
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-promoted oxycarbonylation of unactivated alkenes†
Hefei
Yang
ab,
Yuanrui
Wang
a,
Le-Cheng
Wang
ab and
Xiao-Feng
Wu
 *ab
*ab
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut fuer Katalyse e.V., 18059 Rostock, Germany
First published on 20th August 2024
Abstract
Oxygen-centered radicals are highly reactive and have played a key role in organic transformations since their discovery. Nowadays, the direct difunctionalization of alkenes involving oxygen-centered radicals is still underdeveloped due to the inherent properties of oxygen-centered radicals, especially the intermolecular radical addition of unactivated alkenes. Herein, we report an intermolecular oxygen-centered radical addition carbonylation reaction of unactivated alkenes under visible light irradiation. The transformation was initiated with the direct addition of alkoxycarbonyloxy radicals to alkenes, which then underwent aromatic migration under the intervention of carbon monoxide to achieve the targeted oxycarbonylation products.
Broader contextOxygen-centered radicals are highly reactive and have played a key role in organic transformations since their discovery. The addition of oxygen-centered radicals to unsaturated systems is considered a convenient method for the construction of new C–O bonds. However, the carbonylative version of such transformations has not been developed due to the high reactivity of oxygen radicals. Considering the importance of CO chemistry, which can make use of cheap and abundant carbon monoxide as a C1 source for rapidly producing carbonylated compounds, we developed a new procedure for oxycarbonylation of alkenes, which simultaneously installs an oxygen-centered radical and carbon monoxide to the double bond of unactivated alkenes. Under the assistance of visible light, double functionalization of alkenes was realized and the corresponding oxygen and carbonyl substituted products were obtained in moderate to good yields. |
The efficient and selective assembly of complex structures from simple and inexpensive starting materials is attractive but challenge in synthetic organic chemistry.1 Direct difunctionalization of alkenes is one of the most efficient strategies for constructing complex molecules as alkenes are readily available and highly versatile materials.2 In addition, the use of cheap and abundant carbon monoxide as a C1 source for incorporation into organic molecules also serves as a powerful method for rapidly obtaining carbonylated compounds.3 Carbonylation of alkenes represents an important strategy in chemical synthesis since the combination of alkenes and carbon monoxide in the hydroformylation process pioneered by Otto Roelen4 and also the related Reppe process5 developed by Walter Reppe.6 The carbonylative difunctionalization of alkenes has a very significant synthetic value by adding a carbonyl group and a functional group at each end of the alkene to give β-substituted carbonylated compounds. However, although the carbonylation of alkenes is well advanced, including hydroformylation, borocarbonylation, polyfluoroalkyl carbonylation, arylcarbonylation, etc. (Scheme 1a),7 the oxycarbonylation reaction of alkenes has not been reported yet.
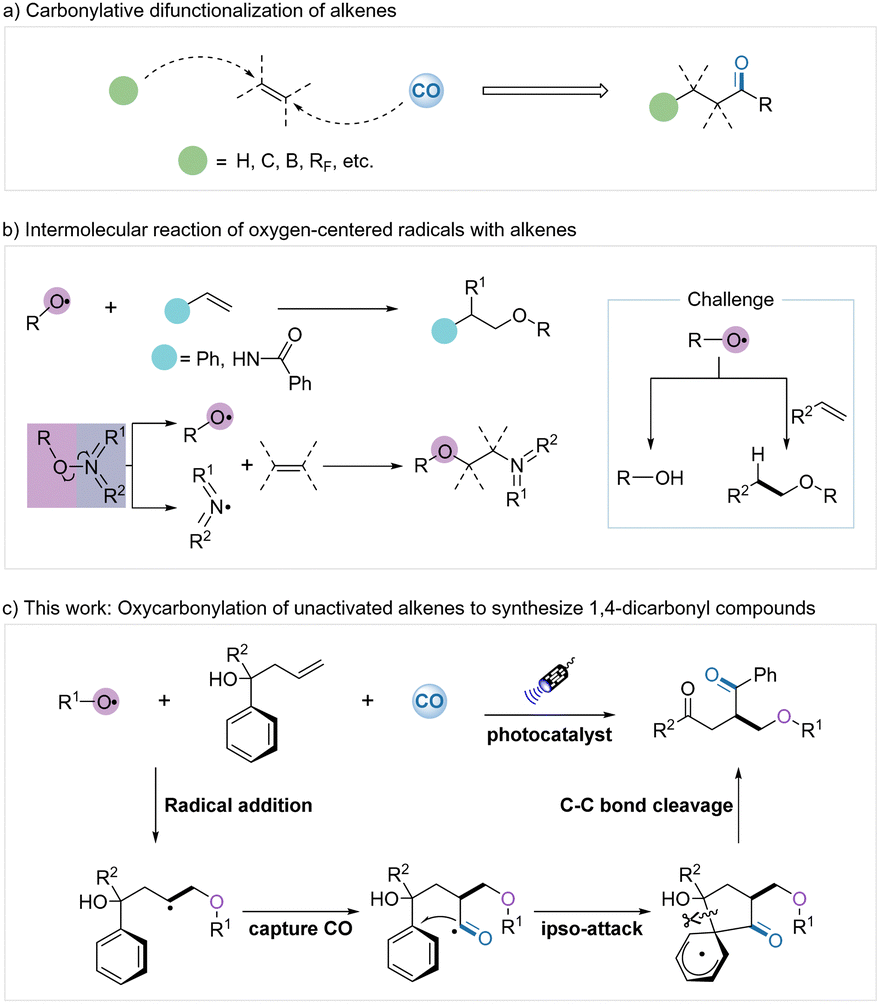 | ||
| Scheme 1 Studies on 1,2-functionalization of alkenes. | ||
Oxygen-centered radicals are highly reactive and have played a key role in organic transformations since their discovery.8 The addition of oxygen-centered radicals to unsaturated systems is considered a convenient method for the construction of new C–O bonds (Scheme 1b). Based on the initiation methods and the specific reactivity of oxygen-centered radicals, a variety of difunctionalizations of alkenes with oxygen-centered radicals were achieved during the past few decades, but mainly intramolecular radical addition.9 In contrast, intermolecular reactions of oxygen-centered radicals with alkenes have been rarely reported.10 Alexanian's group reported the direct dioxygenation of alkenes using hydroxamic acid derivatives as oxygen-centered radical precursors under a molecular oxygen atmosphere.11 Dagousset and coworkers achieved a highly efficient intermolecular alkoxy radical addition of styrenes in the presence of photocatalysis.12 Glorius, Han, et al. developed several novel powerful bifunctional oxygen–nitrogen radical precursors and successfully achieved intermolecular oxygen–nitrogen functionalization of alkenes.13 Xu and co-workers reported an intermolecular alkoxypyridylation of N-vinylbenzamide using N-alkoxypyridinium salts as the bifunctional reagents.14 Nevertheless, the merging of oxygen radical addition and carbonylation of alkenes remains a challenge.
Based on the above discussed backgrounds, we intend to overcome the challenge and directly utilize oxygen-centered radicals and carbon monoxide to achieve oxycarbonylation reaction of unactivated alkenes (Scheme 1c). First, the oxygen-centered radical undergoes an addition process with the unactivated alkene to provide a new carbon radical. The intermediate then traps the carbon monoxide to form the acyl radical and finally undergoes a migratory rearrangement to generate the oxycarbonyl compounds. However, the formation of new carbon radicals from the addition of oxygen-centered radicals to alkenes does not offer any advantage concerning its stability. Regulating the activity of oxygen-centered and carbon radicals becomes the key centrepiece of this reaction. We envisage that these difficulties could be possibly resolved by the following strategies: (1) introducing of suitable polar groups into oxygen-centered radicals to decrease their reactivity; (2) by forming a thermodynamically more favourable β-oxoalkyl radical group to avoid their proton abstraction reactions. As inspired by the pioneering works of Glorius and co-workers,13b,15 we developed a new strategy for the intermolecular oxycarbonylative transformation of alkoxycarbonyloxypyridinium salts with unactivated alkenes.
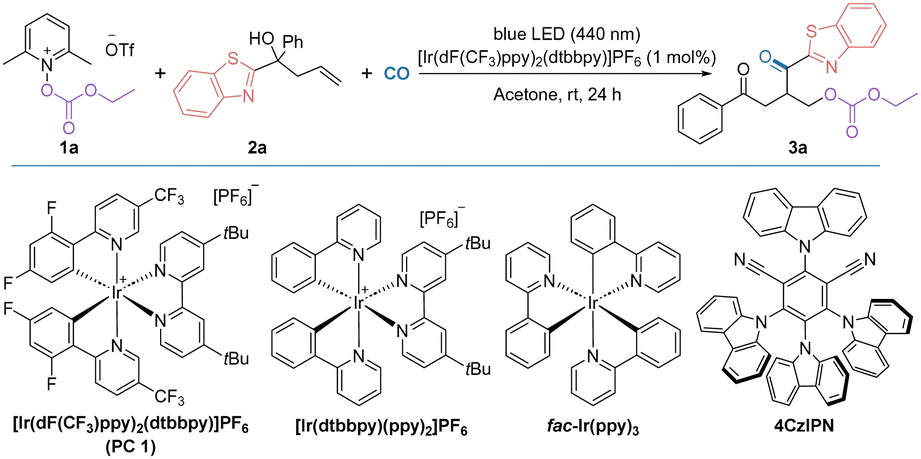
We commenced our research on this visible light-promoted oxycarbonylation by using alkoxycarbonyloxypyridinium salt 1a and unactivated alkenes containing benzothiazole 2a as the model substrates (Table 1). After extensive screening of the reaction parameters, 1 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, acetone (1 mL), 40 bar CO and irradiation under a 15 W blue light source were found to be the optimum combination for the targeted oxycarbonylation and the target product 3a was obtained in 70% isolated yield (Table 1, entry 1). A decrease in the yield of the reaction was observed by changing the solvent, indicating that acetone was still the best choice (Table 1, entries 2–4). Comparing the performance of other photocatalysts such as [Ir(dtbbpy)(ppy)2]PF6, fac-Ir(ppy)3, 4CzIPN and [Ru(bpy)3]Cl2·6H2O, it was observed that their reaction efficiencies were much lower than that of PC 1 (Table 1, entries 5–8; their redox potentials are given in ESI†). Controlled experiments have shown that both the photocatalyst and light source are critical to the success of this reaction, and that if either is absent, the target product cannot be detected (Table 1, entries 9 and 14). It is worth noting that by varying the intensity of the light source and the reaction time, the reaction can still occur, although the yield was reduced. We then tested the effect of pyridine salts with different substituents on the pyridine ring and found that substrate 1a gave the best results (Table 1, entries 12–13; for details, please refer to ESI† for more information). It's also important to mention that the other more common oxygen-centered radicals such as TEMPO and tert-butoxy radical are not working in this transformation. Due to the radical nature of this transformation, no desired reaction occurred when CO gas was replaced with CO sources, such as TFBen, Mo(CO)6, or others. The gap between the yield and conversion was mainly due to the formation of non-carbonylation by-product.
|
|
||
|---|---|---|
| Entry | Variation from the standard reaction conditions | Yield [%] |
a Reaction conditions: 1a (0.4 mmol), 2a (0.2 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (1 mol%), acetone (1 mL) with blue LEDs (15 W, 440 nm) at room temperature for 24 h under CO (40 bar). Isolated yields.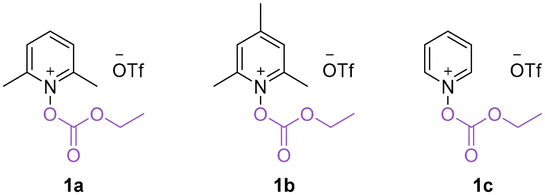
|
||
| 1 | None | 70 |
| 2 | DCM instead of acetone | 56 |
| 3 | EA instead of acetone | 47 |
| 4 | MeCN instead of acetone | 47 |
| 5 | [Ir(dtbbpy)(ppy)2]PF6 instead of PC 1 | 32 |
| 6 | fac-Ir(ppy)3 instead of PC 1 | Trace |
| 7 | 4CzIPN instead of PC 1 | 51 |
| 8 | [Ru(bpy)3]Cl2·6H2O instead of PC 1 | Trace |
| 9 | Without PC | N. D. |
| 10 | 7 W instead of 15 W | 65 |
| 11 | 30 W instead of 15 W | 60 |
| 12 | 1b instead of 1a | 51 |
| 13 | 1c instead of 1a | 20 |
| 14 | Without light | N. D. |
| 15 | 15 h | 56 |
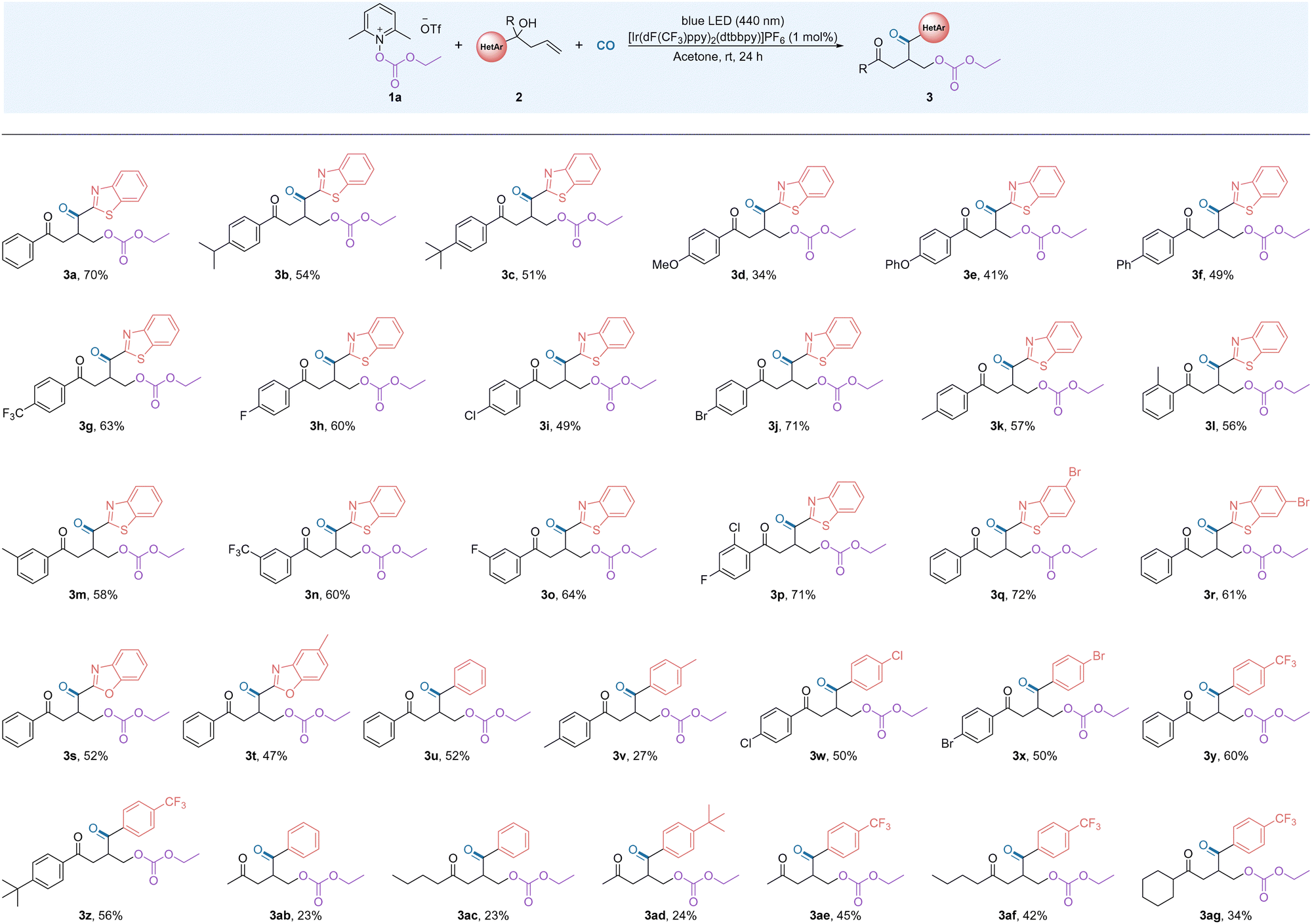
After the optimal conditions for the reaction were established, we began to study the compatibility of a series of unactivated alkenes with different functional groups using alkoxycarbonyloxypyridinium salt 1a (Table 2). In general, the reactivity of the heterocycles is higher than that of the aryl groups and migration occurs faster than for the aryl groups. In more detail, in testing the electronic and steric hindrance effects of the substrate, we found that when electron-donating groups are substituted at the para position of the aromatic ring, such as isopropyl, phenoxy and phenyl, the reaction affords the target products in moderate yields (3b–3f). The presence of electron-withdrawing groups at the para- and meta-positions of the benzene ring promotes the reaction and the corresponding difunctionalized products were obtained in good yields (3g–3j, 3n–3o). When the aryl group has an electron-withdrawing group, the reaction efficiency was better. By installing a methyl group at the ortho-, meta- and para-positions of the aromatic ring of the substrate, the corresponding products were obtained in good yields (3k–3m). This result indicates that the reaction exhibits good tolerance to the steric hindrance effect of substrates. It is worth noting that when the para position of the benzene ring is substituted by a methoxy group, the reaction can still proceed, although the yield was lower (3d). The presence of a methoxy group lead to the decomposition of the substrate and quinone can be detected here. Notably, the excellent tolerance of the halides in the reaction provides a prerequisite for further modification of the products (3h–3j). Interestingly, the aryl substrate substituted with a double electron-withdrawing group showed good compatibility and the target product was obtained in 71% yield. The reaction was also explored with the compatibility of heterocycles other than benzothiazole groups and the corresponding products were obtained in moderate to good yields (3q–3t). Alkenes substituted with the same active group participate in the reaction, for example diphenyl substitution, ditolyl substitution and dihalophenyl substitution. The reaction gave the expected product in moderate to good yields (3u–3x). In these cases, the addition of Lewis acids as a co-additive was tested and no improvement of the desired product yield could be observed. Substrates with electron withdrawing effects are more responsive and the target structure was obtained in good yields when groups with different electrical properties were attached to the diphenyl group (3y–3z). This result shows that the acyl radical, as an electron-rich group, is more likely to react with electron-poor groups. Surprisingly, after adjusting the substituent group on the arene to an alkane, the reaction proceeds as expected, although the yields of the desired products were low (3ab–3ad). However, this defect can be greatly improved by modifying the electrical properties of the phenyl group with functional groups (3ae–3ag).
| a Reaction conditions: 1a (0.4 mmol), 2a (0.2 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (1 mol%), acetone (1 mL) with blue LEDs (15 W, 440 nm) at room temperature for 24 h under CO (40 bar). Isolated yields. |
|---|
|
To gain a deeper understanding of the reaction pathway, we carried out some of the necessary control experiments, as shown in Scheme 2. Firstly, the radical scavengers TEMPO (2,2,6,6-tetramethylpiperidinyl-1-oxide), BHT (butylated hydroxytoluene) and DPE (1,1-diphenylethylene) were added to the reaction under optimum conditions and the results showed that the formation of the target products could not be observed, suggesting that the reaction has a high probability of experiencing radical intermediates (Scheme 2a). However, no radical captured intermediates could be detected either which might due to the low stability of the formed compounds. Next, the reaction also failed to take place under standard conditions without the involvement of carbon monoxide, suggesting that carbon monoxide plays an important role in the reaction (Scheme 2b). The reaction was scaled up to better demonstrate its practical applicability. To our surprise, when the amount of photocatalyst was reduced to half of the standard conditions and the reaction was scaled up to 10 times, the target product was still obtained in a good yield of 69% (Scheme 2c). The product 3a can be further converted to the thiophene derivative 4 in good yield under mild conditions (Scheme 2d). Similarly, alkoxycarbonyloxy as a precursor of alcohol can be easily realized from the substrate 3u to the corresponding alcohol. These transformations may have potential applications in future drug synthesis.
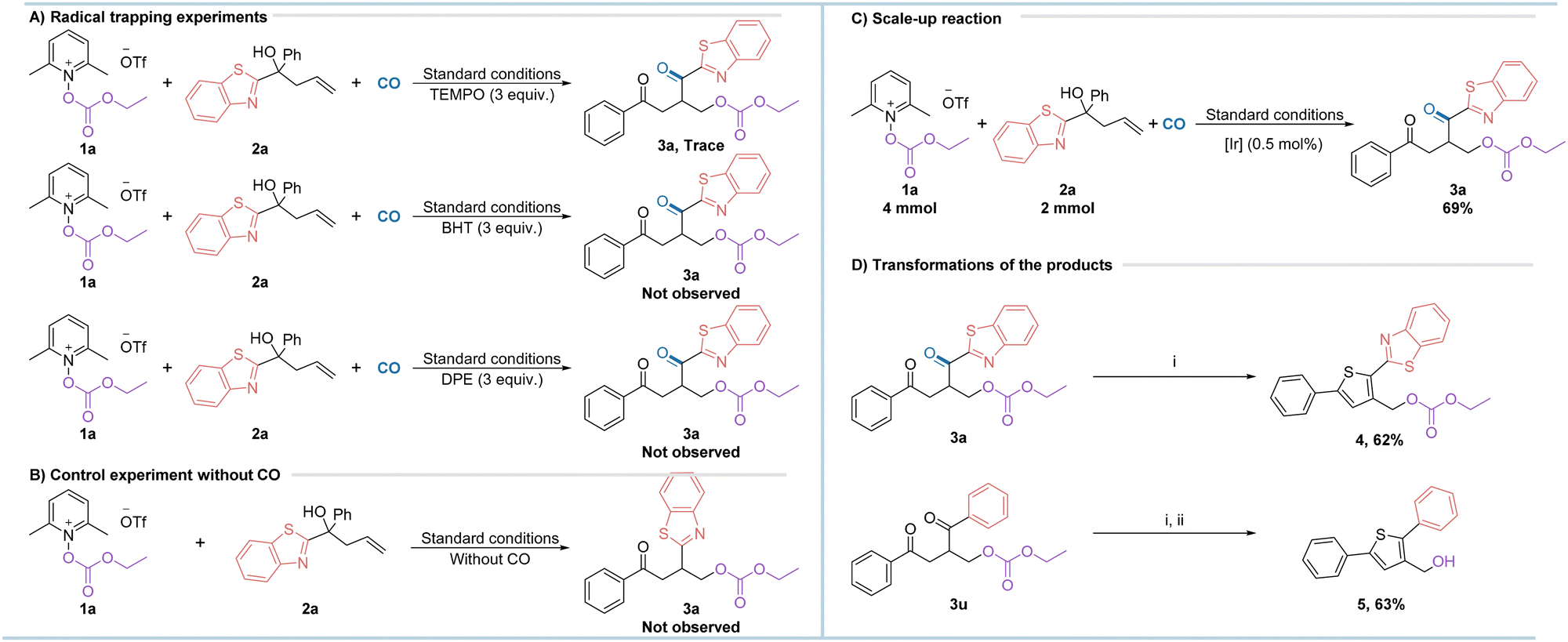 | ||
| Scheme 2 Control experiments and synthetic applications. (i) Lawesson's reagent, DCM, 40 °C, 6 h; (ii) NaOH, 80 °C, 20 h. | ||
Based on the experimental results and previous related reports,15–17 we proposed a possible pathway for this oxycarbonylation of alkenes (Scheme 3). First, the alkoxycarbonyloxypyridinium salt was reduced by the excited photocatalyst to release the alkoxycarbonyloxy radical A, which then reacted with the alkene to form a new carbon radical B. Under the pressure of carbon monoxide, the carbon radical traps the carbon monoxide to obtain the acyl radical C. Subsequent intramolecular ipso attack of the acyl radical yielded the intermediate E. The target product was finally obtained by SET oxidation and deprotonation, while the regenerated photocatalyst continues to participate in the next cycle.
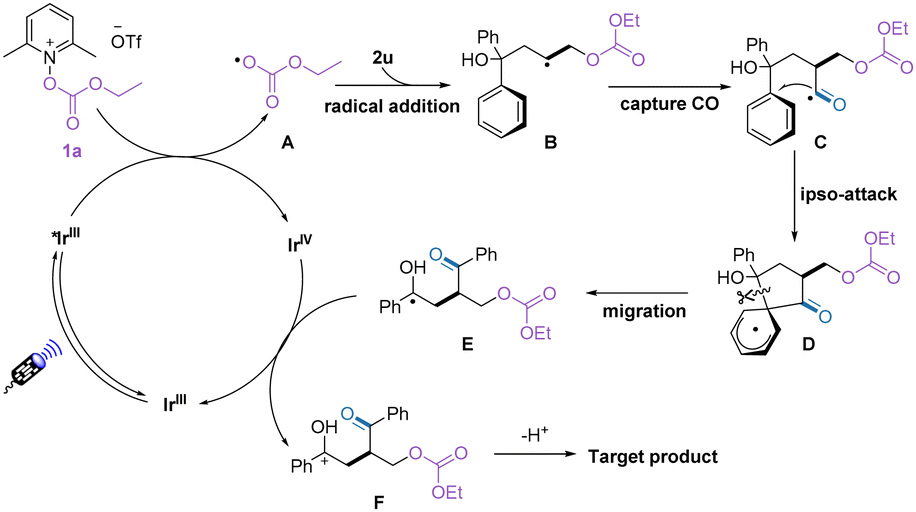 | ||
| Scheme 3 Proposed mechanism. | ||
In conclusion, we have successfully achieved the oxycarbonylation reaction of unactivated alkenes with the simultaneous participation of oxygen-centered radicals and carbon monoxide. The reaction is well tolerated by functional groups and the target products obtained can be used for the construction of bioactive related compounds. The work reported enables a new strategy for intermolecular oxygen functionalisation of alkenes, broadening their scope of application. Meanwhile, we found that carbon monoxide played an important role in the reaction, effectively avoiding the competition of carbon radical hydrogen abstraction reaction, which provided a new idea for the further exploitation of carbon monoxide.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the National Key R&D Program of China (2023YFA1507500) and the International Partnership Program of Chinese Academy of Sciences (Grant No. 028GJHZ2023045FN).References
-
(a) B. M. Trost, Science, 1983, 219, 245–250 CrossRef CAS PubMed
; (b) A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef PubMed
-
(a) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed
-
(a)
B. Gabriele, Carbon monoxide in organic synthesis-Carbonylation chemistry, Wiley-VCH, Weinheim, 2021 Search PubMed
- B. Cornils, W. A. Herrmann and M. Rasch, Angew. Chem., Int. Ed., 2003, 33, 2144–2163 CrossRef
- K. Gabor, Chem. Rev., 2001, 101, 3435–3456 CrossRef
-
(a) I. Fleischer, P. Gehrtz, V. Hirschbeck and B. Ciszek, Synthesis, 2016, 1573–1596 CrossRef
-
(a) Y. Zhang, B. H. Teng and X. F. Wu, Chem. Sci., 2024, 15, 1418–1423 RSC
-
(a) P. Gray and A. Williams, Chem. Rev., 1959, 59, 239–328 CrossRef CAS
-
(a) A. Johns and J. A. Murphy, Tetrahedron Lett., 1988, 29, 837–840 CrossRef CAS
-
(a) X.-F. Xia, Z. Gu, W. Liu, H. Wang, Y. Xia, H. Gao, X. Liu and Y.-M. Liang, J. Org. Chem., 2014, 80, 290–295 CrossRef
- B. C. Giglio, V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 13320–13322 CrossRef CAS PubMed
- A. L. Barthelemy, B. Tuccio, E. Magnier and G. Dagousset, Angew. Chem., Int. Ed., 2018, 57, 13790–13794 CrossRef CAS PubMed
-
(a) S. Q. Lai, B. Y. Wei, J. W. Wang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21997–22003 CrossRef CAS PubMed
- J. Liu, H.-W. Jiang, X.-Q. Hu and P.-F. Xu, Org. Lett., 2024, 26, 3661–3666 CrossRef CAS
- L. Quach, S. Dutta, P. M. Pflüger, F. Sandfort, P. Bellotti and F. Glorius, ACS Catal., 2022, 12, 2499–2504 CrossRef CAS
-
(a) X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS
-
(a) H. Hou, M. Luo, S. Zhai, T. Yuan, M. Zheng and S. Wang, Green Chem., 2024, 26, 1317–1321 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00149d |
| This journal is © The Royal Society of Chemistry 2024 |