
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gastrointestinal tolerance of D-allulose in children: an acute, randomised, double-blind, placebo-controlled, cross-over study†
Davide
Risso
 *a,
Gillian
DunnGalvin
b,
Sameer
Saxena
b,
Andrea
Doolan
b,
Lisa
Spence
c and
Kavita
Karnik
a
*a,
Gillian
DunnGalvin
b,
Sameer
Saxena
b,
Andrea
Doolan
b,
Lisa
Spence
c and
Kavita
Karnik
a
aTate & Lyle PLC, 5 Marble Arch, W1H 7EJ, London, UK. E-mail: davide.risso@tateandlyle.com
bAtlantia Food Clinical Trials, Heron House, Blackpool, Cork, Ireland
cSchool of Public Health, Indiana University Bloomington, Indiana, USA
First published on 14th December 2023
Abstract
D-Allulose, a low-calorie sugar, provides an attractive alternative to added sugars in food and beverage products. There is however limited data on its gastrointestinal (GI) tolerance, with only two studies in adults, and no studies in children to date. We therefore performed an acute, randomised, double-blind, placebo-controlled, cross over study designed to determine, for the first time, the GI tolerance of 2 doses of D-allulose (2.5 g per 120 ml and 4.3 g per 120 ml) in young children. The primary tolerance endpoint was the difference in the number of participants experiencing at least one stool that met a Type 6 or Type 7 description on the Bristol Stool Chart, within 24 hours after study product intake. Secondary endpoints included the assessment of stool frequency, stool consistency, and the presence of GI symptoms. Only one participant in the low dose group experienced a stool type 6 or 7, while no participants experienced a stool type 6 or 7 in the high dose group. A statistically significant difference in the change in stool frequency compared to placebo in the high dose group (p = 0.044) was found, with no significant difference between the groups for stool consistency and no participants experienced unusual stool frequency. All the encountered adverse events were non-serious, either mild or moderate, and there were no serious adverse events. All in all, D-allulose was tolerated well in children, making this ingredient a good candidate to reformulate commercially produced goods by replacing added sugars with lower caloric content.
1. Introduction
A high consumption of free/added sugars (i.e., sugars that are added to food during processing and preparation in any form by the manufacturer, cook or consumer),1 in particular from sugar-sweetened beverages (SSBs), has been strongly linked to poorer diet quality, dental caries, weight gain, obesity, and cardiometabolic diseases such as type 2 diabetes and cardiovascular disease (CVD).2 Considering this, the World Health Organisation (WHO) has recommended a reduction in free sugars in the diet to less than 10% of the total energy intake, with a further reduction to less than 5% for additional health benefits.3 As most of the intake of these added/free sugars derives from packaged and commercially produced foods,4,5 considering not only the health impact but also the economic burdens of added sugars overconsumption, product reformulation has been highlighted as an effective method to reduce less healthful ingredients.6,7Several strategies have been proposed to reduce sugars, depending on the target food product: from technological approaches such as using an inhomogeneous spatial distribution of sucrose or different sugar particle sizes, to the use of specific ingredients that can replace some of the functions of sugars.8 Among these, non-sugar sweeteners (NSS), i.e., synthetic and naturally occurring sweeteners that are not classified as sugars,9 have been widely used to safely replace sugars for energy-reduced, non-cariogenic, or no-added sugars foods. Even if replacing sugar with NSS is a common strategy to lower energy density of beverages, there still are some practical challenges to reproduce a broad range of other qualities, especially in solid foods, like the texture of the full-sugar products. In addition, despite comprehensive safety evaluations by regulatory authorities, some concerns about NSS have recently arisen that challenged their ability to achieving weight control or reducing risk of non-communicable diseases.9 On the other hand, it has been acknowledged that not all sweeteners are the same, as they possess different sweetness intensities and organoleptic properties, metabolic fates, and impact (if any) on the microbiota.9,10 The choice of sweeteners to employ, therefore, is broad and it depends on many factors and aimed applications. Indeed, the use of non-NSS such as sweet proteins, sugar alcohols (polyols) and rare sugars has been recently introduced as a complementary/alternative solution.
Among the latter, D-allulose, a rare ketohexose, is an epimer of D-fructose that was first identified in wheat in the 1930s and has since been found to be naturally occurring in fruits such as dried figs or raisins, and maple syrup.11 It provides an alternative attractive sweetener, as it is approximately 70% as sweet as sugar with a very similar onset, peak and dissipation of sweetness,12 adding bulk and texture, while providing low (i.e., 0.4 kcal g−1) or no calories, depending on the regulation.13 In addition, it is non-cariogenic14,15 and does not raise blood glucose or insulin levels and, when added to sucrose, attenuates both postprandial glucose and insulin responses.16,17D-Allulose has been generally recognized as safe (GRAS) by the United States Food and Drug Administration (FDA) and other organizations worldwide for use as a food and beverage ingredient, that have recognized its safety for all consumers, including children, showing a high tolerability of up to 30 g d−1.18–22 However, dedicated research studies examining its gastrointestinal (GI) tolerance in dedicated populations, are currently lacking in the published literature. There is limited data published on the GI tolerance of D-allulose with, to our knowledge, only two published studies that studied healthy young adults and adults, confirming its high GI tolerance.23,24 However, tolerability has not been assessed in children to date. A confirmation of its tolerance in children is, therefore, needed. The primary objective of this study was therefore to assess the number of participants in a 24-hour period post-consumption of intervention, with at least one loose or watery stool movement. To aid the interpretation of this tolerability measure, secondary objectives of an unusual increase in stool frequency, change in stool consistency and frequency of abnormal and clinically significant GI symptoms were also assessed.
2. Materials & methods
2.1. Study conduct
This study was registered as a clinical trial (https://clinicaltrials.gov; ID: NCT06063096). An Independent Institutional Review Board (i.e., Clinical Research & Ethics Committee of the Cork Teaching Hospitals, Lancaster Hall, 6 Little Hanover Street, Cork, Ireland) reviewed and approved the study. The study was conducted in accordance with the ethical principles that have their origins in the Declaration of Helsinki and in accordance with the International Conference on Harmonisation Good Clinical Practice. A participant information sheet was provided to the parent/guardian of the children, and the overall details of the study were explained to both the parent/guardian and the children at the screening visit at the study site in Cork, Ireland. Informed consent forms were signed by the parents/guardians of the children at the screening visit. The recruitment of the study participant started at the end of 2015 and continued until the end of March 2016.2.2. Participants
The participants were recruited through their parents/guardians, who underwent an initial phone screening that included questions regarding their child's age, weight, and general health. If suitable, the parent/guardian and the child were scheduled for a screening visit. A total of 40 participants were screened, and 30 participants were included in and completed the study. Participants who met the following criteria were considered eligible for enrolment into the study: being able to provide written informed consent provided by a parent/guardian; girls and boys aged 6 to 8 years of age; weight-for-age between the 5th and the 90th percentile as per the Centre for Disease Control and Prevention Growth charts25 and in good overall health, as assessed by their medical history, to increase the homogeneity of the sample population; accustomed to having lunch between 12.00 and 2.30 pm; routinely had up to 3 bowel movements per day or as few as 3 bowel movements per week; able to drink 120 ml of fluid within 30 minutes; parents willing to continue their child's normal food and beverage intake and physical activity throughout the duration of the study; able to attend all 7 visits. Participants were excluded from the study if they met any of the below criteria: any major trauma or surgical event within the 3 months prior to screening; history or presence of clinically significant endocrine or GI disorder including the following hypothyroidism, coeliac disease, functional constipation, irritable bowel syndrome (IBS), organic causes of constipation, i.e., Hirschsprung's disease pseudo-obstruction, spinal cord abnormality, diabetes mellitus, cystic fibrosis, gluten enterophathy, congenital anorectal malformation; functional GI Disorders in accordance with Rome III Diagnostic Questionnaire for Paediatric Functional GI Disorders;26 more than 1 loose stool in the 48 hours preceding dosing, that met a Type 6 or Type 7 description on the Bristol Stool Chart,27 as recorded in the pre-dosing diary (for participants who did experience a loose stool, the screening visit was rescheduled); use of any prescription medication, including antibiotics, laxatives and steroids; regular GI complaints, such as stomach upsets, diarrhoea, constipation, flatulence, abdominal colic; known intolerance or sensitivity to any of the study products; abdominal or anorectal surgery; psychiatric disorders, anxiety, and depression; lactose intolerance; use of supplements that may have affected GI system including laxatives, fibre, and iron supplements; exposure to any non-registered drug product within 30 days prior to screening visit. Participants were able to be withdrawn from the study if: the parent/guardian elected independently to withdraw from the study; the child developed any condition which contravened the original criteria; the child was considered at any point to be unsuitable to continue the study, at the discretion of the investigator.2.3. Investigational products
Participants were provided a preselected option for lunch (shown in ESI Table 1†) and were asked to consume it along with an approximately 120 ml clear, fruit-flavoured drink which contained the study product or placebo. The treatment drink contained D-allulose (Tate & Lyle DOLCIA PRIMA®), while the placebo contained high fructose corn syrup. Each participant received D-allulose at 2.1% inclusion rate (approximately 2.5 g) for Dose 1 and 3.5% inclusion rate (approximately 4.2 g) for Dose 2 in the 120 ml drink. Such doses were calculated based on GRAS notices (i.e., for drinks, a maximum inclusion level of 3.5%) and were below the levels confirmed to be maximum tolerable. Both drinks were composed to be identical in appearance and there was only one batch for all beverages, as all the ingredients were from the same batch and all beverages were made at one time. Full specifications for the drinks are given in ESI Table 2.† The participants were instructed to eat and drink the beverages within a 30-minute period, and could select their preferred flavour, as all doses were available in all flavours. The labelling of the study products according to the study code and participant treatment assignment was performed by a member of the study team who was not involved with the study. The study team remain blinded to the product identity and group allocation.2.4. Study design
This was an acute, randomised, double-blind, placebo-controlled, cross over study to determine the GI tolerance of D-allulose in children. Randomisation was carried out by computer-generated block-randomization lists to one of three treatment sequence groups in equal proportions. An independent statistician prepared the randomization. The data for this study was not unblinded until after the SAP was finalised and the database lock was completed. A total of 40 participants were screened and thirty healthy children entered the study and were randomly assigned to one of three treatment sequences in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio (i.e., 10 participants per sequence) (i.e., Sequence 1: Placebo/Dose 1/Dose 2; Sequence 2: Dose 1/Dose 2/Placebo; Sequence 3: Dose 2/Placebo/Dose 1). The study involved 7 visits over a 3-to-6-week period. Dosing took place at Visits 2, 4 and 6. Visits 3, 5, and 7 occurred 24 hours after Visits 2, 4 and 6, respectively. Dose visits were each separated by 1 week ±7 days. Activities and measurements were carried out as follows:
:1:1 ratio (i.e., 10 participants per sequence) (i.e., Sequence 1: Placebo/Dose 1/Dose 2; Sequence 2: Dose 1/Dose 2/Placebo; Sequence 3: Dose 2/Placebo/Dose 1). The study involved 7 visits over a 3-to-6-week period. Dosing took place at Visits 2, 4 and 6. Visits 3, 5, and 7 occurred 24 hours after Visits 2, 4 and 6, respectively. Dose visits were each separated by 1 week ±7 days. Activities and measurements were carried out as follows:
• Study Visit #1 = Screening Visit (Day −4 to −14). Overall study details explained, informed consent form (ICF), anthropometric measurements (weight and height), inclusion/exclusion criteria, questionnaire on paediatric GI symptoms, medical history, vital signs, prior/concomitant medications, instructions for the diary completion, and appointment for the next visit.
• 48 hours prior to Visit #2. Parent/guardian contacted for: confirmation of child's good health, reminder to complete the pre-study (GI/bowel habit) diary and food diary.
• Study Visit #2 dose visit (within two weeks since Visit #1; Day 0). Baseline/Randomization. Inclusion/exclusion criteria review, study diary review, medical history, Adverse (AEs) and Serious Adverse Events (SAEs), prior/concomitant medications, randomisation, on-site consumption of Dose1/2/placebo according to the treatment arm, instructions for the diary completion.
• Study Visit #3 follow-up visit (24 hours after Visit #2; Day 1). Completed diary recording bowel movements and symptoms, the food diary, stool samples collection, stool samples’ assessment of consistency according to the Bristol Stool Scale, GI Symptoms since Dose1/2/placebo intake, participant's overall general health and wellbeing, AE/SAE, prior/concomitant medications.
• Study Visit #4 dose visit (Day 7 ± 7 days). Crossover according to the treatment sequence. Measurements/activities same as in Visit #2.
• Study Visit #5 follow-up visit (Day 8 ± 7 days). Measurements/activities same as Visit #3.
• Study Visit #6 dose visit (Day 15 ± 7 days). Crossover according to the treatment sequence. Measurements/activities same as in Visit #2.
• Study Visit #7 follow-up visit (Day 16 ± 7 days). Measurements/activities same as Visit #3.
A schematic diagram of the study is provided in Fig. 1.
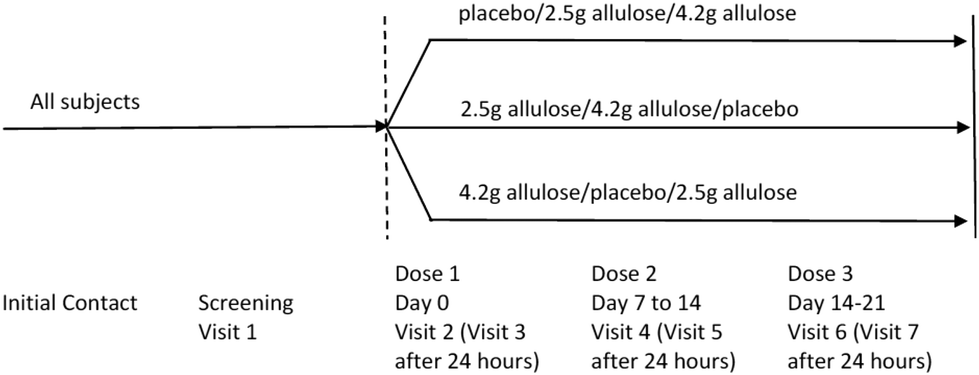 | ||
| Fig. 1 Schematic of study design. | ||
2.5. Tolerance assessments and adverse effects
The assessment of stool consistency for the primary endpoint was completed using the Bristol Stool chart, which includes seven stool forms (i.e., 1 = separate hard lumps, like nuts (difficult defaecation), 2 = sausage shaped but lumpy, 3 = like a sausage or snake but with cracks on its surface, 4 = like a sausage or snake, smooth and soft, 5 = soft blobs with clear cut edge, 6 = fluffy pieces with ragged edges, a mushy stool, 7 = watery, no solid pieces) was used to measure stool consistency.27 The primary objective was to measure the number of participants who experienced, in a 24-hour period post-consumption of intervention, at least one loose or watery stool that met a Type 6 or Type 7 description on the Bristol Stool Chart within 24 hours after study product intake. This was reported as the difference between the baseline (Visit 2, Day 0) and Visit 3 (Day 1), Visit 5 (Day 8 ± 7 days) and Visit 7 (Day 16 ± 7 days) for each of the treatment groups (Placebo, Dose 1, Dose 2), and the difference between the treatment groups. The secondary objective was to measure any unusual increase in stool frequency, defined as greater than 3 bowel movements in the 24-hour period post-consumption. This objective is reported as the difference between baseline and each subsequent timepoint (Visit 3 (Day 1), Visit 5 (Day 8 ± 7 days) and Visit 7 (Day 16 ± 7 days)) as well as the difference between the treatment groups. To aid interpretation of safety and tolerability, (serious) adverse events ((S)Aes) were recorded. The secondary objective focused on GI symptoms including abdominal pain, bloating, cramping, abdominal rumbling (borborygmi), excess flatus and nausea associated with D-allulose consumption. These were reported as the frequency of the event and frequency of participants reporting events by the severity and causality (i.e., related, not related) for each treatment group recorded at Visits 3, 5 and 7, for pre- and post-dose administration. The severity of the event was categorized in three levels. Mild was defined as easily tolerated, causing minimal discomfort and not interfering with normal everyday activities, moderate was defined as sufficiently discomforting to interfere with normal everyday activities, and severe was defined as incapacitating and/or preventing normal everyday activities. Causality of the event was determined in relation to the study product. An event was defined as unrelated if it was clearly related to other factors such as the participant's clinical state, therapeutic interventions, or a concomitant medication administered to the participant and did not follow a known response pattern to the investigational product. It was defined as possibly related to the tested product if it followed a reasonable temporal sequence from the time of investigational product administration and/or follows a known response pattern to the study treatment, but could have been produced by other factors such as the participant's clinical state, therapeutic interventions, or concomitant medications administered to the participant. The event was defined as definitely related if the event followed a reasonable temporal sequence from the time of investigational product administration, followed a known response pattern to the investigational product and could not be reasonably explained by other factors such as the participant's clinical state, therapeutic interventions administered to the participant and either occurs immediately following investigational product, or improves on stopping the investigational product, or reappears on repeat exposure, or there is a positive reaction at the application site.2.6. Statistics
In absence of published tolerance data in children, the sample size estimation was based on recommended sample size for studies in the exploratory phase28 as well as expertise of the study team with regard expected event count. Following the ICH GCP guideline E9,29 power was set at the minimum required level of 80% for the calculation. A sample size of 30 completed participants was calculated in order to be able to have 80% power to detect an increase of 0.25 in the rate of cases of distress with a level of significance of 0.05 for the primary endpoint. Such sample size is also aligned with a recent study from our group, highlighting the tolerance of fiber ingredients in children.30 Additional measures for the secondary endpoint, frequency of bowel movements and the Bristol Scale Score, were also calculated to have power to detect a change of 0.5 units, assuming a standard deviation for each of 1. As this was a tolerability study, all participants were planned to be included in the analysis, regardless of completion status to reduce the risk of potential bias in the analysis if a participant(s) withdrew from the study due to tolerability issues. Therefore, there was not a planned drop-out rate. This was reflected in the definition of the Intention to Treat (ITT) population where completion of study in full is not a requirement.The ITT population included all randomised participants who took at least one dose of treatment, with a valid baseline measurement (Day 0). The Per-Protocol (PP) population included all randomised participants with a valid baseline measurement (Day 0), and valid measurements at each of the study endpoints (Visit 3, 5 and 7). All analyses were performed using both the ITT and PP populations: since very few differences were identified between ITT and PP populations, only the ITT findings are provided. Statistical analyses were performed according to the Statistical analysis plan (SAP) using Rx64 version 3.2.2. Analysis of the primary tolerance endpoint was carried out using a noninferiority analysis (Clopper-Pearson) to calculate 95% confidence intervals (CIs) for events. In addition, equivalence testing with confidence intervals were called using the methodology proposed by.31 A superiority limit of 25% was set for the primary endpoint. If the upper limit of the confidence interval was below the superiority limit, it could be concluded that the participants are able to tolerate the treatment. Such superiority limit was calculated, and presented, as the placebo rate +0.25, therefore confidence intervals for the primary outcome have been calculated for low and high doses, but not for the placebo group, to allow for comparison to the superiority limit to assess tolerability. Secondary endpoints were analysed based on frequency and rate data, which were treated as continuous variables. Primarily mixed effects models were used to better account for the experimental design and the nature of the data. While p-values were reported, they were only for informative basis and were not adjusted for multiplicity. In case of missing data, Last Observation Carried Forward (LOCF) method was applied to conduct the statistical analysis for ITT population.
3. Results
3.1. Participant characteristics
All participants completed the study and there were no discontinuations. Therefore, there were no dropouts in this study. One participant was excluded from the PP population in treatment sequence 1 (placebo/2.5 g/4.2 g) due to a missing endpoint value (i.e., Bristol Stool Chart score missing) (Fig. 2).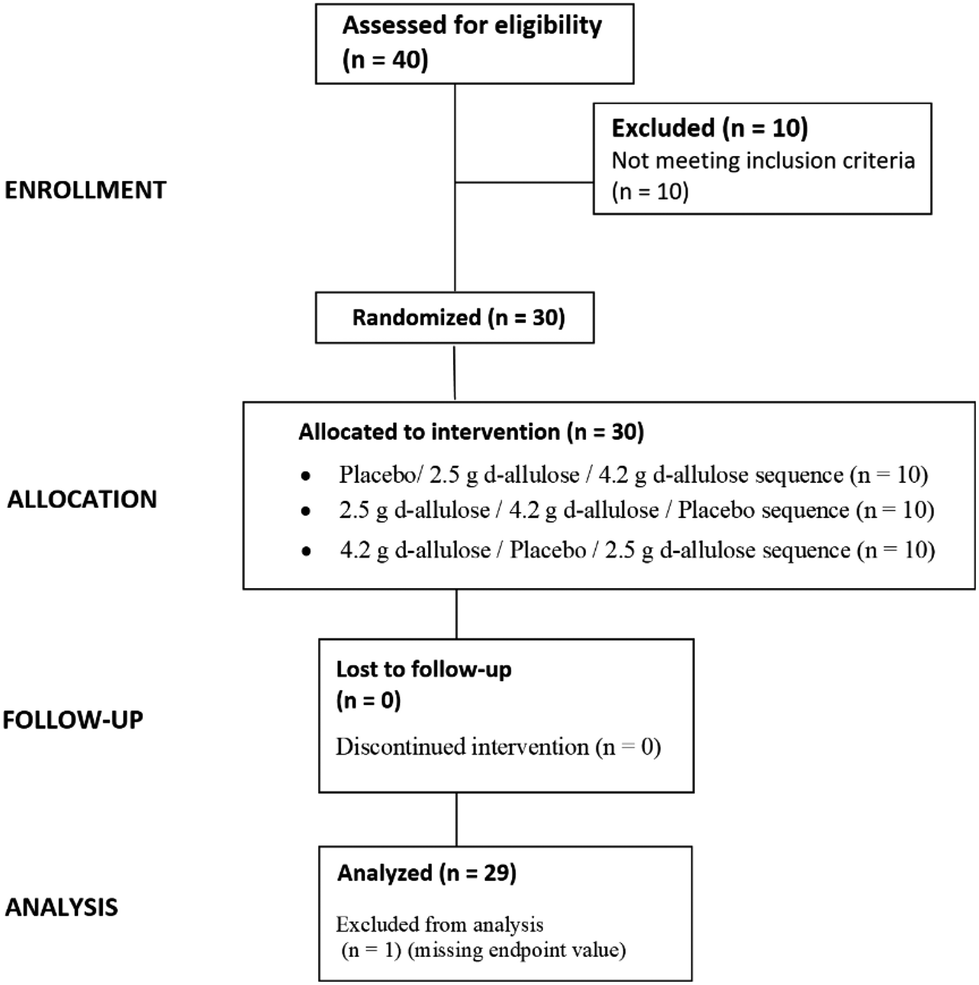 | ||
| Fig. 2 Disposition of study participants. | ||
Selected participant demographic and anthropometric measures at screening are presented in Table 1. ESI Table 3† also illustrates the distribution of weight-for-age percentiles of the screened participants. There were no notable differences between treatment sequences for demographic characteristics. No participant had loose stool, upset stomach, food, or medication allergies, or took concomitant medication at baseline. All participants completed the 24-hour pre food diary and the 48-hour pre study bowel movement diary as required. All participants consumed the lunch drink as required at Visits 2, 4 and 6.
| Characteristic | Statistic/category | Treatment sequence 1a (N = 10) | Treatment sequence 2b (N = 10) | Treatment sequence 3c (N = 10) | Total (N = 30) |
|---|---|---|---|---|---|
| a Placebo/2.5 g D-allulose/4.2 g D-allulose. b 2.5 g D-allulose/4.2 g D-allulose/Placebo. c 4.2 g D-allulose/Placebo/2.5 g D-allulose. d Bristol Stool Chart Type 6 (Fluffy pieces with ragged edges – a mushy stool) or Type 7 (Watery, no solid pieces – entirely liquid) description. | |||||
| Gender | Male | 3 (30%) | 4 (40%) | 5 (50%) | 12 (40%) |
| Female | 7 (70%) | 6 (60%) | 5 (50%) | 18 (60%) | |
| Age (years) | Mean (SD) | 7 (0.94) | 6.8 (0.88) | 6.8 (1.03) | 6.9 (0.93) |
| Median (min, max) | 7 (6,8) | 7 (6,8) | 6 (6,9) | 7 (6, 9) | |
| (Q1, Q3) | 6, 8 | 6, 8 | 6, 7 | 6, 8 | |
| Nationality | African/Irish | 0 (0%) | 0 (0%) | 1 (10%) | 1 (3.3%) |
| Irish | 10 (100%) | 10 (100%) | 9 (90%) | 29 (96.7%) | |
| Height (m) | Mean (SD) | 1.3 (0.05) | 1.3 (0.06) | 1.3 (0.09) | 1.3 (0.07) |
| Median (min, max) | 1.28 (1.17, 1.3) | 1.28 (1.15, 1.37) | 1.25 (1.1, 1.38) | 1.28 (1.1, 1.38) | |
| (Q1, Q3) | 1.23, 1.3 | 1.22, 1.3 | 1.19, 1.34 | 1.21, 1.3 | |
| Weight (kg) | Mean (SD) | 25.9 (2.64) | 26.1 (3.68) | 26.5 (4.64) | 26.1 (3.62) |
| Median (min, max) | 26.6 (21.8, 30.8) | 25.8 (21.2, 31.4) | 26.6 (20.1, 32.6) | 25.8 (20.1, 32.6) | |
| (Q1, Q3) | 24.55, 26.65 | 23.2, 29.25 | 22.6, 30 | 23.2, 29.3 | |
| BMI (kg m−2) | Mean (SD) | 16.2 (0.92) | 16.1 (1.3) | 16.8 (1.29) | 16.4 (1.17) |
| Median (min, max) | 16 (15, 18) | 16 (15, 17) | 17 (15, 18) | 16 (14, 18) | |
| (Q1, Q3) | 16, 16 | 15, 17 | 16, 18 | 16, 17 | |
| Loose stoold | Yes | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| No | 10 (100%) | 10 (100%) | 10 (100%) | 30 (100%) | |
3.2. Gastrointestinal tolerance
| Tolerance endpoint | Placebo (N = 30) | Low dose (N = 30) | High dose (N = 30) | |||
|---|---|---|---|---|---|---|
| N (%) | Superiority limit (%) | n (%) | 95% CI | n (%) | 95% CI | |
| Abbreviations: CI = confidence interval; ITT = intent to treat.a Number of participants experiencing the stool type. Bristol stool types: 1 = separate hard lumps, like nuts (difficult defaecation), 2 = sausage shaped but lumpy, 3 = like a sausage or snake but with cracks on its surface, 4 = like a sausage or snake, smooth and soft, 5 = soft blobs with clear cut edge, 6 = fluffy pieces with ragged edges, a mushy stool, 7 = watery, no solid pieces. | ||||||
| Stool Type 6 or 7 | 0 (0%) | 25% | 1 (3.3%) | 0.1, 17.2 | 0 (0%) | 0, 11.6 |
| Stool Type 1 to 5 | 17 (56.7%) | 81.70% | 22 (73.3%) | 54.1, 87.7 | 25 (83.3%) | 65.3, 94.4 |
| No bowel movement occurred | 13 (43.3%) | 68.30% | 7 (23.3%) | 9.9, 42.3 | 5 (16.7%) | 5.6, 34.7 |
| Placebo | Low dose | High dose | Total | |||||
|---|---|---|---|---|---|---|---|---|
| n (%) | 95% CI | n (%) | 95% CI | n (%) | 95% CI | n (%) | 95% CI | |
| Abbreviations: CI = confidence interval; GI = gastrointestinal. As this was a crossover study, a participant could be counted in more than one dose group, except for the total summary. One participant experienced two GI symptom events, one for both the low dose and the high dose. | ||||||||
| Participants who experienced at least 1 GI symptom event | ||||||||
| Yes | 1 (3.3%) | (0.1, 17.2%) | 4 (13.3%) | (3.8, 30.7%) | 3 (10%) | (2.1, 26.5%) | 7 (23.3%) | (9.9, 42.3%) |
| No | 29 (96.7%) | (82.8, 99.9%) | 26 (86.7%) | (69.3, 96.2%) | 27 (90%) | (73.5, 97.9%) | 23 (76.7%) | (57.7, 90.1%) |
| Number of GI symptom events | ||||||||
| 1 | 1 (3.3%) | (0.1, 17.2%) | 4 (13.3%) | (3.8, 30.7%) | 3 (10%) | (2.1, 26.5%) | 6 (20%) | (7.7, 38.6%) |
| 2 | 0 (0%) | (0, 11.6%) | 0 (0%) | (0, 11.6%) | 0 (0%) | (0, 11.6%) | 1 (3.3%) | (0.1, 17.2%) |
| 3 | 0 (0%) | (0, 11.6%) | 0 (0%) | (0, 11.6%) | 0 (0%) | (0, 11.6%) | 0 (0%) | (0, 11.6%) |
4. Discussion
D-Allulose, a low-caloric rare sugar, provides an attractive alternative to sucrose and added sugars in food and beverage products. It activates the human sweet taste receptor TAS1R2/TAS1R3 heterodimer32 and is ∼70% as sweet as sugar with a very similar onset, peak and dissipation of sweetness, also behaving in a similar way to sugar in recipes.12 In particular, when allulose and sucrose are blended in a 1:1 mixture, this combination achieves a near identical dose–response curve to sucrose.12 It can therefore be used in a variety of products suitable for children such as protein bars, shakes, drinks, ice creams, and yoghurts. Indeed, when asking consumers about their perception of D-allulose inclusion in yogurt formulations, this rare sugar performed similarly to sucrose in liking and purchase intent, and superior to other sweeteners, with fewer off-flavors.33
D-Allulose is GRAS18–22 for use as a food/beverage ingredient for of all ages, including children, and exempt from “Sugars” and “Added Sugars” on the label by the United States FDA.13 This is of particular importance for children, considering that most of their intake of added sugars comes from packaged and commercially produced foods, sweetened beverages in particular,4,5 and in light of the recommendation from the WHO to reduce free sugars in the diet to less than 10%, or even 5% of the total energy intake,3 highlighting the need to limit added sugars at an early age.34
Previous research in healthy and young adults indicated that D-allulose is well tolerated.23,24 Such reports have shown that doses of maximum no-observed-effect level in humans were 0.55 g per kg body weight (BW), when the laxative effect of D-allulose was used as an indicator, or 0.4 g per kg BW as maximum single intake. In absence of published tolerance data in children, and taking into account approved GRAS levels of allulose, such maximum tolerable level in adults have been relevant to identify potentially suitable dosages for children.
This acute, randomised, double-blind, placebo-controlled, cross-over study aimed to verify the tolerance of D-allulose in children, when consumed as a fruit flavoured drink, along with a meal. The doses of D-allulose tested were calculated based on GRAS18–22 and are below the levels confirmed to be maximum tolerable level in adults on g kg−1 basis. The results of this study are aligned with published research in healthy and young adults23,24 and confirmed that, within inclusion levels, D-allulose is tolerated well in children with few, mild or moderate, GI symptom events that all resolved, with no severe symptoms, unusual stool frequency In particular, only one participant in the low dose group experienced a stool type 6 or 7, while no participants experienced a stool type 6 or 7 in the high dose group, confirming no significant difference in terms of stool consistency among the studied arms. It also highlights the potential for D-allulose to be used as a partial replacement of added sugars in foodstuffs aimed at children. This in addition to the previous physiological health benefits attributed to D-allulose such as aiding glucose management16,17 and being non-cariogenic.14,15
Lastly, some limitations need to be considered. As the design of this study was set as an acute trial, we were not able to investigate the presence of any potential chronic effects, such as GI symptoms due to long-term exposure to D-allulose. It is also difficult to extrapolate these results to the more general children population, considering the relatively small sample size (N = 30) and the restricted range of age group analyzed (6 to 8 years old). In addition, we recruited children that were within a healthy range of BMI, being conscious that the results might not be replicable in overweight, or obese, individuals. Nevertheless, the present results fill a gap in the literature regarding the tolerance of allulose in children and the present limitations should be taken into account by future studies addressing this question in a broader, bigger representative sample of the general children population.
5. Conclusions
In conclusion, D-allulose doses of 2.5 g and 4.2 g were tolerated well by healthy 6- to 8-year-old children, making this ingredient a good candidate to be used to reformulate packaged and commercially produced goods by replacing added sugars with lower caloric content.Author contributions
Conceptualization, K.K., A.D., G.D., L.S., S.S.; methodology, K.K., A.D., G.D., L.S., S.S.; formal analysis, A.D., G.D., S.S.; investigation, A.D., G.D., S.S.; resources, K.K., L.S., D.R.; data curation, A.D., G.D., L.S., S.S; writing – original draft preparation, D.R., A.D., G.D., L.S., S.S.; writing – review and editing, D.R., A.D., G.D., L.S., S.S., K.K.; visualization, A.D., G.D., L.S., S.S.; supervision, K.K.; project administration, D.R.; funding acquisition, K.K. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
Davide Risso and Kavita Karnik are employees of Tate & Lyle PLC. At the time of the study, Lisa Spence was an employee of Tate & Lyle PLC. Other authors declare no conflict of interest.References
- G. E. Swan, N. A. Powell, B. L. Knowles, M. T. Bush and L. B. Levy, A definition of free sugars for the UK, Public Health Nutr., 2018, 21(9), 1636–1638 CrossRef.
- Y. Huang, Z. Chen, B. Chen, J. Li, X. Yuan, J. Li, W. Wang, T. Dai, H. Chen, Y. Wang, R. Wang, P. Wang, J. Guo, Q. Dong, C. Liu, Q. Wei, D. Cao and L. Liu, Dietary sugar consumption and health: umbrella review, Br. Med. J., 2023, 381, e071609 CrossRef PubMed.
- World Health Organization, Guideline: Sugars intake for adults and children, in World Health Organization, 2015. https://apps.who.int/iris/handle/10665/149782. Accessed 13 September 2023 Search PubMed.
- V. Azaïs-Braesco, D. Sluik, M. Maillot, F. Kok and L. A. Moreno, A review of total & added sugar intakes and dietary sources in Europe, Nutr. J., 2017, 16, 6 CrossRef PubMed.
- A. Drewnowski and C. D. Rehm, Consumption of added sugars among US children and adults by food purchase location and food source, Am. J. Clin. Nutr., 2014, 100, 901–907 CrossRef CAS PubMed.
- World Health Organization, Eliminating trans fats in europe, in A policy brief. Copenhagen, 2015. https://www.euro.who.int/__data/assets/pdf_file/0010/288442/Eliminating-trans-fats-in-Europe-Apolicy-brief.pdf. Accessed 13 September 2023 Search PubMed.
- K. Trieu, B. Neal, C. Hawkes, E. Dunford, N. Campbell, R. Rodriguez-Fernandez, B. Legetic, L. McLaren, A. Barberio and J. Webster, Salt Reduction Initiatives around the World – A Systematic Review of Progress towards the Global Target, PLoS One, 2015, 10(7), e0130247 CrossRef PubMed.
- J. M. Cooper, The challenges of reformulation for sugars reduction, Food Sci. Technol., 2020, 31(1), 38–41 Search PubMed.
- World Health Organization (WHO), Guideline: use of non-sugar sweeteners, 2023, https://www.who.int/publications/i/item/9789240073616. Accessed 13 September 2023.
- P. Van den Abbeele, J. Poppe, S. Deyaert, I. Laurie, T. K. O. Gravert, A. Abrahamsson, A. Baudot, K. Karnik and D. Risso, Low-no-calorie sweeteners exert marked compound-specific impact on the human gut microbiota ex vivo, Int. J. Food Sci. Nutr., 2023, 74(5), 630–644 CrossRef CAS PubMed.
- H. Daniel, H. Hauner, M. Hornef and T. Clavel, Allulose in human diet: the knowns and the unknowns, Br. J. Nutr., 2022, 128(2), 172–178 CrossRef CAS PubMed.
- M. Wee, V. Tan and C. Forde, A Comparison of Psychophysical Dose-Response Behaviour across 16 Sweeteners, Nutrients, 2018, 10(11), 1632 CrossRef PubMed.
- US Food and Drug Administration (FDA), Guidance for Industry: The Declaration of Allulose and Calories from Allulose on Nutrition and Supplement Facts Labels, 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-declaration-allulose-and-calories-allulose-nutrition-and-supplement-facts-labels. Accessed 13 September 2023.
- H. Hasturk, A clinical study on the effect of allulose, a low-calorie sugar, on in vivo dental plaque pH, Forsyth Institute, Boston, MA, USA, 2018 Search PubMed.
- T. Attin, Confirmation of Toothfriendly Quality, D-Allulose (D-Psicose), Zürich, 2016 Search PubMed.
- T. Iida, Y. Kishimoto, Y. Yoshikawa, N. Hayashi, K. Okuma, M. Tohi, K. Yagi, T. Matsuo and K. Izumori, Acute D-psicose administration decreases the glycemic responses to an oral maltodextrin tolerance test in normal adults, J. Nutr. Sci. Vitaminol., 2008, 54(6), 511–514 CrossRef CAS PubMed.
- F. Au-Yeung, A. L. Jenkins, S. Prancevic, E. Vissers, J. E. Campbell and T. M. S. Wolever, Comparison of postprandial glycemic and insulinemic response of allulose when consumed alone or when added to sucrose: A randomized controlled trial, J. Funct. Foods, 2023, 105, 105569 CrossRef CAS.
- US Food and Drug Administration (FDA), Generally Recognized as Safe (GRAS) Notice (GRN) No. 400, 2012.
- US Food and Drug Administration (FDA), Generally Recognized as Safe (GRAS) Notice (GRN) No. 498, 2014.
- US Food and Drug Administration (FDA), Generally Recognized as Safe (GRAS) Notice (GRN) No. 828, 2020.
- US Food and Drug Administration (FDA), Generally Recognized as Safe (GRAS) Notice (GRN) No. 1024, 2023.
- US Food and Drug Administration (FDA), Generally Recognized as Safe (GRAS) Notice (GRN) No. 1029, 2023.
- Y. Han, B. R. Choi, S. Y. Kim, S. B. Kim, Y. H. Kim, E. Y. Kwon and M. S. Choi, Gastrointestinal Tolerance of D-Allulose in Healthy and Young Adults. A Non-Randomized Controlled Trial, Nutrients, 2018, 10(12), 2010 CrossRef CAS PubMed.
- T. Iida, Y. Kishimoto, Y. Yoshikawa, K. Okuma, K. Yagi, T. Matsuo and K. Izumori, Estimation of Maximum No-effect Level of D-Psicose in Causing Diarrhea in Human Subjects, J. Jpn. Counc. Adv. Food Ingredients Res., 2007, 10, 15–19 CAS (Japanese).
- Centers for Disease Control and Prevention, National Center for Health Statistics. Data Table of Weight-for-age Charts, 2001. https://www.cdc.gov/growthcharts/html_charts/wtage.htm. Accessed 13 September 2023.
- A. Rasquin, C. Di Lorenzo, D. Forbes, E. Guiraldes, J. S. Hyams, A. Staiano and L. S. Walker, Childhood functional gastrointestinal disorders: child/adolescent, Gastroenterology, 2006, 130(5), 1527–1537 CrossRef PubMed.
- S. J. Lewis and K. W. Heaton, Stool form scale as a useful guide to intestinal transit time, Scand. J. Gastroenterol., 1997, 32(9), 920–924 CrossRef CAS PubMed.
- A. L. Whitehead, S. A. Julious, C. L. Cooper and M. J. Campbell, Estimating the sample size for a pilot randomised trial to minimise the overall trial sample size for the external pilot and main trial for a continuous outcome variable, Stat. Methods Med. Res., 2016, 25(3), 1057–1073 CrossRef PubMed.
- J. A. Lewis, Statistical principles for clinical trials (ICH E9): an introductory note on an international guideline, Stat. Med., 1999, 18(15), 1903–1942 CrossRef CAS.
- D. Risso, M. Kaczmarczyk, I. Laurie, E. Mah, T. M. Blonquist, L. Derrig and K. Karnik, Moderate intakes of soluble corn fibre or inulin do not cause gastrointestinal discomfort and are well tolerated in healthy children, Int. J. Food Sci. Nutr., 2022, 73(8), 1104–1115 CrossRef CAS PubMed.
- T. Tango, Equivalence test and confidence interval for the difference in proportions for the paired-sample design, Stat. Med., 1998, 17(8), 891–908 CrossRef CAS.
- Y. Choi, J. A. Manthey, T. H. Park, Y. K. Cha, Y. Kim and Y. Kim, Correlation between in vitro binding activity of sweeteners to cloned human sweet taste receptor and sensory evaluation, Food Sci. Biotechnol., 2021, 30(5), 675–682 CrossRef CAS PubMed.
- M. R. Mora, Z. Wang, J. M. Goddard and R. Dando, Consumers Respond Positively to the Sensory, Health, and Sustainability Benefits of the Rare Sugar Allulose in Yogurt Formulations, Foods, 2022, 11(22), 3718 CrossRef CAS PubMed.
- S. A. Bowman, J. C. Clemens, J. E. Friday, N. Schroeder and R. P. LaComb, Added Sugars in American Children's Diet: What We Eat in America, NHANES 2015–2016. 2019 Dec, in FSRG Dietary Data Briefs [Internet], United States Department of Agriculture (USDA), Beltsville,MD, 2010. Dietary Data Brief No. 26 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fo04210c |
| This journal is © The Royal Society of Chemistry 2024 |