
Three-component coupling reaction of white phosphorus, alcohols and diaryl disulfides: a chlorine-free avenue for accessing phosphorothioates†
Yinwei
Cao
,
Mengpei
Bai
,
Junwei
Huang
,
Fushan
Chen
*,
Yan
Liu
,
Guo
Tang
 * and
Yufen
Zhao
* and
Yufen
Zhao
Department of Chemistry, College of Chemistry and Chemical Engineering, and the Key Laboratory for Chemical Biology of Fujian Province, Xiamen University, Xiamen, Fujian 361005, China. E-mail: 17706925334@163.com; t12g21@xmu.edu.cn
First published on 1st December 2023
Abstract
Synthesis of valuable compounds directly from small and simple molecules is an area of great interest in chemical research. However, the preparation of most organophosphorus compounds from white phosphorus currently relies on inefficient, stepwise procedures that result in significant environmental pollution and poor atom economy. We herein describe a novel and high-yielding route to generate various phosphorothioates with diverse substituents from white phosphorus, disulfides, and alcohols in a single reaction step. This innovative three-component coupling reaction utilizes basic industrial raw materials and offers several advantages, including operational simplicity, mild reaction conditions, a broad substrate scope, and high product selectivity.
Introduction
White phosphorus (P4) is a crucial raw material in the production of phosphorus compounds, with an estimated annual production exceeding 1 million tons.1 The current process for activating P4 primarily involves its oxidation to phosphorus trichloride (PCl3) and subsequent replacement of chloride with suitable nucleophiles.2–4 This process suffers from various issues such as inefficiency, being unsafe, high energy consumption, and significant waste product formation. Hence, green and efficient routes for the direct conversion of P4 into value-added organophosphorus compounds (OPCs) are highly valuable and significant from both economic and environmental perspectives.5–8Phosphorothioate derivatives are essential chemical motifs in organic synthesis, and pharmaceutical, and agricultural chemistry.9,10 Numerous efforts have been made to study P–S–C bond formation, leading to the development of various strategies over the past few decades.11 The most commonly used method is the Michaelis–Arbuzov reaction, which involves nucleophilic substitution of sulfenyl halides ArSX or other sulfur-based reagents with phosphites (Scheme 1a).12,13 The Atherton–Todd-type reaction, typically performed using toxic tetrachloromethane as a halogenating reagent and solvent, provides an alternative route for these scaffolds (Scheme 1b).14–16 However, both of the two prominent reactions have limitations, including the use of moisture-sensitive and foul-smelling regents as well as harsh reaction conditions. In 2016, Han and co-workers conducted a palladium-catalyzed cross-dehydrogenative coupling (CDC) of P(O)H compounds and thiols to produce phosphorothioates, which featured broad substrate scopes and good functional groups compatibility, but required stoichiometric amount of styrene and high reaction temperature (Scheme 1c).17 Subsequently, Jiao et al. disclosed a Cs2CO3-catalyzed, transition-metal-free oxidative CDC reaction (Scheme 1c).18 Other researchers, including our group, have developed phosphorothiolation methods for various substituted aryl-precursors using elemental sulfur as the S-atom source (Scheme 1d).19,20
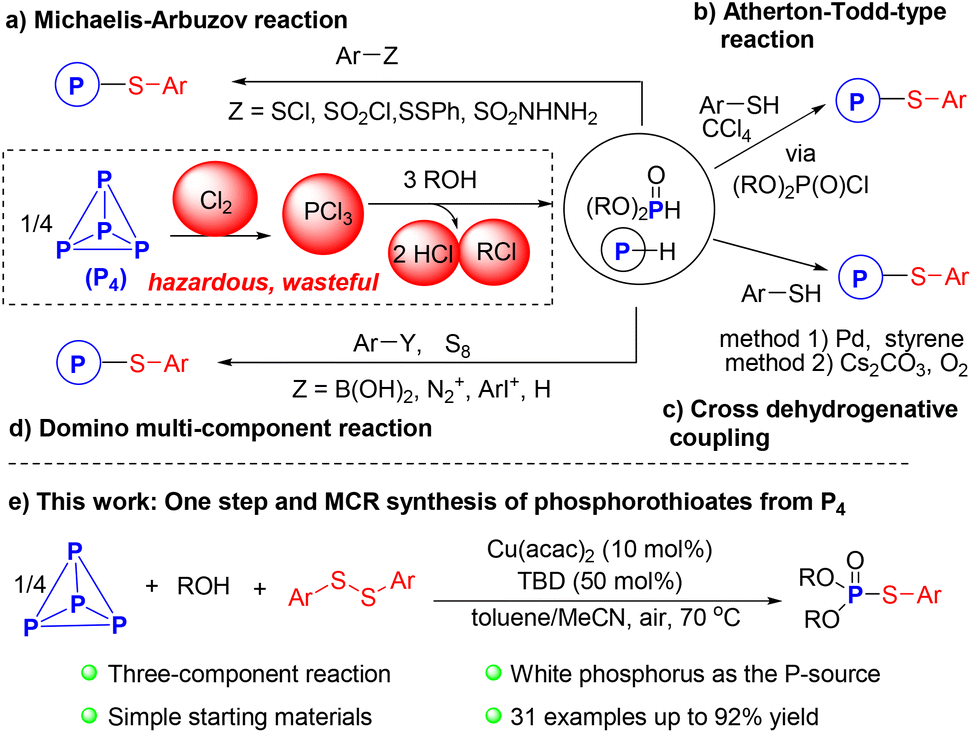 | ||
| Scheme 1 Synthetic methods to the phosphorothioate derivatives. | ||
Despite these impressive advances, many phosphorus reagents used in those strategies must be pre-prepared from PCl3.4 A step-economic reaction utilizing basic industrial raw materials and environmentally friendly building blocks for the preparation of phosphorothioates remains highly desirable.21–25 However, to the best of our knowledge, direct functionalization of P4 for the synthesis of phosphorothioates has not been reported in the literature due to the challenge of cleaving all six P–P bonds of the P4 tetrahedron and orderly installing different atoms at phosphorus center.26–32 As a continuum of our efforts in developing green methodologies for the selective transformation of P4 into useful P1 products without hazardous chlorination steps,33–37 we report the first copper-catalyzed three-component coupling reaction of P4, alcohols, and aryl disulfides for the synthesis of phosphorothioates under mild conditions, exhibiting high phosphorus conversion (Scheme 1e).
Results and discussion
We commenced our project by employing cheap and commercially available ethanol (2a) and diphenyl disulfide (3a) as model substrates to direct functionalization of P4 (1a). When a solution of P4 (0.20 mmol P atom), 2a (2.00 mmol), 3a (0.30 mmol), Cu(acac)2 (0.02 mmol) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 0.10 mmol) in toluene-MeCN (0.4 mL −2.0 mL) was heated at 70 °C under air, the product 4a was formed in 94% yield (entry 1 in Table 1). Other catalysts were tested, such as Fe(acac)2, Co(acac)2, Ni(acac)2, CuCl, CuBr, Cu(CH3CN)PF6, CuSO4, and Cu(OTf)2, but none provided satisfactory outcomes compared to Cu(acac)2 (entries 2–4). To further improve the efficiency of this reaction, several bases were tested as well, with DBU and DABCO giving slightly lower yields, while other bases were less effective (entries 5–7). Subsequently, the reaction with Cu(acac)2 as a catalyst and DBU as a base in other mixed solvents such as toluene-DMSO, -DMF, -DCE, and -THF did not give satisfactory outcomes (entry 8). Temperature control was crucial for the reaction, with any deviation from the optimal temperature resulting in lower yields (entry 9). No desired product 4a was detected by 31P{1H} NMR spectrum in the absence of either the copper catalyst, base, or air (entries 10 and 11).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) |
| a Reaction conditions unless otherwise specified: a mixture of P4 (6.20 mg, 0.20 mmol of P atom, 0.125 M solution of P4 in toluene, 0.40 mL), 2a (2.00 mmol, 10.0 eq.), 3a (0.30 mmol, 1.5 eq.), copper salt (0.02 mmol, 10 mol%) and base (0.10 mmol, 50 mol%) in MeCN (2.0 mL) was stirred under air at 70 °C for 16 h. Yield of the product determined by 31P{1H} NMR analysis of the crude reaction mixture using (C6H5O)3P(O) as an internal standard. b Inorganic bases such as K2CO3, K3PO4, Na2CO3, Cs2CO3, KOH, NaHCO3 and tBuONa were tested. | ||
| 1 | None | 94(90) |
| 2 | Cu(acac)2 → Fe(acac)2, Co(acac)2, Ni(acac)2 | 13–20 |
| 3 | Cu(acac)2 → CuCl, CuBr, Cu(CH3CN)PF6 | 18–41 |
| 4 | Cu(acac)2 → CuSO4,Cu(OTf)2 | 18–26 |
| 5 | TBD → DBU, DABCO | 70–80 |
| 6 | TBD → Et3N, DIPEA, TMEDA, TMG, DBN, MTBD | <70 |
| 7b | TBD → inorganic bases | <28 |
| 8 | Other mixed solvent instead of toluene-CH3CN, such as: toluene-DMSO, -DMF, -DCE or -THF | <18 |
| 9 | 60 °C or 80 °C instead of 70 °C | <65 |
| 10 | Ar instead of dry air | 0 |
| 11 | No copper salt or base | 0 |
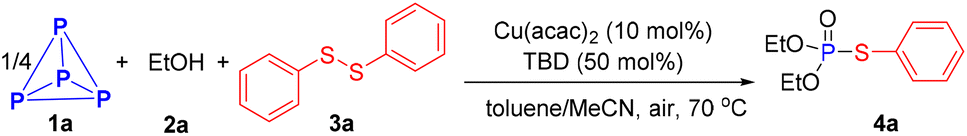
With the optimal reaction conditions in hand, we proceeded to evaluate the scope of this Cu-catalyzed direct functionalization of P4 for synthesizing phosphorothioate derivatives (Scheme 2). It was found that the electronic factor of the aryl disulfides had a significant effect on the present transformation. Aryl disulfides with electro-donating substituents such as methyl, methoxyl, tetrt-butyl, phenyl and even the free amine could be employed in this reaction, readily affording the phosphorylation products in good to excellent yields (4b–4f, 61–87%). Substituted substrates with electron-withdrawing groups on the benzene ring, such as trifloromethyl (4j), nitro (4k and 4l), ester (4m) and carlbamoul (4p), delivering target products in 29–59% yields, which were significantly lower than the yield for those bearing electro-donating groups. Other synthetically useful halogens like fluoro, chloro and bromo groups did not affect the efficiency of this conversion, providing the corresponding products 4g–4i in good yields (76–86%). When substrates with a multisubstituted benzene ring participated in the procedure, 4n and 4o were obtained in yields of 83% and 56%, respectively. Apart from phenyl ring, sterically bulky condensed aromatic ring and reactive heterocyclic substrates were all smoothly converted to respective products (4q–4t, 50–78%). Unfortunately, a mixture of phosphorus-containing compounds was detected by the 31P{1H} NMR spectrum when dialkyl disulfide was applied to this reaction.
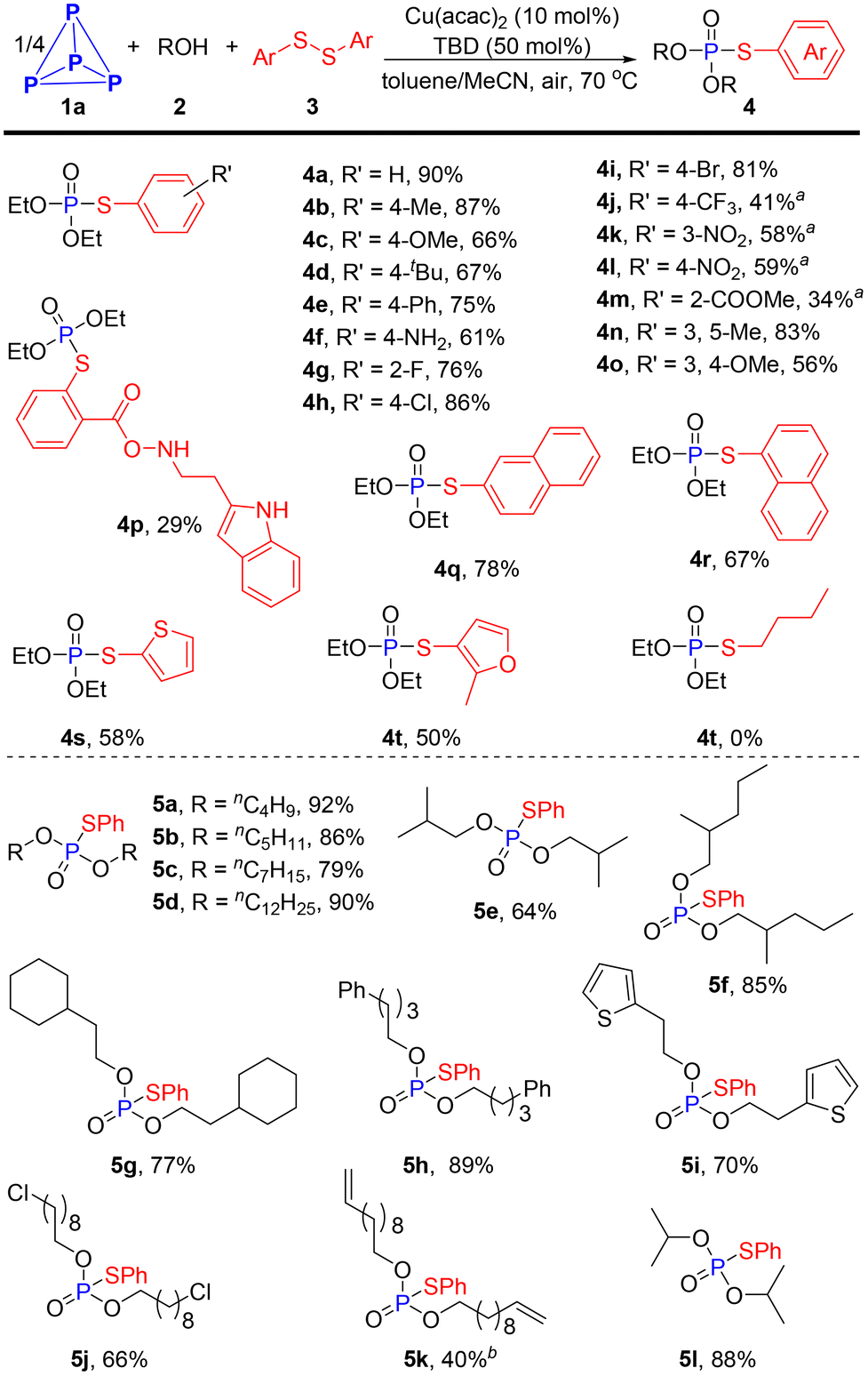 | ||
Scheme 2 Synthesis of phosphorothioate derivatives. 0.2 mmol scale of P atom, isolated yield. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Disulfide (4.0 eq.) was used. bDBU, 24 h. Disulfide (4.0 eq.) was used. bDBU, 24 h. | ||
The scope of alcohols was also examined to further gauge the generality of this methodology. As shown in Scheme 2, various simple primary aliphatic alcohols all successfully yielded the desired phosphorothioates with high efficiency (5a–5i, 64–92%). Functionalized alcohols such as 9-chlorononan-1-ol and 10-undecen-1-ol also reacted well, affording products 5j and 5k in 66% and 40% yields, respectively. Secondary alcohol, iso-propanol, also performed well in this reaction (5l). Obviously, this one-step three-component coupling reaction of P4, alcohols and diaryl disulfides provides a rapid and efficient route to phosphorothioates.
Inspired by our recent work,34 we envisioned that phosphoroselenoate derivatives might be generated when diaryl disulfides were replaced by diphenyl diselenide, and a further nucleophilic substitution would take place with ArOH, eventually affording mixed alkyl/aryl phosphates. Therefore, some experiments using PhSeSePh as a catalyst and phenols as nucleophilic reagents were conducted to test the above hypothesis, and the corresponding mixed phosphonates 7a–7i were isolated in acceptable yields (34–68%, Scheme 3).
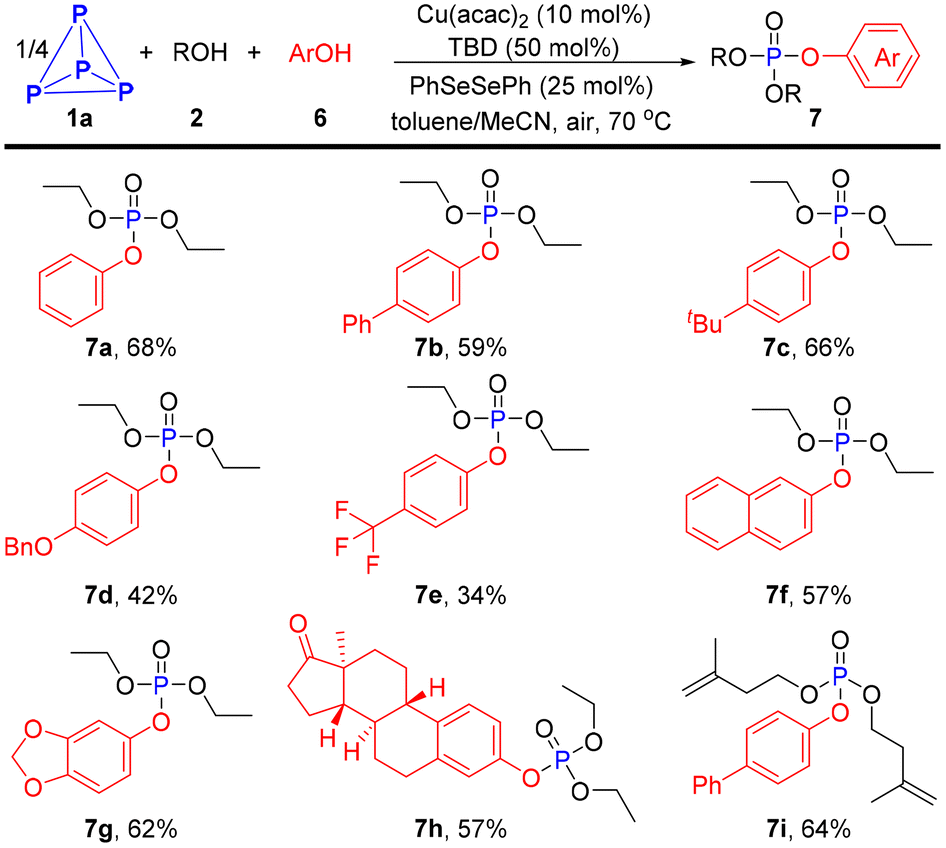 | ||
| Scheme 3 Synthesis of mixed phosphates. P4 (6.2 mg total of P4, a 0.125 M solution of P4 in toluene, 0.40 mL), ROH (2.00 mmol), ArOH (0.40 mmol), Cu(acac)2 (0.02 mmol), TBD (0.10 mmol), PhSeSePh (0.05 mmol) in MeCN (2.0 mL) was stirred at 70 °C under air for 16 h, isolated yield. | ||
A gram scale reaction proceeded smoothly, providing product 4a in 72% yield after extending the reaction time to 72 h. Product 4a can be activated by triflic anhydride to generate a reactive electrophilic P-species.38 Phenol derivatives could then attack the phosphonium intermediate, resulting in the formation of mixed thiophosphates 8a and 8b (Scheme 4).
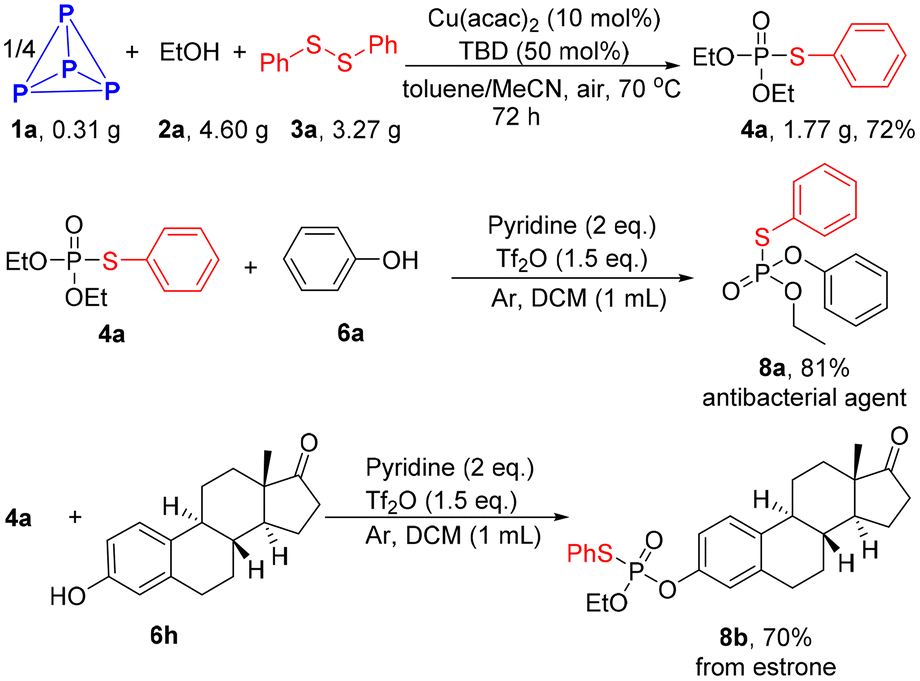 | ||
| Scheme 4 Gram-scale reaction and synthetic applications. | ||
To gain some mechanistic information of this copper-catalyzed three-component reaction, in situ31P{1H} NMP studies were carried out (Fig. 1). When the reaction was performed under standard conditions for 30 min, the signal of the P4 starting material (δ −523.4 ppm) remained and several new signals appeared. A signal at δ 184.7 was assigned to (EtO)2PSPh 11, which could be oxidized by air to afford the final product. Two intermediates were isolated and identified by NMR and HRMS indicating that signal at δ 157.3 and 132.9 ppm corresponds to EtOP(SPh)210 and P(SPh)39, respectively. After 2 h, full consumption of P4 was observed and two distinctive signals at δ 22.3 and 7.7 ppm appeared. The former (δ 22.3 ppm) was assigned to phosphorothioate 4a while the latter (δ 7.7 ppm) is corresponding to (EtO)2P(O)H 12. When the reaction time was extended to 12 h, product 4a was the major phosphorus-containing compound, as monitored by 31P{1H} NMR spectroscopy.
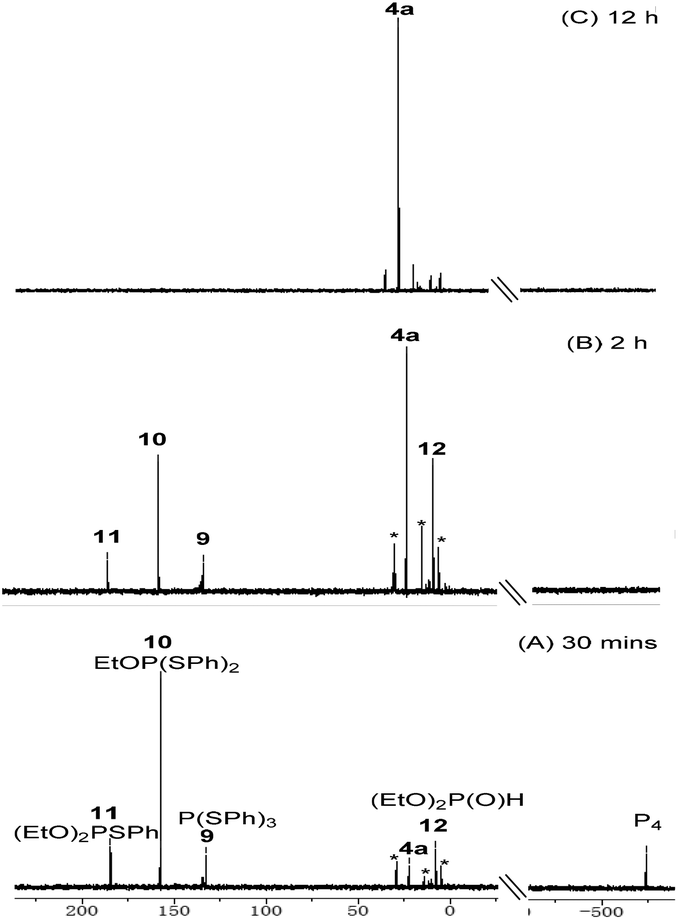 | ||
| Fig. 1 In situ 31P {1H} NMR spectra for the synthesis of 4a. *Unknown compounds. | ||
Some control experiments were also conducted to further investigate the possible reaction mechanism (Scheme 5). A clean transform of P4 into phosphorotrithioite 9 in 80% yield was observed, suggesting this step is promoted by base rather than copper salt (Scheme 5a). Ethanol reacted well with 9 to give final product 4a in 70% yield under standard conditions while 4a was not detected in the absence of TBD (Scheme 5b). In addition, reaction yielded two organophosphorus products 4a and 12 without Cu(acac)2 (Scheme 5c). The treatment of 12 and 3a under identical conditions afford 4a in excellent yield (Scheme 5d).
 | ||
| Scheme 5 Control experiments. | ||
Based on the experimental data above and previous reports, three possible paths for this reaction were proposed (Scheme 6). Initially, the coupling reaction of disulfide 3a and P4 affords triphenyl phosphorotrithioite 9.33,34 The P–O bond formation via P–S bond cleavage in the presence of ethanol 2a as the nucleophile gives intermediate 10. Subsequently, nucleophilic substitution between ethanol 2a and 10 yields intermediate 11, which could be oxidized by air to afford the desired phosphorothioate 4a. Preliminary 31P NMR studies showed (EtO)2P(O)H 12 was involved in this transformation. Therefore, two alternative paths cannot be ruled out. First, H-phosphonates 12 is generated either directly from ethanol breaking the P–P bonds of P4 with the assistance of copper salt39,40 or by nucleophilic substitution of 11 with H2O from air. Then, copper-catalyzed coupling reaction between 12 and disulfide 3a, furnishing the final product 4a.13,41
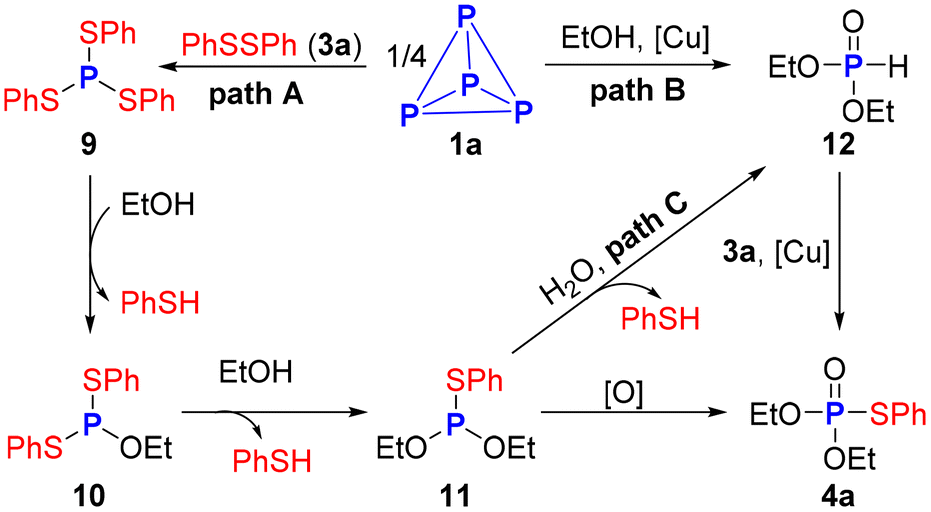 | ||
| Scheme 6 The proposed mechanism. | ||
Conclusions
In conclusion, we have successfully developed the first practical three-component coupling reaction of P4, alcohols, and diaryl sulfides, which provides a rapid way for the synthesis of a structurally diverse array of phosphorothioates along with the construction of P–S, P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and two P–O bonds. The use of commercially available alcohols and disulfides as inexpensive and commercially available starting materials and P4 as the P-atom source represents a prominent advantage of this transformation. This new approach to synthesizing phosphorothioates has the potential to significantly improve the efficiency and sustainability of organophosphorus compound production, making it an attractive option for chemical researchers and the broader chemical industry.
O and two P–O bonds. The use of commercially available alcohols and disulfides as inexpensive and commercially available starting materials and P4 as the P-atom source represents a prominent advantage of this transformation. This new approach to synthesizing phosphorothioates has the potential to significantly improve the efficiency and sustainability of organophosphorus compound production, making it an attractive option for chemical researchers and the broader chemical industry.
Experimental
Synthesis of phosphorothioate (4a–5l)
To an oven-dried schlenk tube with a magnetic stir bar was added Cu(acac)2 (0.02 mmol, 5.3 mg), TBD(1,5,7-triazabicyclo[4.4.0]dec-5-ene) (0.1 mmol, 13.9 mg) and disulfide (0.3 mmol). Then alcohol (2 mmol), acetonitrile (2.00 mL) and P4 (6.2 mg total of P4, a 0.125 M solution of P4 in toluene, 0.40 mL) were sequentially added to the system. Then the system was stirred at 70 °C (oil bath) for 16 h. After competition, the solvent was evaporated by rotary evaporation, the crude reaction mixture was purified by flash chromatography using petroleum-AcOEt [4:1 (v/v)] as the eluent to give the product.
Synthesis of phosphate (7a–7i)
To an oven-dried schlenk tube with a magnetic stir bar was added Cu(acac)2 (0.02 mmol, 5.3 mg), TBD(1,5,7-triazabicyclo[4.4.0]dec-5-ene) (0.1 mmol, 13.9 mg), diselenide (0.05 mmol) and phenol (0.4 mmol). Then alcohol (2 mmol), acetonitrile (2.00 mL) and P4 (6.2 mg total of P4, a 0.125 M solution of P4 in toluene, 0.40 mL) were sequentially added to the system. Then the system was stirred at 70 °C (oil bath) for 16 h. After competition, the solvent was evaporated by rotary evaporation, the crude reaction mixture was purified by flash chromatography using petroleum-AcOEt [4:1 (v/v)] as the eluent to give the product.
Gram-scale preparation of 4a
To an oven-dried schlenk bottle with a magnetic stir bar was added Cu(acac)2 (1 mmol, 261 mg), TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene) (5 mmol, 696 mg) and diphenyl disulfide (15 mmol, 3.275 g). Then ethanol (100 mmol), acetonitrile (100 mL) and P4 (310 mg total of P4, a 0.125 M solution of P4 in toluene, 20 mL) were sequentially added to the system. Then the system was stirred at 70 °C (oil bath) for 72 h. After competition, the solvent was evaporated by rotary evaporation, the crude reaction mixture was purified by flash chromatography using petroleum-AcOEt [4:1 (v/v)] as the eluent to give the product 4a (1.771 g, 72%)
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Key Research and Development Program of China (2020YFA0608300), Space Application System of China Manned Space Program (KJZ-YY-WSM01), and NSFC (21772163, 21778042 and 41876072).References
- W. Gleason, JOM, 2007, 59, 17–19 CrossRef CAS.
- M. B. Geeson and C. C. Cummins, Science, 2018, 359, 1383–1385 CrossRef CAS PubMed.
- Y. Mei, Z. Yan and L. L. Liu, J. Am. Chem. Soc., 2022, 144, 1517–1522 CrossRef.
- J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef.
- M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef.
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef PubMed.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef PubMed.
- D. J. Scott, Angew. Chem., Int. Ed., 2022, 61, e202205019 CrossRef PubMed.
- T. S. Kumar, T. Yang, S. Mishra, C. Cronin, S. Chakraborty, J.-B. Shen, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2013, 56, 902–914 CrossRef.
- R. Xie, Q. Zhao, T. Zhang, J. Fang, X. Mei, J. Ning and Y. Tang, Bioorg. Med. Chem., 2013, 21, 278–282 CrossRef PubMed.
- X. Liu, L. Zhou, R. Yang, X.-R. Song and Q. Xiao, Adv. Synth. Catal., 2023, 365, 2280–2298 CrossRef.
- J. Bai, X. Cui, H. Wang and Y. Wu, Chem. Commun., 2014, 50, 8860–8863 RSC.
- Y.-X. Gao, G. Tang, Y. Cao and Y.-F. Zhao, Synthesis, 2009, 1081–1086 Search PubMed.
- X. Bi, J. Li, F. Meng, H. Wang and J. Xiao, Tetrahedron, 2016, 72, 706–711 CrossRef CAS.
- S. Li, T. Chen, Y. Saga and L.-B. Han, RSC Adv., 2015, 5, 71544–71546 RSC.
- D. J. Jones, E. M. O'Leary and T. P. O'Sullivan, Adv. Synth. Catal., 2020, 362, 2801–2846 CrossRef CAS.
- Y. Zhu, T. Chen, S. Li, S. Shimada and L.-B. Han, J. Am. Chem. Soc., 2016, 138, 5825–5828 CrossRef CAS.
- S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487–2491 CrossRef CAS.
- L. Zhang, S. Yang, L. Chen, S. Yuan, Q. Chen, M.-Y. He and Z.-H. Zhang, Catal. Sci. Technol., 2017, 7, 2356–2361 RSC.
- S. Shi, J. Chen, S. Zhuo, Z. Wu, M. Fang, G. Tang and Y. Zhao, Adv. Synth. Catal., 2019, 361, 3210–3216 CrossRef CAS.
- Y. H. Budnikova, S. A. Krasnov, T. V. Graznova, A. P. Tomilov, V. V. Turigin, I. M. Magdeev and O. G. Sinyashin, Phosphorus Sulfur Relat. Elem., 2008, 183, 513–518 CrossRef CAS.
- U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rödl, R. M. Gschwind and R. Wolf, Nat. Catal., 2019, 2, 1101–1106 CrossRef CAS PubMed.
- V. A. Milyukov, Y. H. Budnikova and O. G. Sinyashin, Russ. Chem. Rev., 2005, 74, 781–805 CrossRef CAS.
- D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Nat. Chem., 2021, 13, 458–464 CrossRef CAS PubMed.
- M. Till, V. Streitferdt, D. J. Scott, M. Mende, R. M. Gschwind and R. Wolf, Chem. Commun., 2022, 58, 1100–1103 RSC.
- H. Grützmacher, Nat. Chem., 2022, 14, 361–364 Search PubMed.
- Y. Xiong, S. Dong, S. Yao, J. Zhu and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202205358 CrossRef CAS PubMed.
- J. Hu, Z. Chai, W. Liu, J. Wei, Z.-J. Lv and W.-X. Zhang, Green Synth. Catal. DOI:10.1016/j.gresc.2022.12.008.
- C. Gendy, J. Valjus, H. M. Tuononen and R. Roesler, Angew. Chem., Int. Ed., 2022, 61, e202115692 CrossRef CAS.
- C. Yang, X. Jiang, Q. Chen, X. Leng, J. Xiao, S. Ye and L. Deng, J. Am. Chem. Soc., 2022, 144, 20785–20796 CrossRef.
- J. Hu, Z. Chai, W. Liu, Z. Huang, J. Wei and W.-X. Zhang, Sci. China: Chem., 2022, 65, 322–327 CrossRef.
- J. Hu, W. Liu and W.-X. Zhang, Phosphorus, Sulfur Silicon Relat. Elem., 2022, 197, 398–407 CrossRef.
- X. Huangfu, Y. Zhang, P. Chen, G. Lu, Y. Cao, G. Tang and Y. Zhao, Green Chem., 2020, 22, 8353–8359 RSC.
- Y. Zhang, Z. Cai, Y. Chi, X. Zeng, S. Chen, Y. Liu, G. Tang and Y. Zhao, Org. Lett., 2021, 23, 5158–5163 CrossRef PubMed.
- Z. Cai, Y. Zhang, Y. Cao, Y. Liu, G. Tang and Y. Zhao, ACS Catal., 2023, 13, 8830–8835 CrossRef.
- G. Lu, J. Chen, X. Huangfu, X. Li, M. Fang, G. Tang and Y. Zhao, Org. Chem. Front., 2019, 6, 190–194 RSC.
- F. Chen, M. Bai, Y. Zhang, W. Liu, X. Huangfu, Y. Liu, G. Tang and Y. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202210334 CrossRef PubMed.
- H. Huang, J. Denne, C.-H. Yang, H. Wang and J. Y. Kang, Angew. Chem., Int. Ed., 2018, 57, 6624–6628 CrossRef PubMed.
- M. Bai, Y. Cao, J. Huang, Y. Liu, G. Tang and Y. Zhao, CCS Chem., 2023 DOI:10.31635/ccschem.023.202303143.
- H. C. Fisher, L. Prost and J.-L. Montchamp, Eur. J. Org. Chem., 2013, 7973–7978 CrossRef.
- H. Huang, J. Ash and J. Y. Kang, Org. Biomol. Chem., 2018, 16, 4236–4242 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis, spectral data and NMR spectra of compounds 4a–4t, 5a–5l, 7a–7i, 8a and 8b. See DOI: https://doi.org/10.1039/d3gc03583b |
| This journal is © The Royal Society of Chemistry 2024 |