DOI:
10.1039/D4EY00194J
(Communication)
EES Catal., 2024,
2, 1253-1262
Embedding the intermetallic Pt5Ce alloy in mesopores through Pt–C coordination layer interactions as a stable electrocatalyst for the oxygen reduction reaction†
Received
12th September 2024
, Accepted 27th September 2024
First published on 1st October 2024
Abstract
Platinum dissolution is one of the primary factors affecting the stability of Pt-based catalysts for the oxygen reduction reaction (ORR) in proton exchange membrane fuel cells (PEMFCs). It is a significant challenge to prevent the dissolution of Pt and enhance the durability of Pt-based catalysts. In this study, we employed a one-step rapid Joule thermal shock method to fabricate a stable ORR catalyst with embedded Pt5Ce alloy (E-Pt5Ce). The strong catalyst-support interactions between the Pt–C layer suppress particle agglomeration and Ostwald ripening, and its steric hindrance effect reduces the electronic density at Pt sites, decreasing the adsorption energy of Pt with oxygen-containing intermediates and preventing Pt dissolution. The Pt–C layer also increases the accessibility of active sites, boosting the ORR activity. In acidic media, E-Pt5Ce shows a mass activity (MA) and specific activity (SA) of 2.86 A mgPt−1 and 2.03 mA cm−2, outperforming the commercial Pt/C by factors of approximately 15 and 5, respectively. When used as a cathode catalyst for a PEMFC, the MA at 0.90 V is almost twice the DOE 2025 target. After stability testing, there is no prominent loss in catalytic activity. Density functional theory calculations confirm that the Pt–C coordination bonds also serve as reactive sites. This work uncovers the mechanism of action of the Pt–C coordination layer, which plays a crucial role in the preparation and performance of ORR catalysts.
Broader context
In acidic media, the dissolution and surface reconstruction of platinum particles directly affect the stability and activity of Pt-based catalysts, leading to a decline in the stability of proton exchange membrane fuel cells. This study presents a one-step rapid Joule thermal shock method to synthesize Pt5Ce alloy catalysts within nitrogen-doped hollow mesoporous carbon spheres. This technique directly leverages the thermal shock effect to swiftly form ordered intermetallic compounds between Pt and Ce atoms while concurrently inducing abundant Pt–C coordination layers between the catalyst and carbon support. In addition, it presents a novel approach for developing efficient, cost-effective cathode catalysts, which yields catalysts with strong catalyst–support interactions facilitated by rich Pt–C coordination layers.
|
1. Introduction
As fossil fuel reserves diminish, the urgency to find sustainable energy solutions grows, particularly in the field of energy conversion where hydrogen energy has become a hot topic of research.1 Proton exchange membrane fuel cells (PEMFCs) are at the forefront of this technology, converting hydrogen into electricity without any CO2 emissions, thus achieving clean energy production.2 The durability of fuel cells hinges on the performance of their catalysts, especially in facilitating the oxygen reduction reaction (ORR) at the cathode.3 Traditionally, platinum-based (Pt-based) catalysts are highly effective for the ORR but face challenges due to their scarcity and high cost.4 To address this issue, researchers have been actively developing catalysts that utilize less Pt while maintaining high catalytic activity.5 However, these low-platinum catalysts often struggle with durability in acidic media.6 At high potentials (>0.70 V), a thermodynamically driven exchange occurs between surface Pt atoms with adsorbed oxygen species (Oads), leading to the growth of surface oxides and irreversible restructuring of the catalyst surface.7 This process disrupts Pt–Pt metal bonds, causing significant dissolution of the outermost Pt layer.8 Therefore, decreasing the adsorption energy of Pt with Oads is crucial for alleviating Pt degradation and enhancing the durability of low-Pt catalysts.
To overcome the problem, there are two primary strategies. One is the development of extended surface catalysts, which enhances the surface area of the catalyst and modifies its topography, thereby diminishing the propensity for Pt particles to aggregate and enlarge.9 Another strategy involves modifying the electronic structure of Pt by forming Pt–transition metal (Pt–TM) alloys.10 This method aims to reduce the adsorption energy of Oads on the catalyst surface. Previous studies have shown that rare earth metal atoms (REs) have a strong regulatory effect on the electronic structure of Pt, for example, Pt5Ce alloy exhibits excellent ORR performance.11 In this study, we synthesized embedded Pt5Ce alloy (E-Pt5Ce) catalysts with rich Pt–C coordination bonds by a one-step rapid Joule thermal shock (RJTS) method. A thermodynamically stable Pt–C coordination layer was constructed to optimize the alloy-support loading. The anchoring effect of the Pt–C layer suppresses the Ostwald ripening of nanoparticles (NPs). Its steric hindrance effect modulates the local electronic structure, inhibiting Pt dissolution, weakening the interaction between Pt and Oads. Additionally, the Pt–C layer also serves as an active site for the ORR, increasing the accessibility of active sites.
The E-Pt5Ce catalyst exhibits outstanding ORR performance. In an acidic environment, E-Pt5Ce demonstrates a half-wave potential (Eh) of 0.92 V, with mass activity (MA) and specific activity (SA) of 2.86 A mgPt−1 and 2.03 mA cm−2 at 0.90 V (versus RHE scale), which are 15 and 5 times higher than that of the commercial Pt/C, respectively. It is noteworthy that E-Pt5Ce also performs excellently as a cathode catalyst for PEMFC single cells. It achieves a peak power density (PPD) of 1.42 W cm−2 at 2 A cm−2. The MA at 0.90 V is 0.83 A mgPt−1, which is almost twice the DOE 2025 target (0.44 A mgPt−1).12 After an accelerated durability test (ADT), there is virtually no loss in cell voltage at 0.80 A cm−2. This reveals the effectiveness of the Pt–C coordination layer and the intrinsic mechanisms for enhancing catalytic activity, which is of great significance for promoting the development and application of hydrogen energy technology.
2. Results and discussion
2.1. Fabrication and physical characterizations
As demonstrated in Fig. 1a, metal salts are homogeneously distributed within the carbon shell of nitrogen-doped hollow mesoporous carbon spheres (N-HMCSs), forming a precursor.13 The bottom-up approach of synthesizing N-HMCS ensures an even distribution of nitrogen groups, enhancing ionomer coverage (Fig. S1, S2 and Table S1, ESI†).14 E-Pt5Ce can be obtained within 100 s through the RJTS, which employs thermal energy to directly reduce metal ions and, under the influence of heat, facilitates the direct synthesis of alloys. The amorphous shell of N-HMCS, approximately 15 nm thick, contains numerous metal dangling bonds with unsaturated electronic configurations (Fig. 1b).15 It is beneficial for constructing a Pt–C coordination layer structure, accelerating charge transfer between active centers. The large-sized N-HMCS features an open surface, interconnected porous channels and abundant defects (Fig. S3, ESI†).16 Metal particles in the precursor aggregate only within the shell (Fig. S4, ESI†). The Pt–C coordination layer formed through the one-step RJTS process serves as an anchor, preventing the aggregation or Ostwald ripening of Pt5Ce NPs during synthesis and subsequent operations. This anchoring effect arises from the thermodynamic stability of the Pt–C coordination bonds, which provide robust interfacial interactions between the Pt5Ce alloy and the carbon support. We obtained uniform E-Pt5Ce with an average particle size of about 3.50 nm, and the catalyst maintains its internal hollow structure (Fig. 1c, d and Fig. S5–S8, ESI†). Fig. 1e displays a regular atomic arrangement structure, while Fig. 1f reveals that these nanocrystals expose (111) and (200) facets. The presence of superlattice electron diffraction spots (110) is consistent with the formation of intermetallic Pt5Ce (Fig. 1g). Detailed experimental sections can be found in the ESI.† The results clearly showcase the uniform and finely dispersed Pt5Ce alloy particles embedded within the N-HMCS shell. These particles are characterized by a highly ordered structure and ideal size, representing an optimal morphology for ORR catalysis.
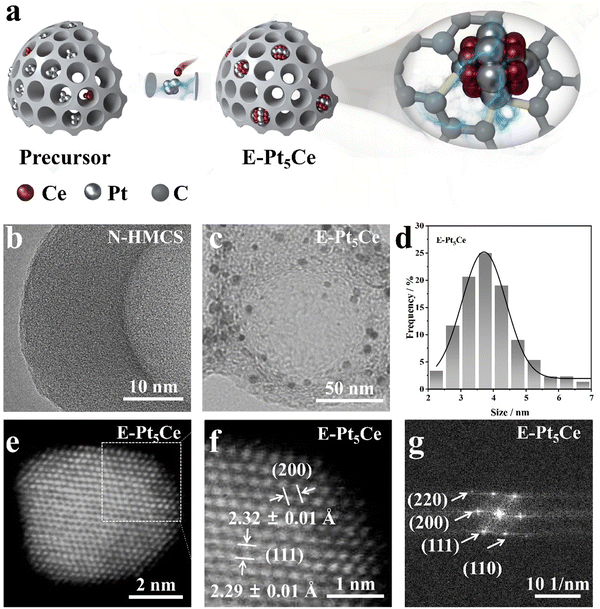 |
| | Fig. 1 (a) Schematic diagram of the synthesis route of E-Pt5Ce. Transmission electron microscopy (TEM) images of (b) N-HMCS and (c) E-Pt5Ce within N-HMCS. (d) Histograms of the statistics of the nanoparticle size distribution of E-Pt5Ce. (e) Aberration-corrected high-angle annular darkfield scanning transmission electron microscopic (AC-HAADF-STEM) image of E-Pt5Ce. (f) Partially enlarged image of (e). (g) The corresponding fast Fourier transform (FTT) pattern of (e). | |
To further investigate the effect of Pt–C coordination layer on the performance of Pt5Ce in the ORR, we prepared Pt5Ce loaded on nitrogen-doped Ketjenblack (L-Pt5Ce) as a reference sample by using the same process (Fig. S9–S12 and Table S1, ESI†).11 Upon scrutiny, it becomes evident that the alloy particles in E-Pt5Ce are fully embedded within the carbon support, whereas in L-Pt5Ce, the alloys exhibit a partial embedding, permitting a clear discernment of the lattice fringes. This observation suggests that E-Pt5Ce might harbor a higher concentration of binding sites at the interface between the alloy and the catalyst, indicating a large number of Pt–C coordination bonds. Two distinct diffraction peaks corresponding to the (002) and (101) crystal planes of amorphous carbon can be observed in Fig. 2a. The typical type-IV curves of the N2 adsorption–desorption isotherm indicate rich mesoporous structures in the carbon supports (Fig. 2b). N-HMCS, with a mesopore diameter of approximately 3.80 nm, exhibits a larger specific surface area compared to N-KB (Fig. S13 and S14, ESI†). Mesopores with diameters ranging from 2.80 to 4.10 nm, integrated with sulfonate functionalities within the ionomer matrix, expedite proton conduction and enhance O2 permeability, thereby facilitating efficient diffusion of O2 towards the catalyst surface.17 A larger DArea/GArea ratio in N-HMCS indicates a higher concentration of defects (Fig. S15, ESI†).18 After loading the Pt5Ce alloy particles, a decrease in the pore volume and cumulative surface area in the mesopore region of E-Pt5Ce is observed, indicating that the alloy particles are indeed incorporated within the mesopores of the carbon support. This finding is consistent with the X-ray diffraction and electron microscopy results, which show that the alloy particles are uniformly dispersed within the mesopores (Fig. 2c and Fig. S16, S17, ESI†). Fig. 2d illustrates a higher M–C ratio in E-Pt5Ce, indicating a stronger Pt–C interaction in E-Pt5Ce.19 In Fig. 2e, M–N denotes the potential presence of Pt–N bonds, where the ratio of Pt–N is relatively low and close. Consequently, the limited impact of the doped N within the support on modulating the electronic structure of Pt and subsequently influencing the catalytic performance is not extensively discussed herein. The similar proportion of pyridinic-N and graphitic-N functional groups indicates that it ensures the uniform dispersion of the ionomer.9a,20Fig. 2f shows more metallic Pt content of E-Pt5Ce.21 The peaks of E-Pt5Ce and L-Pt5Ce shift negatively by 0.45 and 0.32 eV relative to Pt/C (71.52 eV, Fig. S18, ESI†). The binding energy of Ce 3d in the synthesized samples increases by about 0.23 eV (Fig. S19, ESI†).21 Thus, the difference in Pt 4f binding energy is attributed to the varying degrees of Pt–C. This suggests a more pronounced electronic regulation of Pt by Pt–C in E-Pt5Ce. E-Pt5Ce exhibits a Pt L3-edge position and white-line intensity between those of Pt foil and L-Pt5Ce (Fig. 2g). This indicates that the valence state is closer to 0, and thus metallic state Pt is dominant.22 The fitting results presented in Fig. 2h reveal the scattering pathways of Pt–C, Pt–Ce, and Pt–Pt in the synthesized samples. E-Pt5Ce exhibits a more intense shoulder peak for the Pt–C pathway at approximately 1.9 Å, as indicated by the fitting results and Table S3 (ESI†), which show a Pt–C coordination number of 1.8 for E-Pt5Ce.23 However, after anchoring Pt5Ce on the N-KB surface, the Pt–C coordination number decreases to 0.5. Since the support was modified by doping with N pairs, the shoulder peak observed at approximately 1.6 Å could potentially correspond to the Pt–N pathway. Nevertheless, the doping levels of N in both supports are extremely low and comparable, resulting in a minimal and similar number of Pt–N coordination bonds in the catalysts. Therefore, the impact of Pt–N coordination bonds on the ORR performance of the catalysts can be deemed consistent. Fig. 2i–l indicates that the single intensity maximum of E-Pt5Ce is notably shorter than the Pt–C peak in L-Pt5Ce and the Pt–Pt peak in Pt foil. In summary, the presence of Pt–C in the prepared catalysts has been clearly identified, with E-Pt5Ce featuring a richer number of Pt–C coordination bonds, indicating a stronger interaction between Pt5Ce and the carbon support. Pt–C coordination bonds employ an enhanced steric hindrance effect to more effectively modulate the electronic structure of Pt, resulting in decreased electronic density at the Pt sites. This alteration increases the repulsive interactions between adsorbed species, which in turn reduces the adsorption energy of Pt with oxygen-containing intermediates and significantly boosts the performance of the ORR.
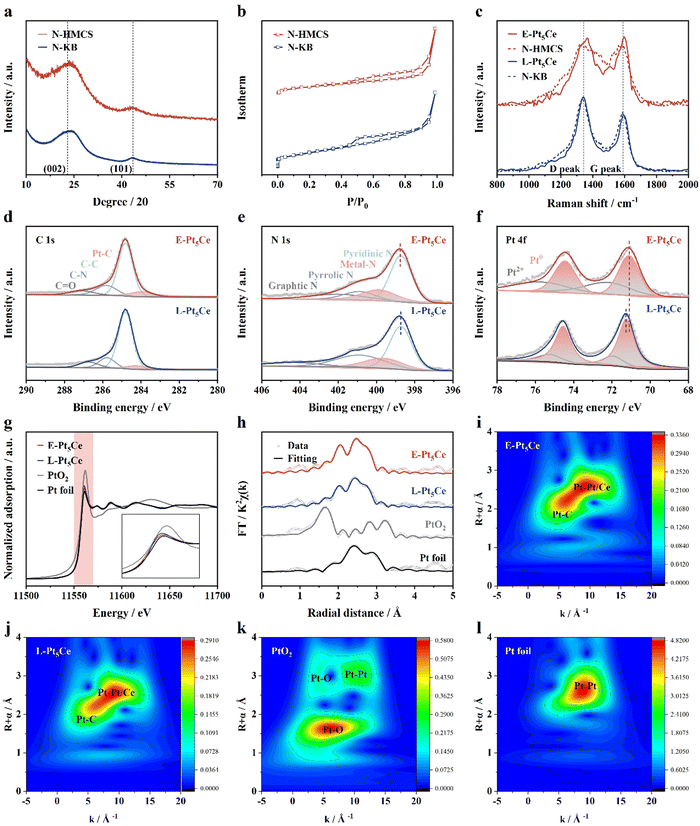 |
| | Fig. 2 (a) XRD patterns, (b) N2 adsorption–desorption isotherm curves, (c) Raman spectra of E-Pt5Ce, L-Pt5Ce, N-HMCS and N-KB. X-ray photoelectron spectroscopy (XPS) spectra of (d) C 1s, (e) N 1s, and (f) Pt 4f of E-Pt5Ce/N-HMCS and L-Pt5Ce/N-KB. (g) The Pt L3-edge X-ray absorption near-edge structure (XANES) spectra and partially enlarged pattern inset, (h) Fourier transformed k2-weighted χ(k)-functions of the extended X-ray absorption fine structure (EXAFS) spectra and fitting structures, (i)–(l) wavelet transformed (WT)-EXAFS of the Pt L3-edge signal of E-Pt5Ce, L-Pt5Ce, PtO2 and Pt foil. | |
2.2. Electrochemical performance
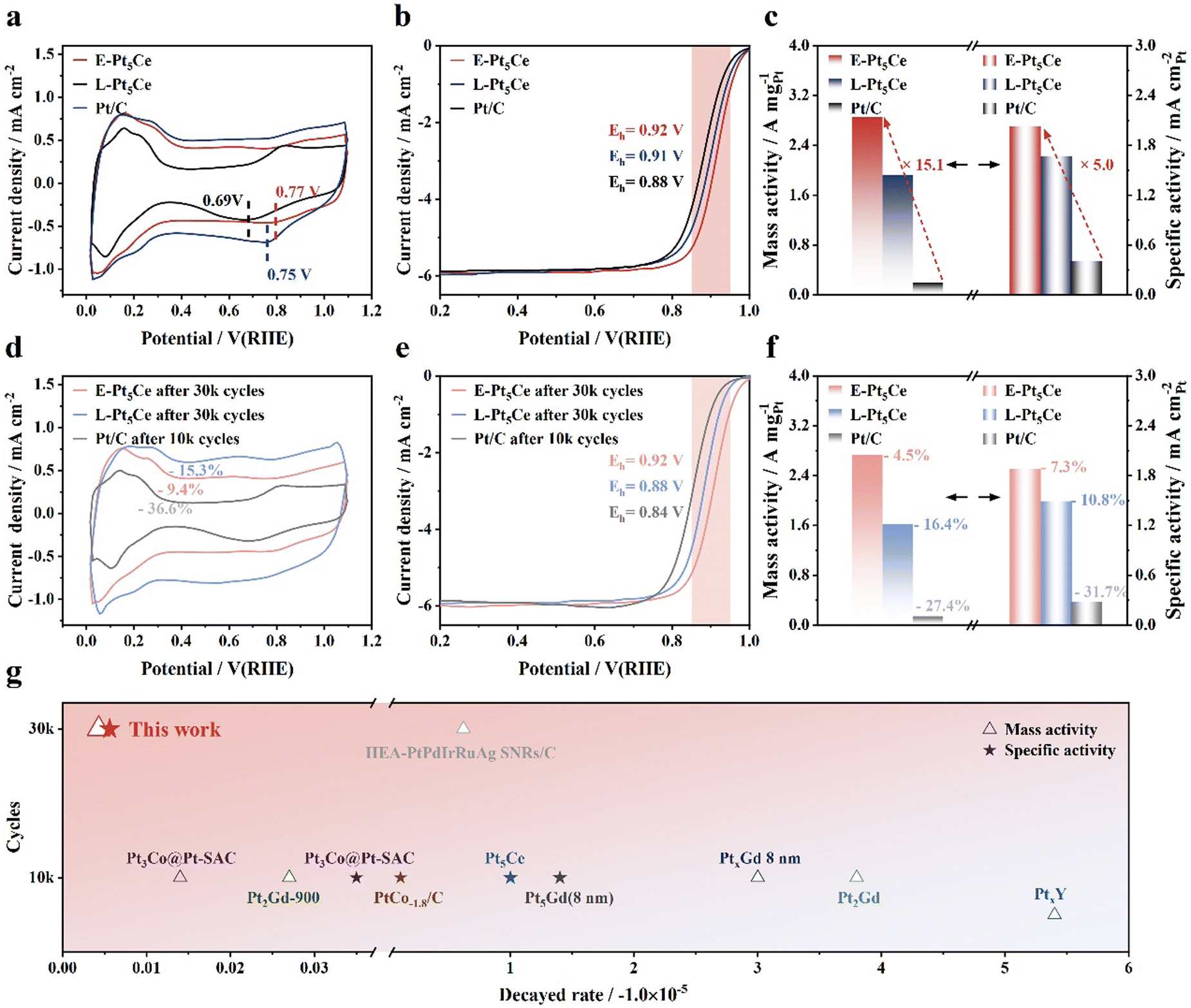
We investigated the electrocatalytic performance of the synthesized catalysts for the ORR using rotating disk electrode (RDE) and membrane electrode assembly (MEA) techniques. Compared to Pt/C, both Pt5Ce catalysts show contractions in the hydrogen adsorption/desorption (HAD) regions, implying that the incorporation of cerium atoms rendered some Pt atoms unusable for HAD (Fig. 3a).10a The prominent peaks observed in the underpotentially deposited hydrogen region in the cyclic voltammetry (CV) curves indicate that E-Pt5Ce exhibits excellent accessibility to electrolyte ions. This improves accessibility, arising from the unique structure and uniform embedding of Pt5Ce within the carbon support, suggesting a favorable surface structure for the ORR.23a The reduction and oxidation peak potentials of E-Pt5Ce are higher than those of the other catalysts, suggesting the weakest surface Pt affinity for O2. In Fig. 3b, the Eh of E-Pt5Ce is 0.92 V, which represents 14 mV positive shift compared to L-Pt5Ce (0.91 V). E-Pt5Ce exhibits a high MA and SA of 2.86 A mgPt−1 and 2.03 mA cm−2 at 0.9 V, which are 15 and 5 times those of Pt/C (0.19 A mgPt−1, 0.41 mA cm−2) (Fig. 3c). L-Pt5Ce exhibits an MA of 1.92 A mgPt−1, and SA of 1.67 mA cm−2. The superior ORR activity of E-Pt5Ce can be attributed to the Pt–C coordination bonds lower *OH binding energy.5b It can also increase the accessibility of reactive active sites. After ADT, the electrochemical active surface areas (ECSA) of E-Pt5Ce remain almost unchanged, while the ECSA retention of Pt/C is the lowest, indicating the best preservation of Pt active sites for E-Pt5Ce (Fig. 3d). Peaks appearing at 0.6 V, potentially attributable to the oxidation of hydroquinone and the reduction of quinone, may suggest that corrosion of the carbon support has occurred in L-Pt5Ce.24 However, after ADT, there are no significant changes observed in the double-layer capacitance region for E-Pt5Ce, indicating high stability. The half-wave potential (Eh) of E-Pt5Ce only decays 4 mV, a quarter of the decay of L-Pt5Ce, and nearly one-tenth of that of Pt/C (Fig. 3e). As shown in Fig. 3f, after ADT, the MA of E-Pt5Ce remains at a high level of 2.73 A mgPt−1, approaching 19.5 times that of Pt/C. The stability advantage of E-Pt5Ce is illustrated in Fig. 3g, where it shows significant superiority compared with the state-of-art catalysts (Table S4, ESI†).2a,19,25 After ADT, aggregation of the E-Pt5Ce particles is inevitable, but its XRD pattern indicates that the crystalline structure of Pt5Ce remains intact (Fig. S20 and S21, ESI†). Even after 60k ADT cycles, E-Pt5Ce still demonstrates super stability (Fig. S22–S24, ESI†). These results reveal the availability and impressive superiority of coordination layer Pt–C in enhancing durability. The Pt–C coordination layer modulates the local electronic structure of Pt, weakens the interaction between Pt and Oads, suppresses Pt dissolution, and enhances the stability of Pt5Ce. It can also be inferred that the rich Pt–C coordination bonds increase the number of active sites.
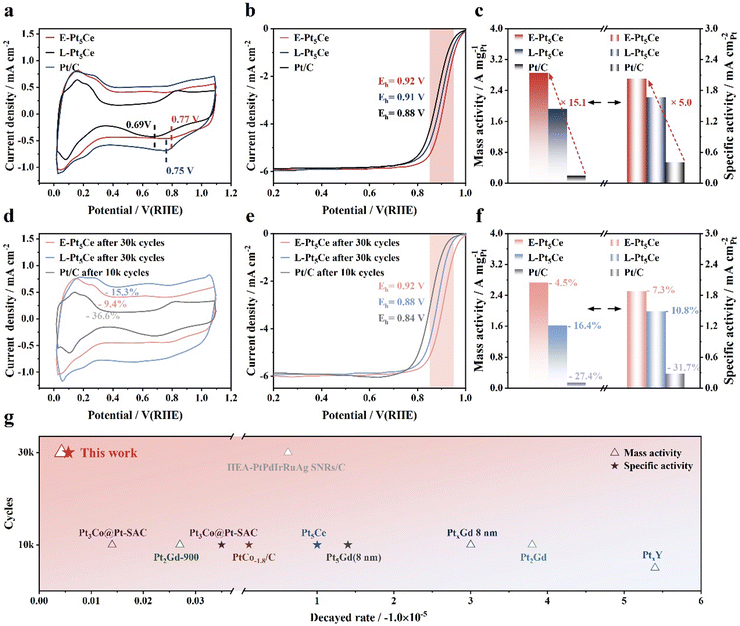 |
| | Fig. 3 Catalytic performance of E-Pt5Ce, L-Pt5Ce and Pt/C. (a) CV curves obtained in a N2-saturated 0.1 M HClO4 solution. (b) Linear sweep voltammetry (LSV) curves and partially enlarged patterns of catalysts recorded in an O2-saturated 0.1 M HClO4 solution at a rotation speed of 1600 rpm and a scan rate of 10 mV s−1. (c) Initial MAs and SAs for catalysts measured at 0.9 V (vs. RHE). (d) CV curves after ADT. ADT was conducted in an O2 atmosphere, using a 0.1 M HClO4 solution, with a voltage range of 0.6–1.0 V (vs. RHE) and a scan rate of 100 mV s−1 at room temperature. (e) LSV curves after ADT and partially enlarged patterns. (f) MAs and SAs after ADT. (g) Comparison of the decayed rate of MA and SA between E-Pt5Ce and the state-of-the-art catalysts. | |
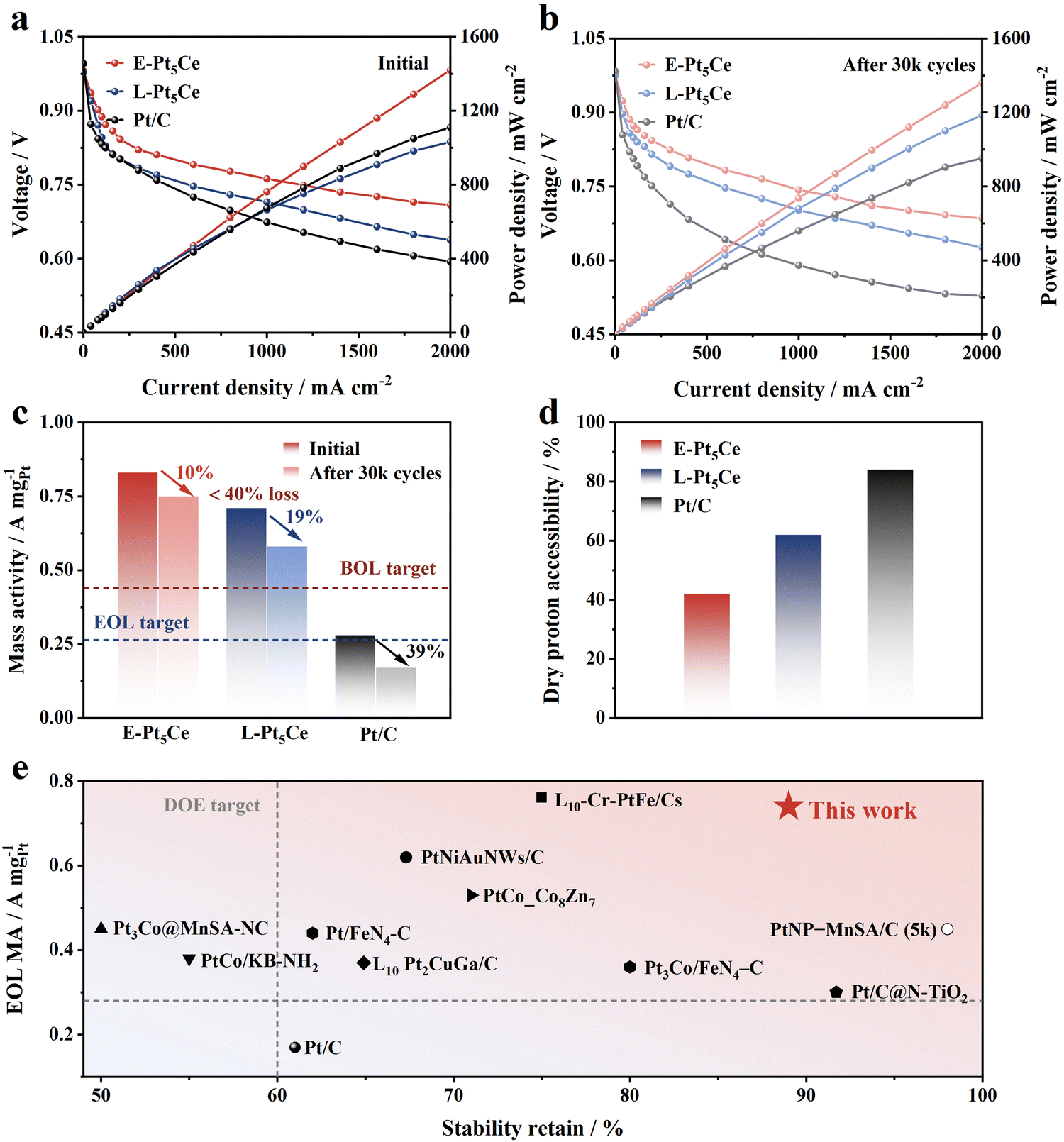
We utilized Pt5Ce alloys as cathodic catalysts in PEMFCs and conducted a single-cell test in H2/O2 fuel cells using a cathode loading of 0.10 mgPt cm−2. The test results under hydrogen air conditions are shown in Fig. S25 (ESI†). The minimal voltage drop of E-Pt5Ce at high current densities under hydrogen air conditions further indicates smooth mass transport.26Fig. 4a shows that E-Pt5Ce performs best, achieving a PPD of 1.42 W cm−2 at 2 A cm−2, while L-Pt5Ce reaches 1.30 W cm−2. Besides, E-Pt5Ce exhibits an exceptionally high current density of 546 mA cm−2 at 0.80 V, which is 1.8 times that of the DOE 2025 target (300 mA cm−2) and even exceeds the DOE 2025 target for heavy-duty vehicles (HDVs) (500 mA cm−2). To mitigate the influence of impedance, we have plotted polarization curves and power density curves subsequent to high-frequency resistance (HFR) correction, as presented in Table S5 and Fig. S26 (ESI†). Notably, under high current densities, the voltage discrepancy between the polarization curves of fuel cells consisting of E-Pt5Ce and commercial Pt/C catalysts becomes more pronounced when compared to those without HFR correction. After 30k cycles, the PPD of E-Pt5Ce only decreases 4% to 1.36 W cm−2, while L-Pt5Ce declines by 10%, and Pt/C by 14% (Fig. 4b). E-Pt5Ce delivers an excellent MA (at 0.90 V) of 0.83 A mgPt−1, better than 0.71 A mgPt−1 of L-Pt5Ce (Fig. 4c). In contrast, Pt/C exhibits a relatively low MA of 0.28 A mgPt−1. Benefiting from the excellent catalyst structure, E-Pt5Ce still maintains such high MA and stability retention percentage, which even surpass most of the advanced Pt alloy catalysts reported at a similar Pt loading to date (Fig. 4e and Table S6, ESI†).10a,19,27 Notably, the dry proton accessibility (DPA) values of the synthesized catalysts are lower than that of Pt/C, indicating hindered proton conduction at low humidity (Fig. 4d and Fig. S27–S29, ESI†). Therefore, the proton conduction of E-Pt5Ce requires the participation of water film because most Pt5Ce are located inside the mesopores of N-HMCS and do not contact the ionomer directly. E-Pt5Ce exhibits hydrophilicity according to the results of contact angle measurements (Fig. S30, ESI†). Suitable mesopores can alleviate water condensation and liquid water accumulation, enhancing O2 permeability in water and reducing oxygen transport resistance at the Pt–water interface.28 The electrode platinum surface area (EPSA) of E-Pt5Ce is higher than that of L-Pt5Ce, indicating a higher Pt utilization in E-Pt5Ce (Tables S7 and S8, ESI†). This further confirms the embedded structure of E-Pt5Ce and not entirely wrapped by carbon, with a significant number of Pt5Ce accessible to protons and O2 through the pore openings. Additionally, the ideal particle size and interparticle spacing significantly enhance the accessible surface of Pt5Ce. This strongly supports the notion that the Pt–C coordination layer can enhance the ORR stability of E-Pt5Ce. E-Pt5Ce is an ORR catalyst with promising practical application prospects.
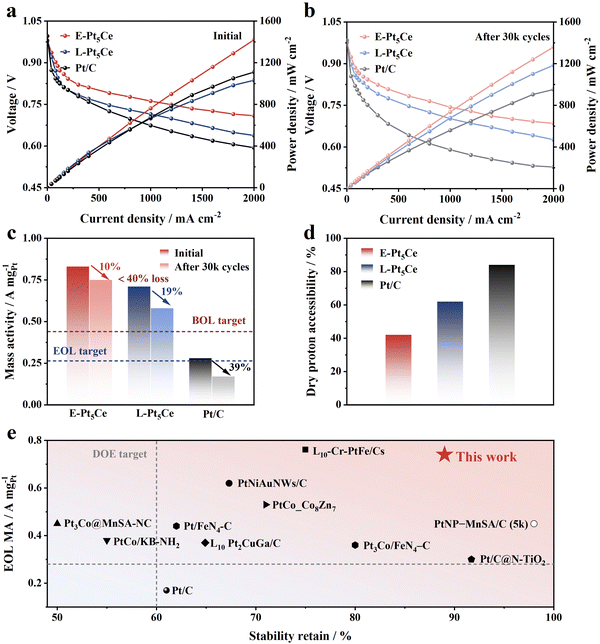 |
| | Fig. 4 Practical performance of E-Pt5Ce, L-Pt5Ce and Pt/C in PEMFC. H2/O2 polarization and power density curves at 80 °C and 100% RH of (a) beginning of life (BOL) and (b) end of life (EOL). (c) MEA MAs at 0.90 V before and after 30k cycles. Note: the red and blue dashed lines indicate DOE 2025 targets for MA at BOL (0.44 A mgPt−1) and EOL (0.264 A mgPt−1 or 40% of the initial value) after 30k cycles, respectively. (d) Comparison of DPA. (e) Comparison of stability retention and EOL MAs between E-Pt5Ce and the state-of-the-art catalysts. | |
2.3. Density functional theory (DFT) calculations
We employed DFT calculations to understand the excellent ORR stability and active sites of E-Pt5Ce. We simulated the structures of two catalysts, Pt5Ce alloy with Pt–C bonds (Pt5Ce (with Pt–C)) and Pt5Ce without Pt–C bonds (Pt5Ce (without Pt–C)). Fig. 5a shows that Oads can be adsorbed on the Pt–C coordination layer, demonstrating that the Pt–C coordination layer increases the number of active sites. In the structure of Pt–C, the Bader charge of Pt is 9.64 e, while in the structure lacking the Pt–C coordination, it is 9.73 e. Combined with Fig. 5b, according to Bader charge analysis, the charge transfer to Pt in the Pt5Ce structure with Pt–C bonds is reduced compared to the Pt5Ce structure without Pt–C bonds. This suggests that the Pt sites in the Pt–C bonded structure exhibit a lower net charge, which may facilitate the adsorption of reactive oxygen intermediates. The C on the defective carbon supports influences the electronic density of Pt. The adsorption of Oads at Pt–C sites is favorable for the ORR process, and the shorter bond length of Pt5Ce (with Pt–C) confirms its better stability. We construct free energy diagrams based on the thermodynamically most favorable paths under conditions of U = 0 V and U = 1.23 V, respectively. According to Fig. 5c, almost all steps can proceed spontaneously at U = 0 V. Under U = 1.23 V, the change in Gibbs free energy indicates that the Pt5Ce (with Pt–C) model has a smaller rate-determining step (RDS) barrier compared to the Pt5Ce (without Pt–C) model (Fig. 5c). For Pt5Ce (without Pt–C), the RDS is the formation of *OOH. The thermodynamic overpotential is at least 1.66 eV.29 In contrast, in Pt5Ce (with Pt–C), the RDS shifts to the desorption of *OH. The thermodynamic overpotential is reduced to 0.41 eV. The desorption of *OH becomes easier, avoiding a decrease in activity due to the blocking of active sites. Encouragingly, due to the introduction of abundant Pt–C, the adsorption of *OOH is enhanced, and the height of the embedded configuration step O2 → *OOH is reduced to 0.15 V. The PDOS analysis in Fig. 5e and f further indicates that compared to Pt5Ce (with Pt–C), the Pt-d band of Pt5Ce (without Pt–C) is narrower and sharper at the Fermi level. On Pt5Ce (with Pt–C), the electron-rich Pt sites show a negatively shifted d-band center, weakening the adsorption of key *OH and *H intermediates on Pt sites, thereby reducing the reaction barrier of the ORR.30 Thus, the PDOS analysis once again confirms the strong electronic coupling between Pt and C, promoting the ORR. Overall, the computational results indicate that the Pt–C coordination layer can also serve as an active site, proving that the adsorption sites for Oads on Pt are altered through the Pt–C coordination layer, effectively preventing surface Pt atoms from exchanging positions with Oads, reducing the adsorption energy of Pt with Oads, and inhibiting Pt dissolution.
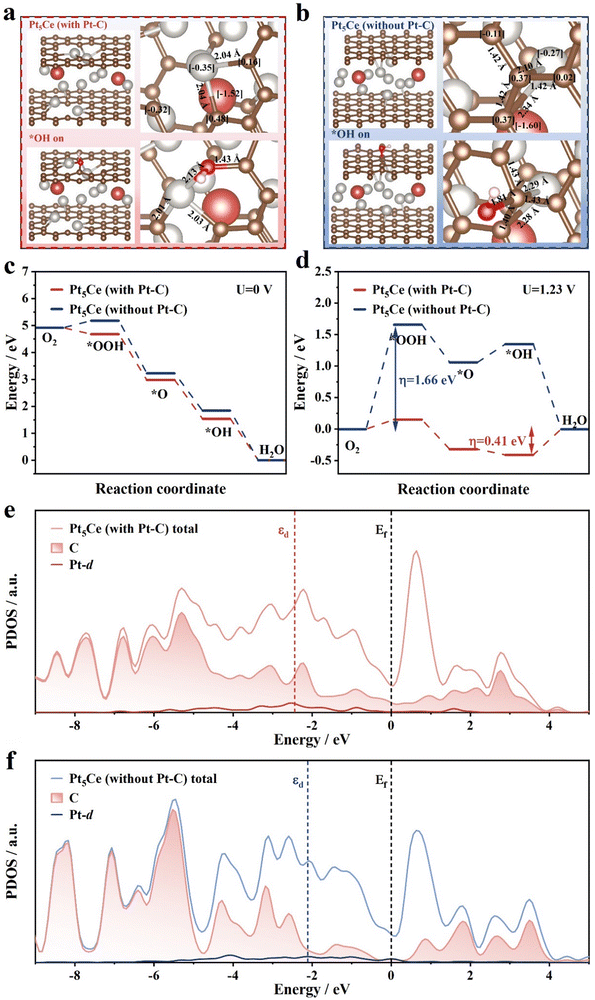 |
| | Fig. 5 DFT calculation results. The models of initial state, with adsorbed OH intermediates on, and Bader charge of (a) Pt5Ce (with Pt–C) and (b) Pt5Ce (without Pt–C). Gibbs free energy profile of the ORR process at (c) U = 0 V and (d) U = 1.23 V. The PDOS results of (e) Pt5Ce (with Pt–C) and (f) Pt5Ce (without Pt–C). | |
3. Conclusion
In summary, we have unveiled the impact of the interaction between the Pt–C coordination layer and Pt5Ce on the performance of Pt5Ce in the ORR. Electron microscopy data indicate that the strong anchoring effect of the Pt–C coordination layer effectively limits the growth of Pt5Ce NPs. XPS analysis shows that E-Pt5Ce exhibits a Pt 4f peak that shifts towards lower binding energy. XAS analysis reveals a higher proportion of Pt–C bonds, confirming the presence of a rich Pt–C coordination structure in E-Pt5Ce. The abundant Pt–C coordination bonds have a significant steric hindrance effect and exert a stronger regulatory influence on the electronic structure of Pt, mitigating Pt dissolution and reducing the adsorption energy of Oads. DFT results further confirm that Pt–C can serve as active sites for the ORR. E-Pt5Ce demonstrates better performance and lower local O2 transport resistance in both RDE and PEMFC tests. The Eh of E-Pt5Ce is 0.92 V, representing 14 mV positive shift in comparison with L-Pt5Ce (0.91 V). E-Pt5Ce exhibits a high MA and SA of 2.86 A mgPt−1 and 2.03 mA cm−2 at 0.90 V. In terms of PEMFC application, E-Pt5Ce achieves an extremely high current density of 546 mA cm−2 at 0.80 V, which even exceeds the DOE 2025 target for HDVs. The voltage drop of E-Pt5Ce is slightest at high current densities. The MA at 0.90 V is 0.83 A mgPt−1, almost twice the DOE 2025 target. After 30k cycles, there is virtually no loss in cell voltage at 0.80 A cm−2. The findings here provide a multifaceted analysis of the mechanisms of action of the Pt–C coordination layer and suggest its broad application in the development of ultra-durable ORR catalysts for harsh electrochemical environments.
Author contributions
Nannan Jiang, Hao Wang, Huihui Jin and Xuwei Liu, participating in the experiment; Nannan Jiang, writing – original draft; Lunhui Guan, writing – review and editing. All authors have given approval to the final version of the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research was supported by the central government guides the local science and technology development special project, Fujian Province (Grant No. 2023L3027).
References
- K. C. H. W. E. Winsche and F. J. SalIzano, Science, 1973, 180, 1325–1332 CrossRef PubMed.
-
(a) M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, U. Gronbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 Search PubMed;
(b) M. K. Debe, Nature, 2012, 486, 43–51 CrossRef PubMed.
-
(a) R. Osborne, BMJ, 2022, 1–2 Search PubMed;
(b) H. Yano, M. Watanabe, A. Iiyama and H. Uchida, Nano Energy, 2016, 29, 323–333 CrossRef.
-
(a) S. Zhang, S. E. Saji, Z. Yin, H. Zhang, Y. Du and C. H. Yan, Adv. Mater., 2021, 33, e2005988 CrossRef PubMed;
(b) M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef.
-
(a) H. C. Yang, N. D. Lu, J. T. Zhang, R. Wang, S. H. Tian, M. J. Wang, Z. X. Wang, K. Tao, F. Ma and S. L. Peng, Carbon Energy, 2023, 5, e337 CrossRef CAS;
(b) J. Zhang, Y. Yuan, L. Gao, G. Zeng, M. Li and H. Huang, Adv. Mater., 2021, 33, e2006494 CrossRef.
-
(a) F. Xiao and M. H. Shao, Matter, 2024, 7, 351–377 CrossRef CAS;
(b) F. Xiao, Q. Wang, G.-L. Xu, X. Qin, I. Hwang, C.-J. Sun, M. Liu, W. Hua, H.-W. Wu, S. Zhu, J.-C. Li, J.-G. Wang, Y. Zhu, D. Wu, Z. Wei, M. Gu, K. Amine and M. Shao, Nat. Catal., 2022, 5, 503–512 CrossRef CAS.
-
(a) X. Sun, X. R. Cao, J. X. Han, C. Ji, H. Varela, V. Del Colle, J. J. Zhang, C. W. Pan and Q. Y. Gao, ACS Catal., 2023, 13, 14753–14762 CrossRef CAS;
(b) V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., 2006, 45, 2897–2901 CrossRef CAS;
(c) J. Li, H.-M. Yin, X.-B. Li, E. Okunishi, Y.-L. Shen, J. He, Z.-K. Tang, W.-X. Wang, E. Yücelen, C. Li, Y. Gong, L. Gu, S. Miao, L.-M. Liu, J. Luo and Y. Ding, Nat. Energy, 2017, 2, 17111 CrossRef CAS.
-
(a) Z. Duan and G. Henkelman, ACS Catal., 2021, 11, 14439–14447 CrossRef CAS;
(b) M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velazquez-Palenzuela, V. Tripkovic, J. Schiotz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS.
-
(a) L. Zhao, J. Zhu, Y. Zheng, M. Xiao, R. Gao, Z. Zhang, G. Wen, H. Dou, Y. P. Deng, A. Yu, Z. Wang and Z. Chen, Adv. Energy Mater., 2022, 12, 2102665 CrossRef CAS;
(b) P. P. Lopes, D. Li, H. Lv, C. Wang, D. Tripkovic, Y. Zhu, R. Schimmenti, H. Daimon, Y. Kang, J. Snyder, N. Becknell, K. L. More, D. Strmcnik, N. M. Markovic, M. Mavrikakis and V. R. Stamenkovic, Nat. Mater., 2020, 19, 1207–1214 CrossRef CAS PubMed.
-
(a) Q. Gong, H. Zhang, H. Yu, S. Jeon, Y. Ren, Z. Yang, C.-J. Sun, E. A. Stach, A. C. Foucher, Y. Yu, M. Smart, G. M. Filippelli, D. A. Cullen, P. Liu and J. Xie, Matter, 2023, 6, 963–982 CrossRef CAS;
(b) G. W. Sievers, A. W. Jensen, J. Quinson, A. Zana, F. Bizzotto, M. Oezaslan, A. Dworzak, J. J. K. Kirkensgaard, T. E. L. Smitshuysen, S. Kadkhodazadeh, M. Juelsholt, K. M. O. Jensen, K. Anklam, H. Wan, J. Schafer, K. Cepe, M. Escudero-Escribano, J. Rossmeisl, A. Quade, V. Bruser and M. Arenz, Nat. Mater., 2021, 20, 208–213 CrossRef CAS PubMed.
- N. Jiang, B. Huang, M. Wang, Y. Chen, Q. Yu and L. Guan, Adv. Sci., 2024, 11, e2305110 CrossRef PubMed.
- Hydrogen and Fuel Cell TechnologiesOffice Multi-Year Program Plan, https://www.energy.gov/sites/default/files/2024-05/hfto-mypp-2024.pdf.
- J. R. Cheng, C. Lyu, H. R. Li, J. W. Wu, Y. Hu, B. Han, K. L. Wu, M. Hojamberdiev and D. S. Geng, Appl. Catal., B, 2023, 327, 122470 CrossRef CAS.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hubner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- H. Lan, J. Wang, L. Cheng, D. Yu, H. Wang and L. Guo, Chem. Soc. Rev., 2024, 53, 684–713 RSC.
- M. N. Islam, A. B. Mansoor Basha, V. O. Kollath, A. P. Soleymani, J. Jankovic and K. Karan, Nat. Commun., 2022, 13, 6157 CrossRef CAS PubMed.
- B. Zheng, J. Fan, B. Chen, X. Qin, J. Wang, F. Wang, R. Deng and X. Liu, Chem. Rev., 2022, 122, 5519–5603 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef.
- C. Griesser, H. Li, E. M. Wernig, D. Winkler, N. Shakibi Nia, T. Mairegger, T. Gotsch, T. Schachinger, A. Steiger-Thirsfeld, S. Penner, D. Wielend, D. Egger, C. Scheurer, K. Reuter and J. Kunze-Liebhauser, ACS Catal., 2021, 11, 4920–4928 CrossRef.
- Z. Qiao, C. Wang, C. Li, Y. Zeng, S. Hwang, B. Li, S. Karakalos, J. Park, A. J. Kropf, E. C. Wegener, Q. Gong, H. Xu, G. Wang, D. J. Myers, J. Xie, J. S. Spendelow and G. Wu, Energy Environ. Sci., 2021, 14, 4948–4960 RSC.
- J. Liu, M. Jiao, B. Mei, Y. Tong, Y. Li, M. Ruan, P. Song, G. Sun, L. Jiang, Y. Wang, Z. Jiang, L. Gu, Z. Zhou and W. Xu, Angew. Chem., Int. Ed., 2019, 58, 1163–1167 CrossRef PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D. J. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 15938 CrossRef.
-
(a) B. Liu, R. Feng, M. Busch, S. Wang, H. Wu, P. Liu, J. Gu, A. Bahadoran, D. Matsumura, T. Tsuji, D. Zhang, F. Song and Q. Liu, ACS Nano, 2022, 16, 14121–14133 CrossRef PubMed;
(b) Y. Li, Z. Wu, C. Wang, X. Yu, W. Gao, B. Wang, C. Wu, Y. Yao, J. Yang and Z. Zou, Adv. Funct. Mater., 2024 DOI:10.1002/adfm.202310428.
- R. Sharma, S. Gyergyek and S. M. Andersen, Appl. Catal., B, 2022, 311, 121351 CrossRef CAS.
-
(a) S. Zhu, M. Sun, B. Mei, L. Yang, Y. Chu, Z. Shi, J. Bai, X. Wang, Z. Jiang, C. Liu, B. Huang, J. Ge and W. Xing, Natl. Sci. Rev., 2023, 10, nwad162 CrossRef CAS;
(b) L. Zhao, C. Fu, L. Luo, J. You, L. An, X. Yan, S. Shen and J. Zhang, Appl. Catal., B, 2022, 318, 121831 CrossRef CAS;
(c) P. Malacrida, M. Escudero-Escribano, A. Verdaguer-Casadevall, I. E. L. Stephens and I. Chorkendorff, J. Mater. Chem. A, 2014, 2, 4234–4243 RSC;
(d) A. Velázquez-Palenzuela, F. Masini, A. F. Pedersen, M. Escudero-Escribano, D. Deiana, P. Malacrida, T. W. Hansen, D. Friebel, A. Nilsson, I. E. L. Stephens and I. Chorkendorff, J. Catal., 2015, 328, 297–307 CrossRef;
(e) Y. Hu, J. O. Jensen, L. N. Cleemann, B. A. Brandes and Q. Li, J. Am. Chem. Soc., 2020, 142, 953–961 CrossRef CAS PubMed;
(f) Y. Jiang, T. Fu, J. Liu, J. Zhao, B. Li and Z. Chen, RSC Adv., 2022, 12, 4805–4812 RSC;
(g) L. Tao, M. Sun, Y. Zhou, M. Luo, F. Lv, M. Li, Q. Zhang, L. Gu, B. Huang and S. Guo, J. Am. Chem. Soc., 2022, 144, 10582–10590 CrossRef CAS PubMed.
- L.-C. Lin, C.-H. Kuo, Y.-H. Hsu, L.-C. Hsu, H.-Y. Chen, J.-L. Chen and Y.-T. Pan, Appl. Catal. B, 2022, 317, 121767 CrossRef CAS.
-
(a) Y. Zeng, J. Liang, C. Li, Z. Qiao, B. Li, S. Hwang, N. N. Kariuki, C. W. Chang, M. Wang, M. Lyons, S. Lee, Z. Feng, G. Wang, J. Xie, D. A. Cullen, D. J. Myers and G. Wu, J. Am. Chem. Soc., 2023, 145, 17643–17655 CrossRef CAS;
(b) X. Liu, Z. Zhao, J. Liang, S. Li, G. Lu, C. Priest, T. Wang, J. Han, G. Wu, X. Wang, Y. Huang and Q. Li, Angew. Chem., Int. Ed., 2023, 62, e202302134 CrossRef CAS;
(c) L. Gao, T. Sun, X. Chen, Z. Yang, M. Li, W. Lai, W. Zhang, Q. Yuan and H. Huang, Nat. Commun., 2024, 15, 508 CrossRef CAS PubMed;
(d) Z. Y. Chen, C. Hao, B. Yan, Q. Chen, H. Feng, X. Mao, J. Cen, Z. Q. Tian, P. Tsiakaras and P. K. Shen, Adv. Energy Mater., 2022, 12, 2201600 CrossRef CAS;
(e) X. Wei, S. Song, W. Cai, Y. Kang, Q. Fang, L. Ling, Y. Zhao, Z. Wu, X. Song, X. Xu, S. M. Osman, W. Song, T. Asahi, Y. Yamauchi and C. Zhu, ACS Nano, 2024, 18, 4308–4319 CrossRef CAS;
(f) H. Liu, Q. Lu, Y. Gao, C. Huang, A. Zhang, F. Liu, H. Xu, X. Liu, B. Shan and R. Chen, Chem. Eng. J., 2023, 463 Search PubMed;
(g) X. Liu, Y. Wang, J. Liang, S. Li, S. Zhang, D. Su, Z. Cai, Y. Huang, L. Elbaz and Q. Li, J. Am. Chem. Soc., 2024, 146, 2033–2042 CrossRef.
- T. Mashio, H. Iden, A. Ohma and T. Tokumasu, J. Electroanal. Chem., 2017, 790, 27–39 CrossRef.
- A. Han, X. Wang, K. Tang, Z. Zhang, C. Ye, K. Kong, H. Hu, L. Zheng, P. Jiang, C. Zhao, Q. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19262–19271 CrossRef PubMed.
- L. Gong, J. Zhu, F. Xia, Y. Zhang, W. Shi, L. Chen, J. Yu, J. Wu and S. Mu, ACS Catal., 2023, 13, 4012–4020 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a