL Vice-Chancellor's Scholarship Scheme for their generous support. We sincerely thank NCATS and the Sanford-Burnham Medical Research Institute for performing the bioassays and PubChem for providing access to their database. We thank the anonymous peer reviewers and the editor for helpful comments.Notes and references
- C. Poulard, L. M. Noureddine, L. Pruvost and M. Le Romancer, Life, 2021, 11, 1082 CrossRef CAS PubMed.
- M. Mowbray, T. Savage, C. Wu, Z. Song, B. Anye Cho, E. A. Del Rio-Chanona and D. Zhang, Biochem. Eng. J., 2021, 172, 108054 CrossRef CAS.
- L. F. Salas-Nuñez, A. Barrera-Ocampo, P. A. Caicedo, N. Cortes, E. H. Osorio, M. F. Villegas-Torres and A. F. González Barrios, Metabolites, 2024, 14, 154 CrossRef PubMed.
- S. Goldman, R. Das, K. K. Yang and C. W. Coley, PLoS Comput. Biol., 2022, 18(2), e1009853 CrossRef CAS PubMed.
- S. L. Robinson, M. D. Smith, J. E. Richman, K. G. Aukema and L. P. Wackett, Syst. Biol., 2020, 5(1), ysaa004 CAS.
- G. Tsagkogeorga, H. Santos-Rosa, A. Alendar, D. Leggate, O. Rausch, T. Kouzarides, H. Weisser and N. Han, Commun. Biol., 2022, 5, 868 CrossRef CAS PubMed.
- T. I. Aravena, E. Valdés, N. Ayala and V. A. D'Afonseca, Cancer Inf., 2023, 29, 1176935123116148 Search PubMed.
- A. Spadaro, A. Sharma and I. Dehzangi, Methods, 2024, 226, 127–132 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2023, 51, D1373–D1380 CrossRef PubMed.
- PubChem Data Count https://pubchem.ncbi.nlm.nih.gov/docs/statistics, accessed April 2024 Search PubMed.
- Bioassay Record https://pubchem.ncbi.nlm.nih.gov/bioassay/504332, accessed April 2024 Search PubMed.
- A. M. Quinn, A. Allali-Hassani and M. Vedadi, Mol. Biosyst., 2010, 6, 782–788 RSC.
- Bioassay Record https://pubchem.ncbi.nlm.nih.gov/bioassay/1996, accessed April 2024 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot, E. Duchesnay and M. Braun, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan and S. Chintala, in Advances in Neural Information Processing Systems 32, Curran Associates, Inc., 2019, pp. 8024–8035 Search PubMed.
- T. Akiba, S. Sano, T. Yanase, T. Ohta, and M. Koyama, Proceedings of the 25th International Conference on Knowledge Discovery and Data Mining, Anchorage, 2019 Search PubMed.
- M. Zaharia, R. S. Xin, P. Wendell, T. Das, M. Armbrust, A. Dave, X. Meng, J. Rosen, S. Venkataraman, M. J. Franklin, A. Ghodsi, J. Gonzalez, S. Shenker and I. Stoica, Commun. ACM, 2016, 59, 56–65 CrossRef.
- N. Artrith, K. T. Butler, F. Coudert, S. Han, O. Isayev, A. Jain and A. Walsh, Nat. Chem., 2021, 13, 505–508 CrossRef CAS PubMed.
- S. Nematzadeh, F. Kiani, M. Torkamanian-Afshar and N. Aydin, Comput. Biol. Chem., 2022, 97, 107619 CrossRef CAS PubMed.
- W. D. Ihlenfelldt, Y. Takahahashi, H. Abe and S. Sasaki, Cactvs (Version 3.4.8.18), J. Chem. Inf. Comput. Sci., 1994, 34, 109–116 CrossRef.
- PubChem https://pubchem.ncbi.nlm.nih.gov/accessed June 2024 Search PubMed.
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714–3717 CrossRef CAS PubMed.
- T. Cheng, Y. Zhao, L. Xun, L. Fu, X. Young, X. Zhang, L. Yan, R. Wang and L. Luhua, J. Chem. Inf. Model., 2007, 47, 2140–2148 CrossRef CAS PubMed.
- J. Zhu, Y. Xia, L. Wu, S. Xie, T. Qin, W. Zhou, H. Li, and T. Liu, in Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining (KDD '22), ed. A. Zhang and H. Rangwala, Association for Computing Machinery, New York, 2022, pp. 2626–2636 Search PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- H. Kaur, H. S. Pannu and A. K. Malhi, ACM Comput. Surv., 2019, 53, 1–36 Search PubMed.
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, J. Artif. Life Res., 2002, 16, 321–357 Search PubMed.
- R. Mohammed, J. Rawashdeh and M. Abdullah, 11th International Conference on Information and Communication Systems (ICICS), Irbid, 2020, pp. 243–248 Search PubMed.
- D. Singh and B. Singh, Appl. Soft Comput., 2020, 97, 105524, DOI:10.1016/j.asoc.2019.105524.
- V. G. Costa and C. E. Pedreira, Artif. Intell. Rev., 2023, 56, 4765–4800 CrossRef.
- A. Parmar, R. Katariya and V. Patel, in International Conference on Intelligent Data Communication Technologies and Internet of Things (ICICI), ed. J. Hemanth, H. Fernando, P. Lafata and Z. Baig, Springer, Cham, 2018 Search PubMed.
- C. Bentejac, A. Csorgo and G. A. Martinez-Munoz, Artif. Intell. Rev., 2021, 54, 1937–1967 CrossRef.
- Z. A. Ali, Z. H. Abduljabbar, H. A. Taher, A. B. Sallow and S. M. Almufti, Acad. J. Nawroz U., 2023, 12, 320–334 CrossRef.
- J. Cervantes, F. Garcia-Lamont, L. Rodriguez-Mazahua and A. Lopez, Neurocomputing, 2020, 408, 180–215 CrossRef.
- P. Refaeilzadeh, L. Tang and H. Liu, in Encyclopedia of Database Systems, ed. L. Liu and M. T. Ozsu, Springer, Boston, 2009 Search PubMed.
- J. Schmidt, Testing for Overfitting, Johns Hopkins University, Applied Physics Laboratory, Cornell University, arXiv:2305, Ithaca, 2023 Search PubMed.
- J. M. Perkel, Nature, 2018, 563, 145–146 CrossRef CAS PubMed.
- Accuracy, precision, specificity & sensitivity, https://labtestsonline.org.uk/articles/accuracy-precision-specificity-sensitivity#:%7E:text=Atestmethodcanbe,revealatest'sbasicreliability, accessed April 2024 Search PubMed.
- F. K. Ewald, L. Bothmann, M. N. Wrigth, B. Bischil. G. Casalicchio and G. Koning, arXiv, 2024, preprint, DOI:10.48550/arXiv.2404.12862.
- A. Hapfelmeier, R. Hornung and B. Haller, Comput. Stat. Data Anal., 2023, 181, 107689 CrossRef.
- C. Y. Zhai, W. Song, X. Liu, L. Liu and X. Zhao, in 2018 IEEE 9th International Conference on Software Engineering and Service Science (ICSESS), IEEE, Beijing, China, 2018, pp. 160–163 Search PubMed.
- W. Jia, M. Sun, J. Lian and S. Hou, Complex Intell. Syst., 2022, 8, 2663–2693 CrossRef.
- G. Van Rossum and J. F. Drake, Python (Version 3.12.3), Centrum voor Wiskunde en Informatica Amsterdam, 1995 Search PubMed.
- G. D. J. Davis and A. H. R. Vasanthi, Eur. J. Pharmaceut. Sci., 2015, 76, 110–118 CrossRef CAS PubMed.
- S. M. Lundberg, G. Erion and H. Chen, et al. , Nat. Mach. Intell., 2020, 2, 56–67 CrossRef PubMed.
Footnotes |
| † This article is dedicated to Luben Ivanov. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00101j |
| This journal is © The Royal Society of Chemistry 2024 |