
The mechanochemical synthesis of environmentally benign fully biobased 4th generation benzoxazines and their polymers: mechanistic insights into the catalytic activity of latent catalysts†
Vaishaly
Duhan
a,
Shivani
Yadav
a,
Christophe
Len
 bc and
Bimlesh
Lochab
*a
bc and
Bimlesh
Lochab
*a
aMaterials Chemistry Laboratory, Department of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence, Gautam Buddha Nagar, Uttar Pradesh 201314, India. E-mail: bimlesh.lochab@snu.edu.in
bSchool of Chemistry, Xi'an Jiaotong University, 28 Xianning West Road, Xi'an, China
cInstitute of Chemistry for Life and Health Sciences, Chimie ParisTech, PSL Research University, 11 rue Pierre et Marie Curie, Paris, France
First published on 11th December 2023
Abstract
Sustainable methodologies, such as mechanochemical mixers, have revolutionized the way compounds are synthesized in minimal time and with better yields. In this work, a series of latest 4th generation benzoxazine monomers were synthesized via a facile, highly scalable, efficient mechanochemical ball-milling process, making them more viable than those synthesized via traditional synthesis route. The 4th generation benzoxazine (BZ) monomers contained 100% biosynthons (vanillin, ortho-vanillin and furfuryl amine) and were purified without strenuous separation techniques. We found that the variation in the nature of aryl substitution (with and without phenolic–OH) at the reactive oxazine C2 centre governs the polymerization temperature, volatilization of monomers, kinetics of polymerization, thermal and mechanical properties. As expected, the control monomer without inherent phenolic–OH showed a high polymerization temperature and the advantages offered by vanillin regioisomers facilitated the polymerization with minimal volatile release. The latent catalytic effect in the monomer was confirmed by both temperature-dependent NMR and SCXRD studies. Interestingly, distal phenolic–OH was found to be more labile and polymerized easily at a lower temperature than when it was present at the ortho-position. Additionally, the former monomer structure led to a well-defined polymer network with an appreciable Tg (116 °C) and thermal stability (Tmax of 348 °C and char yield of 36%). Furthermore, it also revealed an excellent storage modulus and good adhesion properties compared to many classical petroleum-based polybenzoxazines. Overall, we showcased the viability of employing the benefits of copolymerizing two generations of benzoxazines at low temperature and explored this new class of latest benzoxazine monomers as greener adhesives with improved stability to enable future designing and open up the possibility of using them in several practical and innovative applications.
1. Introduction
Polybenzoxazines (PBZs) represents a promising class of polymers owing to their attractive properties, such as low shrinkage during polymerization, a high glass transition temperature, thermal stability, good chemical resistance, low humidity uptake and desirable mechanical properties.1,2 Additionally, the ease of synthesis, along with extensive opportunities for structural modifications and the ability to undergo catalyst-free ring-opening polymerization (ROP), is further appealing from both synthetic and industrial perspectives.Currently, the practical applications of PBZs have remained elusive to some extent because of the requirement of a very high polymerization temperature, accounting for a concomitant significant mass loss, especially in earlier benzoxazine (BZ) monomer generations, during polymerization. To enable the polymerization of BZ at lower temperatures, various curing accelerators, catalysts, or initiators have been reported, ranging from inorganic salts to organic compounds. These external aids, including phenols,3 carboxylic acids,4p-toluenesulfonic acid and its derivatives,5 amines,6–8 methylimidazole,9 inorganic salts,10–12 organocatalysts, metal–organic frameworks,13 nanoparticles14 and polymers15 led to fast rates of polymerization. However, their use affects the nature of linkages in the polymer structure and often suffers from incompatibility issues, demanding additional processing steps. Alternatively, the introduction of active acidic functionalities, such as phenolic hydroxyls, thiols, and hydroxyethyl groups, in BZ monomers can also lower the ROP temperature.16–19
However, the issue of the short shelf life of BZs persists. To mitigate this limitation, the incorporation of functionalities with an inherent latent catalytic effect in the phenolic and amine counterparts of BZ has been attempted.20–24 Incorporating inactive structural motifs within the BZ monomer, which become activated upon an external trigger, is an interesting approach. In particular, functionalities involving intramolecular H-bonding lock the acidic labile hydrogen atoms and render them available once heat is applied to accelerate the polymerization reaction without compromising the shelf life of monomers.
The utilization of renewable feedstocks has led to a paradigm shift in reducing overdependence on the widespread use of petroleum-based feedstocks, aiming to maximize waste utilization and reduce environmental hazards arising from polymer usage. Several desirable functionalities are naturally integrated into renewable resources, and introducing them synthetically in petro-based starting materials is often challenging. Over the past few decades, the production of sustainable feedstock–derived polymeric materials including polybenzoxazines,25 gained significant interest to overcome the challenges associated with the depletion of fossil reserves, fluctuation in petroleum prices, and environmental concerns. A wide variety of renewable phenolics and amines are explored in polybenzoxazines, including cardanol,26,27 magnolol,28 urushiol,29 coumarin,30 eugenol,31 vanillin,32,33 guaiacol,34 isoeugenol, sesamol,35 bisguaiacol-F,36 resveratrol,37 catechol,38 catechin,23 chavicol,39 carvacrol,40 ferulic acid,41 peterostilbene,42 chrysin,43 anethole,44 furfurylamine, stearylamine,34 chitosan,15 isomannide,45 rosin,46 and tyramine.47 Biomass-derived derivatives of phenols and amines have proven successful as renewable resources, offering excellent opportunities to synthesize sustainable polymers capable of partially or fully replacing petroleum-based PBZs.22,25,30,34,48,49 Biobased PBZs have demonstrated their utility in several applications including high-performance anticorrosive coatings,50 superparamagnetic materials,51 cathode materials in next-generation batteries beyond Li-ions,52,53 antibacterials,54,55 and as potential flame-retardant materials.36,56,57 Recently, the replacement of formaldehyde with other aldehydes has also been employed to form 4th generation BZs.58–62 More recently, in this latest generation of BZs with oxazine-ring substitution found to empower structural tailoring close to the oxazine-ring reactive centre and accounted as a strategy to overcome mass loss issues and facilitated polymerization at lower temperature requirements than unsubstituted monomers.63,64 The progression of structural changes in benzoxazine monomers over the years is illustrated in Fig. 1.
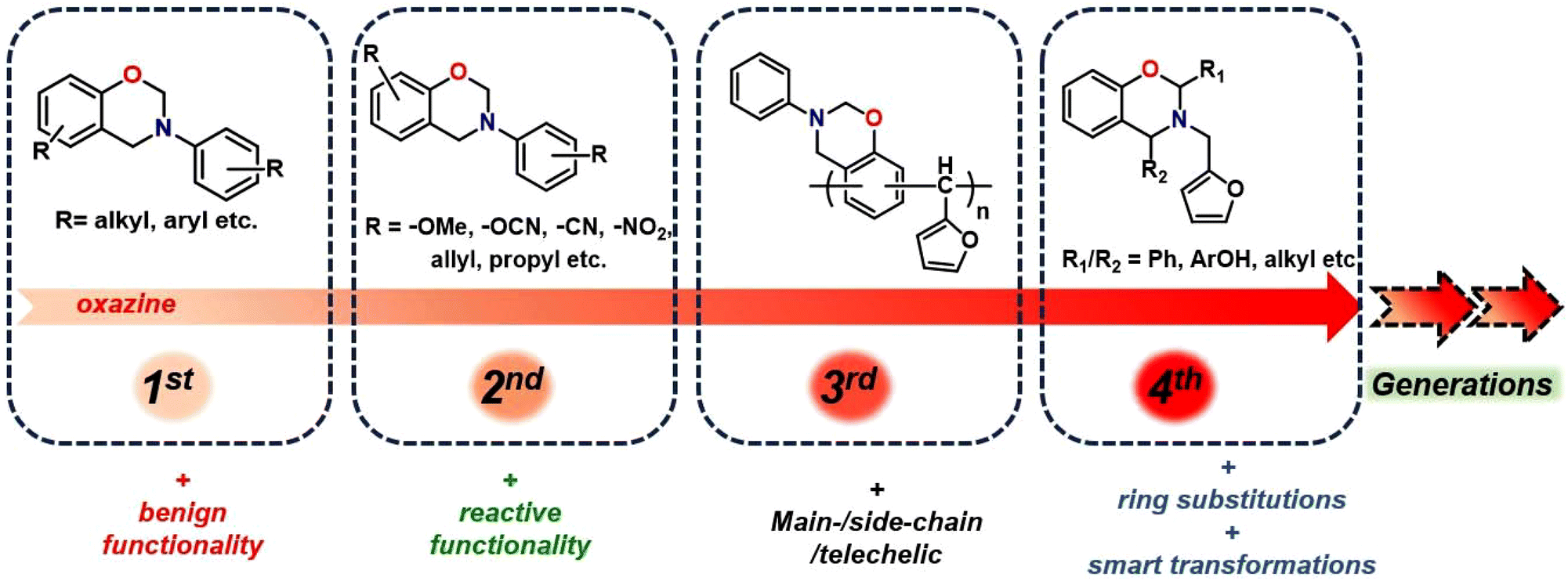 | ||
| Fig. 1 The evolution of benzoxazine monomers; illustrated with representative structures. | ||
The first generation is the most primitive and the monomer bears benign substituents. The second-generation monomers possess additional functionalities, which can self-/co-polymerize, and crosslink via other mechanisms, and may aid oxazine ring-opening reactions. In the third generation, oxazine groups are present in the main-chain, side-chain, and as terminal groups in the polymer to provide properties of both thermoplastics and thermosets. The fourth-eneration monomers possess smart architecture features and are subclassified into two types. This typically involves either in situ transformation of oxazine functionalities to others during polymerization65 or substitutions at the 2- and/or 4-position of the oxazine ring are explored.66
Vanillin, a biomass-derived feedstock phenolic aldehyde extracted from the vanilla bean, has been extensively explored as a phenol feedstock in unsubstituted BZs.32 Naturally, vanillin exists in two regioisomers, where the aldehyde is either in the ortho- or para-position relative to the phenolic–OH (Hphenolic), as shown in Fig. 2.
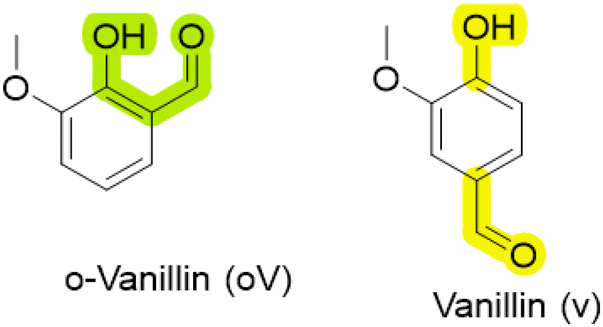 | ||
| Fig. 2 The structures of natural occurring regioisomers of vanillin. | ||
However, the exploration of a complete environmentally friendly process right from the synthetic methods, use of biosynthons, and latent catalytic effects at the oxazine-ring-substituted 4th generation benzoxazines have not been investigated and may provide a combined benefits in one go. The utility of the aldehyde functionality in vanillin as a replacement for formaldehyde has not been reported in BZ monomer synthesis. It is interesting to explore vanillin as a dual feedstock, serving as both phenolic and aldehyde starting materials, to synthesize oxazine-ring-substituted vanillin-based BZs. Furthermore, it is interesting to study the influence of this Hphenolic, at the 2- and 4-positions in vanillin, on polymerization characteristics in the latest generation of BZ. Still, although polybenzoxazines show great promise in many applications, their scalability is limited by their impractical industrially viable synthetic method. In the past few decades, there has been significant interest in alternative, cleaner, less energy-intensive, and sustainable chemical synthesis approaches compared to traditional conventional heating methods. Among the notable green techniques, both microwave-assisted and mechanochemical methods67 have emerged as highly efficient and environmentally friendly processes for the preparation of various molecules.68 They allowed accessibility to certain structures with a high product selectivity, which are difficult to obtain by other methods.69,70 Notably, the mechanical activation of molecules has enabled solvent-free, unprecedented mechanochemical “parallel synthesis”, with scalability ranging from milligrams to multikilogram, encouraging their usage for industrial polymer applications.
Current work presents a mechanochemical synthesis as a green, fast, and efficient process for producing fully biobased 4th generation BZs. Natural feedstock, vanillin, has been cleverly chosen to showcase an inbuilt latent catalytic effect, eliminating the need for the usual additional synthetic and processing steps. The polymerization behavior, mechanical properties, and thermal stability were studied and compared with the control structure.
2. Experimental
2.1. Materials
o-Vanillin (oV, 99%), vanillin (v, 99%), benzaldehyde (reagent plus, ≥99%), and furfuryl amine (fa, ≥99%) were purchased from Sigma-Aldrich, toluene, dichloromethane, and hexane from Rankem, and ethanol (absolute) from Analytical CS Reagent. Aniline (a) was procured from Qualigens, cardanol (C, technical grade) from Satya Cashew Chemicals Pvt. Ltd, India, and formalin from Fisher Scientific.2.2. Measurements
The synthesis of biobased BZ monomers was performed both in a microwave reactor (Anton Paar, Monowave 200) and a ball mill (planetary, RETSCH PM 200; vibrating mechanochemical mixer, RETSCH MM 400). In a microwave reactor, the temperature was controlled by an IR sensor and the power limited to 800 W. The structures of the monomers were confirmed using proton (1H) and carbon (13C) nuclear magnetic resonance, 13C distortionless enhancement by polarization transfer (DEPT), 2D NMR, namely, 1H–1H COSY (correlation spectroscopy), 1H–13C heteronuclear single quantum correlation (HSQC), and heteronuclear multiple bond correlation (HMBC) experiments performed using a Bruker 400 MHz using chloroform-d (CDCl3) as a solvent and tetramethyl silane (TMS) as an internal standard. Dimethyl sulphoxide-d6 (DMSO-d6) was used as a solvent in the temperature-dependent NMR study. Proton signals are labelled in lower case letters, while carbon signals are represented with the same in capital case. FTIR spectra were recorded using a Nicolet iS20 mid-infrared FTIR spectrometer equipped with an interferometer with a KBr/Ge-coated beam splitter and dynamic alignment, a thermoelectrically cooled (TEC) DTGS detector, and an attenuated total reflectance diamond (iD5-ATR) accessory. Spectra were recorded in the 4000–400 cm−1 range with a resolution of 0.25 cm−1, and each spectrum was co-added with 32 scans. Mass spectrometry analysis was performed using an Agilent HRMS Q-ToF 6540 Series in the electrospray ionization (ESI) mode. The crystal data of the monomer were collected from X-ray diffraction data acquired using a D8 Venture Bruker AXS single-crystal X-ray diffractometer equipped with a CMOS PHOTON 100 detector with monochromatized microfocus sources (Mo Kα = 0.71073 Å) at different temperatures (16, 40 and 70 °C). The crystal structure was determined using the SHELX software, which was integrated in APEX 3. The non-H atoms were found using different Fourier syntheses and refined with anisotropic thermal parameters. All the hydrogen atoms were placed in their calculated places and refined using a riding model and HFIX instructions. A single crystal of monomer was obtained using a co-solvent mixture (hexane/dichloromethane 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). GC-MS analyses of monomers were performed using an Agilent 5977B GC/MSD with a HP-5 MS column (30 m × 0.25 mm × 0.25 μm) with septum purge flow (3 mL min−1), split ratio of 30:1, and split flow of 30 mL min−1 at a thermal aux temperature of 250 °C. The polymerization behaviour of monomers was determined using a DSC-3 differential scanning calorimeter (Star System, Mettler Toledo). Samples (4 ± 1 mg) were placed in hermetic sealed aluminium pans and heated from 25 to 350 °C at 10 °C min−1 under a constant flow rate of nitrogen at 50 mL min−1. Thermogravimetric analysis (TGA) of the monomers and polymers was performed using a Mettler Toledo thermogravimetric analyzer (TGA) with a built-in gas controller (TGA2 SF/1100) and fitted using an XP1U TGA balance (ultramicrobalance) at a flow rate of 50 mL min−1 of nitrogen and a heating rate of 10 °C min−1 in the temperature range of 35–800 °C. The limiting oxygen index (LOI) of the polymers was calculated from char yield (CY) obtained in the TGA data using van Krevelen and Hoftyzer71eqn (1):
:1 v/v). GC-MS analyses of monomers were performed using an Agilent 5977B GC/MSD with a HP-5 MS column (30 m × 0.25 mm × 0.25 μm) with septum purge flow (3 mL min−1), split ratio of 30:1, and split flow of 30 mL min−1 at a thermal aux temperature of 250 °C. The polymerization behaviour of monomers was determined using a DSC-3 differential scanning calorimeter (Star System, Mettler Toledo). Samples (4 ± 1 mg) were placed in hermetic sealed aluminium pans and heated from 25 to 350 °C at 10 °C min−1 under a constant flow rate of nitrogen at 50 mL min−1. Thermogravimetric analysis (TGA) of the monomers and polymers was performed using a Mettler Toledo thermogravimetric analyzer (TGA) with a built-in gas controller (TGA2 SF/1100) and fitted using an XP1U TGA balance (ultramicrobalance) at a flow rate of 50 mL min−1 of nitrogen and a heating rate of 10 °C min−1 in the temperature range of 35–800 °C. The limiting oxygen index (LOI) of the polymers was calculated from char yield (CY) obtained in the TGA data using van Krevelen and Hoftyzer71eqn (1):| LOI = 17.5 + 0.4 × CY | (1) |
2.3. Solvent-free synthesis of intermediates and benzoxazine monomers
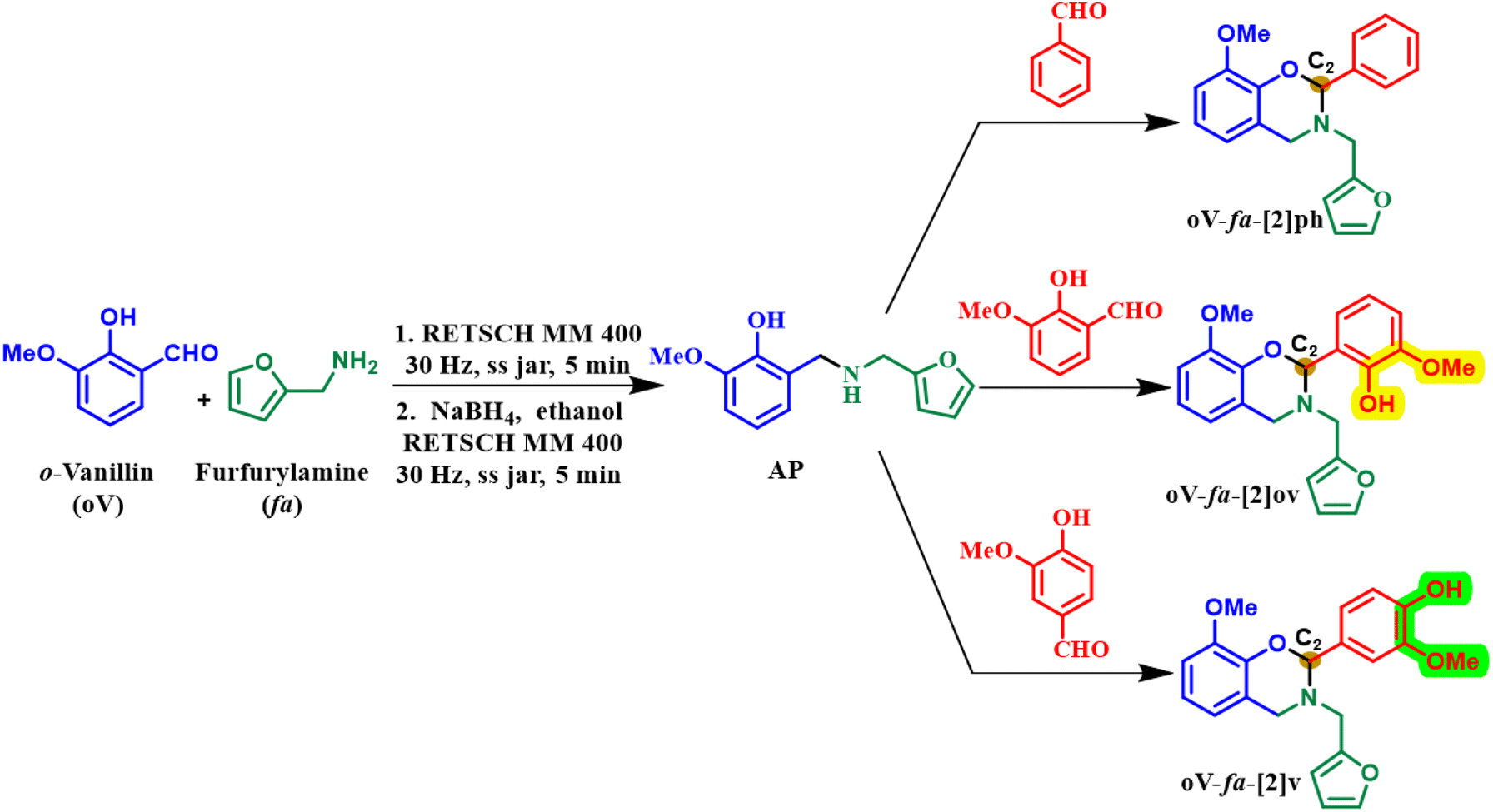 | ||
| Scheme 1 A generalized synthetic scheme of oxazine-ring-substituted BZ monomers using microwave (MW), mixer mill (MM) and planetary mill (PM). | ||
| Monomer | Aldehyde | Conventional heating | Microwave reactor (MW) | Mechanochemical synthesis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mixer mill (MM) | Planetary mill (PM)a | |||||||||
| Time (h) | Yield (%) | Power (W) | Time (min) | Yield (%) | Time (min) | Yield (%) | Time (min) | Yield (%) | ||
| a 12 mmol of aldehyde was used. | ||||||||||
| oV-fa-[2]ph | Benzaldehyde | 6 | 75 | 100 | 30 | 76 | 15 | 72 | — | — |
| oV-fa-[2]ov | o-Vanillin | 3 | 53 | — | 10 | 60 | 5 | 56 | — | — |
| oV-fa-[2]v | Vanillin | 12 | 47 | 500 | 20 | 53 | 30 | 20 | 25 | 52 |
2.3.2.1. Conventional heating method. The reactants aminophenol (AP, 1 mmol) and respective aldehyde (0.7 mmol) were taken in a 25 mL round-bottom flask and stirred at 65 °C for x h (Table 1). The crude product was then washed with warm hexane (3 × 5 mL) and ethanol (3 × 5 mL) to yield the pure BZ monomers.
2.3.2.2. Microwave (MW) method. Reactants were taken in a G10 microwave reaction vial (with operational volume: 2–6 mL) and the reaction was performed under MW irradiation at a constant power and a temperature of 45 °C.
2.3.2.3. Mechanochemical method. In a mechanochemical mixer mill (MM), reactants were taken in a 5 mL reaction vial, along with three stainless steel (ss) balls (two of size 5 mm and one of 7 mm), and kept at 25 Hz, whereas, in the case of planetary mill (PM), the synthesis was performed at 8 Hz in a 250 mL stainless steel (ss) jar with 60 ss balls of diameter 0.5 cm.
oV-fa-[2]ph. 1H NMR (400 MHz, CDCl3, ppm): δ 7.5822 (d, 2H), 7.4093 (s, 1H), 7.3475 (t, 2H), 7.2985 (d, 1H), 6.8095 (m, 2H), 6.5202 (qd, 1H), 6.3275 (qd, 2H), 6.2527 (d, 1H), 6.0782 (s, 1H), 3.9591 (s, 3H), 3.9365 (d, 2H), 3.8261 (s, 2H). 13C NMR (100 MHz, CDCl3, ppm): δ 152.39, 148.00, 142.56, 142.48, 138.84, 128.54, 128.12, 126.68, 120.15, 119.58, 110.29, 109.92, 108.76, 90.52, 56.12, 47.79, 46.11. HRMS (M + H)+ calcd for C20H19NO3, 322.1365; found, 322.1442. FTIR (cm−1): oxazine ring skeletal C–H bending out-of-plane vibration (926 cm−1), CAr–O–C symmetric (1015 cm−1) and anti-symmetric (1220 cm−1) stretching bands, and C–N–C symmetric stretching vibrations (1269 cm−1).
oV-fa-[2]ov. 1H NMR (400 MHz, CDCl3, ppm): δ 9.9342 (s, 1H), 7.4210 (s, 1 H), 6.8894 (s, 1 H), 6.8501 (s, 1 H), 6.8322 (s, 1 H), 6.7129 (s, 1 H), 6.4765 (s, 1 H), 6.3128 (d, 1H), 6.2864 (d, 1H), 6.0850 (s, 1H), 4.1055 (d, 2 H), 3.8767 (s, 6 H), 3.8121 (s, 2 H). 13C NMR (100 MHz, CDCl3, ppm): δ 150.29, 148.18, 148.05, 144.45, 143.20, 141.00, 122.35, 121.02, 119.46, 118.94, 118.60, 112.23, 110.36, 110.14, 109.62, 87.36, 56.07, 48.47, 44.71. HRMS (M + H)+ calcd for C21H21NO5, 368.1420; found, 368.1492.
FTIR (cm−1): oxazine ring skeletal C–H bending out-of-plane vibration (918 cm−1), CAr–O–C symmetric (1003 cm−1) and anti-symmetric (1233 cm−1) stretching bands, and C–N–C symmetric stretching vibrations (1265 cm−1).
oV-fa-[2]v. 1H NMR (400 MHz, CDCl3, ppm): δ 7.3926 (s, 1H), 7.1388 (t, 2H), 6.8968 (m, 1H), 6.8873 (m, 2H), 6.5430 (dd, 1H), 6.3278 (t, 1H), 6.2265 (d, 1H), 5.9990 (s, 1H), 5.5971 (s, 1H), 3.9411 (s, 2H), 3.9213 (s, 2H), 3.8767 (s, 6H). 13C NMR (100 MHz, CDCl3, ppm): δ 152.43, 147.9936, 146.69, 145.53, 142.42, 130.60, 120.21, 119.83, 119.60, 114.36, 110.28, 109.92, 109.35, 108.70, 90.47, 56.10, 56.05, 50.93, 47.19, 46.56. HRMS (M + H)+ calcd for C21H21NO5, 368.1420; found, 368.1489. FTIR (cm−1): oxazine ring skeletal C–H bending out-of-plane vibration (918 cm−1), CAr–O–C symmetric (1029 cm−1) and anti-symmetric (1210 cm−1) stretching bands, and C–N–C symmetric stretching vibrations (1259 cm−1).
2.4. Hydrogen bonding studies by 1H NMR
A concentration-dependent NMR study was performed to determine the nature of H-bonding, intra- or inter-molecular, in the oV-fa-[2]ov monomer. 1H NMR of a known concentration of oV-fa-[2]ov (5, 10, 15, and 20 mM) in CDCl3 was recorded. A temperature-dependent study was performed to estimate the strength of H-bonding by recording NMR of oV-fa-[2]ov (10 mM in DMSO-d6) at different temperatures (25 to 70 °C).2.5. Preparation and polymerization cycle for homopolymers and copolymers
In a Teflon container, a solution of 4th generation BZ monomer (oV-fa-[2]ph or oV-fa-[2]ov or oV-fa-[2]v) in THF (1 g mL−1) was separately prepared and physically blended with a 1st generation (C–a) monomer to obtain a blend of three weight ratios (x:y as 1:1, 1:3 and 3:1). Upon homogenization, the solvent was slowly evaporated in a vacuum oven at 50 °C. The copolymers are abbreviated as OpxCay, OoxCay, and OvxCay, where x and y are the weight percentage ratio of the 4th generation (Op = oV-fa-[2]ph, Oo = oV-fa-[2]ov and Ov = oV-fa-[2]v) and 1st generation (Ca) monomers, respectively. The copolymers are represented as Op1Ca1, Op1Ca3, Op3Ca1; Oo1Ca1, Oo1Ca3, Oo3Ca1; Ov1Ca1, Ov1Ca3 and Ov3Ca1.
The polymerization of monomers and blends was performed in an air oven by sequential heating for 1 h each to obtain homopolymers and copolymers.
:1 w/w and additional heating at 210 °C for 1:1 and 1:3 w/w.
2.7. Dynamic mechanical analysis using a rheometer
An MCR302 (Anton Paar) rheometer fitted with a parallel plate geometry and a convection temperature device (CTD – 450) was used to perform dynamic mechanical analysis (DMA). A sample was placed on a disposable lower measuring plate (25 mm) and sandwiched with the upper plate (8 mm) and heated from room temperature to a specific temperature (180 °C for oV-fa-[2]ph; 170 °C for oV-fa-[2]ov, and 160 °C for oV-fa-[2]v) at a heating rate of 2 °C min−1, and further kept at this temperature (neat) or 210 °C (blends) for 1.5 h to ensure complete polymerization. The crosslinked polymer was subjected to an oscillatory mode with amplitude γ = 0.5% and a frequency of 1 Hz.2.8. Preparation of thermosetting resins for Lap Shear Strength (LSS) and mechanical properties
Single-lap-joint adhesively bonded metal specimens with monomers as neat or blends were subjected to tension loading (metal-to-metal, mild steel coupons). These tests were conducted using an INSTRON 3366 equipped with a 10 kN load cell at a strain rate of 0.002 s−1, following the procedure outlined in ASTM D1002-10. Solvent casting of a monomer solution (40 mg mL−1) in ethyl acetate onto the pre-marked region (15 × 15 mm2) of mild steel coupons (65 × 15 mm2) with an average roughness of 0.16 ± 0.02 μm was performed. The plates were secured with binder clips at the coated overlap area and thermally polymerized in an air oven using the above-mentioned polymerization cycle. The bonded overlap area was measured using digital calipers before testing. The adhesion strength, represented as the lap shear strength (τ), was calculated using eqn (2), and the adhesive failure mode was assessed visually.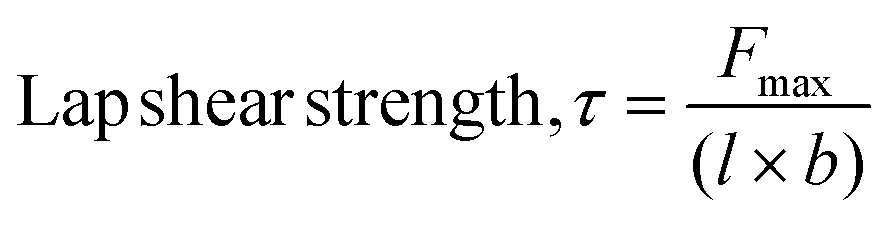 | (2) |
The mechanical properties were evaluated using a Universal Testing Machine (UTM Instron-3366, USA) mechanical tester and rectangular polymeric specimens were made with a sample size of 65 × 1 × 0.1 mm3, at a strain rate of 0.5 mm s−1.
2.9. Swelling studies
The samples were individually soaked in different solvents at 25 °C to ascertain the solvent resistance for the obtained polymers. At different time intervals, the samples were removed from the solvent and after every 24 h, the solvent was replaced. Kimwipes were used to gently remove the adsorbed solvent on the surface. The samples are immediately weighed to a 10−4 g precision on a balance. The swelling ratio was calculated using eqn (3). After soaking the polymers in solvents, the polymer was wiped with a clean tissue paper to remove excess solvents from the surface. The samples were weighed immediately in a weighing balance with an accuracy of 10−4 g.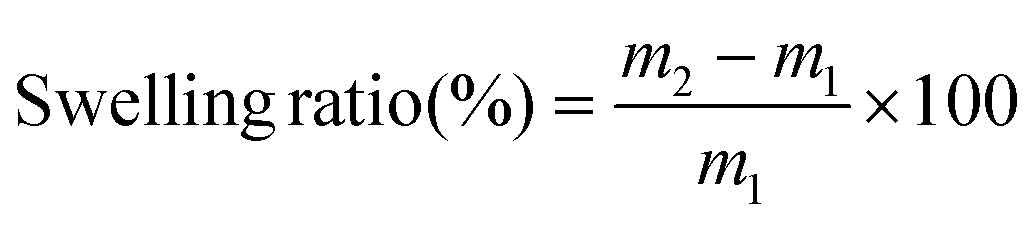 | (3) |
Statistical analysis was performed using the GraphPad Prism Version 7.04 software. Student's two-tailed t-test was performed using the mean from the minimum of 3–6 independent observed values, and the respective p values are reported.
3. Results and discussion
3.1. Synthesis and characterization of monomers
Solventless sustainable techniques were used to prepare three monomers using biobased feedstocks via a common aminophenol (AP) derivative, Scheme 1, to study the effect of the phenolic–OH (abbreviated as Hphenolic) on the polymerization reaction. Reference monomer, oV-fa-[2]ph, has no Hphenolic, while the other two fully biobased monomers oV-fa-[2]ov and oV-fa-[2]v, bear Hphenolic either located proximal or distal from the reactive oxazine centre (C2).A telescoping mechanochemical procedure of reductive amination of o-hydroxy aldehyde derivative, o-vanillin, and furfuryl amine was performed involving an initial condensation reaction to form the Schiff base, followed by in situ reduction to form the aminophenol (AP) derivative. A series of C2-substituted BZ monomers were prepared in one pot by solvent-free condensation of AP with three different aldehydes (benzaldehyde, vanillin, and o-vanillin) for the ring closure reaction. The methodologies adopted for this step were conventional heating, microwave and mechanochemical. Without the need for bulk dissolution of reactants via typical heating of reactants, both milling and microwave irradiation assisted in achieving easy chemical transformation to form BZ monomers in good yields. Synthesis via conventional heating occurred in 3–12 h (47–75%), where the yield is the highest for benzaldehyde. Similarly, microwave reactions followed the same trend with the reaction occurring relatively fast (10–30 min) with good conversion observed in a mechanochemical mixer (MM) process (5–30 min). Despite several attempts, the condensation of AP with vanillin in MM resulted in a poor yield (20%, 30 min). An easier transformation with an improved yield (53%, 25 min) is achieved in planetary mill (PM) ascribed to the difference in the application of mechanical force, “planetary” motion of the jar with rotation and spinning around a central and own axis, compared with MM. Such creation of centrifugal forces provides the gravitational effects which is similar to roller mills with an industrial scale-up potential.
The structure of the synthesized intermediate and monomers was confirmed by FTIR, 1H and 13C NMR spectroscopy, and high-resolution electrospray ionization mass spectrometry. BZ monomers revealed the characteristic FTIR peaks due to the oxazine ring, CAr–O–C2 anti-symmetric (1212–1245 cm−1) and symmetric (1075 cm−1) stretching bands, and the oxazine ring skeletal C–H bending out-of-plane vibrations (912–922 cm−1) (Fig. S1†). A broad band at 3333 cm−1 was observed in the case of the oV-fa-[2]v monomer corresponding to the Hphenolic, while it remain unnoticed in oV-fa-[2]ov indicating involvement in intramolecular H-bonded formation. From 1H NMR, the number of protons clearly match the integral value of the NMR signals, confirming the successful formation of monomers (Fig. 3a, S2a, and S3a†). The characteristic methine proton O–C2(Ph)–N, “a” signal, at the C2 centre of the oxazine-ring-substituted BZ monomers, was observed in the range of 5.99–6.08 ppm as a singlet. The methylene protons due to N–CH2–Ar “b” and N–CH2–fa “c” are expected to appear as a doublet of doublet (dd) due to geminal coupling with a prochiral centre at C2.63
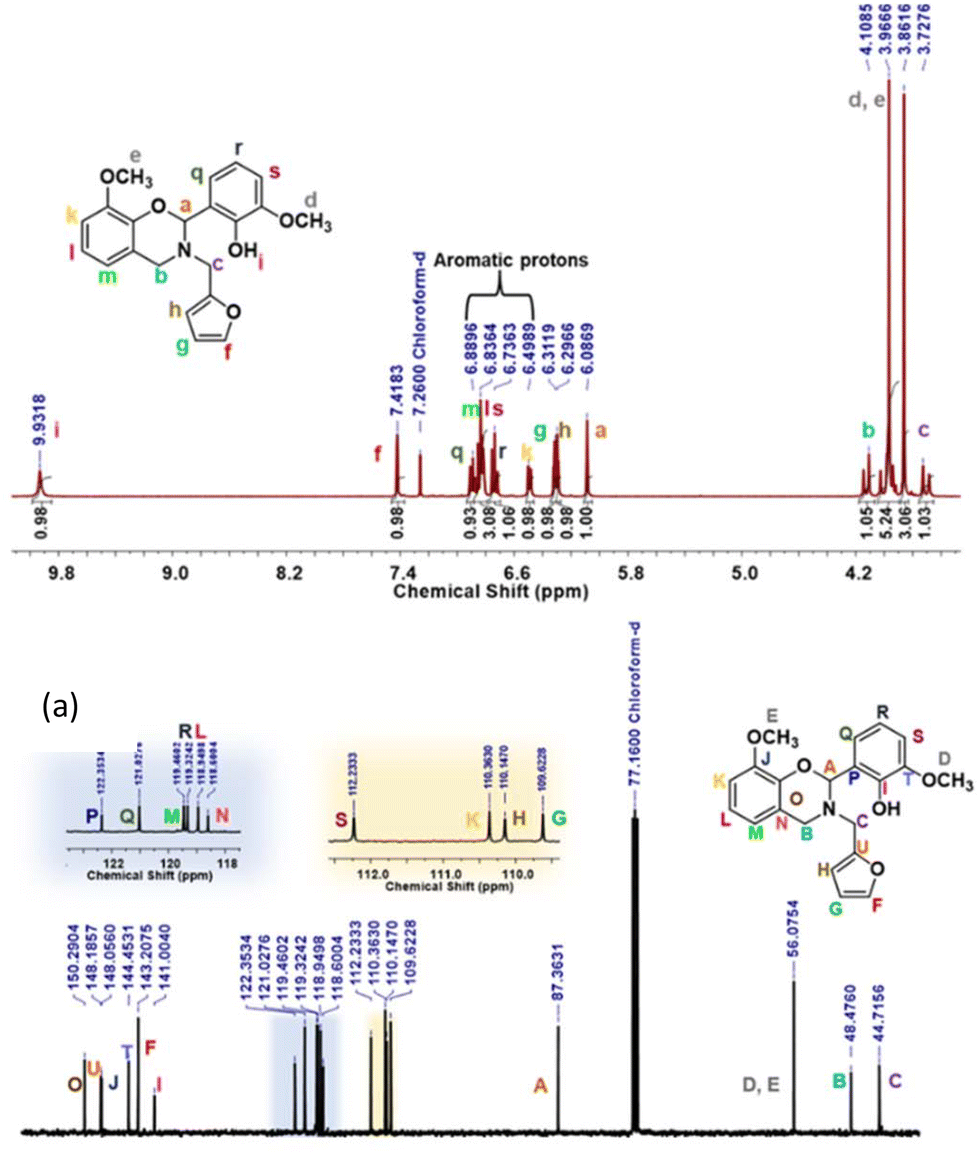 | ||
| Fig. 3 The NMR spectra of oV-fa-[2]ov: (a) 1H and (b) 13C. | ||
However in all the synthesized monomers, methylene proton signals N–CH2–Ar (b, 3.85–4.1 ppm) and N–CH2–fa (c, 3.72–3.94 ppm) appeared as multiplets due to the co-existence of –OCH3 signal(s) (d and e) in the similar range. The structural characterization of monomers was also confirmed by 13C NMR spectra (Fig. 3b, S2b, and S3b†). The characteristic carbons due to O–CH(Ph)–N, N–CH2–Ar, N–CH2–fa and –OCH3 are also successfully assigned as A (87.36 to 90.52 ppm), B (44.71 to 46.60 ppm), C (48.47 to 56.12 ppm) and D and E (56.12 ppm), confirming the successful formation of monomers. This assignment of protons and carbons was assisted by 2D and DEPT NMR analyses (Fig. S4–S6†). Mass spectrometry confirmed the formation of the monomers (Fig. S7†).
3.3. Intramolecular hydrogen bonding and the latent-catalytic effect
There are no reports that discuss the in-built latent effect of Hphenolic in lowering the polymerization temperature of fully biobased 4th generation BZs. It is anticipated that, besides the stability of iminium ion intermediates due to oxazine-ring substitution at C2, the presence of acidic hydrogen may endow an inherent additional capability to catalyze the polymerization reaction.73 Both acidity and availability of Hphenolic in the monomer is expected to catalyze the polymerization reactions at low temperatures. To gain a better insight into the nature of the possible hydrogen bonding within the oV-fa-[2]ov monomer, a series of designed NMR spectroscopy experiments and SCXRD analyses were performed, and the results are shown in Fig. 4. Understanding the chemical shift of the Hphenolic proton signal is influenced by the nature of the solvent, and to estimate the relative strength of hydrogen bonding, 1H NMR spectra in CDCl3 (weak hydrogen-bonding donors and acceptors) and DMSO-d6 (strong hydrogen-bonding acceptors) were recorded. A signal of integral intensity of one proton at δ = 9.942 and 9.422 ppm in CDCl3 and DMSO-d6, respectively was noticed. To confirm and assign Hphenolic in the monomer (Fig. 4a), a drop of D2O in the NMR tube was added, and the contents were vigorously shaken. Prior to re-recording NMR, D2O was easily removed, as it is immiscible and less dense than CDCl3. The disappearance of the signal confirmed the exchange of labile Hphenolic with deuterium. The concentration-dependent NMR study revealed that the chemical shift of the Hphenolic signal remains unchanged, supporting its participation in the intramolecular hydrogen bonding.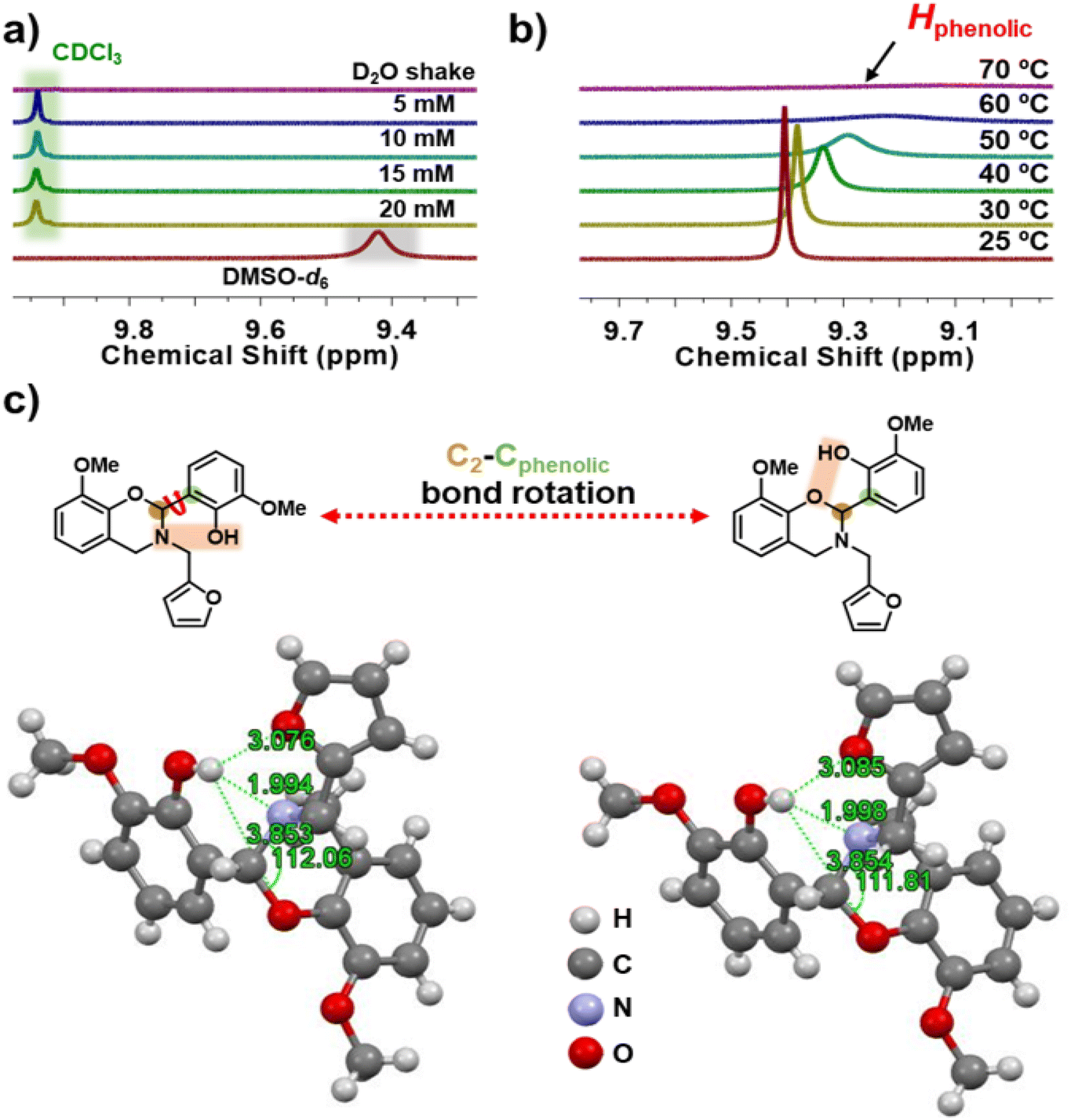 | ||
| Fig. 4 The effect on H-bonding. Stacked zoomed-in 1H NMR spectra for oV-fa-[2]ov: (a) concentration-dependent study without and with D2O shake; recorded in CDCl3. (b) A temperature-dependent study with shifting of the phenolic proton resonance value; recorded in DMSO-d6. (c) A representative chemical structure of possible conformers of oV-fa-[2]ov mediated by temperature. An ORTEP diagram of oV-fa-[2]ov recorded at 16 °C (left) and 70 °C (right) [ORTEP diagram at 40 °C, is provided in Fig. S8†]. | ||
The key parameters to estimate the relative strength of hydrogen bonding is the difference in chemical shift [Δδ = δ(DMSO-d6) − δ(CDCl3)] and hydrogen bond acidity [ANMR = 0.0065 + 0.133 × (Δδ)] values, which are calculated as 0.52 and 0.069, respectively, which confirmed Hphenolic involvement in a very strong intramolecular H-bonding interaction.22,74 From a variable temperature NMR study (Fig. 4b), a well-defined Hphenolic signal upfield clearly shifted (lower ppm) with significant signal broadening upon increase in the temperature-supported temperature-driven weakening of intramolecular hydrogen bonding, and ultimately, the signal vanished at 70 °C. Undoubtedly, the hydrogen bonding becomes sufficiently weak with temperature to assist the release of the bonded proton “trapped-state” as in the six-membered ring to the “free-state”.
Learning from the above-mentioned solution-based NMR study and correlating with bulk polymerization, we were able to perform a similar study in solid state using single crystals obtained for one of the monomers along with the confirmation of the full 3D structure. The asymmetric unit, ORTEP diagram for oV-fa-[2]ov of formula C21H21NO5 is represented in Fig. 4c. The symmetry of space group P−1 generates a triclinic unit cell [a = 7.9414(6), α = 71.148°(3) b = 9.2292(7), β = 82.961°(3) c = 13.4539(11), γ = 81.232°(3)]. Complete data can be found in the CIF file, freely available with the CCDC number 2291158 (16 °C), 2291159 (40 °C) and 2291694 (70 °C)† at the Cambridge Crystallographic Data Centre. The successful claim of a strong 6-membered hydrogen bonding, with the bond distance between hydrogen and nitrogen (Hphenolic⋯Noxazine) determined as 1.994 Å lies within the range of the covalent interactions, as confirmed by SCXRD. The characteristic O–C2(Ph)–N angle which is prone to heterolytic cleavage to assist ROP got reduced from 112.06, which is less than that of the earlier unsubstituted and substituted BZ structures,45,64 supporting prudential role of functionalities at the 2-substituted oxazine-ring of the monomer. To gain better insights of the temperature dependency on the strength of hydrogen bonding for the same crystal, SCXRD data were recorded at three different temperatures (16, 40 and 70 °C). Due to the limitation of acquiring SCXRD data at elevated temperatures (>70 °C) for prolonged time and a low melting temperature of the monomer, the analysis was performed up to 70 °C. The ORTEP diagram for oV-fa-[2]ov at 40 and 70 °C is presented in Fig. S8† and Fig. 4c, respectively. Table S1† reports the main crystal data and refinement parameters for oV-fa-[2]ov at each temperature. As expected, with the increase in temperature, the bond distances Hphenolic⋯Noxazine, Hphenolic⋯Ooxazine, and Hphenolic⋯Ofuran were altered. Interestingly, the Hphenolic⋯Noxazine distance increased from 1.994 to 1.998 Å, while the Hphenolic⋯Ooxazine distance decreased from 3.853 to 3.850 Å and the characteristic O–C2(Ph)–N angle got reduced from 112.06 to 111.81°. Even though the changes are infinitesimal, they clearly indicate an increase in ring strain at the O–C2(Ph)–N angle and approach of Hphenolic to Ooxazine. Eventually, temperature-guided geometric conformations may offer a possibility of the C2–Cphenolic bond rotation, enabling the proximity of Hphenolic to the other neighbouring proton acceptor (Ooxazine) to create intramolecular O–Hphenolic⋯Ooxazine hydrogen bonds via a quasi-chelating ring to assist ROP in the dual mode, both due to the increase in angle strain and latent catalytic effects.62 As per earlier reported data, the latent catalyst can be activated by simply breaking the hydrogen bonding upon heating and can facilitate the acid-catalyzed polymerization of the BZs.
3.2. Thermal behaviour of the monomers
To determine the polymerization characteristics and mass loss during polymerization of the monomers, DSC, GC-MS and TGA were performed. From Fig. 5 (summarized in Table 2), the existence of two events in DSC thermograms was noticed, a first sharp endothermic peak represents a sharp melting point (Tm), supporting the high purity of each monomer and the second exotherm peak is associated with the ring-opening polymerization temperature (Tp). oV-fa-[2]ph, oV-fa-[2]ov, and oV-fa-[2]v showed Tm as 105, 119, and 128 °C and Tp as 214, 189, and 181 °C, respectively. Clearly, the observed temperature characteristics support the effect and nature of secondary bond interactions with (intra- vs. inter-) and without H-bonding within the structures. The ring-opening reaction occurs by the cleavage of O–C2(Ph)–N position and facilitated by the protonation of oxazine-ring heteroatoms. The existence of inherent acidic Hphenolic induced catalysis of ROP reactions, thus led to a lower polymerization temperature than the one without it. Such intramolecular proton transfer process is expected to be relatively facile and favoured due to the proximity of ortho Hphenolic to the reactive C2-centre of the oxazine ring. Unexpectedly, although Hphenolic is remotely located from the reaction centre at para-position, it still showed the lowest Tp value. This could be ascribed to the extent of proton transfer in the equilibria and strength of H-bonding, which is clearly higher for a six-membered ring, enabling a slight restriction for the migration of the bound acidic protons from one centre Hphenolic to the other heteroatoms in the oxazine ring as compared to the relatively free intermolecularly bound Hphenolic. Earlier work reported that unsubstituted BZ suffers from significant mass losses during polymerization which is highly undesired as both the yield and quality of the polymer network are affected. Mukherjee et al.63 reported that PH-fa shows a mass loss of 80%, out of which 64% occurred prior to polymerization.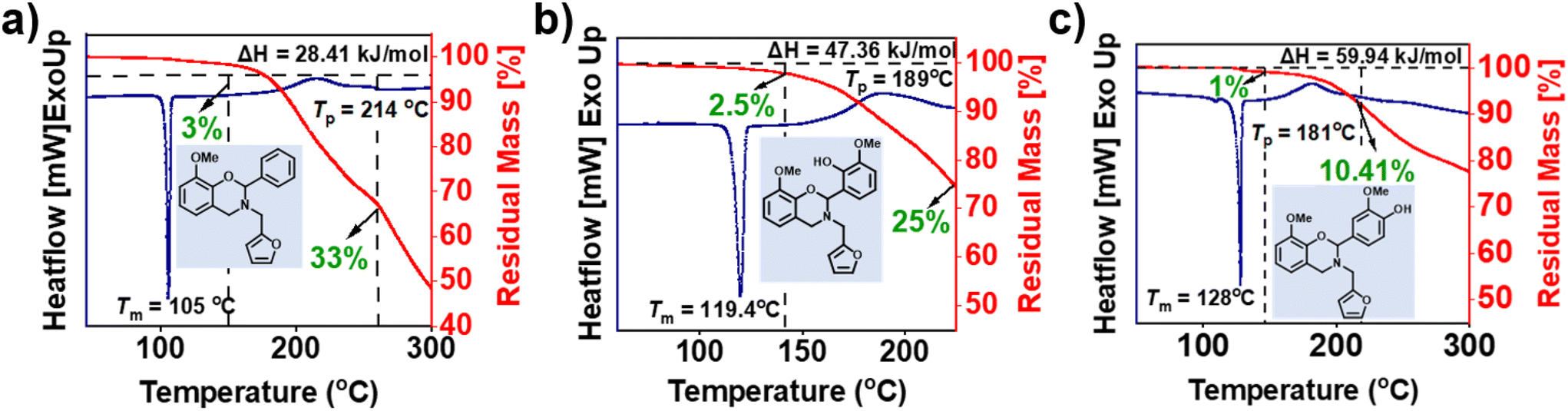 | ||
| Fig. 5 (a–c) The DSC thermograms of the monomers at a heating rate of 10 °C min−1 under an N2 atmosphere. | ||
| Monomer | Monomer | Polymer | Char yielda (%) | LOI | |||||
|---|---|---|---|---|---|---|---|---|---|
| T polymerization (°C) | ΔH (kJ mol−1) | E a modified Ozawa (kJ mol−1) | T decomposition (°C) | ||||||
| T o | T p | T 5% | T 10% | T max | |||||
| a At 800 °C. b Value in parenthesis is obtained from the Kissinger–Akahira–Sunose (KAS) method. | |||||||||
| oV-fa-[2]ph | 149 | 214 | 28.41 | 100.7 (103.7)b | 299 | 318 | 357 | 40 | 33.5 |
| oV-fa-[2]ov | 142 | 189 | 47.36 | 134.4 (135.84)b | 306 | 323 | 361 | 42 | 34.3 |
| oV-fa-[2]v | 147 | 181 | 59.94 | 88.29 (99.17)b | 307 | 326 | 348 | 47 | 36.3 |
From the TGA analysis of monomers (Fig. 5), it was observed that both fully biobased monomers, oV-fa-[2]ov and oV-fa-[2]v, showed a lower overall mass and eventually reduced by three-fold than that observed in oV-fa-[2]ph. Thus, the introduction of a phenyl group in the oxazine ring at C2 is quiet promising to minimize such mass loss issues. In general, the higher the ΔH value, the higher the ease of polymerization. In our case, ΔH and %mass loss of monomers followed a reverse trend ascribed to the concomitant mass loss during polymerization. Thus, the obtained ΔH values require a careful interpretation. If we assume that the presence of –OH and –OMe groups in the phenyl ring at C2 in both oV-fa-[2]ov and oV-fa-[2]v reduces the number of available reaction sites for electrophilic aromatic substitution polymerization reactions at this benzene ring, then a reduced ΔH value is expected. Conversely, higher ΔH values are obtained in both than oV-fa-[2]ph, supporting the higher susceptibility of these monomers towards electrophilic aromatic substitution. Among oV-fa-[2]ov and oV-fa-[2]v, the lower ΔH value of the former could be an induction of steric interference due to substituents and neighbouring furan ring at the C2 reactive centre for polymerization, consequently making it prone to higher volatilization.
To gain mechanistic insights into the mass loss behaviour and to investigate the effect of a monomer structure on the nature of volatiles liberated during polymerization, GC-MS analysis was performed. The method applied and spectra obtained are presented in Fig. 6 and Table S2,† respectively. The monomers undergo dissociation, and the fragments liberated were captured and detected. Interestingly, the first chromatogram appeared at only 6 min and a much lower temperature of 97 °C for oV-fa-[2]ph, while it was observed at a delayed time of ∼11.5 min and a higher temperature of 150 °C for both oV-fa-[2]ov and oV-fa-[2]v. This indicates the appreciable thermal stability of the latter monomer structures in congruence to TGA of monomers. The volatiles are rich in phenolic and benzene fragments in oV-fa-[2]ph, while oV-fa-[2]ov liberated all possible different mass fragments, and oV-fa-[2]v showed predominance of substituted benzene species. Notably, a higher abundance of furan fragments was observed at 12 min in oV-fa-[2]ov than in oV-fa-[2]v, indicating the dangling nature and steric congestion towards the polymer network growth due to the proximity of –OH/OMe groups in the former monomer. At 222 °C, a broad vs. sharp elution chromatogram was obtained in phenyl vs. vanillin-based monomers, respectively.
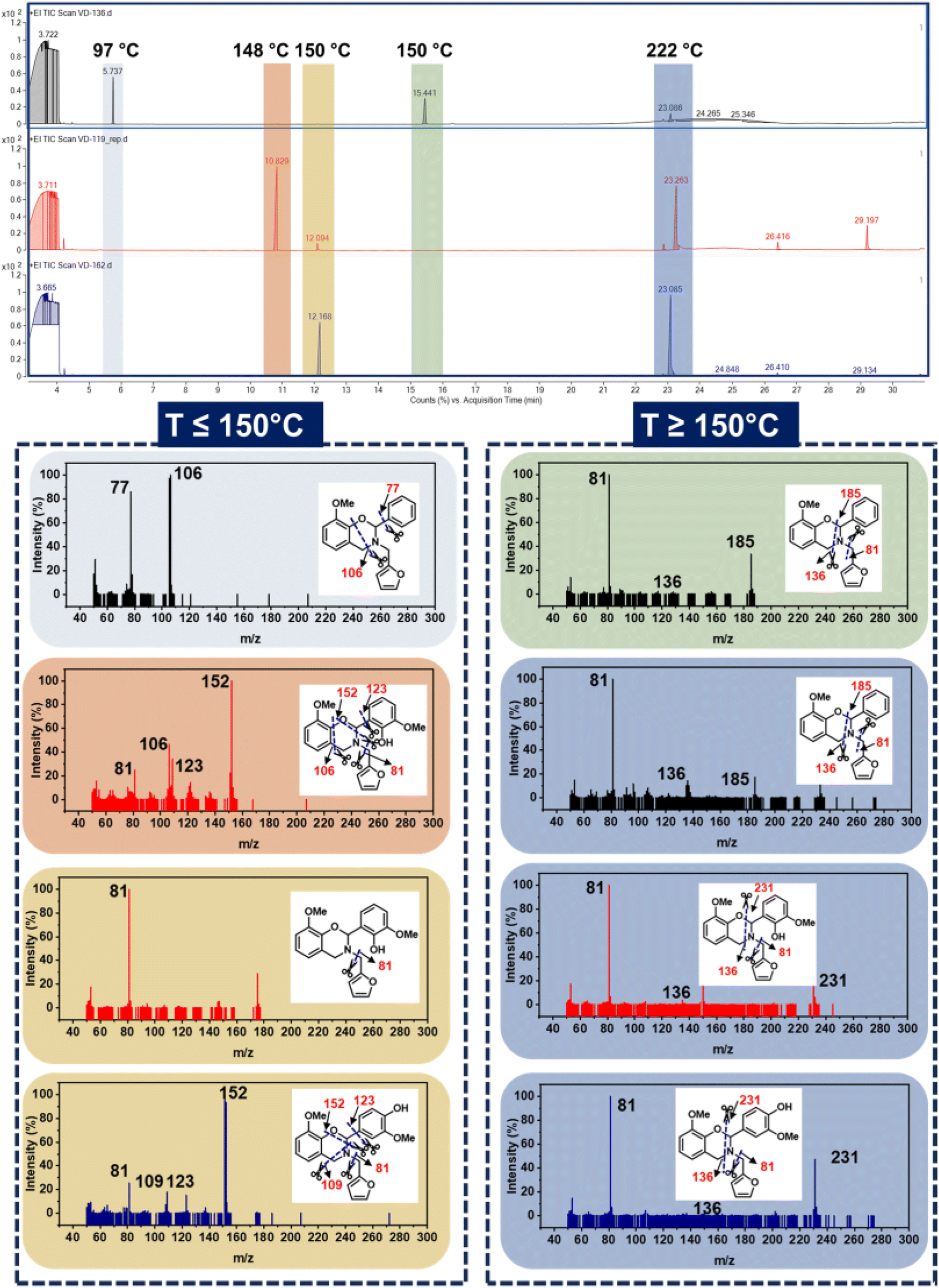 | ||
| Fig. 6 The GC/MS chromatogram of the monomers and the corresponding fragmentation pattern and m/z (Da) values. | ||
The liberation of furan-based imines in each monomer type is mass driven, 185 Da being lighter showcased relatively higher volatilization than the imine (231 Da) derivative from the other two monomers, i.e., oV-fa-[2]ov/v. Consequently the residual mass left to polymerize is seemingly rich in furan units, retain monomer features, furan and substituted benzene units for oV-fa-[2]ph, oV-fa-[2]ov, and oV-fa-[2]v, respectively. Clearly, this indicates the promising thermal stability and resistance to volatilization offered by fully biobased monomers.
FTIR kinetics was further performed to determine the temperature at which the characteristic oxazine absorption bands in the synthesized monomers are affected due to their participation in the ROP. Fig. S9† shows significant broadening of the absorption bands (1212–1245 cm−1, C–O–C antisymmetric stretching mode and 912–922 cm−1 BZ related mode) at 130 °C in both oV-fa-[2]ov and oV-fa-[2]v, while a similar effect is noticed at 160 °C in oV-fa-[2]ph, inferring the catalyzing effect induced in the ring-opening reaction due to Hphenolic. Temperature-dependent SCXRD studies supported the mapping of phenolic–OH proximity more to the oxazine N than to O; however, to investigate whether Hphenolic transfers fast intra- or intermolecularly, the calculations of activation energy (Ea) of polymerization were performed using the Kissinger–Akahira–Sunose (KAS)75 and modified Ozawa methods. A sequence of DSC traces at different heating rates of 5, 7.5, 10, 15, and 20 °C min−1 were recorded. From the slope of the plots of ln(β/Tp2) and ln(β) against 1/Tp, the activation energy of polymerization was calculated using eqn (4) and (5) for the KAS and modified Ozawa methods:
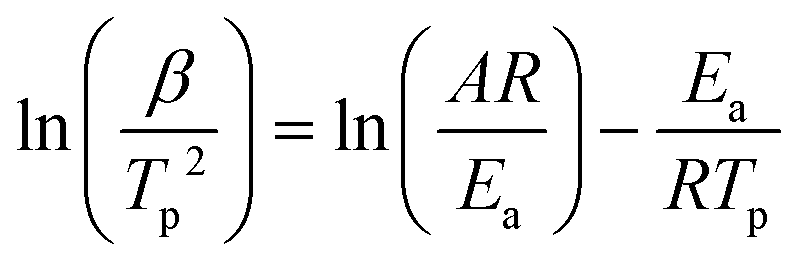 | (4) |
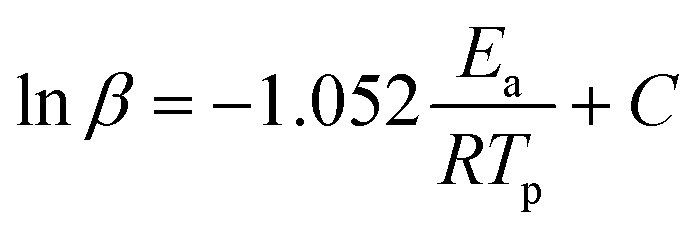 | (5) |
Fig. 7a–c shows the plots obtained for the KAS and modified Ozawa methods using respective DSC thermograms of the monomers (details are provided in Table S3†). The Ea values for polymerization were calculated from the slope, which follows the order oV-fa-[2]ov (134 kJ.mol−1) > oV-fa-[2]ph (101 kJ.mol−1) > oV-fa-[2]v (88 kJ.mol−1). From this trend, it can be interpreted that kinetically, the ROP is most favoured in the monomer having Hphenolic at the para-position than the one having no additional functional groups attached and followed by the monomer having Hphenolic at the ortho-position of the C2(Ph) entity. This is accounted to the availability of more propagation sites free from steric congestion at the reactive centre and resonance stabilization of the intermediate iminium ion (Fig. 7a′–c′) to dictate the extension of the polymer network.
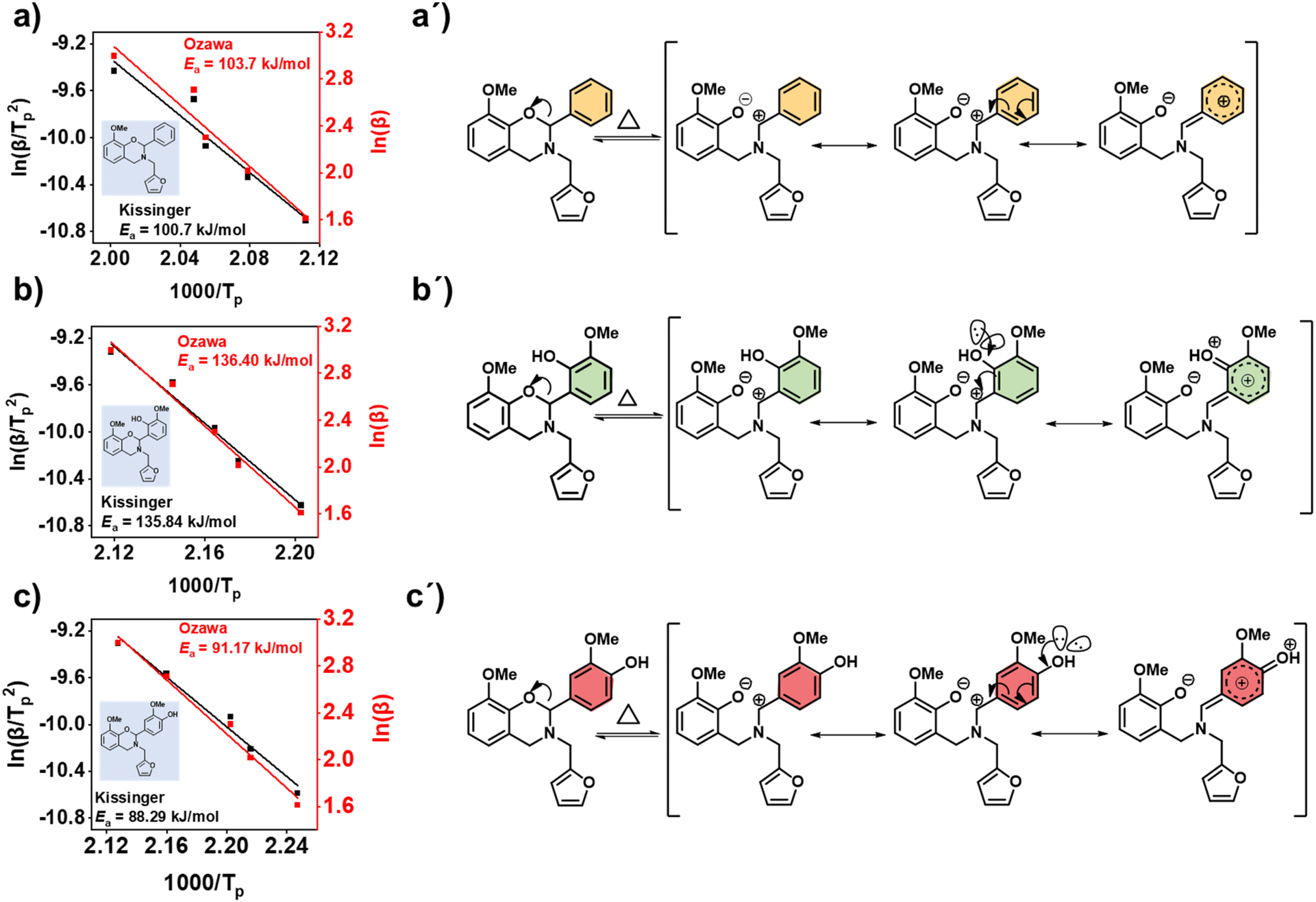 | ||
| Fig. 7 (a–c) The plots of activation energy Ea calculated by the Kissinger–Akahira–Sunose (KAS) (black) and modified Ozawa (red) methods for the polymerization reaction. (a′–c′) The stability of the ring-opened iminium ion intermediate due to charge delocalization in the functionalised un/substituted benzene structure in the respective monomer. For the sake of comparable analysis Hphenolic at C2 of benzene ring is not shifted to the hetero-atoms in the oxazine-ring. | ||
3.4. Thermal properties of the polybenzoxazine
Monomers were thermally polymerized in a convection oven prior to thermal stability analysis. Post confirmation of complete polymerization by IR, DSC, and solubility tests in different solvents, TGA was recorded, and thermograms are presented in Fig. 8. The characteristic parameters of thermal stability such as initial thermal stability, temperatures at 5% mass loss (Td5% = 299–307 °C) and 10% mass loss (Td10% = 318–326 °C), maximum mass loss (Tmax = 348–361 °C), and char yield (CY = 40–47%) were obtained, and the results are presented in Table 2. The synthesized PBZs, poly(oV-fa-[2]ph/ov/v), displayed very high thermal stability and appreciable char yield (CY) and limiting oxygen index (LOI) comparative of other first- and fourth-generation PBZs.58,60,76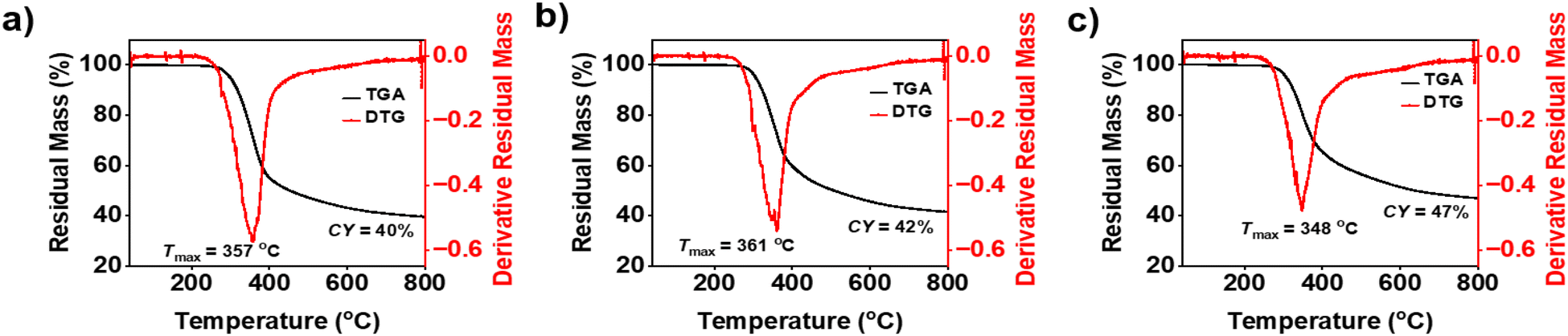 | ||
| Fig. 8 TGA thermograms for (a) poly(oV-fa-[2]ph), (b) poly(oV-fa-[2]ov) and (c) poly(oV-fa-[2]v) at a heating rate of 10 °C min−1 under a N2 atmosphere. | ||
Besides benzene rings, the participation of a furan ring in cross-linking reactions accounted for improved char yields.77,78 High decomposition temperatures, char yields and LOI values are indicative of the effectiveness of bio-synthons to form halogen-free flame-resistant sustainable polymers.79
To ensure crosslinking, we performed swelling studies of neat polymers and copolymers in different solvents (acetone, hexane, water, dimethyl sulfoxide, ethanol, toluene and dichloromethane). The variation in the mass of samples when soaked in different solvents was monitored after every 24 h for four consecutive days. The relative swelling ratios are shown in Fig. S11.† It was observed that the swelling ratio of neat polymers and copolymers remains nearly unaffected in water, hexane, and ethanol. A lower swelling ratio in water and ethanol followed by DMSO suggests the highly hydrophobic nature of the samples. The homopolymers of the 4th generation swelled up significantly in dichloromethane and revealed insignificant colouration of the solvent after 3 days, which is not evident in case of copolymers with the 1st generation monomer, confirming their improved solvent resistance.
3.5. Mechanical properties
PBZs are exciting candidates for adhesive applications owing to the existence of polar functionalities, which extend the favourable temperature for the polymerization of the synthesized BZs, and the high thermal stability of the so-formed polymers is worthy of exploring interactions at the interfaces in this direction. The requirement of low temperature for polymerization of the synthesized BZs, and high thermal stability of the so-formed polymers is worthy to explore in this direction. In general, adhesive bearing both soft and hard segments empower higher lap shear strength (LSS), a measure of adhesive capability. Another natural abundant renewable phenol sourced 1st generation oxazine-ring unsubstituted primitive BZ monomer, cardanol–aniline (C–a), was chosen as the comonomer to bulk copolymerize with the synthesized 4th generation BZs to enable soft segments within copolymers, poly(1st generation-co-4th generation), due to its low-crosslink density, flexible-nature, and low viscosity. Unsubstituted primitive BZ, C–a, was synthesized using a usual one-step multicomponent condensation (details are provided in ESI†) reaction of cardanol (C), aniline (a), and formaldehyde.The adhered coupons using various homo- and copolymers based on bare BZ monomers and their monomer blends (1:1 or 1:3 or 3:1 w/w) are analyzed using an Instron instrument. The pictorial representation of the assembly used to measure the adhesive strength along with the LSS values obtained is presented in Fig. 9a and b. Expectedly, all the neat 4th generation PBZs showed poor adhesion strength (LSS = 0.1 kg cm−2) ascribed to the formation of a rigid and brittle polymer network. On the contrary, C–a based PBZ showed LSS = 20 kg cm−2. The copolymers with C–a with oV-fa-[2]ph/ov/v showed a much higher adhesive strength of 51 kg cm−2 and 37 kg cm−2 at the respective optimum comonomer feed-in ratio, signifying the benefits gained due to the co-existence of hard and soft segments. The thermo-mechanical properties of 4th generation homopolymers and their copolymers was assessed by rheological studies. From Fig. S12,† it is clear that the storage modulus of poly(oV-fa-[2]v) is highest compared to poly(oV-fa-[2]ph) followed by poly(oV-fa-[2]ov). No rubbery plateau is observed in poly(oV-fa-[2]ph), indicating that the molecular weight of polymers is low involving an assemblage of units with broad molecular weight distribution. Usually, for a low crosslinked thermoset expected to show a low Tg value, and for such polymers, the storage modulus begins to decrease at much low temperature.
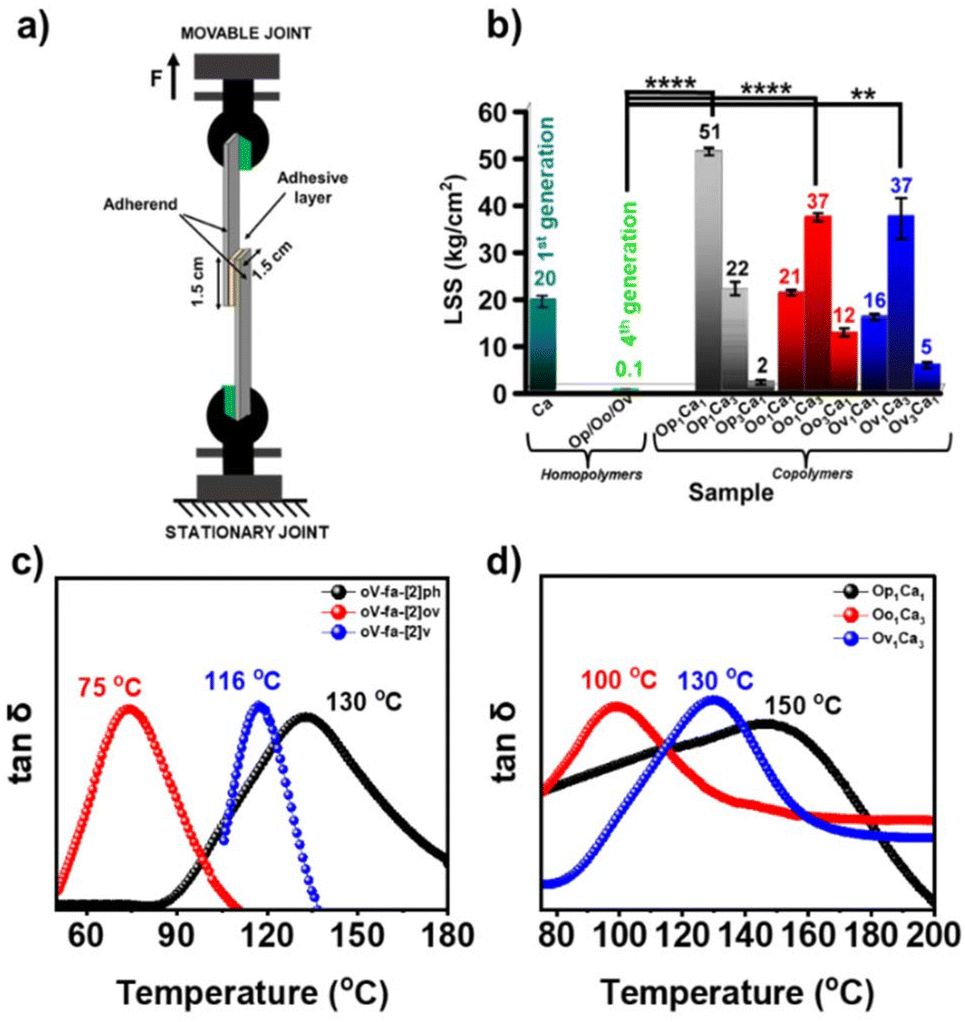 | ||
| Fig. 9 Mechanical properties. (a) Adhesive strength measurements. A schematic illustration of the LSS measurements using a polymer as the adhesive. (b) The adhesive strength of the homo- and copolymers determined at 35 °C. Bars display mean ± S.E. ****p < 0.0001 and **p < 0.0024. The dynamic mechanical thermal analysis study of samples. Damping factor (tanδ) plots of (c) homopolymers and (d) copolymers. | ||
The observed poly(oV-fa-[2]ov) has low storage modulus than poly(oV-fa-[2]ph) and poly(oV-fa-[2]v), but instead of the decreasing storage modulus, it began to increase with the temperature, indicating the reorganization of a quasi-network to more stable network structure. Expectedly, poly(oV-fa-[2]v) showed a significant high storage modulus and a sharp tanδ peak indicating the formation of a well-defined polymer network. The reason why tanδ is high despite a broad peak observed in the case of poly(oV-fa-[2]ph) is a separate matter and not very well understood. Certainly, the reactivity and stability of iminium ion intermediates to propagate into a crosslinked network vs. the stability of the iminium ion, electrophilic centre, via the respective phenyl derivative (substituted vs. unsubstituted) is another governing parameter. However, a high network heterogeneity indicate “partial” cross-linking and broadening of the tanδ plot (Fig. 9c). An increase in the glass transition temperature (Tg), which is determined as the temperature at the tanδ peak, increases for each polymer in the following order copolymer poly(oV-fa-[2]ov) < poly(oV-fa-[2]v) < poly(oV-fa-[2]ph). The same trend is observed in their respective analyzed copolymers with C–a, with an increased Tg value (Fig. 9d).
The absence of independent polymerizing exothermic peaks due to individual monomers (4th generation and 1st generation C–a, Tp = 260 °C) in the DSC plot (Fig. S13†) confirmed the successful copolymerization reaction. Furthermore, the beneficial effect of the ring-opened structures of these C2-substituted monomers enabled a lowering in C–a copolymerization temperature. The knitting-in of C–a units within the 4th generation polymer network may have led to reinforcement of the copolymeric network, altering the molecular structure with restricting mobility of polymer chains, thus improving the overall thermal (Fig. S14†) and mechanical properties. Moreover, this is due to increased viscoelastic dissipation and simultaneously the highest and broad tanδ accounting for the strongest adhesion characteristics in case of oV-fa-[2]ph and its copolymer.
Following the same optimized composition of samples used for adhesive strength measurements, bulk mechanical properties were also determined. The samples were cured without any reinforcing fillers at atmospheric pressure in a Teflon mould in an air oven, which accounted for their low mechanical properties. The samples showed a nearly blister-free composite (Fig. S15†), indicating good processability and nearly zero mass loss during polymerization. The tensile stress–strain curves of polybenzoxazines, prepared from pure cardanol–aniline (C–a) and its different blends with other 4th generation benzoxazine monomers, revealed an elastic response with minimal plastic deformation and finally terminated by brittle failure (Fig. S16†), typical of a thermoset-associated rigid network. Except Ov1Ca3, which additionally showed a relatively sustained plateau of deformation at a high strain, likely a result of bucking plastic yielding before brittle fracture. The tensile strength, modulus, and elongation at the break of samples is presented in Fig. S16c and d.† C–a being unsubstituted mono-oxazine (compared to 4th generation monomers) produced a polymer with low cross-linking densities, and therefore, it showed overall inferior mechanical properties. The comonomer not only allowed low-temperature polymerization of C–a, but also provided densification of the network structure that empowered a steady increase in stress and stiffness. A more plasticizing effect of Oo1Ca3 than that of Ov1Ca3 due to dangling methoxy and hydroxy functionalities near the oxazine ring C2 centre in the former comonomer contributed to relatively high tensile strength and elongation at break.
4. Conclusions
The present work demonstrated a practical and scalable mechanochemical method for the synthesis of a series of 4th generation benzoxazines via a simple high-yielding fast low-energy consumption reaction. We further demonstrated the viability of vanillin as a dual-functional reactant as both the phenol and aldehyde to form C2 aryl-substituted monomers, showcasing the benefits of functionality-enriched natural molecules in benzoxazine chemistry. Without any need for external catalysts, the ROP of benzoxazines was achieved fast and at low temperatures due to the existence of inherent acidic phenolic–OH functionalities within the monomer, enabling further greening of the classical ROP of benzoxazines. Mechanistic insights into the latent catalyst effect provide opportunities to structurally design new monomers unravelling a new direction to this class of low temperature polymerizable systems. Additionally, we further demonstrated the viability of these monomers as effective comonomers and catalysts for earlier generation monomers with better thermal, mechanical, and adhesive properties. By demonstrating a facile synthesis procedure and showcasing the versatility of these greener monomers, we further advance the design of inherent latent catalyst comonomers to form superior performing copolymers for many upcoming practical applications.Author contributions
The manuscript was written with the contributions of all authors. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VD, SY, and BL would like to acknowledge the financial support from the Shiv Nadar Foundation and the SERB-Power Research Grant (Grant No. SPG/2021/002537). The authors thank Satya Cashew Chemicals Pvt. Ltd (India) for gifting cardanol for the research work.References
- H. Ishida and D. J. Allen, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 1019–1030 CrossRef CAS.
- B. Lochab, M. Monisha, N. Amarnath, P. Sharma, S. Mukherjee and H. Ishida, Polymers, 2021, 13, 1260 CrossRef CAS PubMed.
- H. Ishida and Y. Rodriguez, J. Appl. Polym. Sci., 1995, 58, 1751–1760 CrossRef CAS.
- J. Dunkers and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1913–1921 CrossRef CAS.
- Y. Wang and H. Ishida, Polym. Mater.: Sci. Eng., 1999, 81, 114–115 CAS.
- J. Sun, W. Wei, Y. Xu, J. Qu, X. Liu and T. Endo, RSC Adv., 2015, 5, 19048–19057 RSC.
- J. Wang, Y. Z. Xu, Y. F. Fu and X. D. Liu, Sci. Rep., 2016, 6, 38584 CrossRef.
- A. Kocaarslan, B. Kiskan and Y. Yagci, Polymer, 2017, 122, 340–346 CrossRef.
- A. Sudo, R. Kudoh, H. Nakayama, K. Arima and T. Endo, Macromolecules, 2008, 41, 9030–9034 CrossRef.
- C. Liu, D. Shen, R. M. Sebastián, J. Marquet and R. Schönfeld, Macromolecules, 2011, 44, 4616–4622 CrossRef.
- C. Wang, C. Zhao, J. Sun, S. Huang, X. Liu and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2016–2023 CrossRef.
- T. Zhang, L. Bonnaud, J.-M. Raquez, M. Poorteman, M. Olivier and P. Dubois, Polymers, 2020, 12, 415 CrossRef PubMed.
- P. Sharma, M. Srivastava, B. Lochab, D. Kumar, A. Ramanan and P. K. Roy, ChemistrySelect, 2016, 1, 3924–3932 CrossRef.
- I. Biru, C. Damian, S. Gârea and H. Iovu, Eur. Polym. J., 2016, 83, 244–255 CrossRef.
- M. Monisha, N. Yadav and B. Lochab, ACS Sustainable Chem. Eng., 2019, 7, 4473–4485 CrossRef.
- K. Zhang, P. Froimowicz, L. Han and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3635–3642 CrossRef.
- G. Kaya, B. Kiskan and Y. Yagci, Macromolecules, 2018, 51, 1688–1695 CrossRef CAS.
- X. Y. He, T. Wang, Z. C. Pan, A. Q. Dayo, J. Wang and W. B. Liu, J. Appl. Polym. Sci., 2021, 138, 50131 CrossRef CAS.
- M. Monisha, S. Sahu and B. Lochab, Biomacromolecules, 2021, 22, 4408–4421 CrossRef CAS PubMed.
- P. Froimowicz, K. Zhang and H. Ishida, Chem. – Eur. J., 2016, 22, 2691–2707 CrossRef CAS PubMed.
- M. Arslan, React. Funct. Polym., 2019, 139, 9–16 CrossRef CAS.
- K. Zhang, Y. Liu, M. Han and P. Froimowicz, Green Chem., 2020, 22, 1209–1219 RSC.
- S. Mukherjee, N. Amarnath, M. Ramkumar and B. Lochab, Macromol. Chem. Phys., 2022, 223, 2100458 CrossRef CAS.
- M. Stępień, A. Marszałek-Harych, M. Gazińska, A. Gągor, Ł. John and J. Ejfler, Macromolecules, 2023, 56, 5730–5742 CrossRef.
- Y. Lyu and H. Ishida, Prog. Polym. Sci., 2019, 99, 101168 CrossRef CAS.
- E. Calò, A. Maffezzoli, G. Mele, F. Martina, S. E. Mazzetto, A. Tarzia and C. Stifani, Green Chem., 2007, 9, 754–759 RSC.
- L. R. V. Kotzebue, F. W. M. Ribeiro, V. G. Sombra, J. P. Feitosa, G. Mele, S. E. Mazzetto and D. Lomonaco, Polymer, 2016, 92, 189–200 CrossRef CAS.
- N. Teng, S. Yang, J. Dai, S. Wang, J. Zhao, J. Zhu and X. Liu, ACS Sustainable Chem. Eng., 2019, 7, 8715–8723 CrossRef CAS.
- H. Xu, W. Zhang, Z. Lu and G. Zhang, RSC Adv., 2013, 3, 3677–3682 RSC.
- P. Froimowicz, C. R. Arza, L. Han and H. Ishida, ChemSusChem, 2016, 9, 1921–1928 CrossRef CAS.
- L. Dumas, L. Bonnaud, M. Olivier, M. Poorteman and P. Dubois, J. Mater. Chem. A, 2015, 3, 6012–6018 RSC.
- N. Sini, J. Bijwe and I. K. Varma, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 7–11 CrossRef CAS.
- N. Yadav, M. Monisha, R. Niranjan, A. Dubey, S. Patil, R. Priyadarshini and B. Lochab, Carbohydr. Polym., 2021, 254, 117296 CrossRef PubMed.
- C. Wang, J. Sun, X. Liu, A. Sudo and T. Endo, Green Chem., 2012, 14, 2799–2806 RSC.
- M. A. L. Salum, D. Iguchi, C. R. Arza, L. Han, H. Ishida and P. Froimowicz, ACS Sustainable Chem. Eng., 2018, 6, 13096–13106 CrossRef.
- T. Periyasamy, S. P. Asrafali, S. Muthusamy and S.-C. Kim, New J. Chem., 2016, 40, 9313–9319 RSC.
- K. Zhang, M. Han, Y. Liu and P. Froimowicz, ACS Sustainable Chem. Eng., 2019, 7, 9399–9407 CrossRef.
- L. R. Kotzebue, J. S. R. de Oliveira, J. B. da Silva, S. E. Mazzetto, H. Ishida and D. Lomonaco, ACS Sustainable Chem. Eng., 2018, 6, 5485–5494 CrossRef.
- L. Dumas, L. Bonnaud, M. Olivier, M. Poorteman and P. Dubois, Eur. Polym. J., 2016, 81, 337–346 CrossRef.
- J. Liu, Y. Wuliu, J. Dai, J. Hu and X. Liu, Eur. Polym. J., 2021, 157, 110671 CrossRef.
- M. Comí, G. Lligadas, J. C. Ronda, M. Galia and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4894–4903 CrossRef.
- Y. Lu and K. Zhang, Eur. Polym. J., 2021, 156, 110607 CrossRef.
- W. Zhao, B. Hao, Y. Lu and K. Zhang, Eur. Polym. J., 2022, 166, 111041 CrossRef.
- F. Li, W. Zhang and J. Wang, Eur. Polym. J., 2023, 199, 112466 CrossRef.
- N. Amarnath, S. Shukla and B. Lochab, ACS Sustainable Chem. Eng., 2019, 7, 18700–18710 CrossRef.
- X. Liu, R. Zhang, T. Li, P. Zhu and Q. Zhuang, ACS Sustainable Chem. Eng., 2017, 5, 10682–10692 CrossRef.
- Q. Chen, D. Ren, W. Sheng and K. Zhang, J. Appl. Polym. Sci., 2023, 141(2), e54792 CrossRef.
- L. Dumas, L. Bonnaud, M. Olivier, M. Poorteman and P. Dubois, Green Chem., 2016, 18, 4954–4960 RSC.
- J. Dai, N. Teng, Y. Peng, Y. Liu, L. Cao, J. Zhu and X. Liu, ChemSusChem, 2018, 11, 3175–3183 CrossRef.
- J. Dai, S. Yang, N. Teng, Y. Liu, X. Liu, J. Zhu and J. Zhao, Coatings, 2018, 8, 88 CrossRef.
- M. Monisha, N. Yadav, S. B. Srivastava, S. P. Singh and B. Lochab, J. Mater. Chem. A, 2018, 6, 2555–2567 RSC.
- A. Ghosh, S. Shukla, M. Monisha, A. Kumar, B. Lochab and S. Mitra, ACS Energy Lett., 2017, 2, 2478–2485 CrossRef.
- O. Bayram, B. Kiskan, E. Demir, R. Demir-Cakan and Y. Yagci, ACS Sustainable Chem. Eng., 2020, 8, 9145–9155 CrossRef.
- S. Sahu, R. Niranjan, R. Priyadarshini and B. Lochab, Chemosphere, 2023, 328, 138587 CrossRef.
- X. Yuan, X. Su, Y. Wang, L. Liu, R. Li and C. Wang, ACS Appl. Polym. Mater., 2023, 5, 5650–5661 CrossRef.
- H. Yan, C. Sun, Z. Fang, X. Liu, J. Zhu and H. Wang, Polymer, 2016, 97, 418–427 CrossRef.
- R. Yang, M. Han, B. Hao and K. Zhang, Eur. Polym. J., 2020, 131, 109706 CrossRef.
- S. Ohashi, F. Cassidy, S. Huang, K. Chiou and H. Ishida, Polym. Chem., 2016, 7, 7177–7184 RSC.
- I. Machado, I. Hsieh, E. Rachita, M. L. Salum, D. Iguchi, N. Pogharian, A. Pellot, P. Froimowicz, V. Calado and H. Ishida, Green Chem., 2021, 23, 4051–4064 RSC.
- R. C. Pereira, L. R. Kotzebue, D. Zampieri, G. Mele, S. E. Mazzetto and D. Lomonaco, Mater. Today Commun., 2019, 21, 100629 CrossRef.
- R. Tavernier, L. Granado, G. Foyer, G. David and S. Caillol, Polymer, 2021, 216, 123270 CrossRef.
- R. Tavernier, L. Granado, M. Tillard, L. Van Renterghem, T.-X. Métro, F. Lamaty, L. Bonnaud, J.-M. Raquez, G. David and S. Caillol, Polym. Chem., 2022, 13, 5745–5756 RSC.
- S. Mukherjee, N. Amarnath and B. Lochab, Macromolecules, 2021, 54, 10001–10016 CrossRef.
- S. Mukherjee and B. Lochab, Chem. Commun., 2022, 58, 3609–3612 RSC.
- T. Agag, J. Liu, R. Graf, H. W. Spiess and H. Ishida, Macromolecules, 2012, 45, 8991–8997 CrossRef CAS.
- R. Tavernier, L. Granado, G. Foyer, G. David and S. Caillol, Macromolecules, 2020, 53, 2557–2567 CrossRef CAS.
- K. Martina, L. Rotolo, A. Porcheddu, F. Delogu, S. R. Bysouth, G. Cravotto and E. Colacino, Chem. Commun., 2018, 54, 551–554 RSC.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- K. S. McKissic, J. T. Caruso, R. G. Blair and J. Mack, Green Chem., 2014, 16, 1628–1632 RSC.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef.
- D. Van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- S. Shukla, A. Mahata, B. Pathak and B. Lochab, RSC Adv., 2015, 5, 78071–78080 RSC.
- K. Zhang, L. Han, P. Froimowicz and H. Ishida, Macromolecules, 2017, 50, 6552–6560 CrossRef CAS.
- M. H. Abraham, R. J. Abraham, W. E. Acree Jr., A. E. Aliev, A. J. Leo and W. L. Whaley, J. Org. Chem., 2014, 79, 11075–11083 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- C. R. Arza, P. Froimowicz, L. Han, R. Graf and H. Ishida, Macromolecules, 2017, 50, 9249–9256 CrossRef CAS.
- Y. L. Liu and C. I. Chou, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5267–5282 CrossRef CAS.
- X. Shen, J. Dai, Y. Liu, X. Liu and J. Zhu, Polymer, 2017, 122, 258–269 CrossRef CAS.
- A. Van, K. Chiou and H. Ishida, Polymer, 2014, 55, 1443–1451 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of C-a, NMR (1H, 2D (HMBC, HSQC, COSY), 13C, and DEPT of monomers), FTIR and DSC data of monomers, FTIR kinetics of polymerization, ORTEP diagram of oV-fa-[2]ov at 40 °C, crystal data for compound at variable temperatures, GC-MS method, swelling studies, DMA analysis and tensile stress–strain plots. CCDC 2291158 (16 °C), 2291159 (40 °C) and 2291694 (70 °C). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc03522k |
| This journal is © The Royal Society of Chemistry 2024 |