
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteractions of peroxy radicals from monoterpene and isoprene oxidation simulated in the radical volatility basis set†
Meredith
Schervish
 ab,
Martin
Heinritzi
c,
Dominik
Stolzenburg
de,
Lubna
Dada
f,
Mingyi
Wang
ag,
Qing
Ye
ah,
Victoria
Hofbauer
a,
Jenna
DeVivo
a,
Federico
Bianchi
ij,
Sophia
Brilke
e,
Jonathan
Duplissy
ij,
Imad
El Haddad
f,
Henning
Finkenzeller
k,
Xu-Cheng
He
ijl,
Aleksander
Kvashnin
m,
Changhyuk
Kim
no,
Jasper
Kirkby
cp,
Markku
Kulmala
ij,
Katrianne
Lehtipalo
ij,
Brandon
Lopez
q,
Vladimir
Makhmutov
rs,
Bernhard
Mentler
t,
Ugo
Molteni
fu,
Wei
Nie
ijv,
Tuuka
Petäjä
ij,
Lauriane
Quéléver
ij,
Rainer
Volkamer
k,
Andrea C.
Wagner
cw,
Paul
Winkler
e,
Chao
Yan
ijv and
Neil M.
Donahue
*aq
ab,
Martin
Heinritzi
c,
Dominik
Stolzenburg
de,
Lubna
Dada
f,
Mingyi
Wang
ag,
Qing
Ye
ah,
Victoria
Hofbauer
a,
Jenna
DeVivo
a,
Federico
Bianchi
ij,
Sophia
Brilke
e,
Jonathan
Duplissy
ij,
Imad
El Haddad
f,
Henning
Finkenzeller
k,
Xu-Cheng
He
ijl,
Aleksander
Kvashnin
m,
Changhyuk
Kim
no,
Jasper
Kirkby
cp,
Markku
Kulmala
ij,
Katrianne
Lehtipalo
ij,
Brandon
Lopez
q,
Vladimir
Makhmutov
rs,
Bernhard
Mentler
t,
Ugo
Molteni
fu,
Wei
Nie
ijv,
Tuuka
Petäjä
ij,
Lauriane
Quéléver
ij,
Rainer
Volkamer
k,
Andrea C.
Wagner
cw,
Paul
Winkler
e,
Chao
Yan
ijv and
Neil M.
Donahue
*aq
aCarnegie Mellon University, Department of Chemistry, Pittsburgh, PA, USA. E-mail: nmd@andrew.cmu.edu; Tel: +1 412 268-4415
bUniversity of California, Irvine Department of Chemistry, Irvine, CA, USA
cInstitute for Atmospheric and Environmental Sciences, Goethe University Frankfurt, 60438 Frankfurt Am Main, Germany
dInstitute of Materials Chemistry, TU Wien, 1060 Vienna, Austria
eFaculty of Physics, University of Vienna, 1090 Vienna, Austria
fLaboratory of Atmospheric Chemistry, Paul Scherrer Institute, 5232 Villigen, Switzerland
gUniversity of Chicago, Department of the Geophysical Sciences, Chicago, IL, USA
hAtmospheric Chemistry Observations and Modeling Laboratory, U.S. National Science Foundation National Center for Atmospheric Research (NSF NCAR), Boulder, Colorado 80301, USA
iInstitute for Atmospheric and Earth System Research/Physics, University of Helsinki, Helsinki 00014, Finland
jHelsinki Institute of Physics, University of Helsinki, 00014 Helsinki, Finland
kDepartment of Chemistry, CIRES, University of Colorado Boulder, Boulder, CO 80309-0215, USA
lYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK
mLebedev Fisysical Institute, 119991, Moscow, Leninsky Prospect 53, Russia
nSchool of Civil and Environmental Engineering, Pusan National University, Busan, 46241, Republic of Korea
oDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA
pCERN, The European Organization for Nuclear Research, Geneve 23 CH-1211, Switzerland
qCarnegie Mellon University Department of Chemical Engineering, Pittsburgh, PA, USA
rLebedev Physical Institute of the Russian Academy of Sciences, 119991 Moscow, Russia
sMoscow Institute of Physics and Technology (National Research University), 141701 Moscow, Russia
tIon Molecule Reactions & Environmental Physics Group Institute of Ion Physics and Applied Physics Leopold-Franzens University, Innsbruck Technikerstraße 25, A-6020 Innsbruck, Austria
uSwiss Federal Research Institute WSL, Plant Regeneration Ecology, Birmensdorf, CH-8903, Switzerland
vJoint International Research Laboratory of Atmospheric and Earth System Research, School of Atmospheric Sciences, Nanjing University, Nanjing, China
wAerosol Physics Laboratory, Physics Unit, Tampere University, FI-33014 Tampere, Finland
First published on 24th June 2024
Abstract
Isoprene affects new particle formation rates in environments and experiments also containing monoterpenes. For the most part, isoprene reduces particle formation rates, but the reason is debated. It is proposed that due to its fast reaction with OH, isoprene may compete with larger monoterpenes for oxidants. However, by forming a large amount of peroxy-radicals (RO2), isoprene may also interfere with the formation of the nucleating species compared to a purely monoterpene system. We explore the RO2 cross reactions between monoterpene and isoprene oxidation products using the radical Volatility Basis Set (radical-VBS), a simplified reaction mechanism, comparing with observations from the CLOUD experiment at CERN. We find that isoprene interferes with covalently bound C20 dimers formed in the pure monoterpene system and consequently reduces the yields of the lowest volatility (Ultra Low Volatility Organic Carbon, ULVOC) VBS products. This in turn reduces nucleation rates, while having less of an effect on subsequent growth rates.
Environmental significanceIsoprene and monoterpenes are critical components of the atmosphere in all regions influenced by biogenic emissions. Their relative contributions to secondary organic aerosol and new particle formation and growth play important roles in aerosol climate interactions and likely human health effects from aerosols, but the interactions between them are less studied and highly dependent on environmental conditions. Building a proper understanding of these interactions is essential for accurate modeling of changes since the industrial revolution and also anticipated changes during future decarbonization and biosphere alteration due to climate change. |
1 Introduction
Monoterpene (C10H16) emissions from trees (mostly confers) are sufficient to sustain boundary-layer mixing ratios of ten to hundreds of parts per trillion (ppt),1,2 with an estimated global flux of c.f. 90 Tg C year−1.3 Isoprene (C5H8) emissions from trees (mostly deciduous, concentrated in the tropics), are much higher than monoterpene emissions, sustaining boundary-layer mixing ratios up to several parts per billion (ppb),1,4,5 with an estimated global flux of c.f. 460 Tg C year−1. Together, monoterpenes and isoprene are thought to dominate total biogenic emissions. Because the relative emissions of monoterpenes vs. isoprene are highly species (and thus ecosystem) dependent, the concentration ratio of monoterpenes to isoprene varies widely across the globe.The oxidation products of biogenic organic compounds contribute to new particle formation in remote areas of the atmosphere.6–8 Some of these oxidation products, called highly-oxygenated organic molecules (HOMs),9 can have low enough vapor pressures to contribute to growth of existing particles even at the smallest sizes (1–2 nm diameter); the lowest volatility among them may even nucleate with or without inorganic companions. Specifically, the production rate of HOMs from pure monoterpene oxidation is sufficient to substantially contribute to new-particle formation.7,10 Modeling has confirmed their contribution,11,12 and this “pure biogenic” new-particle formation may have been a dominant pathway for cloud condensation nucleus (CCN) formation in the pre-industrial continental atmosphere.11 It should be noted that not all HOMs have such low vapor pressures and many highly oxidized species, especially those formed from small precursor molecules, may not even reach the LVOC volatility range.13
Added isoprene suppresses nucleation even with high concentrations of monoterpenes both in chamber studies and ambient measurements.4,14–17 The cause of this suppression is, however, debated, and model simulations of both chamber experiments and ambient conditions can help resolve the debate.
One proposed cause of the suppression is OH depletion by the isoprene and consequent suppression of α-pinene oxidation rates, leading to a lower production rate of low-volatility products, a lower steady state saturation ratio, and thus lower nucleation rates.14,18,19 However, isoprene contributes to atmospheric recycling of OH and so OH concentrations can still remain high in isoprene-rich environments.20,21 Furthermore, monoterpene ozonolysis products contribute heavily to HOM formation and can produce similar yields even when OH concentrations are low.7,22,23
A second proposed cause of the suppression is direct entanglement of terpene and isoprene oxidation mechanisms. Recent experimental work suggests instead that isoprene suppression of nucleation is due to the suppression of the class known as ultra-low volatility organic compounds (ULVOCs) within the Volatility Basis Set (VBS).23,24 These are, nominally, the “nucleators.” Isoprene oxidation products can suppress the covalently bound C20 dimers that form from α-pinene oxidation chemistry.21 ULVOCs govern nucleation,25 but only a subset of the C20 dimers in monoterpene oxidation even extend into this range.23 While isoprene oxidation does lead to condensible, low-volatility products that can contribute to secondary organic aerosol mass, few if any of these products reach the ULVOC range.26–29 The proposed coupling in this case is direct via the oxidation mechanisms rather than indirect via oxidation rates. Specifically, peroxy radicals derived from isoprene can react with peroxy radicals derived from α-pinene, generating C15 heterodimers at the expense of C20 dimers in proportion to the relative abundance (really oxidation rate) of isoprene.
The C20 (and C18 and C19) dimers have been measured in chamber experiments on α-pinene ozonolysis. While particle-phase reaction mechanisms to produce these species have been proposed and experimental evidence for these mechanisms has been observed, direct measurement of gas-phase dimer species confirms a gas-phase mechanism to generate them.30–35 The proposed gas-phase mechanism for dimer formation involves the reaction of two peroxy radicals forming a tetroxide intermediate that decomposes to form the dimer species.36 The rate coefficients for these reactions are highly dependent on the structures of the reacting peroxy radical species and are faster when the peroxy radicals are highly oxidized due to the presence of electron-withdrawing groups near the peroxy moiety.37–39 Based on direct volatility measurements, some dimers have volatilities in the range of ULVOCs and extremely low volatility organic compounds (ELVOCs) and thus can play a role in nucleation and growth.40–43 While most studies that have measured dimer species have focused on a single precursor, there is experimental evidence of precursor-cross-product dimer formation in systems with multiple precursors.21,44 Measurements by Heinritzi et al.21 also link the suppression of nucleation to the suppression of the C20 dimers, confirming that the nucleating species of α-pinene oxidation are in fact the C20 dimers.
Here we shall explore the interactions between monoterpene and isoprene oxidation in a model employing the radical two-dimensional Volatility Basis Set (radical-VBS), which has been described in Schervish and Donahue.23 The radical-VBS extends earlier VBS development24,45 by explicitly treating peroxy-radical (RO2) formation from organic precursors within a gas-phase photo-oxidation mechanism. The RO2 chemistry includes autoxidation reactions, in which RO2 undergo internal H-atom transfers and subsequently re-form RO2 species with additional oxygen-containing functional groups (largely –OOH). In the radical-VBS, RO2 volatility is treated explicitly, and successive autoxidation steps reduce volatility by forming “OxnRO2”, where n represents the number of sequential steps of autoxidation.9 RO2 termination chemistry is treated in termination “kernels” via standard termination reactions (unimolecular termination, HO2 reaction, NO reaction, etc.). Those kernels distribute products in the 2D-VBS space (defined by volatility, c°, and oxygenation, O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C). In this way the radical-VBS can represent changing yields of condensible organic products caused by changing photochemical reaction conditions (temperature, NOx, RO2:HO2, etc.).23,46–48
:C). In this way the radical-VBS can represent changing yields of condensible organic products caused by changing photochemical reaction conditions (temperature, NOx, RO2:HO2, etc.).23,46–48
The RO2 reactions in the radical-VBS also include RO2 + R′O2 cross reactions, which introduce a rich reaction space including radical propagation (formation of RO radicals), termination (formation of carbonyls and alcohols) and dimerization (termination via formation of peroxides, here called ROOR). Because of this, the radical-VBS can treat the interactions of different organic precursors such as α-pinene and isoprene. Our objective is to explore those interactions, and also to explore the extent to which chamber experiments can be readily applied to atmospheric conditions. This is not necessarily straightforward, as the RO2 + RO2 reactions are intrinsically nonlinear, and laboratory (chamber) experiments seldom, if ever, reproduce ambient atmospheric conditions exactly.
2 The model
We represent terpene oxidation with a reduced mechanism described earlier for α-pinene;23,47 it isolates RO2 reactions and here we extend it to treat isoprene as well. Here we introduce the general chemistry scheme as well as the specific implementation for isoprene, but specific information on the α-pinene implementation can be found in Schervish and Donahue.23 The potential reaction pathways for RO2 from α-pinene and isoprene are the same.2.1 Peroxy radical chemistry
There are comprehensive atmospheric oxidation mechanisms for isoprene.49,50 However, our goal is to focus on the RO2 interactions and so we employ a highly simplified oxidation scheme that includes the key elements of RO2 isomerization and thus autoxidation, including its strong temperature dependence. We represent the isoprene chemistry in a similar way to the α-pinene chemistry described in Schervish and Donahue23 but with a few differences described here. One notable assumption in the mechanism is that only a fraction of the RO2 radicals formed from either precursor will readily undergo autoxidation. The first-generation RO2 from a precursor, “prec”, (“ap” or “ip”) are labeled “precOx0RO2” and “precRO2” for those with and without the capacity for rapid isomerization. They are otherwise identical in reactivity. Here, as in Schervish and Donahue,47 we use an αOH = 0.25 for the fraction of RO2 formed from OH oxidation of α-pinene that can autoxidize based on recent experimental findings.51,52The propensity of first-generation RO2 to isomerization via H-atom transfer will depend on the structure of the RO2. For example RO2 from α-pinene that retain the C4 ring are inflexible and recalcitrant,53 and the OO location in isopreneRO2 strongly influences subsequent isomerization.54 For this reason we split the first-generation RO2 into prec0RO2, which we assume has negligibly slow isomerization, and a fraction labeled precOx0RO2, which can isomerize. Here, for each precursor we assume this fraction is 0.25. This is uncertain but consistent with that used in Xu et al.55 for global HOM simulations and with experimental and molecular dynamic simulations on the H-shift potential of initial terpene peroxy radicals.51–53,56
We employ a highly simplified (yet still very rich) scheme designed to isolate RO2 chemistry, and here we consider only NOx free conditions. While this is representative of the chamber experiments we will simulate in this work, it should be noted that these conditions favor the autoxidation pathway and dimer formation and even trace NOx will influence the mechanisms.23,25 Both α-pinene and isoprene oxidation leads to peroxy radicals, apRO2 and ipRO2. Further, a fraction of each can produce a succession of functionalized RO2via autoxidation:
 | (R1) |
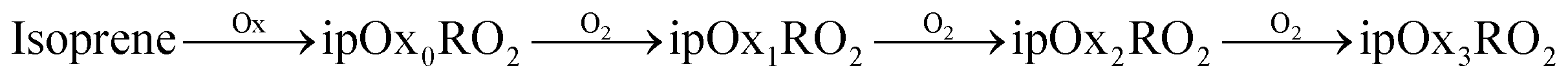 | (R2) |
Once formed, both isoprene and α-pinene RO2 can terminate via unimolecular or bimolecular reaction. The only available pathways in this NOx-free simulation are HO2 or another organic peroxy radical. Overall the scheme for any given peroxy radical is as follows:
| RO2 → monomer | (R3) |
| RO2 + HO2 → ROOH | (R4) |
| RO2 + R′O2 → ROOOOR′ | (R5) |
| ROOOOR′ → RO + R′O + O2α | (R6) |
ROOOOR′ → R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + R′OH + O2β O + R′OH + O2β | (R7) |
| ROOOOR′ → ROOR′ + O2γ | (R8) |
| RO → monomer | (R9) |
The RO2 may decompose or react with HO2, or it may react with other R′O2via a “tetroxide” intermediate. As a substantial simplification we terminate the ROx radical chemistry in reactions (R3) and (R9); this is to focus on the immediate consequences of RO2 branching, and we do not claim that it fully represents HOx–ROx chemistry. We treat three RO2 pathways – the radical, the molecular, and the dimer – with branching ratios α, β, and γ, but here we assume β = 0. As we do not treat explicit alkoxy radical (RO) chemistry, the first pathway immediately resolves into molecular products, as do all other alkoxy radicals. Therefore, in this work, the radical and molecular channels would resolve into functionally the same products. As the branching between these pathways is uncertain, future work will focus on separating these reactions and representing the rich space of alkoxy radical chemistry, especially in its ability to further propagate the radical chemistry investigated here.
Because we care about volatility, each ROOR′ product has a different volatility and thus must be represented separately. This breaks the conventional method for treating RO2 with “reactive families” (i.e. RO2 + {RiO2} → RO + O2, where {RiO2} is either the sum of all RO2 or a subset such as secondary RO2).57 Unfortunately, this means that the number of reactions increases with the square of the number of RO2 species in the mechanism, rather than linearly, and so the mechanism gets very large, very quickly. In the future we shall add treatment of NOx, the molecular RO2 reaction channel, and more sophisticated treatment of the rich RO chemistry.58,59
The precursors and all RO2 have 300 K saturation concentrations that anchor a Volatility Basis Set shown in Fig. 1, and all reaction products are distributed in the VBS according to these anchor points, as described in Schervish and Donahue.23 The molecular products are distributed according to kernels in the VBS because they are surrogates representing an ensemble of isomers and related products. When a specific reaction terminates a specific RO2, the appropriate reaction kernel is anchored to that RO2 to determine where in the 2D-VBS space the products will lie. Thus the reaction products of Ox1RO2 and Ox2RO2 with HO2 are determined via the same kernel but fall in different bins in the 2D-VBS due to the different properties of the peroxy radicals to which the kernels are anchored. Similarly, due to the differences in the reaction kernels (given in the SI of Schervish and Donahue23), an Ox3RO2 that terminates to a dimer product via reaction with another Ox3RO2 will have products at much lower volatility (in the ULVOC range) than the same peroxy radical terminating with HO2.
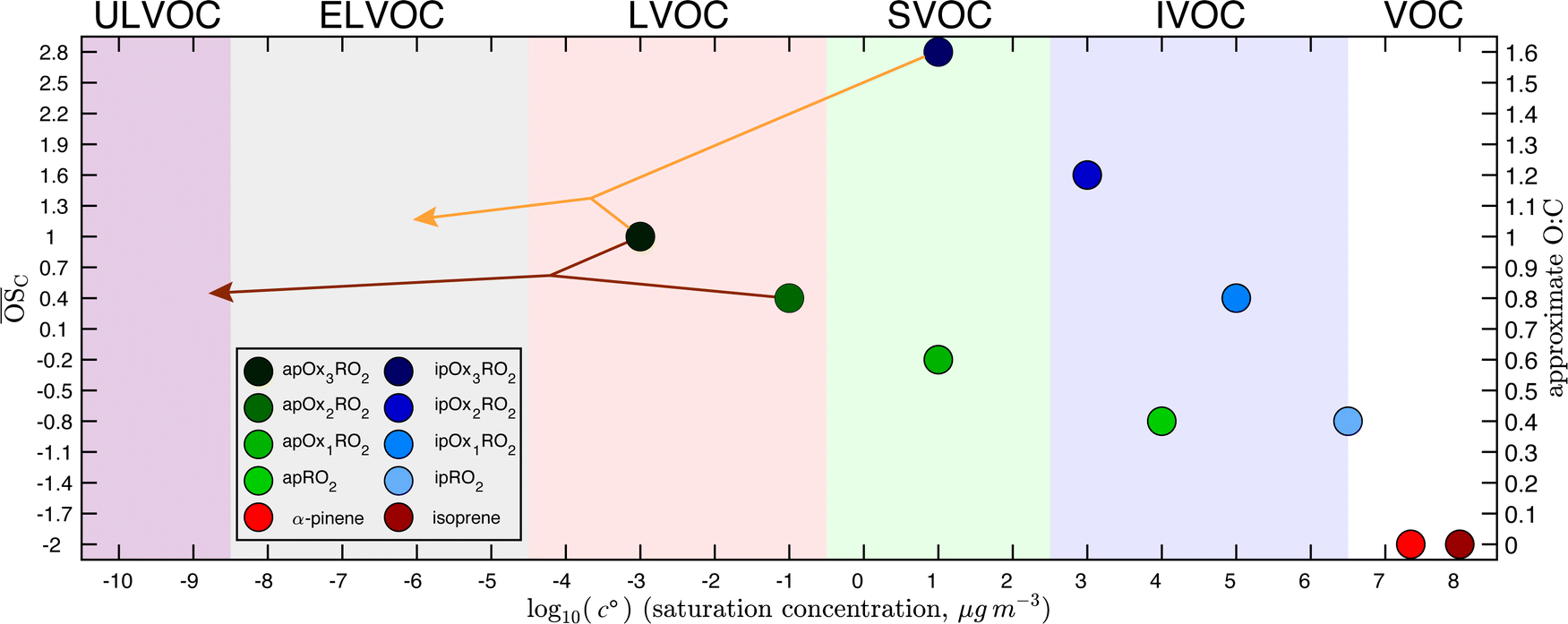 | ||
| Fig. 1 Isoprene and α-pinene peroxy radicals (RO2) in the two-dimensional volatility basis set (2D-VBS), with log c°(300 K) volatility on the x-axis and carbon oxidation state (and approximate O:C) on the y-axis. Volatility classes are indicated with background colors and labeled above the plot. Isoprene and α-pinene are shown in red. Peroxy radicals from α-pinene are shown in green with the more autoxidized peroxy radicals in progressively darker shades. Peroxy radicals from isoprene peroxy are shown in blue, again with the more autoxidized peroxy radicals in progressively darker shades. Nucleation is driven principally by ULVOC products, which in this system are formed by highly autoxidized association products from α-pinene alone. Only products in the LVOC range and lower substantially contribute to subsequent particle growth under typical atmospheric conditions, but cross reactions among the various RO2 shown here can populate much of that range, depending strongly on the specific RO2 involved. | ||
The full details of the mechanism are in the ESI.† The kinetic parameters for autoxidation and association reaction are provided in Table S1.† The complete list of isoprene reactions in the model along with their rate coefficients is provided in Table S2.† The isoprene–α-pinene cross reactions are provided in Table S3.† These reactions along with the inorganic chemistry in Table S4† and α-pinene chemistry in Table S5† comprise the total ensemble of reactions represented in this version of the model.
Here, each oxidant, Ox will react with isoprene to produce some RO2 that can undergo autoxidation as well as some OH.
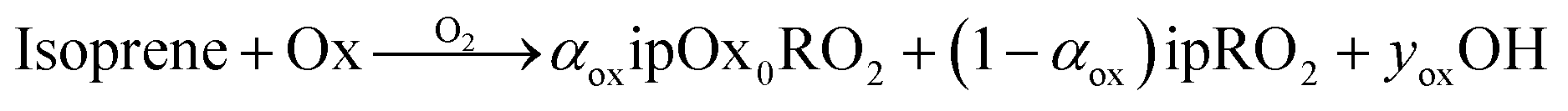
In these simulations, ozonolysis (largely of α-pinene) drives the chemistry; this produces OH radicals, inducing secondary oxidation. The rate coefficient for the reaction of OH with α-pinene is roughly half the rate coefficient for the reaction of OH with isoprene, and so as the isoprene concentration rises up to and past the α-pinene concentration with increasing isoprene input flow, isoprene becomes the dominant OH sink.
We base our scheme for autoxidation of isoprene on our scheme for α-pinene. The internal RO2 isomerization at the heart of autoxidation is an H-atom transfer, which will have an activation energy causing a strong temperature dependence. This is clear in the progressively reduced O:C and higher intrinsic volatility of α-pinene ozonolysis products observed as temperature drops in CLOUD.40,64 Here we use a slightly lower activation energy (temperature) of 7300 K for both α-pinene and isoprene autoxidation to weaken the temperature dependence, counterbalanced by higher pre-factors to achieve similar HOM concentrations and growth rates to those measured experimentally.21 This gives an H-atom transfer rate coefficient for Ox0RO2 of 0.02 s−1 at 298 K, which is very similar to that used in Schervish and Donahue.23 For isoprene, lower pre-factors cause slightly longer autoxidation lifetimes.
These parameter updates for the interactions between RO2 autoxidation and cross reactions reflect new constraints provided by observed particle formation and growth rates in lieu of detailed temperature-dependent kinetics for the elementary reactions. The higher pre-factors used in our prior work for α-pinene allow for significant amounts of ELVOC and LVOC isoprene products to form even at low isoprene concentrations, which is inconsistent with observations for pure isoprene chemistry.65,66 We present the kinetic parameters in Table S1.† As with α-pinene, the autoxidation parameters used here are uncertain; sensitivity analyses on these parameters are warranted, but experimental kinetics constraints are urgently needed.
2.2 C10 dimers and C15 heterodimers
The key species comprising ULVOC (and thus new-particle formation) are the ROOR dimers. We assume that the yield, γ, is a function of the (log) average volatility of the reacting RO2 as described in Schervish and Donahue.23 This is because dimer formation is spin forbidden and thus requires that the weakly avoided crossing between the spin states be located at a lower energy than the reactants, and the cluster formation energy of the reacting RO2 is related to their volatility.36,70,71 We thus calculate the dimer yield based on the geometric mean volatility of the reacting RO2,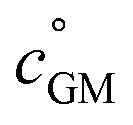 and a critical reference volatility,
and a critical reference volatility, 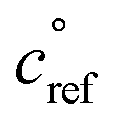 ,
,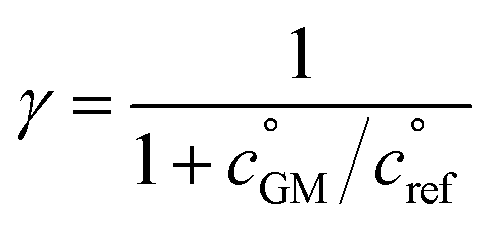 | (1) |
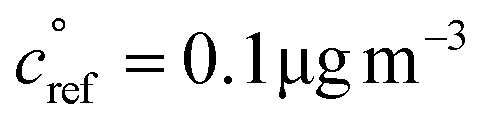 . This is informed by the results from Schervish and Donahue23 showing a stronger suppression of dimers at cold temperatures and high NOx conditions than observed in chamber studies of those conditions.25,64 We use this
. This is informed by the results from Schervish and Donahue23 showing a stronger suppression of dimers at cold temperatures and high NOx conditions than observed in chamber studies of those conditions.25,64 We use this 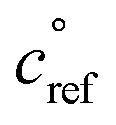 for both isoprene and α-pinene association reactions. Electron-withdrawing groups, like –OOH groups from sequential autoxidation, stabilize the tetroxide intermediate leading to dimer formation. As volatility decreases with increasing –OOH groups, a volatility dependence is appropriate, however it only indicates the likelihood the electron-withdrawing groups are close to the radical on a single size carbon backbone. Thus, in the future, a higher
for both isoprene and α-pinene association reactions. Electron-withdrawing groups, like –OOH groups from sequential autoxidation, stabilize the tetroxide intermediate leading to dimer formation. As volatility decreases with increasing –OOH groups, a volatility dependence is appropriate, however it only indicates the likelihood the electron-withdrawing groups are close to the radical on a single size carbon backbone. Thus, in the future, a higher 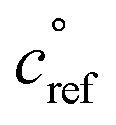 for isoprene might be appropriate. In this work, we simply use one value to avoid expanding the parameter space. We do allow for different association rate coefficients for ipRO2 and apRO2, which could capture some of this effect via higher association rate coefficient for ipRO2. However, we still find that the same rate coefficients for each similarly oxidized RO2 from isoprene and α-pinene produces the best results when compared to experimental measurements.
for isoprene might be appropriate. In this work, we simply use one value to avoid expanding the parameter space. We do allow for different association rate coefficients for ipRO2 and apRO2, which could capture some of this effect via higher association rate coefficient for ipRO2. However, we still find that the same rate coefficients for each similarly oxidized RO2 from isoprene and α-pinene produces the best results when compared to experimental measurements.
Oxidation of α-pinene produces a variety of C20 dimers and in a similar fashion isoprene chemistry produces a variety of C10 dimers. Because we represent the branching of these association reactions toward dimers as being volatility dependent, the likelihood of forming a dimer from a reaction between two isoprene peroxy radicals is lower than their similarly oxidized α-pinene counterparts. Therefore, while we expect to see C10 dimers forming in our simulations, their yields will likely be lower than the C20 dimers when similar amounts of isoprene and α-pinene are present. This is difficult to test with experimental data as current instrumentation cannot easily distinguish the formation pathway of each C10 species measured and separate products of an accretion reaction between two C5 ipRO2 from a monomer product of an apRO2.
For the rate coefficients of the RO2 cross reactions we continue to follow a scheme based on Madronich and Calvert37 as described in Schervish and Donahue.23 The individual rate coefficients are highly uncertain, and building on our earlier work we adjusted the rate coefficients of the RO2 association reactions from the base case to reproduce trends described in Heinritzi et al.;21 however, no individual rate coefficient was adjusted by more than an order of magnitude and all are within the range of measured peroxy radical association rate coefficients. We also adjusted the rate coefficients for isoprene RO2 to reproduce HOM and dimer concentrations in Heinritzi et al.21. These rate coefficients are provided in Table S1.†
2.3 Kernels
Stable RO2 products are resolved into VBS distributions using kernels that are anchored to the peroxy radical that produced them. These kernels allow us to represent a wide variety of stabilization pathways producing a wide variety of different products through one surrogate species. The variety of species that the surrogate represents are instead mapped to a distribution of products within the VBS defined by a transformation relative to the surrogate apRO2 (or ipRO2) volatility and O:C. We fully resolve the products of the RO2 chemistry described above and take the final concentration for each species and map those to a distribution of products in the 2D-VBS using the appropriate kernel for each type of product (e.g. ROOH, ROOR, etc.). The kernels are described in more detail in Schervish and Donahue23 and given in the ESI† of that work.
3 Results
To explore this chemistry using the radical-VBS we simulate a Continuously Stirred Tank Reactor (CSTR) as described in Schervish and Donahue47 with a steady flow in of organic precursors and ozone and a flushing timescale of 3 h, which is reflective of the flushing timescale of the CLOUD chamber at CERN.72 Organic precursors are added at a constant flow rate to achieve a desired steady-state concentration. Simulations are run with flows achieving steady-state α-pinene mixing ratios of approximately 700 ppt and steady-state isoprene mixing ratios between 0 and approximately 10 ppb. The temperature in all simulations in this work was fixed at 298 K.Nucleation and growth are driven by the actual concentrations of condensible vapors. Those concentrations in turn are governed by the balance of production and loss; in chambers as well as the real atmosphere, loss via condensation dominates for these vapors. In the atmosphere, the condensation sink for H2SO4 and condensible vapors varies between 1 h−1 and 1 s−1,73,74 but 10 h−1 is typical of remote locations. This corresponds to a condensation lifetime of 6 minutes, and this condensation sink consists almost entirely of accumulation mode particles.
In chambers with fairly low aerosol loading (such as CLOUD), the major loss of condensible vapors is (presumably irreversible) deposition to the chamber walls. Under typical mixing fan speeds in CLOUD, the H2SO4 deposition timescale is roughly 4 minutes.72 The organic vapors are heavier, with correspondingly lower diffusion constants, and so have deposition timescales of order 10 min.75,76 This correspondence of vapor deposition timescales between CLOUD and remote ambient conditions is a design feature, though caution is required because turbulent deposition to the chamber walls scales as 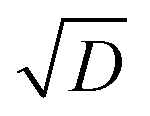 whereas laminar condensation to particles scales as D (or molecular speed in the kinetic regime).77,78 In order to compare modeled UVLOC collision frequencies and modeled size-dependent growth rates with chamber data, we therefore add a wall-loss term to our simulation of the CSTR affecting all of the closed shell products and RO2 formed from precursor oxidation. Here that is 0.0017 s−1 (a 10 min lifetime).
whereas laminar condensation to particles scales as D (or molecular speed in the kinetic regime).77,78 In order to compare modeled UVLOC collision frequencies and modeled size-dependent growth rates with chamber data, we therefore add a wall-loss term to our simulation of the CSTR affecting all of the closed shell products and RO2 formed from precursor oxidation. Here that is 0.0017 s−1 (a 10 min lifetime).
3.1 General results
Fig. 2 shows the yields from simulations with the same flow rate of α-pinene, but varying the flow rates of isoprene to achieve different steady-state concentrations. These yields are given with respect to one α-pinene carbon so the total yields increase from the bottom to the top plot as isoprene oxidation products are added in. Different classes of products are shown with different colors in the histograms, as shown in the legend. As the isoprene concentration rises, more C5 products form; most are SVOCs, IVOCs, and VOCs, consistent with the expected volatility of isoprene oxidation products. Some C10 and C15 dimers form with increased isoprene, reaching into the LVOC range of the VBS, but with low yields. As discussed previously, these low yields may be a consequence of using the same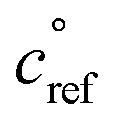 for both α-pinene and isoprene RO2, leading to less favorable C15 and C10 dimer formation than if isoprene RO2 were assigned a higher
for both α-pinene and isoprene RO2, leading to less favorable C15 and C10 dimer formation than if isoprene RO2 were assigned a higher 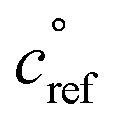 .
.
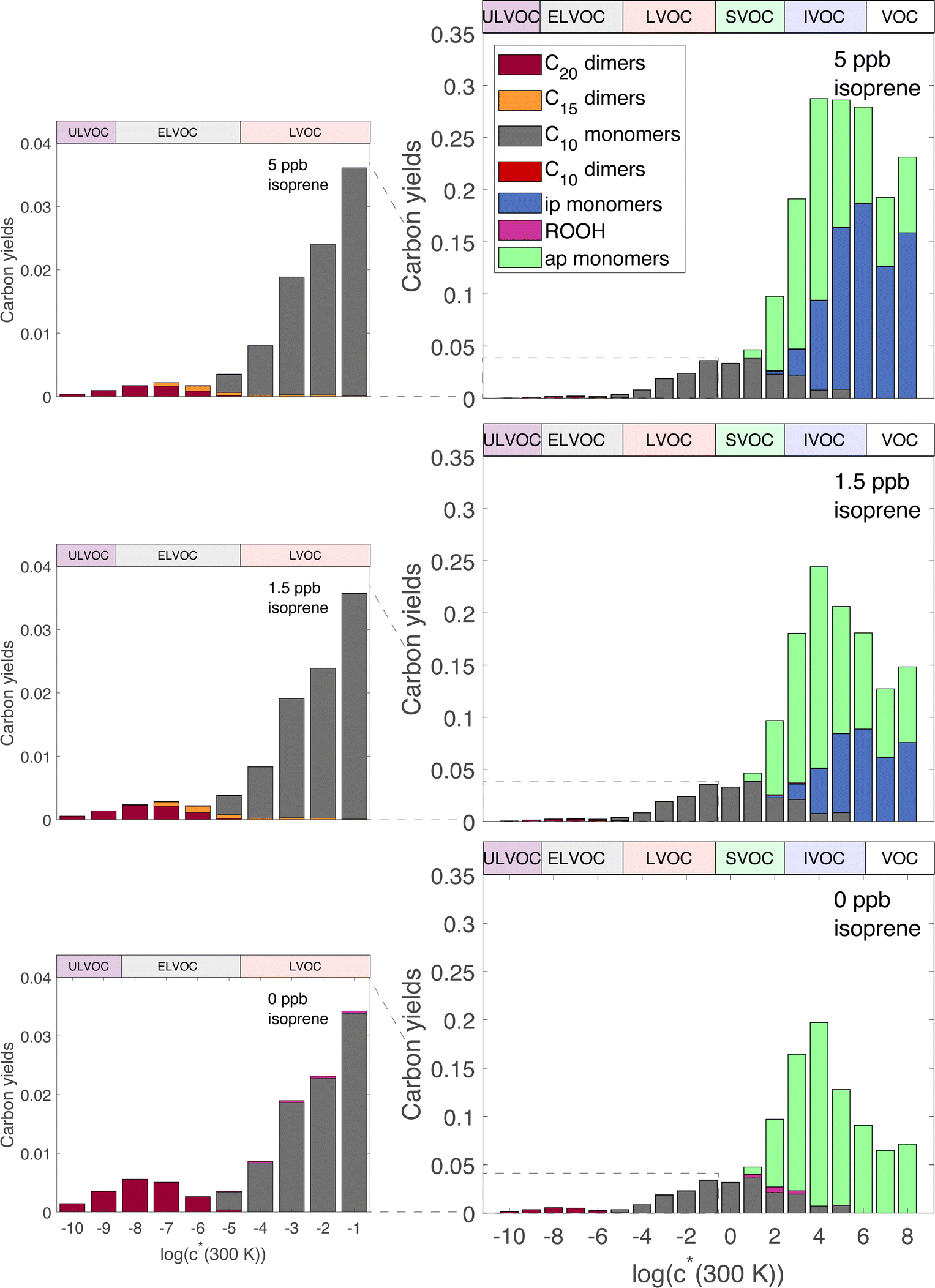 | ||
| Fig. 2 Yields (vs. oxidized α-pinene) at steady state of product classes vs. volatility for simulations of mixed α-pinene and isoprene oxidation in an oxidation chamber. Simulations are for constant α-pinene at 700 ppt with isoprene at: (bottom) 0 ppb; (middle) 1.5 ppb; and (top) 5 ppb. Left-hand panels zoom in on the low-volatility range with a maximum yield of 4%. Yields are with respect to amount of oxidized α-pinene; thus, the total yields increase with increasing isoprene, most notably in the right-hand panels. However, the low-volatility yields in the left-hand panels remain constant or decrease with increasing isoprene, despite the total yield increase. Yields are colored by product types. Products labeled “ap monomers” are the products that have not undergone autoxidation, while products labeled “C10 monomers” are HOM monomers from α-pinene that have undergone autoxidation. The C20 dimers are the only products in the ULVOC range while the C15 and C10 dimers are mostly in the ELVOC and LVOC range. | ||
The most dramatic effect of increasing isoprene is a sharp decrease in the C20 dimers. The overall effect of isoprene on these distributions is thus to decrease the ULVOC and ELVOC yields, but to mostly maintain the LVOC yields because of the added carbon from the isoprene and the shortening of the autoxidation chain due to higher levels of RO2. Additionally, total C10 decreases with increasing isoprene, leading to approximately stable LVOC yields despite decreasing LVOC concentrations (Fig. 3). This is consistent with Heinritzi et al.,21 where a reduction of approximately 40% was observed in the C10 compounds when 4.9 ppb of isoprene was added with 771 ppt of α-pinene compared to pure α-pinene experiments. As shown in Fig. S1,† this reduction in α-pinene C10 compounds is due to lowered α-pinene reactivity due to lower OH concentrations with increasing isoprene. However, as α-pinene's reactivity with ozone in this work accounts for the majority of it's reactivity, the reduction in reactivity with increasing isoprene is much smaller than if the only oxidant present were OH. The large decrease in C20 dimers, but smaller decrease of LVOCs, affects new-particle formation by suppressing nucleation and early growth, but has a smaller effect on growth of larger particles.
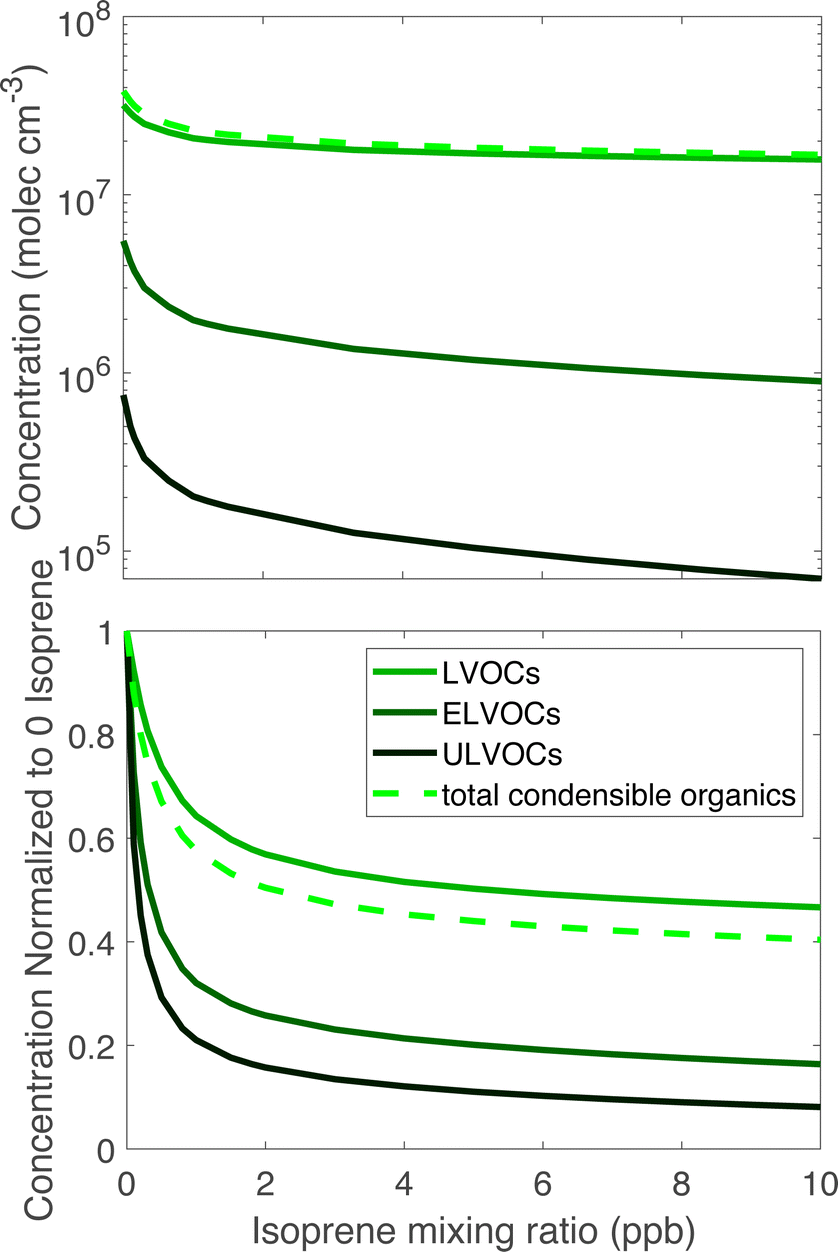 | ||
| Fig. 3 ULVOC, ELVOC, and LVOC classes vs. isoprene. The top panel shows absolute concentrations and the bottom panel shows values relative to those at zero isoprene. The sum (condensible organics) is shown with a dashed curve. The isoprene effect on the ULVOCs is greatest, the ELVOCs are also suppressed to a lesser extent, and the LVOCs are least sensitive. | ||
In these simulations very few peroxy radicals from either isoprene or α-pinene react with HO2 because the HOx concentration is low due to less efficient HOx recycling from isoprene ozonolysis. Unimolecular isoprene chemistry can recycle HOx, and CO in the chamber may also convert some OH to HO2.47 This is not included in our simplified mechanism. However even a simulation assuming the most abundant peroxy radical produced 0.7 HO2 per unimolecular reaction as recommended in Peeters et al.,67 the vast majority of products of any RO2 + HO2 end up in the SVOC to IVOC range and thus will not contribute to nucleation or growth of the smallest particles.
Fig. 3 shows ULVOC, ELVOC, and LVOC classes vs. steady-state isoprene concentrations. Taken together, we refer to these as “condensible organics”, this total is shown with the lighter dashed curve. The most obvious feature is that the ULVOCs decrease the most as isoprene rises, the ELVOCs decrease to a lesser extent, and the LVOCs decrease the least. The ULVOCs at the highest steady-state isoprene concentration here decrease to about one tenth of their value in the absence of isoprene. The ELVOCs decrease by about 80% because the C20 dimers in this range are also suppressed, but that is also the range where the most of the C15 dimers appear. The LVOCs decrease as well, but are not as dramatically affected as the ULVOCs or ELVOCs, as some C15 dimers will end up in that range, but some just displace LVOC α-pinene monomer products. The increase is also due to C10 dimers from two isoprene peroxy radicals, most of which have yields in the LVOC range, but the yields of these products are small. Because most of the condensible material is in the LVOC range, the condensible organic concentration decreases, but only by about 60%, less than the decrease of the ELVOC and ULVOC classes.
3.2 Comparison with CLOUD observations
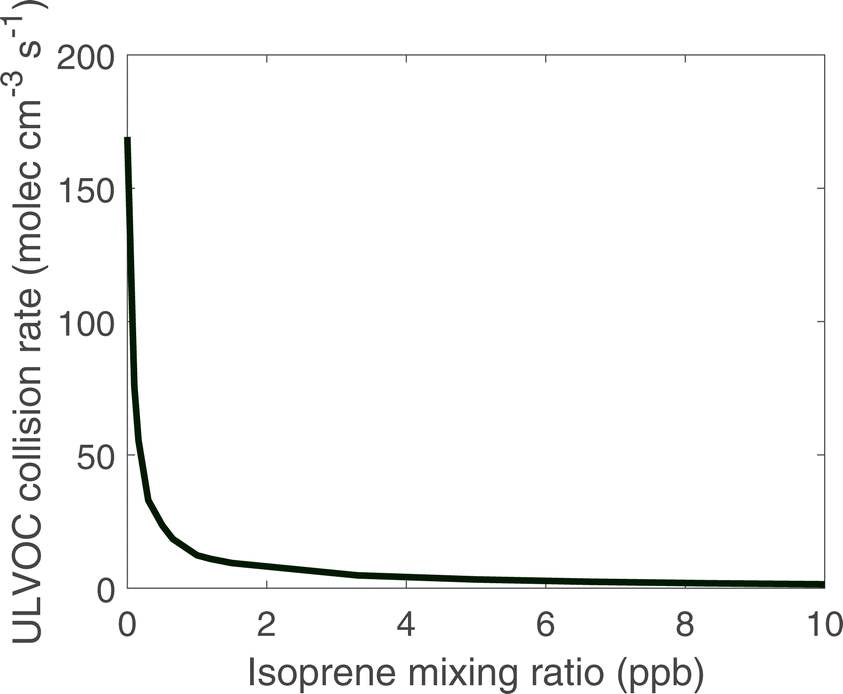 | ||
| Fig. 4 ULVOC collision rate as a function of steady-state isoprene concentration in a simulation including chamber wall losses for condensible vapors. The collision rate is kkin[ULVOC]2, where kkin is the gas-kinetic collisional rate coefficient. ULVOC collisions drop dramatically with isoprene even well below 1 ppb. | ||
We can also approximate the nucleation rate once ULVOC concentrations are known.46 While in this work we use only ULVOCs as the nucleating species, we expect that the ability of a species to participate in nucleation depends on the effective supersaturation of that species, which is a function of the saturation concentration (log c°), the gas-phase concentration of that species, and the overall abundance of less volatile species.79 This means that species in the ELVOC range may participate in nucleation when their gas-phase concentrations are sufficiently high. The actual new particle formation rates are lower than the ULVOC collision frequency; the observed nucleation rates (J1.7) for the conditions simulated here were between 1 and 10 particles cm−3 s−1 (in the presence of ions produced by galactic cosmic rays),21 indicating an average ULVOC nucleation efficiency of roughly 0.01, or that the effective nucleating vapor concentration is roughly 10% of the ULVOC concentration, with nucleation at the kinetic limit. This ∼1% ULVOC nucleation efficiency is consistent across ULVOC produced from isoprene, monoterpene, and sesquiterpene oxidation.80 However, whatever the nucleation efficiency, the added isoprene clearly reduces the ULVOC collision frequency and will consequently reduce the nucleation rate proportionately.7
Our model simulations thus confirm that adding isoprene to a system driven by α-pinene ozonolysis will significantly suppress the new-particle formation rate but for the most part preserve or even enhance growth rates. This is exactly what is observed. Future work will include a parameterization of the nucleation efficiency based on the geometric mean of a cluster (i.e. the average log c° of a pair of ULVOCs in the VBS); this presumably rises from a very low value outside of the ULVOC range to near unity deep into that range.
In Fig. 5, we show the growth rates normalized to the growth rate with no isoprene present vs. isoprene at dp = 1.6, 2.5, 5.6, and 10 nm. We show these values as they are the middle of the range reported in Heinritzi et al.21. The growth rates simulated here range from approximately 15 to 3 nm h−1 which is comparable to the approximately 20 to 2 nm h−1 seen in Heinritzi et al.21. The relative reduction from 0 to ∼4 ppb of isoprene is also similar for all sizes, but we do not present a direct comparison as the measured α-pinene and isoprene mixing ratios were not constant at every size where a growth rate was measured. All simulated cases had a steady-state concentration of approximately 1100 ppt of α-pinene. Growth is suppressed at all (physical) diameters between 1.6 and 10 nm, consistent with the CLOUD observations reported by Heinritzi et al.21. However, the suppression is greatest at the smallest sizes (about a factor of 3 from no isoprene to 4 ppb of isoprene) and weaker at the larger sizes (about a factor of 2 at 10 nm) all due to the reduction in ULVOC and LVOC concentrations with increasing isoprene. The growth rates we simulate here as well as the reduction effect by isoprene are similar to those observed in CLOUD.21
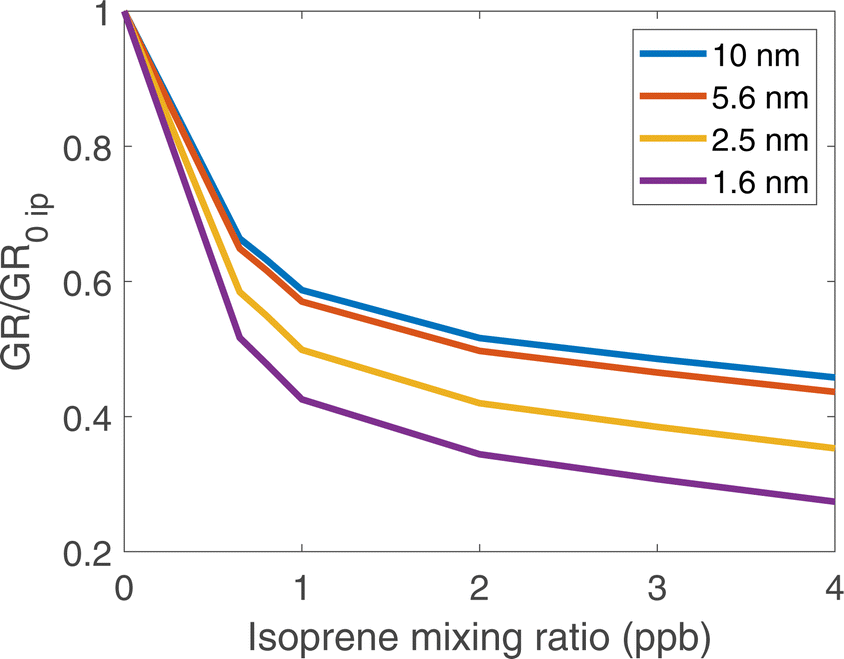 | ||
| Fig. 5 Growth rates (normalized to the growth rate with no isoprene present) vs. isoprene at different particle diameters for ozonolysis of (constant) α-pinene and isoprene. Added isoprene suppresses particle growth for all sizes below 10 nm but most dramatically at the smallest sizes. | ||
A direct comparison with the observations described in Heinritzi et al.21 is the formation rate (fluxes) at any given particle size. Formally, Jdp = sG(dn/ddp), the growth rate multiplied by the size distribution, and it reflects the overall formation rate affected by particle losses, including condensation (including wall loss) and coagulation, once the system is at steady state. The formation rate thus inevitably declines with size (at steady state) and reflects the survival probability.78 Here the condensation sink was much smaller than the wall loss frequency, and so wall losses dominate.
Fig. 6 shows the formation rates of particles from 1.7 to 7 nm for different α-pinene and isoprene flow rates chosen to match steady-state concentrations from Heinritzi et al.21 at 298 K. Nucleation is
| J1.7 = knuc[ULVOC]2; knuc = 0.01kkin | (2) |
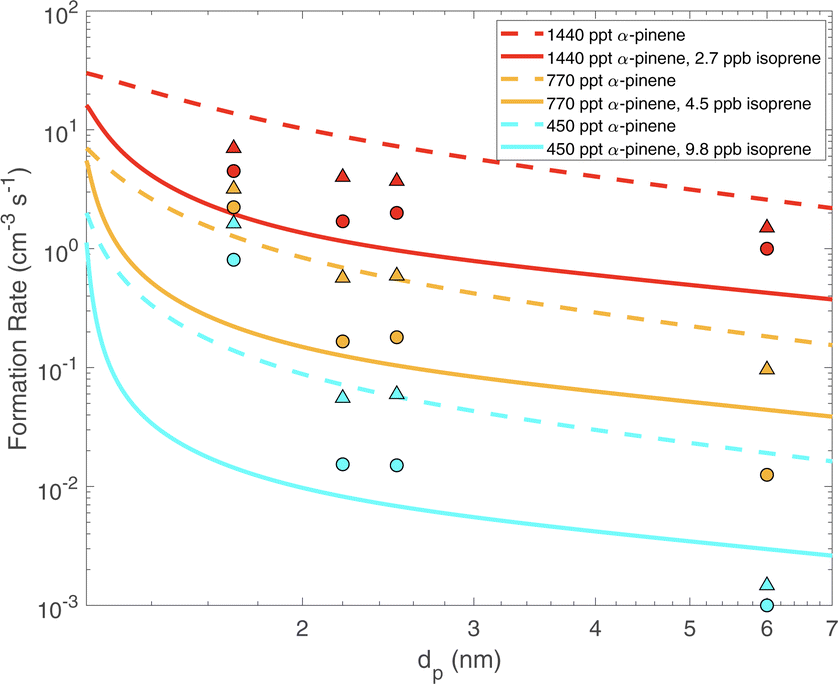 | ||
| Fig. 6 Formation rates of particles (flux across a given size cut) vs. diameter (dp) for different α-pinene and isoprene levels. Steady state α-pinene was 450, 770 and 1440 ppt for the three colored sets of curves, without (dashed) and with (3–10 ppbv, solid) isoprene. Symbols are data from Heinritzi et al.21 without (circles) and with (3–10 ppbv, triangles) isoprene. Isoprene suppresses formation at all (very small) sizes but this is most pronounced for dp ≲ 2.5 nm. | ||
Specifically, three different α-pinene flows give concentrations of 450, 770 and 1440 ppt, with zero isoprene shown as dashed curves and high isoprene as solid curves. Symbols are experimental measurements from Heinritzi et al.21. A strong reduction in the formation rate can be seen for dp ≲ 2.5 nm with a more gradual decline at larger sizes. This is due to the stronger suppression of ULVOCs by isoprene; these contribute most to growth at smaller sizes, whereas at larger sizes higher volatility classes, which are less affected by isoprene, can contribute to growth. The model broadly agrees with the observations from CLOUD.21
4 Conclusion
We have explored the effect of isoprene on the products of α-pinene ozonolysis, specifically investigating the effect on the species that participate in nucleation and growth, the ULVOCs and E/LVOCs respectively. In this work, we use a relatively simple scheme for RO2 chemistry to investigate the effect of isoprene on the products of α-pinene ozonolysis. In the future, we will explore a more detailed model to simulate mixtures of additional precursors, including a richer alkoxy radical (RO) scheme and more detailed RO2 association reaction representations, as well as constraining the wide host of uncertain parameters discussed in this work.ULVOCs are suppressed with increasing isoprene, due to the increased competition for C10 peroxy radicals by C5 peroxy radicals. ELVOCs are also suppressed but to a lesser extent because the C20 dimers in this range are reduced with increasing isoprene concentration, but this is compensated for somewhat as many of the C15 dimers appear in this range, depending on their yields. The LVOCs are less affected than ULVOCs or ELVOCs by isoprene because C15 and C10 dimers are both formed in these volatility ranges, but some of these only replace C10 monomers. While total C10 products decrease, likely in part due to competition for oxidants, the decrease in C20 dimers, the nucleating species, is stronger. This indicates that their decrease during ozonolysis experiments is not solely caused by competition for OH.
We model the dependency on growth rate of 1–10 nm particles and compare to Heinritzi et al.21. For plausible kinetics and dimer yields, we found a reduction in growth rates at all particle sizes, but this reduction is most prominent at the smallest sizes, which agrees with experimental results and theoretical arguments that only ULVOCs and ELVOCs can contribute to growth of the smallest particles.
The stoichiometric yields of these nucleating species are very low, in the per mil range, and our model confirms that these yields are very likely sensitive to the full RO2 branching chemistry including formation of covalently bound dimers. This means that the full ensemble of organic compounds being oxidized in a given air mass (or experiment) may interact directly with each other, and not just through their influence on the underlying oxidant and inorganic radical chemistry.
Data availability
The data unique to this work are the rate coefficients and reaction sequence contained in the ESI.†Author contributions
MS and NMD conceived the project and designed the overall scope. MS implemented and executed the radical VBS model. MS and NMD wrote the manuscript. FB, LD, SB, JD, IEH, HF, XCH, MH, AK, CK, JK, MK, KL, BL, VM, BM, UM, WN, TP, LQ, DS, RV, AW, MW, PW, CY and QY prepared the CLOUD facility and instruments and engaged in scientific discussion regarding the paper.Conflicts of interest
The authors declare no conflicts.Acknowledgements
This work is dedicated to the memory of Astrid Kiendler-Scharr. It was supported by: Grant CHE2336463 from the U.S. National Science Foundation; the Vienna Science and Technology Fund (WWF) through project VRG22-003; Grant number PZ00P2_216181 from the Swiss National Science Foundation Ambizone Scheme; Grant number 200021_213071 from the Swiss National Foundation; and the European Research Council project NANODYNAMITE (ERC 616075).Notes and references
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- A. B. Jardine, K. J. Jardine, J. D. Fuentes, S. T. Martin, G. Martins, F. Durgante, V. Carneiro, N. Higuchi, A. O. Manzi and J. Q. Chambers, Geophys. Res. Lett., 2015, 42, 1576–1583 CrossRef CAS.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- S. Lee, J. Uin, A. Guenther, J. de Gouw, F. Yu, A. Nadykto, J. Herb, N. Ng, A. Koss, W. Brune, K. Baumann, V. Kanawade, F. Keutsch, A. Nenes, K. Olsen, A. Goldstein and Q. Ouyang, J. Geophys. Res.: Atmos., 2016, 121(14), 621–635 Search PubMed.
- S. Martin, M. Andreae, P. Artaxo, D. Baumgardner, Q. Chen, A. Goldstein, A. Guenther, C. Heald, O. Mayol-Bracero, P. McMurry, T. Pauliquevis, U. Poeschl, K. Prather, G. Roberts, S. Saleska, M. Silva Dias, D. Spracklen, E. Swietlicki and I. Trebs, Rev. Geophys., 2010, 48, RG2002 CrossRef.
- F. Riccobono, S. Schobesberger, C. E. Scott, J. Dommen, I. K. Ortega, L. Rondo, J. Almeida, A. Amorim, F. Bianchi, M. Breitenlechner, A. David, A. Downard, E. Dunne, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, A. Hansel, H. Junninen, M. Kajos, H. Keskinen, A. Kupc, O. Kupiainen, A. Kürten, T. Kurtén, A. N. Kvashin, A. Laaksonen, K. Lehtipalo, V. Makhmutov, S. Mathot, T. Nieminen, T. Olenius, A. Onnela, T. Petäjä, A. P. Praplan, F. D. Santos, S. Schallhart, J. H. Seinfeld, M. Sipilä, D. V. Spracklen, Y. Stozhkov, F. Stratmann, A. Tomé, G. Tsagkogeorgas, P. Vaattovaara, H. Vehkamäki, Y. Viisanen, A. Vrtala, P. E. Wagner, E. Weingartner, H. Wex, D. Wimmer, K. S. Carslaw, J. Curtius, N. M. Donahue, J. Kirkby, M. Kulmala, D. R. Worsnop and U. Baltensperger, Science, 2014, 344, 717–721 CrossRef CAS PubMed.
- J. Kirkby, J. Duplissy, K. Sengupta, C. Frege, H. Gordon, C. Williamson, M. Heinritzi, M. Simon, C. Yan, J. Almeida, J. Tröstl, T. Nieminen, I. K. Ortega, R. Wagner, A. Adamov, A. Amorim, A.-K. Bernhammer, F. Bianchi, M. Breitenlechner, S. Brilke, X. Chen, J. Craven, A. Dias, S. Ehrhart, R. C. Flagan, A. Franchin, C. Fuchs, R. Guida, J. Hakala, C. R. Hoyle, T. Jokinen, H. Junninen, J. Kangasluoma, J. Kim, M. Krapf, A. Kürten, A. Laaksonen, K. Lehtipalo, V. Makhmutov, S. Mathot, U. Molteni, A. Onnela, O. Peräkylä, F. Piel, T. Petäjä, A. P. Praplan, K. Pringle, A. Rap, N. A. Richards, I. Riipinen, M. P. Rissanen, L. Rondo, N. Sarnela, S. Schobesberger, C. E. Scott, J. H. Seinfeld, M. Sipilä, G. Steiner, Y. Stozhkov, F. Stratmann, A. Tomé, A. Virtanen, A. L. Vogel, A. Wagner, P. E. Wagner, E. Weingartner, D. Wimmer, P. M. Winkler, P. Ye, X. Zhang, A. Hansel, J. Dommen, N. M. Donahue, D. R. Worsnop, U. Baltensperger, M. Kulmala, K. S. Carslaw and J. Curtius, Nature, 2016, 530, 521–526 CrossRef PubMed.
- J. Tröstl, W. K. Chuang, M. Heinritzi, C. Yan, U. Molteni, L. Ahlm, C. Frege, F. Bianchi, R. Wagner, M. Simon, K. Lehtipalo, C. Williamson, J. S. Craven, J. Duplissy, A. Adamov, J. Almeida, A.-K. Bernhammer, M. Breitenlechner, S. Brilke, A. Dias, S. Ehrhart, R. C. Flagan, A. Franchin, C. Fuchs, H. Gordon, R. Guida, M. Gysel, A. Hansel, C. R. Hoyle, T. Jokinen, H. Junninen, J. Kangasluoma, H. Keskinen, J. Kim, M. Krapf, A. Kürten, A. Laaksonen, M. Lawler, M. Leiminger, S. Mathot, O. Möhler, T. Nieminen, A. Onnela, T. Petäjä, F. M. Piel, P. Miettinen, M. P. Rissanen, L. Rondo, N. Sarnela, S. Schobesberger, K. Sengupta, M. Sipilä, J. N. Smith, G. Steiner, A. Tomè, A. Virtanen, A. C. Wagner, E. Weingartner, D. Wimmer, P. M. Winkler, P. Ye, K. S. Carslaw, J. Curtius, J. Dommen, J. Kirkby, M. Kulmala, I. Riipinen, D. R. Worsnop, N. M. Donahue and U. Baltensperger, Nature, 2016, 530, 527–531 CrossRef PubMed.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. Rissanen, R. Pontus, T. Berndt, J. Crounse, P. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. Worsnop, J. Thornton, N. M. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- D. Stolzenburg, L. Fischer, A. L. Vogel, M. Heinritzi, M. Schervish, M. Simon, A. C. Wagner, L. Dada, L. R. Ahonen, A. Amorim, A. Baccarini, P. S. Bauer, B. Baumgartner, A. Bergen, F. Bianchi, M. Breitenlechner, S. Brilke, S. Buenrostro Mazon, D. Chen, A. Dias, D. C. Draper, J. Duplissy, I. El Haddad, H. Finkenzeller, C. Frege, C. Fuchs, O. Garmash, H. Gordon, X. He, J. Helm, V. Hofbauer, C. R. Hoyle, C. Kim, J. Kirkby, J. Kontkanen, A. Kürten, J. Lampilahti, M. Lawler, K. Lehtipalo, M. Leiminger, H. Mai, S. Mathot, B. Mentler, U. Molteni, W. Nie, T. Nieminen, J. B. Nowak, A. Ojdanic, A. Onnela, M. Passananti, T. Petäjä, L. L. J. Quéléver, M. P. Rissanen, N. Sarnela, S. Schallhart, C. Tauber, A. Tomé, R. Wagner, M. Wang, L. Weitz, D. Wimmer, M. Xiao, C. Yan, P. Ye, Q. Zha, U. Baltensperger, J. Curtius, J. Dommen, R. C. Flagan, M. Kulmala, J. N. Smith, D. R. Worsnop, A. Hansel, N. M. Donahue and P. M. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9122–9127 CrossRef CAS PubMed.
- H. Gordon, J. Kirkby, U. Baltensperger, F. Bianchi, M. Breitenlechner, J. Curtius, A. Dias, J. Dommen, N. M. Donahue, E. M. Dunne, J. Duplissy, S. Ehrhart, R. C. Flagan, C. Frege, C. Fuchs, A. Hansel, C. R. Hoyle, M. Kulmala, A. Kürten, K. Lehtipalo, V. Makhmutov, U. Molteni, M. P. Rissanen, Y. Stozkhov, J. Tröstl, G. Tsagkogeorgas, R. Wagner, C. Williamson, D. Wimmer, P. M. Winkler, C. Yan and K. S. Carslaw, J. Geophys. Res.: Atmos., 2017, 122, 8739–8760 CrossRef.
- B. Zhao, S. Wang, N. M. Donahue, S. H. Jathar, X. Huang, W. Wu, J. Hao and A. L. Robinson, Sci. Rep., 2016, 6, 28815 CrossRef CAS PubMed.
- T. Kurtén, K. Tiusanen, P. Roldin, M. Rissanen, J.-N. Luy, M. Boy, M. Ehn and N. Donahue, J. Phys. Chem. A, 2016, 120, 2569–2582 CrossRef PubMed.
- A. Kiendler-Scharr, J. Wildt, M. Dal Maso, T. Hohaus, E. Kleist, T. F. Mentel, R. Tillmann, R. Uerlings, U. Schurr and A. Wahner, Nature, 2009, 461, 381–384 CrossRef CAS PubMed.
- D. Wimmer, S. Buenrostro Mazon, H. E. Manninen, J. Kangasluoma, A. Franchin, T. Nieminen, J. Backman, J. Wang, C. Kuang, R. Krejci, J. Brito, F. Goncalves Morais, S. T. Martin, P. Artaxo, M. Kulmala, V.-M. Kerminen and T. Petäjä, Atmos. Chem. Phys., 2018, 18, 13245–13264 CrossRef CAS.
- V. P. Kanawade, B. T. Jobson, A. B. Guenther, M. E. Erupe, S. N. Pressley, S. N. Tripathi and S.-H. Lee, Atmos. Chem. Phys., 2011, 11, 6013–6027 CrossRef CAS.
- H. Yu, J. Ortega, J. N. Smith, A. B. Guenther, V. P. Kanawade, Y. You, Y. Liu, K. Hosman, T. Karl, R. Seco, C. Geron, S. G. Pallardy, L. Gu, J. Mikkilä and S.-H. Lee, Aerosol Sci. Technol., 2014, 48, 1285–1298 CrossRef CAS.
- A. Kiendler-Scharr, S. Andres, M. Bachner, K. Behnke, S. Broch, A. Hofzumahaus, F. Holland, E. Kleist, T. F. Mentel, F. Rubach, M. Springer, B. Steitz, R. Tillmann, A. Wahner, J.-P. Schnitzler and J. Wildt, Atmos. Chem. Phys., 2012, 12, 1021–1030 CrossRef CAS.
- G. McFiggans, T. F. Mentel, J. Wildt, I. Pullinen, S. Kang, E. Kleist, S. Schmitt, M. Springer, R. Tillmann, C. Wu, D. Zhao, M. Hallquist, C. Faxon, M. Le Breton, A. M. Hallquist, D. Simpson, R. Bergstrom, M. E. Jenkin, M. Ehn, J. A. Thornton, M. R. Alfarra, T. J. Bannan, C. J. Percival, M. Priestley, D. Topping and A. Kiendler-Scharr, Nature, 2019, 565, 587–593 CrossRef CAS PubMed.
- H. Fuchs, A. Hofzumahaus, F. Rohrer, B. Bohn, T. Brauers, H.-P. Dorn, R. Häseler, F. Holland, M. Kaminski, X. Li, K. Lu, S. Nehr, R. Tillmann, R. Wegener and A. Wahner, Nat. Geosci., 2013, 6, 1023–1026 CrossRef CAS.
- M. Heinritzi, L. Dada, M. Simon, D. Stolzenburg, A. C. Wagner, L. Fischer, L. R. Ahonen, S. Amanatidis, R. Baalbaki, A. Baccarini, P. S. Bauer, B. Baumgartner, F. Bianchi, S. Brilke, D. Chen, R. Chiu, A. Dias, J. Dommen, J. Duplissy, H. Finkenzeller, C. Frege, C. Fuchs, O. Garmash, H. Gordon, M. Granzin, I. E. Haddad, X. He, J. Helm, V. Hofbauer, C. R. Hoyle, J. Kangasluoma, T. Keber, C. Kim, A. Kürten, H. Lamkaddam, J. Lampilahti, T. M. Laurila, C. P. Lee, K. Lehtipalo, M. Leiminger, H. Mai, V. Makhmutov, H. E. Manninen, R. Marten, S. Mathot, R. L. Mauldin, B. Mentler, U. Molteni, T. Müller, W. Nie, T. Nieminen, A. Onnela, E. Partoll, M. Passananti, T. Petäjä, J. Pfeifer, V. Pospisilova, L. Quéléver, M. P. Rissanen, C. Rose, S. Schobesberger, W. Scholz, K. Scholze, M. Sipilä, G. Steiner, Y. Stozhkov, C. Tauber, Y. J. Tham, M. Vazquez-Pufleau, A. Virtanen, A. L. Vogel, R. Volkamer, R. Wagner, M. Wang, L. Weitz, D. Wimmer, M. Xiao, C. Yan, P. Ye, Q. Zha, X. Zhou, A. Amorim, U. Baltensperger, A. Hansel, M. Kulmala, A. Tomé, P. M. Winkler, D. R. Worsnop, N. M. Donahue, J. Kirkby and J. Curtius, Atmos. Chem. Phys. Discuss., 2020, 2020, 1–18 Search PubMed.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipila, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurten, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard, M. Canagaratna, M. D. Maso, T. Berndt, T. Petäjä, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- M. Schervish and N. M. Donahue, Atmos. Chem. Phys., 2020, 20, 1183–1199 CrossRef CAS.
- N. M. Donahue, S. A. Epstein, S. N. Pandis and A. L. Robinson, Atmos. Chem. Phys., 2011, 11, 3303–3318 CrossRef CAS.
- C. Yan, W. Nie, A. L. Vogel, L. Dada, K. Lehtipalo, D. Stolzenburg, R. Wagner, M. P. Rissanen, M. Xiao, L. Ahonen, L. Fischer, C. Rose, F. Bianchi, H. Gordon, M. Simon, M. Heinritzi, O. Garmash, P. Roldin, A. Dias, P. Ye, V. Hofbauer, A. Amorim, P. S. Bauer, A. Bergen, A.-K. Bernhammer, M. Breitenlechner, S. Brilke, A. Buchholz, S. B. Mazon, M. R. Canagaratna, X. Chen, A. Ding, J. Dommen, D. C. Draper, J. Duplissy, C. Frege, C. Heyn, R. Guida, J. Hakala, L. Heikkinen, C. R. Hoyle, T. Jokinen, J. Kangasluoma, J. Kirkby, J. Kontkanen, A. Kürten, M. J. Lawler, H. Mai, S. Mathot, R. L. Mauldin, U. Molteni, L. Nichman, T. Nieminen, J. Nowak, A. Ojdanic, A. Onnela, A. Pajunoja, T. Petäjä, F. Piel, L. L. J. Quéléver, N. Sarnela, S. Schallhart, K. Sengupta, M. Sipilä, A. Tomé, J. Tröstl, O. Väisänen, A. C. Wagner, A. Ylisirniö, Q. Zha, U. Baltensperger, K. S. Carslaw, J. Curtius, R. C. Flagan, A. Hansel, I. Riipinen, J. N. Smith, A. Virtanen, P. M. Winkler, N. M. Donahue, V.-M. Kerminen, M. Kulmala, M. Ehn and D. R. Worsnop, Sci. Adv., 2020, 6, eaay4945 CrossRef CAS PubMed.
- J. E. Krechmer, M. M. Coggon, P. Massoli, T. B. Nguyen, J. D. Crounse, W. Hu, D. A. Day, G. S. Tyndall, D. K. Henze, J. C. Rivera-Rios, J. B. Nowak, J. R. Kimmel, R. L. Mauldin, H. Stark, J. T. Jayne, M. Sipilä, H. Junninen, J. M. St. Clair, X. Zhang, P. A. Feiner, L. Zhang, D. O. Miller, W. H. Brune, F. N. Keutsch, P. O. Wennberg, J. H. Seinfeld, D. R. Worsnop, J. L. Jimenez and M. R. Canagaratna, Environ. Sci. Technol., 2015, 49, 10330–10339 CrossRef CAS PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- R. M. Kamens, M. W. Gery, H. E. Jeffries, M. Jackson and E. I. Cole, Int. J. Chem. Kinet., 1982, 14, 955–975 CrossRef CAS.
- M. Riva, S. H. Budisulistiorini, Z. Zhang, A. Gold, J. A. Thornton, B. J. Turpin and J. D. Surratt, Atmos. Environ., 2017, 152, 314–322 CrossRef CAS.
- C. M. Kenseth, Y. Huang, R. Zhao, N. F. Dalleska, J. C. Hethcox, B. M. Stoltz and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8301–8306 CrossRef CAS PubMed.
- C. M. Kenseth, N. J. Hafeman, S. P. Rezgui, J. Chen, Y. Huang, N. F. Dalleska, H. G. Kjaergaard, B. M. Stoltz, J. H. Seinfeld and P. O. Wennberg, Science, 2023, 382, 787–792 CrossRef CAS PubMed.
- M. Wang, L. Yao, J. Zheng, X. Wang, J. Chen, X. Yang, D. R. Worsnop, N. M. Donahue and L. Wang, Environ. Sci. Technol., 2016, 50, 5702–5710 CrossRef CAS PubMed.
- K. Kristensen, K. L. Enggrob, S. M. King, D. R. Worton, S. M. Platt, R. Mortensen, T. Rosenoern, J. D. Surratt, M. Bilde, A. H. Goldstein and M. Glasius, Atmos. Chem. Phys., 2013, 13, 3763–3776 CrossRef.
- F. Yasmeen, R. Vermeylen, R. Szmigielski, Y. Iinuma, O. Böge, H. Herrmann, W. Maenhaut and M. Claeys, Atmos. Chem. Phys., 2010, 10, 9383–9392 CrossRef CAS.
- K. Kristensen, Å. K. Watne, J. Hammes, A. Lutz, T. Petäjä, M. Hallquist, M. Bilde and M. Glasius, Environ. Sci. Technol. Lett., 2016, 3, 280–285 CrossRef CAS.
- V.-T. Salo, R. Valiev, S. Lehtola and T. Kurtén, J. Phys. Chem. A, 2022, 126, 4046–4056 CrossRef CAS PubMed.
- S. Madronich and J. G. Calvert, J. Geophys. Res.: Atmos., 1990, 95, 5697 CrossRef CAS.
- Y. Zhao, J. A. Thornton and H. O. T. Pye, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12142–12147 CrossRef CAS PubMed.
- T. Berndt, W. Scholz, B. Mentler, L. Fischer, H. Herrmann, M. Kulmala and A. Hansel, Angew. Chem., Int. Ed., 2018, 57, 3820–3824 CrossRef CAS PubMed.
- M. Simon, L. Dada, M. Heinritzi, W. Scholz, D. Stolzenburg, L. Fischer, A. C. Wagner, A. Kürten, B. Rörup, X.-C. He, J. Almeida, R. Baalbaki, A. Baccarini, P. S. Bauer, L. Beck, A. Bergen, F. Bianchi, S. Bräkling, S. Brilke, L. Caudillo, D. Chen, B. Chu, A. Dias, D. C. Draper, J. Duplissy, I. El Haddad, H. Finkenzeller, C. Frege, L. Gonzalez-Carracedo, H. Gordon, M. Granzin, J. Hakala, V. Hofbauer, C. R. Hoyle, C. Kim, W. Kong, H. Lamkaddam, C. P. Lee, K. Lehtipalo, M. Leiminger, H. Mai, H. E. Manninen, G. Marie, R. Marten, B. Mentler, U. Molteni, L. Nichman, W. Nie, A. Ojdanic, A. Onnela, E. Partoll, T. Petäjä, J. Pfeifer, M. Philippov, L. L. J. Quéléver, A. Ranjithkumar, M. Rissanen, S. Schallhart, S. Schobesberger, S. Schuchmann, J. Shen, M. Sipilä, G. Steiner, Y. Stozhkov, C. Tauber, Y. J. Tham, A. R. Tomé, M. Vazquez-Pufleau, A. Vogel, R. Wagner, M. Wang, D. S. Wang, Y. Wang, S. K. Weber, Y. Wu, M. Xiao, C. Yan, P. Ye, Q. Ye, M. Zauner-Wieczorek, X. Zhou, U. Baltensperger, J. Dommen, R. C. Flagan, A. Hansel, M. Kulmala, R. Volkamer, P. M. Winkler, D. R. Worsnop, N. M. Donahue, J. Kirkby and J. Curtius, Atmos. Chem. Phys. Discuss., 2020, 2020, 1–42 Search PubMed.
- K. Lehtipalo, C. Yan, L. Dada, F. Bianchi, M. Xiao, R. Wagner, D. Stolzenburg, L. R. Ahonen, A. Amorim, A. Baccarini, P. S. Bauer, B. Baumgartner, A. Bergen, A.-K. Bernhammer, M. Breitenlechner, S. Brilke, A. Buckholz, S. B. Mazon, D. Chen, X. Chen, A. Dias, J. Dommen, D. C. Draper, J. Duplissy, M. Ehn, H. Finkenzeller, L. Fisher, C. Frege, C. Fuchs, O. Garmash, H. Gordon, J. Hakala, X. C. He, L. Heikkinen, M. Heinrizi, J. C. Helm, V. Hofbauer, C. R. Hoyle, T. Jokinen, J. Kangasluoma, V.-M. Kerminen, C. Kim, J. Kirkby, J. Kontkanen, A. Kürten, M. J. Lawler, H. Mai1, S. Mathot, R. L. Mauldin III, U. Molteni, L. Nichman, W. Nie, T. Nieminen, A. Ojdanic, A. Onnela1, M. Passananti, T. Petäjä, F. Piel, V. Pospisilova, L. L. J. Quéléver, M. P. Rissanen, C. Rose, N. Sarnela, S. Schallhart, K. Sengupta, M. Simon, C. Tauber, A. Tomé, J. Tröst, O. Väisänen, A. L. Voge, R. Volkamer, A. C. Wagner, M. Wang, L. Weitz, D. Wimmer, P. Ye, A. Ylisirniö, Q. Zha, K. Carslaw, J. Curtius, N. Donahue, R. C. Flagan, A. Hansel, I. Riipinen, A. Virtanen, P. M. Winkler, U. Baltensperger, M. Kulmala and D. R. Worsnop, Sci. Adv., 2018, 4, 1–9 CrossRef PubMed.
- C. Mohr, F. D. Lopez-Hilfiker, T. Yli-Juuti, A. Heitto, A. Lutz, M. Hallquist, E. L. D'Ambro, M. P. Rissanen, L. Hao, S. Schobesberger, M. Kulmala, R. L. Mauldin, U. Makkonen, M. Sipilä, T. Petäjä and J. A. Thornton, Geophys. Res. Lett., 2017, 44, 2958–2966 CrossRef CAS.
- X. Zhang, R. C. McVay, D. D. Huang, N. F. Dalleska, B. Aumont, R. C. Flagan and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14168–14173 CrossRef CAS PubMed.
- D. Thomsen, L. D. Thomsen, E. M. Iversen, T. N. Björgvinsdóttir, S. F. Vinther, J. T. Skønager, T. Hoffmann, J. Elm, M. Bilde and M. Glasius, Environ. Sci. Technol., 2022, 56, 16643–16651 CrossRef CAS PubMed.
- W. K. Chuang and N. M. Donahue, Atmos. Chem. Phys., 2016, 16, 123–134 CrossRef CAS.
- B. Zhao, M. Shrivastava, N. M. Donahue, H. Gordon, M. Schervish, J. E. Shilling, R. A. Zaveri, J. Wang, M. O. Andreae, C. Zhao, B. Gaudet, Y. Liu, J. Fan and J. D. Fast, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25344–25351 CrossRef CAS PubMed.
- M. Schervish and N. M. Donahue, Environ. Sci.: Atmos., 2021, 1, 79–92 CAS.
- B. Zhao, J. Fast, M. Shrivastava, N. M. Donahue, Y. Gao, J. E. Shilling, Y. Liu, R. A. Zaveri, B. Gaudet, S. Wang, J. Wang, Z. Li and J. Fan, Geophys. Res. Lett., 2022, 49, e2022GL100940 CrossRef.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed.
- F. Wiser, B. K. Place, S. Sen, H. O. T. Pye, B. Yang, D. M. Westervelt, D. K. Henze, A. M. Fiore and V. F. McNeill, Geosci. Model Dev., 2023, 16, 1801–1821 CrossRef CAS.
- T. Berndt, S. Richters, T. Jokinen, N. Hyttinen, T. Kurtén, R. V. Otkaer, H. G. Kjaergaard, F. Stratmann, H. Herrmann, M. Sipilä, M. Kulmala and M. Ehn, Nat. Commun., 2016, 7, 13677 CrossRef CAS PubMed.
- T. Berndt, S. Richters, R. Kaethner, J. Voigtländer, F. Stratmann, M. Sipilä, M. Kulmala and H. Herrmann, J. Phys. Chem. A, 2015, 119, 10336–10348 CrossRef CAS PubMed.
- T. Kurtén, M. P. Rissanen, K. Mackeprang, J. A. Thornton, N. Hyttinen, S. Jørgensen, M. Ehn and H. G. Kjaergaard, J. Phys. Chem. A, 2015, 119, 11366–11375 CrossRef PubMed.
- J. D. Crounse, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Phys. Chem. Chem. Phys., 2011, 13, 13607–13613 RSC.
- R. Xu, J. A. Thornton, B. H. Lee, Y. Zhang, L. Jaeglé, F. D. Lopez-Hilfiker, P. Rantala and T. Petäjä, Atmos. Chem. Phys., 2022, 22, 5477–5494 CrossRef CAS.
- S. Richters, H. Herrmann and T. Berndt, Environ. Sci. Technol., 2016, 50, 2354–2362 CrossRef CAS PubMed.
- N. M. Donahue and R. G. Prinn, J. Geophys. Res.: Atmos., 1990, 95, 18387–18411 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and I. Subcommittee, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- Y. B. Lim and P. J. Ziemann, Environ. Sci. Technol., 2009, 43, 2328–2334 CrossRef CAS PubMed.
- A. P. Teng, J. D. Crounse and P. O. Wennberg, J. Am. Chem. Soc., 2017, 139, 5367–5377 CrossRef CAS PubMed.
- J. H. Kroll, T. F. Hanisco, N. M. Donahue, K. L. Demerjian and J. G. Anderson, Geophys. Res. Lett., 2001, 28, 3863–3866 CrossRef CAS.
- T. L. Malkin, A. Goddard, D. E. Heard and P. W. Seakins, Atmos. Chem. Phys., 2010, 10, 1441–1459 CrossRef CAS.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- Q. Ye, M. Wang, V. Hofbauer, D. Stolzenburg, D. Chen, M. Schervish, A. Vogel, R. L. Mauldin, R. Baalbaki, S. Brilke, L. Dada, A. Dias, J. Duplissy, I. El Haddad, H. Finkenzeller, L. Fischer, X. He, C. Kim, A. Kürten, H. Lamkaddam, C. P. Lee, K. Lehtipalo, M. Leiminger, H. E. Manninen, R. Marten, B. Mentler, E. Partoll, T. Petäjä, M. Rissanen, S. Schobesberger, S. Schuchmann, M. Simon, Y. J. Tham, M. Vazquez-Pufleau, A. C. Wagner, Y. Wang, Y. Wu, M. Xiao, U. Baltensperger, J. Curtius, R. Flagan, J. Kirkby, M. Kulmala, R. Volkamer, P. M. Winkler, D. Worsnop and N. M. Donahue, Environ. Sci. Technol., 2019, 53, 12357–12365 CrossRef CAS PubMed.
- L. Xing, M. Shrivastava, T.-M. Fu, P. Roldin, Y. Qian, L. Xu, N. L. Ng, J. Shilling, A. Zelenyuk and C. D. Cappa, Environ. Sci. Technol., 2018, 52, 9225–9234 CrossRef CAS PubMed.
- T. Jokinen, T. Berndt, R. Makkonen, V.-M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther, D. R. Worsnop, M. Kulmala, M. Ehn and M. Sipilä, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7123–7128 CrossRef CAS PubMed.
- J. Peeters, T. L. Nguyen and L. Vereecken, Phys. Chem. Chem. Phys., 2009, 11, 5935–5939 RSC.
- T. L. Nguyen, L. Vereecken and J. Peeters, ChemPhysChem, 2010, 11, 3996–4001 CrossRef CAS PubMed.
- M. E. Jenkin, J. C. Young and A. R. Rickard, Atmos. Chem. Phys., 2015, 15, 11433–11459 CrossRef CAS.
- G. Hasan, V.-T. Salo, R. R. Valiev, J. Kubečka and T. Kurtén, J. Phys. Chem. A, 2020, 124, 8305–8320 CrossRef CAS PubMed.
- C. D. Daub, I. Zakai, R. Valiev, V.-T. Salo, R. B. Gerber and T. Kurtén, Phys. Chem. Chem. Phys., 2022, 24, 10033–10043 RSC.
- J. Duplissy, J. Merikanto, A. Franchin, G. Tsagkogeorgas, J. Kangasluoma, D. Wimmer, H. Vuollekoski, S. Schobesberger, K. Lehtipalo, R. C. Flagan, D. Brus, N. M. Donahue, H. Vehkamäki, J. Almeida, A. Amorim, P. Barmet, F. Bianchi, M. Breitenlechner, E. M. Dunne, R. Guida, H. Henschel, H. Junninen, J. Kirkby, A. Kürten, A. Kupc, A. Määttänen, V. Makhmutov, S. Mathot, T. Nieminen, A. Onnela, A. P. Praplan, F. Riccobono, L. Rondo, G. Steiner, A. Tome, H. Walther, U. Baltensperger, K. S. Carslaw, J. Dommen, A. Hansel, T. Petäjä, M. Sipilä, F. Stratmann, A. Vrtala, P. E. Wagner, D. R. Worsnop, J. Curtius and M. Kulmala, J. Geophys. Res.: Atmos., 2016, 212, 1752–1775 CrossRef.
- D. M. Westervelt, J. R. Pierce, I. Riipinen, W. Trivitayanurak, A. Hamed, M. Kulmala, A. Laaksonen, S. Decesari and P. J. Adams, Atmos. Chem. Phys., 2013, 13, 7645–7663 CrossRef.
- N. M. Donahue, L. N. Posner, D. M. Westervelt, Z. Li, M. Shrivastava, A. A. Presto, R. C. Sullivan, P. J. Adams, S. N. Pandis and A. L. Robinson, in Airborne Particulate Matter: Sources, Atmospheric Processes and Health, The Royal Society of Chemistry, 2016, pp. 35–71 Search PubMed.
- J. E. Krechmer, D. Pagonis, P. J. Ziemann and J. L. Jimenez, Environ. Sci. Technol., 2016, 50, 5757–5765 CrossRef CAS PubMed.
- P. Ye, X. Ding, J. Hakala, V. Hofbauer, E. S. Robinson and N. M. Donahue, Aerosol Sci. Technol., 2016, 50, 822–834 CrossRef CAS.
- A. Kürten, C. Williamson, J. Almeida, J. Kirkby and J. Curtius, Atmos. Chem. Phys., 2015, 15, 4063–4075 CrossRef.
- N. M. Donahue, W. Chuang and M. Schervish, ch. Gas-phase organic oxidation chemistry and atmospheric particles, in Advances in Chemistry of the Contemporary Atmosphere, ed. J. R. Barker, A. Steiner and T. J. Wallington, World Scientific, 2019, pp. 199–317 Search PubMed.
- N. M. Donahue, I. K. Ortega, W. Chuang, I. Riipinen, F. Riccobono, S. Schobesberger, J. Dommen, U. Baltensperger, M. Kulmala, D. R. Worsnop and H. Vehkamaki, Faraday Discuss., 2013, 16, 91–104 RSC.
- L. Dada, D. Stolzenburg, M. Simon, L. Fischer, M. Heinritzi, M. Wang, M. Xiao, A. L. Vogel, L. Ahonen, A. Amorim, R. Baalbaki, A. Baccarini, U. Baltensperger, F. Bianchi, K. R. Daellenbach, J. DeVivo, A. Dias, J. Dommen, J. Duplissy, H. Finkenzeller, A. Hansel, X.-C. He, V. Hofbauer, C. R. Hoyle, J. Kangasluoma, C. Kim, A. Kürten, A. Kvashnin, R. Mauldin, V. Makhmutov, R. Marten, B. Mentler, W. Nie, T. Petäjä, L. L. J. Quéléver, H. Saathoff, C. Tauber, A. Tome, U. Molteni, R. Volkamer, R. Wagner, A. C. Wagner, D. Wimmer, P. M. Winkler, C. Yan, Q. Zha, M. Rissanen, H. Gordon, J. Curtius, D. R. Worsnop, K. Lehtipalo, N. M. Donahue, J. Kirkby, I. E. Haddad and M. Kulmala, Sci. Adv., 2023, 9, eadi5297 CrossRef CAS PubMed.
- D. Stolzenburg, M. Wang, M. Schervish and N. M. Donahue, J. Aerosol Sci., 2022, 166, 106063 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00056k |
| This journal is © The Royal Society of Chemistry 2024 |