
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemistry in a graph: modern insights into commercial organic synthesis planning†
Claudio
Avila
 *,
Adam
West
,
Anna C.
Vicini
,
William
Waddington
,
Christopher
Brearley
,
James
Clarke
and
Andrew M.
Derrick
*,
Adam
West
,
Anna C.
Vicini
,
William
Waddington
,
Christopher
Brearley
,
James
Clarke
and
Andrew M.
Derrick
Pfizer UK R&D Ltd, Pharmaceutical Sciences Small Molecule (PSSM), Chemical Research and Development (CRD), Discovery Park House, Ramsgate Road, CT13 9NJ, Sandwich, UK. E-mail: Claudio.Avila@science.cl
First published on 24th July 2024
Abstract
Across the chemical sciences, synthesis planning is a key aspect for defining synthesis routes, starting from idea generation, combining literature searches and laboratory experimentation, and including scaling-up considerations for large scale manufacturing. This iterative process, which relies heavily on information sharing, is crucial in pharmaceutical development, where drug candidates are transformed into commercially viable Active Pharmaceutical Ingredients (APIs), impacting the access to medicines for billions of people. In this work, we demonstrate that by capturing chemical pathway ideas digitally, at the point of conception, we can systematically merge these ideas with synthetic knowledge derived from predictive algorithms. This serves as a preliminary step for further route evaluation. To achieve this, we introduce a new method for storing, analysing, and displaying chemical information using graph databases and graph representations, illustrated with the commercial synthesis planning of the GLP-1 inhibitor Lotiglipron. Compared to traditional methods, graph databases naturally fit the substrate-arrow-product model traditionally used by chemists, offering a modern alternative to store and access chemical knowledge. This framework facilitates a universal chemistry approach, allowing to share and combine data from many different sources and organisations, and enabling new ways to optimise the complete route selection process.
Introduction
In the field of synthetic organic chemistry, synthesis planning has always been an important step to produce materials on various scales. A structured approach to conceptualise new routes was pioneered by Nobel laureate E. Corey in 1957, known today as retrosynthetic analysis.1 In most cases this analysis is still orchestrated by humans, often focusing on invention and demonstration of individual creativity or intellect.2 Even for small molecules, multiple retrosynthetic suggestions can lead to viable routes to synthesise a given compound, each with its own advantages and drawbacks.3 Selecting the best route remains a complex process, often plagued by unconscious bias due to the human limitation of handling large amounts of data. A novel digital approach enabling collaboration and unbiased decision making is presented in this paper, exemplified in the context of pharmaceutical development. The same principles can be applied across all chemical sciences where new synthetic routes are needed.In pharmaceutical development, the selection of a synthetic route for commercial manufacturing of an Active Pharmaceutical Ingredient (API) can be a long iterative process.4 The search for an API (or target molecule) begins by identifying the ‘pharmacophore’, defined by the IUPAC as “an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response”.5 Starting from this, research groups propose a diversity of targets (an array of analogous molecules with similar therapeutic potential), which are used during the testing phases of the drug discovery process,6 and elaborate a simple synthetic route to rapidly access them. From this process, an enabling chemistry route emerges, often used to deliver early campaigns to fund clinical and toxicological trials, typically in the kilogram production range and under intense time constraints.
Once the API moves into the later stages of clinical development (human trials), this route is seldom orientated toward the objectives of a commercial manufacturing process, which involves achieving commercial feasibility often at multi-tonne scales and meeting the quality attributes required by regulatory agencies.7,8 As a result, the synthesis planning for the commercial manufacturing stage starts with scarce data relevant to the final objective.3,9
To progress towards a more optimal synthetic route, all possible theoretical ideas need to be gathered. This may include full synthetic routes, fragments of routes, or individual reactions that may be of use later. This step is often called ‘idea generation’ or ‘brainstorming’,10 and it requires a large number of contributors to achieve a diverse set of ideas, to ensure the optimal solution is amongst this initial set.11 The number of ideas generated at this stage is usually large (50 or more routes), this hinders how data is displayed and makes decision making difficult. Effective idea visualisation is essential to proceed to the next step.
The ideas are then organised and triaged, serving as the basis for the execution of the synthesis plan in the laboratory. The consequences of decisions made during this triage, where limited experimental data is accessible, propagate through all stages of development and even to the final commercial API manufacturing process.12
In pharmaceutical development, a common approach used for route selection is to apply the SELECT12,13 criteria (or similar standards used across the chemical industry14). SELECT includes a series of factors to account for Safety: including process safety, and exposure to substances harmful to health; Environmental: the volume of natural resources consumed, and the generation of substances harmful to the environment;15Legal: intellectual property rights to produce the drug, and legal requirements for control and use of intermediates and reagents; Economics: meeting the cost of goods targeted for commercialisation, and the investment required to support the desired production; Control: including the chemistry and physical process parameters (PP) and the control of the quality attributes (QA);16 and Throughput: availability of raw materials, and timescale of the manufacturing process.13
The practical implementation follows a series of logical steps, in which a multidisciplinary team is required to aggregate multiple types and sources of information (theoretical, semantic, qualitative, quantitative, etc.). Traditionally, the input of experienced process scientists is needed to cover process chemistry, process safety, regulatory compliance, process engineering, and manufacturing considerations.9,12 Their input is combined with initial laboratory trials, literature references, and any other accessible source of information, such as results from modelling tools regarding process safety17 and physical property predictions like solubility.18 The objective is to determine the most promising routes, prioritising those with the highest value and likelihood of success. Following a comprehensive scrutiny of these factors, directed by SELECT, a few routes emerge as promising choices for subsequent investigation in the laboratory. Ultimately, a singular route is selected as the commercial route for large scale manufacturing.
After the initial route selection, the work focuses on the process development i.e. identifying the necessary unit operations and the process conditions for each step. During this stage, the individual steps of the selected route may be refined, this can include step reordering or the decision to telescope one or more steps.19 However, more often changes are limited to unit operations, and reaction conditions (solvents, catalysts, and reagent selection, temperature, etc.). Very occasionally, a major drawback is identified that requires revisiting the route selection process. In such instances, all the data collected up to that point are not discarded but are utilised to (re)evaluate alternative synthesis paths.
Fig. 1A depict a flow diagram illustrating the complete development process, while Fig. 1B shows the data flow of a random molecule synthesis, illustrating the steps considered by the described traditional approach. These network diagrams show individual routes appearing as branches from the main target molecule, formed by a group of single reaction steps and the corresponding intermediate molecules.
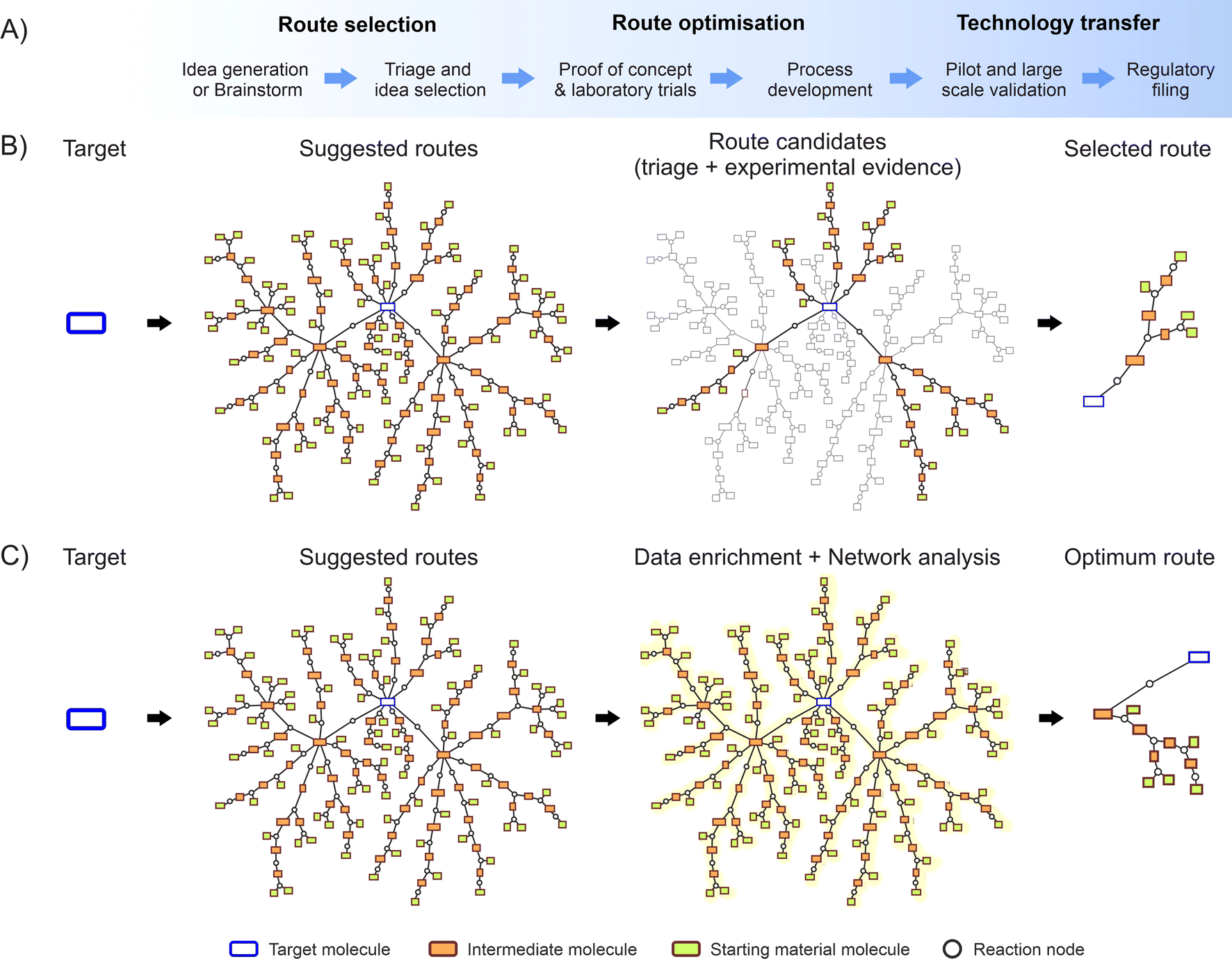 | ||
| Fig. 1 (A) Traditional workflow from initial concept up to the commercial production of a pharmaceutical API; (B) the current methods usually start from the target molecule, gathering a large number of ideas (retrosynthetic analysis), narrowing this down to fewer feasible routes, up to selecting the most feasible by a panel of experts; (C) envisioned process: a data rich approach collating many synthesis ideas from different sources, subsequently enriching the entire network with experimental and modelling data, and using an algorithmic approach to identify the optimum route for commercial synthesis. | ||
The determination of an optimum route is a multi-factor problem, and the human-led solution is vulnerable to bias, even when applying the SELECT criteria rigorously.3,20 Lack of supporting information or chemistry knowledge leads to some ideas being left aside without adequate assessment and decisions can align behind a single local optimum just because more is known about this route.21 One key barrier for the traditional approach is the inherent human bias to gravitate towards familiar methodologies and well-established procedures.22 Decisions often draw from the experiences and successes of the scientists involved in the process, leading to a potential lack of diversity in the exploration of new synthetic pathways. This gap is confirmed by the emergence of generics into the market, appearing as soon as key patents have expired, with alternative viable commercial routes in other branches of the chemical space.23 Branches that may have even been present in the original synthesis planning stage but were not explored.
Several challenges contribute to perpetuate this problem. For instance, the lack of centralised data systems or access to individual applications acting in isolation, and the absence of common data formats and repositories,24 currently act as a barrier preventing a systematic analysis of the entire collection of ideas and data.25,26 In addition, the lack of utilisation of equivalent information from past projects poses a significant obstacle in chemical research.27 Despite differences in target molecules, steps, and transformations, the potential for transferrable knowledge remains largely untapped due to the absence of supportive systems.28,29 This results in missed opportunities for cross-project insights, leading to redundant efforts and resource inefficiencies.
To overcome these barriers, there is a need to implement supportive systems to enable the systematic capture, organisation, and dissemination of knowledge across various projects.30,31 Such systems could take the form of centralised databases, collaborative platforms, or knowledge management tools designed to promote information sharing and collaboration.32,33 In theory, as Fig. 1C illustrates, for synthesis route planning these ideas could be programmatically enriched with additional data e.g. data from additional experimental sources, literature references, theoretical or predictive information, etc.; subsequently enabling the calculation of metrics intrinsic to the entire dataset.34,35 Moreover, algorithms could be applied across the complete information network,36–38 potentially unveiling unprecedented insights beyond the scope of conventional human capabilities.
This paper illustrates how introducing key digitalisation elements can evolve the traditional route selection methods into a more advanced approach. Storing chemical knowledge directly on graph databases enables direct digitalisation of human inputs, real-time access to scientists, holistic overviews for decision makers, and the enrichment of the metadata by direct application of software and algorithms on the database. Finally, implementing advanced network metrics will facilitate identification of the global optimum route, which in turn could facilitate access and lower cost of medicines for patients globally.
Next-gen approaches to commercial organic synthesis planning
Graph databases for universal chemistry
Storing and accessing multi-layered process and chemical information in a systematic manner remains an unresolved challenge. For route design, the number of factors to consider are too diverse and with different levels of abstraction that are almost impossible to be organised and connected in a hierarchical or nested structure. Partial attempts have captured specific layers,39 but not yet defining a universal database schema that can represent and maintain more complex multidimensional relationships, particularly considering the dynamic nature of chemical R&D.Today, graph representations are emerging in a variety of scientific fields.40–42 In chemistry, graph representations have been used to visualise synthesis paths,36,43,44 and to create representations of large chemical networks.36,45,46 These representations have proved useful to tackle problems in risk management of chemical threats,31 and to organise results delivered by predictive retrosynthesis software,45,47 in an accessible way for chemists and data scientists.48 Building upon these insights, this paper proposes advancing beyond mere visualisation to leveraging graph databases as comprehensive data management systems.
A graph database is a management system that stores information in the form of nodes (objects containing properties or attributes that describe the data it represents) and relationships or edges (arrows defining connections between nodes, indicating the nature of the relationship and its direction),49 providing a powerful new method of storing highly complex and variable information.50–52 Defining a graph database for chemistry is straightforward because the traditional representation of chemical reactions, conceptually describing the transformation of chemical substances from reactants to products as a diagram, aligns naturally with a graph structure (this symbolic translation is depicted in Fig. 2A). For instance, a starting material (a molecule node) is transformed into a product (another molecule node) by a chemical reaction (a reaction node), in which the edges provide the relationship (if a substrate is a precursor or a product).
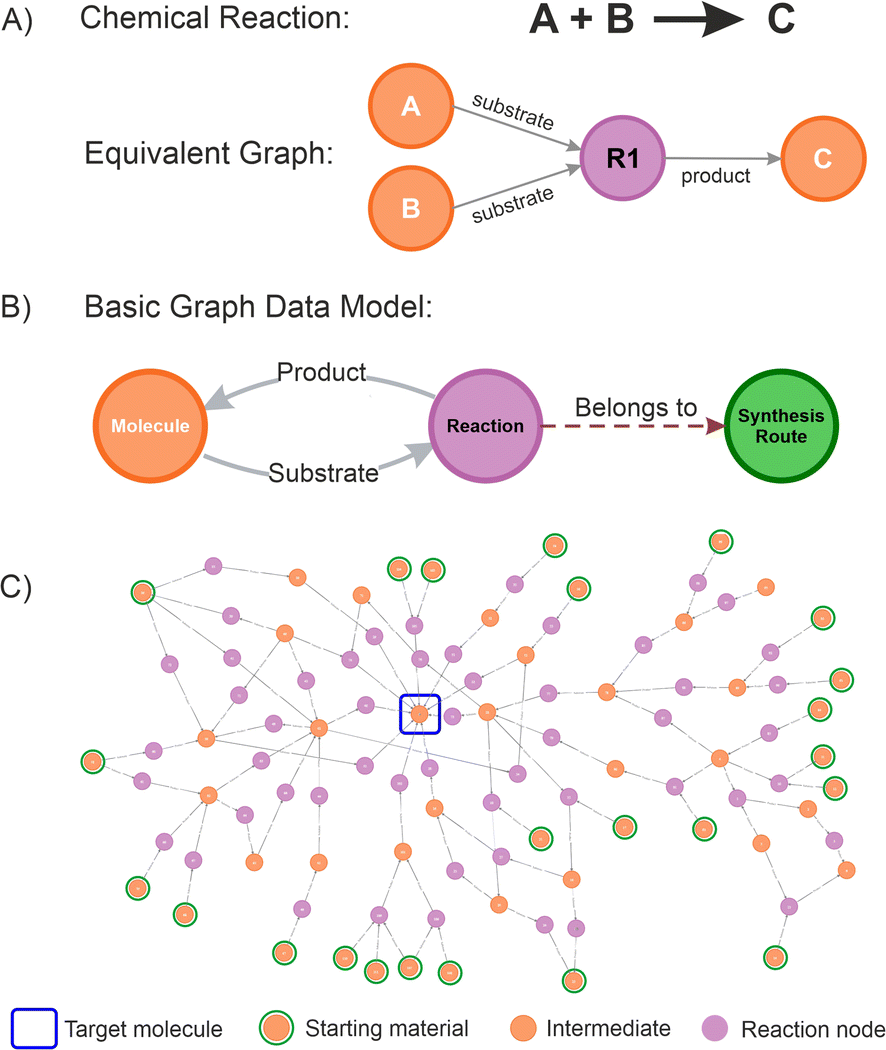 | ||
| Fig. 2 (A) Chemical representation of a single reaction (substrates A and B react to produce C) and its equivalent graph representation in the context of graph databases; (B) basic data model showing the relationships between molecule, reaction, and route nodes. In the graph database, nodes can be used to store specific domain information, creating a multidimensional data structure; (C) example of large network of transformations (omitting route nodes), illustrating possible paths from the starting materials to the target molecule, as observed from a graph database. | ||
Furthermore, in a graph database, molecule nodes can contain specific properties such as names or identifiers (SMILES representations, INCHI keys, IUPAC names, etc.), as well as other fields capturing specific physical or chemical information. In the same way, a reaction node can contain specific properties such as rection conditions, yields, selectivity, purity, and other scores. Additional nodes can be created representing different objects without interfering with any of the data and relationships established. In this work, a route node was also created to be able to store individual route attributes and introduce more advanced network analysis. Extra nodes can be added to the graph database containing users, projects, ideas information, etc.; which might be helpful to add additional management layers, but we omitted them in this paper for clarity.
Some rules need to be established to preserve the logic of the graph database. For instance, a molecule node must be unique, which means if different representations are available for the same molecule, all of these should be gathered under a single node. This enables consistency and facilitate to implement efficient searching strategies. Conversely, reaction nodes are unlimited, allowing to capture all the possible options to do a transformation.
Currently, most of the retrosynthesis software packages available use this type of graph representation,53 and translating this into a graph database schema results in a basic data model (Fig. 2B). A graph data model could become extremely complex depending on the numbers of nodes and relationships defined, the extent of the layers, and the processes modelled.54 Applying the basic data model proposed allows for connecting different molecule nodes to various reaction nodes, potentially resulting in large interaction networks (an example is given in Fig. 2C), from which the path from any molecule to any desired product can be established. The proposed schema was successfully used in a number of internal projects at Pfizer. An example is given for the route selection of Lotiglipron (PF-07081532), a GLP-1 (Glucagon-Like Peptide-1) receptor agonists developed for indications including type-2 diabetes and obesity.55 The results obtained are used to illustrate some of the concepts introduced, and specific details are presented in the ESI Section.†
In the context of commercial route selection, node properties were customised to include specific aspects concerning the SELECT criteria. A summary table for each of the nodes defined for this work is presented in Fig. 3A. Each type of node contains specific properties associated to the particular domain it represents (molecule, reactions, routes). During the data capture process, the scientists can provide insights in any form: chemical drawings, captured by the molecule nodes; reaction conditions and metadata captured by the reaction nodes; rankings, scores, comments, and suggestions, all of them channelled accordingly to the most appropriate graph structure aligned with any of the SELECT aspects.
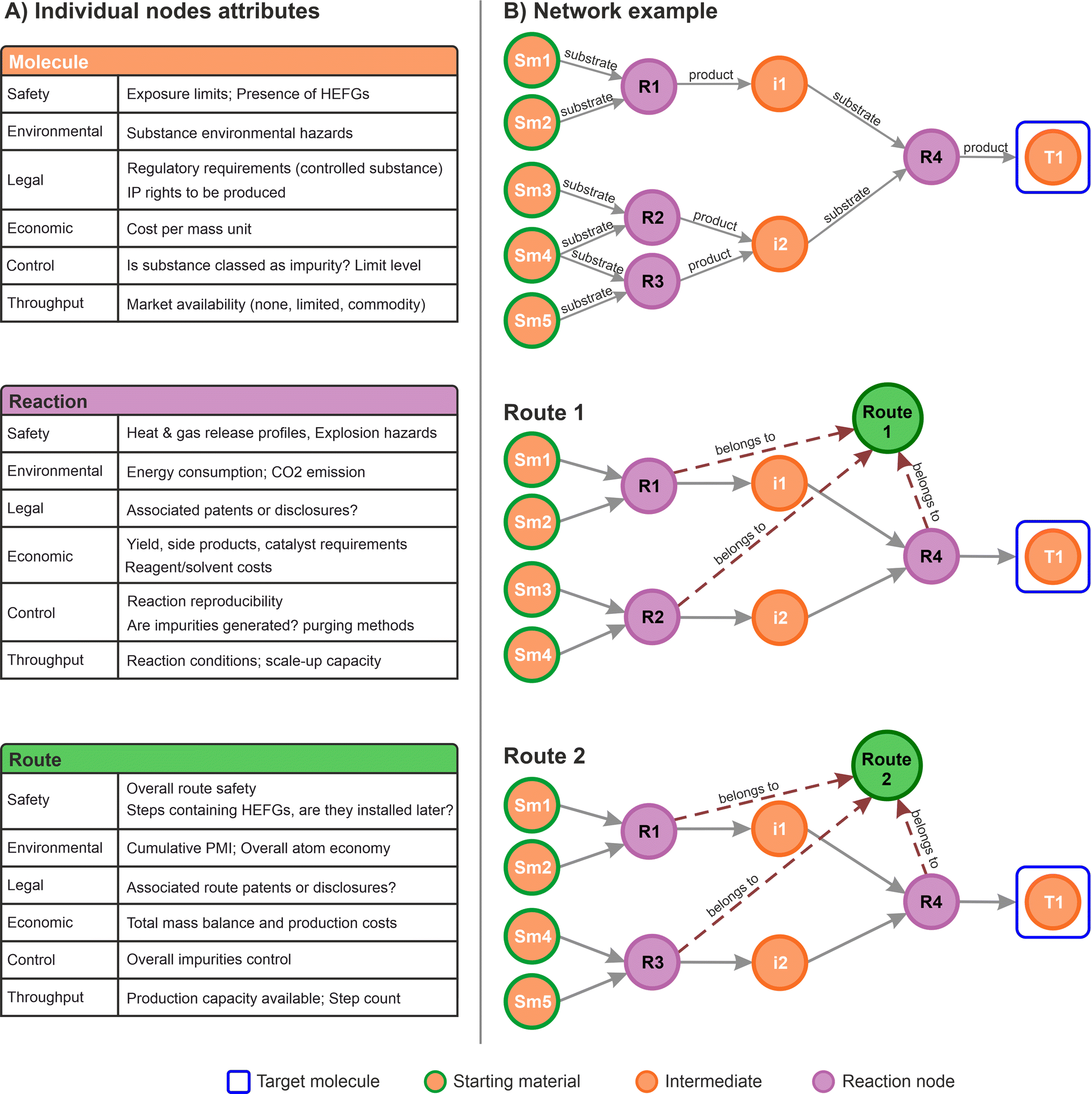 | ||
| Fig. 3 (A) Example of nodes attributes for molecules, reactions, and routes, aligned with the SELECT criteria. Other attributes can also be stored such as names, identifiers, etc. without interfering with the data model and other elements already stored (HEFGs stands for high-energy functional groups); (B) example of a small graph database network, illustrating the route identification with two possible paths linked to a route node (green). Some edges labels (arrows) were removed for clarity. | ||
Subsequently, the types of nodes defined can capture any information relevant to the process. For instance, labels for impurities and side products can be incorporated within the relationships or the reaction nodes. While impurities and side products are strictly molecules, their role within a specific reaction determines their labelling. Occasionally, an impurity may serve as the desired product in a different process, and vice versa. Therefore, capturing this information should focus on the specific chemical transformation rather than the molecules themselves. This example aligns with the proposed data model.
Fig. 3B shows a small network from which two routes can be identified, illustrating how specific information can be stored in the corresponding route nodes. From a higher-level perspective this approach allows connections to all the different multidisciplinary aspects in a single data structure, in a similar way as applied in a variety of other graph databases applications.52,56
Human retrosynthetic analysis and digital idea capture
Across the chemical sciences, retrosynthetic analysis is a collaborative effort unfolded through an iterative process, where initial ideas serve as the foundation for generating new ones. Chemists usually engage in a dynamic exchange of concepts, building upon existing strategies and fostering creativity to develop viable routes, ensuring that the alternatives are not only innovative but also strategically sound.12 This human-driven approach generates a substantial volume of ideas, and the complexity made imperative to implement an effective knowledge management system, ensuring that valuable insights are captured, preserved, and available for future reference and refinement.In this context, once a suitable storage solution was identified (the graph database), the focus was placed on capturing the information directly from the scientists, and at the same time, enhancing the process by allowing them to construct over other scientist's contributions. In order to achieve this dynamic construction, we proposed a standardised three step procedure including: (a) the idea capture at source (using a scientist user interface, or by accessing insights already digitalised); (b) a translation layer to enrich and transform the captured data to fit the graph data model (algorithm layer); and (c) the storage of this information into the graph database. Fig. 4A illustrates the complete process.
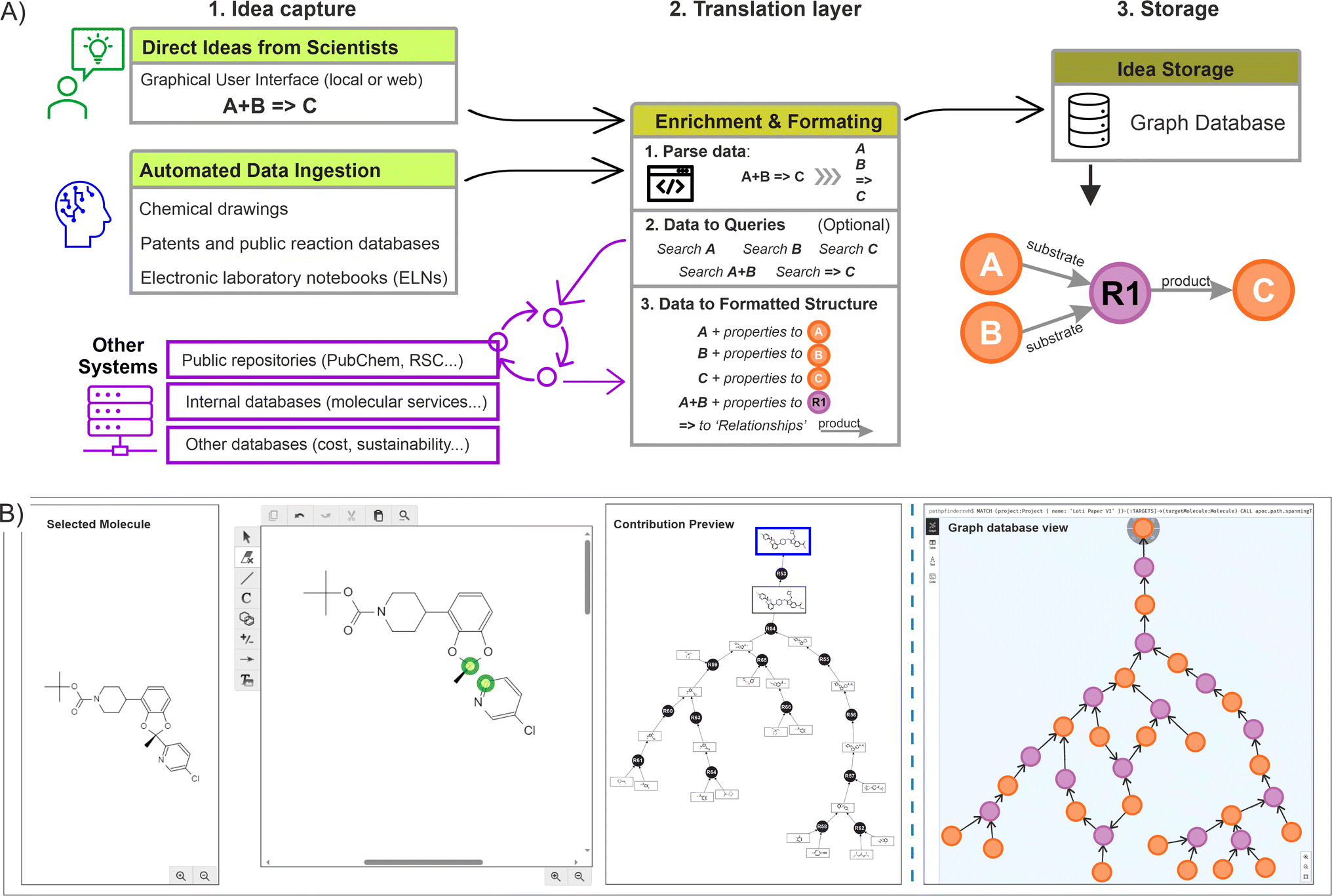 | ||
| Fig. 4 (A) Idea capture process starts with ideas obtained directly from the scientist or ingested from other data systems. Next, a translation layer parses and query the data in other databases to capture additional metadata (enrichment), with the results categorised to fit the data model. Finally, the data is queried for consistency into the graph database before being stored permanently. (B) Example of graphical user interface (GUI), with an embedded drawing canvas to introduce chemical ideas directly from the scientist (left). The resulting fragments are extracted and queried programmatically into the graph database, building a dynamic graph visible to the scientist (contribution preview). Once the idea is submitted, the data is registered into the graph database (Neo4J backend representation on right side). | ||
The creation of a basic rendering tool was required to capture direct ideas from scientists (using commercial drawing packages such as Chemdraw or Biovia draw), producing outputs that were channelled to the graph database across an intranet network. In this case, a drawing canvas was embedded, automatically loading a molecule selected by the scientist from the network being created (which served as the basis for the upcoming idea). Within this canvas, the scientist could make any change or disconnection, adjusting fragments or synthons into molecules feasible to exist (without undefined atoms). Upon submission, the different fragments were incorporated into the graph database and the relationships created (an example of such rendering interface is shown in Fig. 4B). If the desired starting point was not present already in the graph, disconnections should be made from any other suitable molecule linked to the main target, ensuring all the ideas are connected (no isolated nodes are allowed). If a more appropriate connection is introduced later, this will be automatically reflected on the graph structure.
From a system perspective, when the scientist proceeds to submit, an algorithm picks all the individual fragments from the canvas (extracted as SMILES or INCHI keys), individually searching for them in the graph database. If the fragment is not found, a new molecule(s) and reaction nodes are created, with the corresponding ‘relationships’ established (substrate to reaction node, reaction to product node, or any other edge). Conversely, if the fragment is available already in the database, only the reaction node is created with the corresponding relationships. While this process takes place, an additional data enrichment step can be implemented by using the individual identifiers and querying them in other accessible systems (Fig. 4A, centre).
In addition of capturing the scientist rendering, a form was deployed alongside to include additional metadata, such as reaction conditions, chances of success, scalability, etc. Similarly, this information was parsed, channelled, and stored directly into the properties of the corresponding nodes (an example is provided in ESI Section 1).† Moreover, additional automated mechanisms could also be implemented to gather ideas from various sources, including ingesting data from other database systems, extracting ideas from literature references, or retrieving previously registered ideas in the chemistry sections of ELNs. After performing validation checks to preserve the graph integrity, such as confirming that the molecules remain unique in the database and that the corresponding encoding is correct (done by a preliminary cross-validation search), as well as including the potential enrichment of the nodes and relationships, the data is subsequently written into the graph database. Fig. 4B shows a resulting network (small) as seen from a graph database (in this case using Neo4J).
Depositing the data in a centralised graph allowed other scientists to visualise the contributions as they were created, being able to add on top of them (introducing additional metadata in already created nodes), or use this information to generate new ideas. For Lotiglipron, a summary is provided in ESI Section 2,† also showing the raw visualisation of the data in the graph database.
Integrating predictive algorithms
Over the last ten years, cross-disciplinary research involving chemists, cheminformatics, chemical engineers, and data engineers has resulted in a variety of algorithms to predict not just steps but completed synthesis routes. These efforts have been captured in variety of commercial and open-source software packages such as IBM Rxn for chemistry, ASKCOS, Chemairs, Synthia, Reaxis, SciFinderN, amongst others, easily available to the whole chemistry community.57,58 While these packages have been primarily designed for helping during the drug development phases, predicted routes can also be used to increase the idea diversity in commercial route selection. In terms of format, graphs generated using these packages share similar principles for capturing chemical information. The only missing elements are standardisation and transfer of these into the graph database.For Lotiglipron, a first attempt to enrich the human-generated network of ideas consisted in capturing some interesting routes generated by predictive software, and adding them manually into the graph. For this, we used the software package ASKCOS,59 which is currently under development by the Machine Learning for Pharmaceutical Discovery and Synthesis (MLPDS) consortium.60 ASKCOS is a retrosynthesis package designed to generate machine learning-driven synthesis routes, allowing users to input a target molecule, and then generating potential synthetic routes based on models that have been trained with a variety of different chemical databases. Implementation details can be found on ESI Section 3.† In this case, ideas were manually transferred to the graph database, and we anticipate this process will be straightforward in the future by ingesting data automatically from any of those tools.
Additional manual filtering steps were needed to remove the noise accompanying suitable predictions. Usually, these algorithms work well with simpler molecules, but when facing complex transformations, as those found in the pharmaceutical industry including heterocycle formation, bypassing unwanted or unsafe chemistry, or providing alternatives to generate desired chiral species (as in Lotiglipron case), these still tend to fail. This noise can also affect the human creativity and decision making by overwhelming the graph, and masking areas of interest that could be expanded. For this reason, we proposed a separate process, gathering and filtering synthetic ideas in parallel, and merging with human contributions only at the end of a brainstorm cycle. The direct value of these algorithms is still under scrutiny,53 and current failures are justified by the lack of appropriated data training repositories. Current datasets available are reactions stored in ELNs (still requiring individual curation), information captured in public repositories such as patents (for instance, datasets created from USPO data), or extracted automatically from literature sources. Most of these cases still contain biases towards positive data (only successful cases reported). We suggest this situation could be completely overturned by using the graph database itself as a source of curated data for algorithm training.
Fig. 5 shows the resulting network obtained for Lotiglipron. In Fig. 5A, the ‘scientist view’ integrates both human and ASCKOS predicted ideas. It is designated ‘scientist view’ since this shows how the data is presented to the scientist on a user interface, where duplicate molecules are allowed for clarity, aiding in the identification of individual branches of the tree structure. Below, Fig. 5B shows the corresponding native graph database representation, illustrating how the data is organised and stored within the database. While the information presented in both views is the same, the scientist visualisation differs from the graph database representation, where molecules nodes are unique.
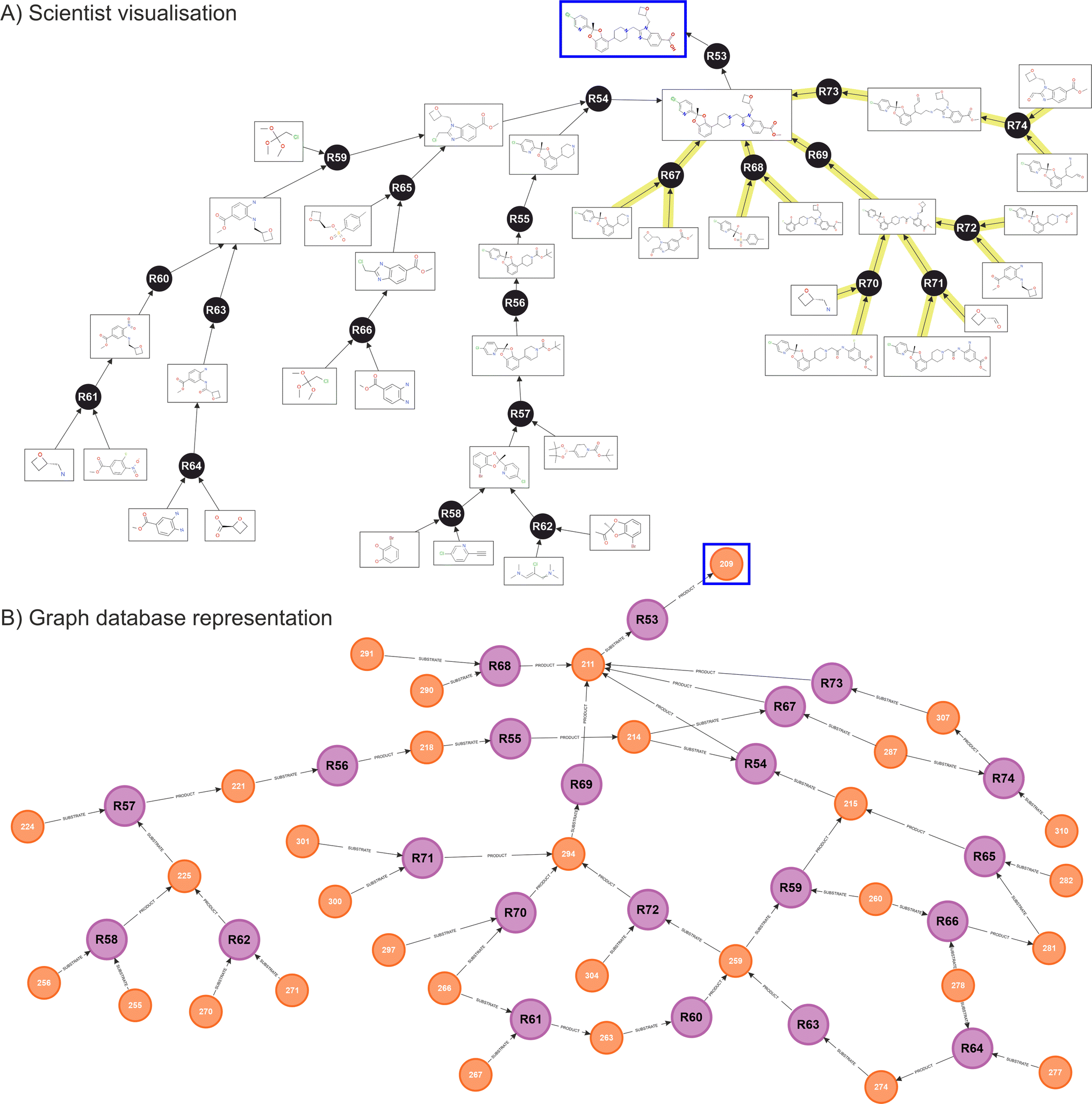 | ||
| Fig. 5 GLP1 (Lotiglipron) Network – (A) scientist view, combining human ideas (left side, no overlaying colour), with synthetic ideas from predictive software ASKCOS (right side, yellow overlaid). The resulting network observed from the scientist interface shows linear branches with duplication of substrates and products for user clarity. (B) Equivalent back-end graph database representation obtained from native graph database (Neo4J), showing unique molecule nodes. In both cases, the target molecule is indicated by a square. | ||
When the database contains a significant number of transformations, specific graph features begin to appear such as cycling recurrences. In these, a substrate A feeds into a reaction to produce substrate B, which then feeds back into substrate A. Also, key reactions and key intermediates can be identified by observing ‘hubs’ nodes, where many relationships are pointing (an example of this are intermediates 211 and 294 in the graph database diagram, Fig. 5B).
The Lotiglipron graph was subjected to an automated algorithm designed to identify individual synthesis routes. This algorithm works by transforming the initial cyclic graph, which represents all possible synthesis pathways, into an acyclic type of graph (this operation is performed outside the database, on a duplicated dataset). In the acyclic form, pathways do not loop back on themselves, simplifying the structure and making it easier to analyse. By converting the graph, straightforward and well-known methods such as depth-first search (DFS) and breadth-first search (BFS),61 are applied to identify and extract the individual synthesis routes efficiently. These methods systematically explore the nodes and edges, traversing the entire network and assigning specific nodes identifiers as suggested in Fig. 2 and 3.
As Fig. 6 shows, the algorithm identified six routes obtained from human suggestions, and six additional routes obtained from artificial suggestions. These nodes were subsequently enriched with further annotations and data concerning to the specific route properties, and this higher-level layer allowed the optimisation of the route selection. After this step, the graph database was ready to be interrogated (ESI Section 4† provides instructions to reconstruct the full Lotiglipron network in a graph database).
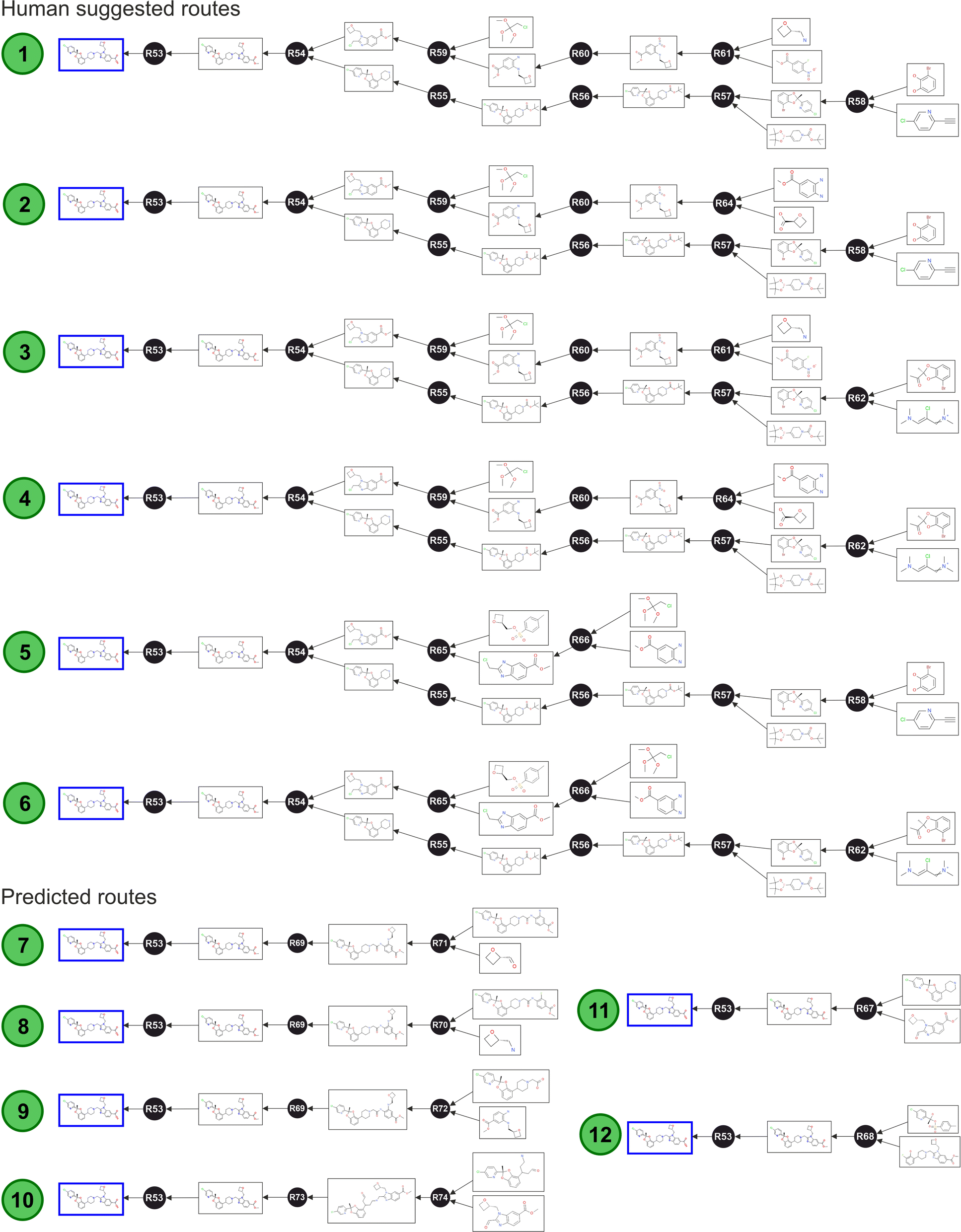 | ||
| Fig. 6 GLP1 (Lotiglipron) Network – automated route identification algorithm returned twelve routes: 1–6 human generated; 7–12 from predictive algorithm. Route 1 corresponds to the enabling chemistry route. The shortest path query implemented without any constraint returned routes 11 and 12. These two transformations are possible but without real commercial value since the routes are partially developed (the starting materials cannot be purchased). Overlaying additional data, including layers aligned to the SELECT criteria, can help to determine an optimum commercial route. | ||
The native Neo4J language Cypher was directly used for querying the database. This approach differs to others suggested in literature, where programming languages have been created to interact with chemical information.62 Decoupling the query language from the chemistry language introduce robustness and allow experts from other fields to manipulate the data without any prior chemistry knowledge.
An example query that the graph database is optimised to answer is ‘find the shortest route from the target molecule up to the starting materials under specific constrains’. This is also known as Dijkstra's algorithm. This query can deliver the route with the minimum number of steps, which at the same time minimise the weight of any of the constrains imposed. Elaborating the queries and constraints goes beyond the scope of this paper, but we envisioned the calculation of additional metrics cascaded across the entire network during the enrichment phase, and using those values for resolving the query. As an example, some of the network metrics considered are listed below:
- Environmental metrics,34,63 for instance estimating the process mass intensity (PMI) of each step and determining the routes with the lowest cumulative PMI value (cPMI).
- Safety,26 determining the potential flammability and explosiveness of all the substrates and intermediates, and obtaining the average route safety by pondering the accumulated hazards across all the route steps.
- Cost of syntheses,36 determining the costs of the individual steps (observed or estimated), and applying rules such as excluding the use of precious metals that would make the large scale process unviable.
- Legal right to operate,25 identifying disconnections in the network which are common to industrial patents.
The shortest path query implemented without any constrain returned routes 11 and 12 (Fig. 6), using the Dijkstra's algorithm. However, this result only considered the number of steps and not the full SELECT criteria. Upon careful examination, both suggested transformations are theoretically possible but there is a lack of scalability information. For instance, route 11 is impractical because the starting materials are not commercially available in the required form. A similar problem is observed with route 12. In these cases, the predictive algorithm did not further expand the starting materials, only adding partial information. This highlights an important point: queries like this are useful only if they include information on commodities or commercially available materials. To make meaningful comparisons, all routes should be traced back to compounds of similar complexity to assess their respective potential effectively. The same point is valid for all the parameters covered by SELECT criteria.
Discussion and outlook
Impacting the production of modern medicines
The strategy suggested in this paper can be used directly to identify a global optimum synthetic route by taking in to account many conflicting priorities. A significant impact to the global access of medicines could be achieved by enabling more cost-effective production methods and by accelerating their development. Additionally, the strict regulation of the pharmaceutical industry requires the manufacturer to implement a sound control strategy to limit the presence of undesired compounds in the API. The design of the synthetic route is a fundamental aspect of this strategy. Thus, it is imperative to incorporate impurity control filters as one of the fundamental layers throughout the decision-making process, and the proposed approach is a step towards this.Another area where this holistic approach could aid is in the environmental and carbon footprint of the process. Incorporating factors such as chemical hazards, waste minimisation, and energy consumption as part of the decision-making process will help processes move towards net zero and further lower production costs. Some aspects of Green Chemistry could be applied today across an entire graph network, such as PMI as previously described.
Regarding material costs and availability, accuracy during the synthesis planning is difficult to achieve. Often decision makers turn to non-specialised catalogue companies for a guide price and then apply an economy of scale factor. Here is another opportunity of connected graph database, historical and up-to-date pricing for raw materials, pre-GMP7 intermediates and transformations8 can be built into the graph allowing predictive algorithms to make better cost estimates of these new entities. This layer enables the optimisation of realistic productions costs, which may raise the priority of other route options often overlooked based purely on synthetic characteristics. Realising new synthetic routes based on cost and synthetic novelty offers companies options either to protect intellectual property or to allow freedom to operate. Thus, protecting or disclosing different routes is based on different weightings of the design parameters. A weighting that may change over the lifecycle of the product as new information becomes available and is added to the graph.
Just considering the API alone, large pharmaceutical companies typically cover a significant proportion of the business space. This operational model requires large resource investments to operate. To become more agile some companies are becoming more modular and outsourcing activities to contract research organisations (CROs), universities and research institutes. In this paradigm, a key limitation is the efficiency of data and knowledge transfer between organisations. Domain specific information lacks standardisation and its application is not systematic. Inevitably data is lost, transferred in a non-digital manner, or not shared at all. In the proposed approach, specific graphical user interfaces could be designed to allow controlled portions of the graph to be shared with these third parties, allowing enrichment of the chemical information directly in the database. Access control would be exercised based on centralised polices of data protection.
Besides, chemical process development traditionally relies on inputs from chemists, scientists, and engineers following well-established workflows. However, decisions are still subjective and lacking algorithmic support. Critical data from large-scale manufacturing and other sources remain disconnected. Our approach introduces a method to define commercial synthesis routes. Furthermore, it can be expanded to encompass various engineering and operational aspects, including unit operations and physical transformations, effectively bridging the gap between route formulation and manufacturing.
Towards the design of an ideal chemistry platform
It is difficult to provide a clear assessment about the future possibilities. The number of tools reported in literature is so vast and diverse that it is only possible to focus on specific aspects of the ideal requirements. Firstly, there is the scientist's perspective, they need specialised tools to cater for the unique needs specific to their workflow, for example experimental data handling, analytical data, procedural information and equipment data. Secondly, there is the business needs, where tools that help to prioritise, enable the generation of metrics, and facilitate decision making using simple user-friendly interfaces are required. Thirdly, there is the perspective of platform and software providers who develop and supply these systems. In this case, the market tends to segment itself based on user needs; targeting niche markets where the demand for specialised features is high. Conversely, platforms focusing on business-oriented data and decision-making target a broader audience, including managers and executives. As a result, a holistic integration that bridges the gap between technical details and high-level business decisions seems almost unrealistic.The irruption of graph databases is opening completely new horizons for chemical sciences. At the molecular level traditional chemistry representations can be directly represented as a graph. Graph databases excel in representing and storing complex relationships between entities, making them well-suited for capturing the intricate structures of molecules, and at the same time, providing a suitable place to store individual domain properties. At the synthetic route level the ability to identify relationships between various chemical entities, reactions, and properties, and to enable simple query and data retrieval would satisfy to a large extent many scientist's needs.
Capturing chemical data in graph databases empowers data reusability, offering access to knowledge from past projects. It transforms the dynamics of the decision-making by providing a contemporaneous picture, which changes with new information. Such platforms emerge as a way of training advanced AI models, addressing one of the main problems these tools are facing today.
Finally, a long-term proposal involves creating a universal chemistry data framework centred around graph database technology, accessible to all. Identifying the most suitable agency, forum, or legal body to establish these standards poses challenges. IUPAC stands out as a natural candidate due to its international recognition and expertise in chemical sciences. However, implementation is complex, requiring data sharing and cooperation among organisations. Commercial entities can also contribute significantly by providing necessary infrastructure. For instance, platforms similar to Wikipedia, which are privately funded but open to the public, could serve as models for facilitating widespread data sharing and collaboration. Alternatively, a consortium of companies, universities, and institutions could financially support the creation of a global centralised repository, resembling the features described in this paper. This repository could host public and private layers, allowing each company private access while enabling disclosure of legally protected or obsolete parts of their explored chemical space to the public. Such centralised repository could bring an unprecedented level of collaboration across the chemical sciences and bridge the gap to many other disciplines.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.† There is no code required and only specific Neo4J queries are used to create a graph database instance (using neo4j available at https://www.neo4j.com). For the specific demonstration provided in the paper, all the commands and queries to reproduce step-by-step the graph database (using the specific network of molecules supplied) can be found in Section 4 of the ESI Section.†Author contributions
Claudio Avila: conceptualization, investigation, methodology, writing – original draft; Adam West: conceptualization, investigation, methodology, writing – original draft; Anna Vicini: conceptualization, investigation, methodology, writing – review & editing; William Waddington: conceptualization, investigation, methodology, writing – review & editing; Christopher Brearley: conceptualization, investigation, methodology, writing – review & editing; James Clarke: conceptualization, investigation, methodology, writing – review & editing; Andrew Derrick: conceptualization, investigation, methodology, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our gratitude to Charles Santa Maria and Stephane Caron for their business support in realising this project. Special thanks to the Pfizer Digital Insights team, led by Jonathan Lowe and supported by Abby Garreth, Lina Tian, Michelle Ong, Mariano Coutada, and all the developers involved, for their assistance in building the user interfaces and implementing the graph database. We also want to thank Rajesh Mishra, Jason Mustakis and Vijay Bulusu from Pfizer PSSM Data Sciences, for collaborating with us to build the initial proof of concepts. Finally, we express our gratitude to all the process chemists from CRD Pfizer Sandwich for their valuable feedback, expertise, and dedication to the field of chemistry.References
- E. J. Corey, Angew. Chem., Int. Ed. Engl., 1991, 30, 455–465, DOI:10.1002/anie.199104553.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew Chem. Int. Ed. Engl., 2001, 40, 2004–2021, DOI:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5.
- J. Studley, in Complete Accounts of Integrated Drug Discovery and Development: Recent Examples from the Pharmaceutical Industry, 2022, vol. 4, pp. 333–355, DOI:10.1021/bk-2022-1423.ch009.
- H. J. Federsel, Acc. Chem. Res., 2009, 42, 671–680, DOI:10.1021/ar800257v.
- S. A. Busby, S. Carbonneau, J. Concannon, C. E. Dumelin, Y. Lee, S. Numao, N. Renaud, T. M. Smith and D. S. Auld, ACS Chem. Biol., 2020, 15, 2636–2648, DOI:10.1021/acschembio.0c00495.
- J. P. Hughes, S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249, DOI:10.1111/j.1476-5381.2010.01127.x.
- Good manufacturing practice (GMP), European Medicines Agency, available at https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compliance-research-development/good-manufacturing-practice.
- M. Faul, C. Busacca, M. Eriksson, F. Hicks, W. Kiesman, M. Smulkowski, J. Orr and S. Pfeiffer, Org. Process Res. Dev., 2014, 18, 594–600, DOI:10.1021/op5000607.
- N. G. Anderson, in Practical Process Research and Development, 2012, pp. 47–87, DOI:10.1016/b978-0-12-386537-3.00003-4.
- N. E. Bodé and A. B. Flynn, J. Chem. Educ., 2016, 93, 593–604, DOI:10.1021/acs.jchemed.5b00900.
- S. Caille, S. Cui, M. M. Faul, S. M. Mennen, J. S. Tedrow and S. D. Walker, J. Org. Chem., 2019, 84, 4583–4603, DOI:10.1021/acs.joc.9b00735.
- P. Cornwall, L. J. Diorazio and N. Monks, Bioorg. Med. Chem., 2018, 26, 4336–4347, DOI:10.1016/j.bmc.2018.06.006.
- M. Butters, D. Catterick, A. Craig, A. Curzons, D. Dale, A. Gillmore, S. P. Green, I. Marziano, J. P. Sherlock and W. White, Chem. Rev., 2006, 106, 3002–3027, DOI:10.1021/cr050982w.
- R. B. Leng, M. V. M. Emonds, C. T. Hamilton and J. W. Ringer, Org. Process Res. Dev., 2012, 16, 415–424, DOI:10.1021/op200264t.
- K. Rossen, J. Org. Chem., 2019, 84, 4580–4582, DOI:10.1021/acs.joc.9b00344.
- A. Q. Mohammed, P. K. Sunkari, P. Srinivas and A. K. Roy, Org. Process Res. Dev., 2015, 19, 1634–1644, DOI:10.1021/op500295a.
- A. D. Allian, N. P. Shah, A. C. Ferretti, D. B. Brown, S. P. Kolis and J. B. Sperry, Org. Process Res. Dev., 2020, 24, 2529–2548, DOI:10.1021/acs.oprd.0c00226.
- S. Boobier, D. R. J. Hose, A. J. Blacker and B. N. Nguyen, Nat. Commun., 2020, 11, 5753, DOI:10.1038/s41467-020-19594-z.
- J. Magano, Org. Process Res. Dev., 2022, 26, 1562–1689, DOI:10.1021/acs.oprd.2c00005.
- D. J. Griffin, C. W. Coley, S. A. Frank, J. M. Hawkins and K. F. Jensen, Org. Process Res. Dev., 2023, 27, 1868–1879, DOI:10.1021/acs.oprd.3c00229.
- Y. F. Lin, Z. R. Zhang, B. Mahjour, D. Wang, R. Zhang, E. Shim, A. McGrath, Y. N. Shen, N. Brugger, R. Turnbull, S. Trice, S. Jasty and T. Cernak, Nat. Commun., 2021, 12, 7327, DOI:10.1038/s41467-021-27547-3.
- I. E. Dror, Anal. Chem., 2020, 92, 7998–8004, DOI:10.1021/acs.analchem.0c00704.
- T. Laird, Org. Process Res. Dev., 2012, 16, 365, DOI:10.1021/op300038x.
- H. Gurulingappa, A. Mudi, L. Toldo, M. Hofmann-Apitius and J. Bhate, RSC Adv., 2013, 3, 16194–16211, 10.1039/c3ra40787j.
- K. Molga, P. Dittwald and B. A. Grzybowski, Chem, 2019, 5, 460–473, DOI:10.1016/j.chempr.2018.12.004.
- H. B. B. Anuradha, M. Y. Gunasekera and O. Gunapala, Process Saf. Environ. Prot., 2020, 133, 358–368, DOI:10.1016/j.psep.2019.11.001.
- M. Fialkowski, K. J. M. Bishop, V. A. Chubukov, C. J. Campbell and B. A. Grzybowski, Angew. Chem., Int. Ed., 2005, 44, 7263–7269, DOI:10.1002/anie.200502272.
- S. Kanza, C. Willoughby, N. J. Knight, C. L. Bird, J. G. Frey and S. J. Coles, Digital Discovery, 2023, 2, 602–617, 10.1039/d2dd00121g.
- O. Kononova, T. J. He, H. Y. Huo, A. Trewartha, E. A. Olivetti and G. Ceder, Iscience, 2021, 24(3), 102155, DOI:10.1016/j.isci.2021.102155.
- K. M. Jablonka, L. Patiny and B. Smit, Nat. Chem., 2022, 14, 365, DOI:10.1038/s41557-022-00910-7.
- P. E. Fuller, C. M. Gothard, N. A. Gothard, A. Weckiewicz and B. A. Grzybowski, Angew Chem. Int. Ed. Engl., 2012, 51, 7933–7937, DOI:10.1002/anie.201202210.
- J. R. Bai, L. W. Cao, S. Mosbach, J. Akroyd, A. A. Lapkin and M. Kraft, JACS Au, 2022, 2, 292–309, DOI:10.1021/jacsau.1c00438.
- P. L. Turtscher and M. Reiher, J. Chem. Inf. Model., 2023, 63, 147–160, DOI:10.1021/acs.jcim.2c01136.
- J. Li and M. D. Eastgate, React. Chem. Eng., 2019, 4, 1595–1607, 10.1039/c9re00019d.
- J. M. Weber, Z. Guo and A. A. Lapkin, ACS Eng. Au, 2022, 2, 333–349, DOI:10.1021/acsengineeringau.2c00002.
- T. Badowski, K. Molga and B. A. Grzybowski, Chem. Sci., 2019, 10, 4640–4651, 10.1039/c8sc05611k.
- W. Wang, Y. W. Liu, Z. Wang, G. F. Hao and B. A. Song, Chem. Sci., 2022, 13, 12604–12615, 10.1039/d2sc04419f.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, ACS Cent. Sci., 2016, 2, 131–138, DOI:10.1021/acscentsci.6b00015.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle, S. D. Dreher, J. M. Hawkins, K. F. Jensen and C. W. Coley, J. Am. Chem. Soc., 2021, 143, 18820–18826, DOI:10.1021/jacs.1c09820.
- T. Gimadiev, R. Nugmanov, D. Batyrshin, T. Madzhidov, S. Maeda, P. Sidorov and A. Varnek, J. Chem. Inf. Model., 2021, 61, 554–559, DOI:10.1021/acs.jcim.0c01280.
- D. Jiang, C.-Y. Hsieh, Z. Wu, Y. Kang, J. Wang, E. Wang, B. Liao, C. Shen, L. Xu, J. Wu, D. Cao and T. Hou, J. Med. Chem., 2021, 64, 18209–18232, DOI:10.1021/acs.jmedchem.1c01830.
- R. Xue, J. Liao, X. Shao, K. Han, J. Long, L. Shao, N. Ai and X. Fan, Chem. Res. Toxicol., 2019, 33, 202–210, DOI:10.1021/acs.chemrestox.9b00238.
- G. Zahoránszky-Kőhalmi, N. Lysov, I. Vorontcov, J. Wang, J. Soundararajan, D. Metaxotos, B. Mathew, R. Sarosh, S. G. Michael and A. G. Godfrey, J. Chem. Inf. Model., 2022, 62, 2226–2238, DOI:10.1021/acs.jcim.1c01202.
- S. Pablo-García, R. Pérez-Soto, A. Sabadell-Rendón, D. Garay-Ruiz, V. Nosylevskyi and N. López, Digital Discovery, 2024 10.1039/d4dd00087k.
- Y. Mo, Y. Guan, P. Verma, J. Guo, M. E. Fortunato, Z. Lu, C. W. Coley and K. F. Jensen, Chem. Sci., 2020, 12, 1469–1478, 10.1039/d0sc05078d.
- M. Pasquini and M. Stenta, J. Cheminf., 2023, 15, 41, DOI:10.1186/s13321-023-00714-y.
- W. Gao and C. W. Coley, J. Chem. Inf. Model., 2020, 60, 5714–5723, DOI:10.1021/acs.jcim.0c00174.
- K. Lee, J. Woo Kim and W. Youn Kim, ChemSystemsChem, 2020, 2, e1900057, DOI:10.1002/syst.201900057.
- Neo4J, What is a Graph Database?, https://neo4j.com/developer/graph-database/.
- L. Wang, C. Sun, C. Zhang, W. Nie and K. Huang, Inf. Software Technol., 2023, 164, 107327, DOI:10.1016/j.infsof.2023.107327.
- H. Matter, C. Buning, D. D. Stefanescu, S. Ruf and G. Hessler, J. Chem. Inf. Model., 2020, 60, 6120–6134, DOI:10.1021/acs.jcim.0c00947.
- M. J. Statt, B. A. Rohr, D. Guevarra, J. N. Breeden, S. K. Suram and J. M. Gregoire, Digital Discovery, 2023, 2, 909–914, 10.1039/d3dd00067b.
- A. Thakkar, S. Johansson, K. Jorner, D. Buttar, J. L. Reymond and O. Engkvist, React. Chem. Eng., 2021, 6, 27–51, 10.1039/d0re00340a.
- A. Santos, A. R. Colaco, A. B. Nielsen, L. Niu, M. Strauss, P. E. Geyer, F. Coscia, N. J. W. Albrechtsen, F. Mundt, L. J. Jensen and M. Mann, Nat. Biotechnol., 2022, 40, 692–702, DOI:10.1038/s41587-021-01145-6.
- Lotiglipron, Pfizer Provides Update on GLP-1-RA Clinical Development Program for Adults with Obesity and Type 2 Diabetes Mellitus, https://www.pfizer.com/news/press-release/press-release-detail/pfizer-provides-update-glp-1-ra-clinical-development, accessed 01 February, 2024.
- S. Timón-Reina, M. Rincón and R. Martínez-Tomás, Database, 2021, 2021, 1–22, DOI:10.1093/database/baab026.
- S. Johansson, A. Thakkar, T. Kogej, E. Bjerrum, S. Genheden, T. Bastys, C. Kannas, A. Schliep, H. Chen and O. Engkvist, Drug Discovery Today: Technol., 2019, 32–33, 65–72, DOI:10.1016/j.ddtec.2020.06.002.
- Z. Wang, W. Zhao, G. Hao and B. Song, Org. Chem. Front., 2021, 8, 812–824, 10.1039/d0qo00946f.
- MLPDS, ASKCOS Software, https://askcos.mit.edu/.
- T. J. Struble, J. C. Alvarez, S. P. Brown, M. Chytil, J. Cisar, R. L. DesJarlais, O. Engkvist, S. A. Frank, D. R. Greve, D. J. Griffin, X. J. Hou, J. W. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. A. Nicolaou, A. D. Palmer, D. J. Price, R. I. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. H. Green, R. Barzilay, C. W. Coley and K. F. Jensen, J. Med. Chem., 2020, 63, 8667–8682, DOI:10.1021/acs.jmedchem.9b02120.
- R. Scheffler, Theor. Comput. Sci., 2022, 936, 116–128, DOI:10.1016/j.tcs.2022.09.018.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson, D. Angelone and L. Cronin, Science, 2019, 363, 144, DOI:10.1126/science.aav2211.
- J. Li, J. Albrecht, A. Borovika and M. D. Eastgate, ACS Sustainable Chem. Eng., 2018, 6, 1121–1132, DOI:10.1021/acssuschemeng.7b03407.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00120f |
| This journal is © The Royal Society of Chemistry 2024 |