
Green and sustainable synthesis of TPD-based donor–acceptor-type conjugated polymer photocatalysts for hydrogen production under visible light†
Menghan
Chang‡
,
Xinjuan
Zhang‡
,
Lin
Wang
,
Di
Wang
,
Qiang
Zhang
 * and
Yan
Lu
*
* and
Yan
Lu
*
Key Laboratory of Display Materials & Photoelectric Devices, Ministry of Education, Tianjin Key Laboratory for Photoelectric Materials and Devices, School of Materials Science & Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: zhangqiang@email.tjut.edu.cn; luyan@tjut.edu.cn
First published on 29th November 2023
Abstract
Donor (D)–acceptor (A) structured conjugated polymers (CPs) have attracted increased attention owing to their application in efficient photocatalytic hydrogen production from water, benefiting from their unique advantages of high charge mobility, strong light-harvesting ability, and tunable band gaps. As a typical “A” building block to construct D–A type CPs, thieno[3,4-c]pyrrole-4,6-dione (TPD) may bind co-catalyst, similar to Pt though the σ–π anchor of its carbonyl group with Pt to enhance the performance of solar-driven hydrogen production. Herein, we developed three TPD-based D–A-type CPs, namely, TPD2T, TPDTT, and TPDBDT, using 2,2′-bithiophene (2T), cyclopenta[2,1-b:3,4-b]dithiophene (TT), and benzo[1,2-b:4,5-b]dithiophene (BDT) as “D” units, via direct C–H/C–H coupling polycondensation, offering an atom-economic and eco-friendly way to achieve new CP materials for highly effective hydrogen evolution. Among them, TPDBDT-based nanoparticles exhibited the highest photocatalytic activity with remarkable hydrogen evolution rates of 1819.4 μmol h−1 g−1 and 7420.0 μmol h−1 g−1 in the absence and presence of co-catalyst Pt, respectively. The mechanism of the hydrogen production of TPD-based polymers was discussed based on DFT calculations. This work not only demonstrates the first example of CPs synthesized via C–H/C–H cross-coupling polymerization for application in water splitting for hydrogen production but also helps to understand the relationship between polymer structure and photocatalytic hydrogen production performance.
Introduction
Hydrogen energy is one of the most effective and ideal energy carriers for solving increasingly severe energy and environmental problems.1–4 Currently, hydrogen is mainly produced by steam reforming methane, with large amounts of carbon dioxide as by-products.5,6 Recently, visible-light-driven water splitting for hydrogen production has become an important and popular pathway for clean energy production and energy storage because this process directly utilizes solar energy (the most abundant and cleanest energy source) to produce hydrogen without greenhouse gas emissions.7–10 In particular, organic semiconductor material-based water splitting has attracted much attention owing to the advantages of high charge mobility, strong light-harvesting ability, and tunable band gaps.11–13 In 1985, Yanagida and coworkers reported for the first time the production of hydrogen from water using oligo-/poly(p-phenylene)s as photocatalysts.14 After that, many conjugated polymer (CP) materials have been developed for this purpose, mainly including dibenzothiophene-S,S-dioxide and its derivatives,15–18 as well as benzothiadiazole (BTH)-containing CPs.19–21Most recently, it has been demonstrated that CPs with donor–π–acceptor (D–π–A) structure can significantly enhance photocatalytic performance, benefitting from the larger π-electron delocalization to extend the carrier migration distance, thereby boosting the exciton separation efficiency and reducing the exciton binding energy.22–25 For instance, Cooper et al. developed a D–A type polymer P7 by introducing dibenzo[b,d]thiophene sulfone structures into poly(p-phenylene), which showed excellent hydrogen evolution rate (HER) with a 45 times higher HER than the parent poly(p-phenylene).16 In 2017, Tian and coworkers reported a D–A polymer PFODTTBT that showed significantly red-shifted absorption by around 100 nm, deeper reduction potentials and excited state (−0.90 V and 1.08 V for PFODTTBT, respectively) compared with the reference polymer F8T2 (−0.78 and 1.68 V for F8T2, respectively). PFODTTBT dots achieved a higher HER of up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μmol h−1 g−1 (>420 nm without cocatalyst), while F8T2 dots did not show any consistent reactivity to light-driven proton reduction under the same conditions.20 Although some remarkable results have been achieved based on these CP materials, their overall photo-catalytical performance, especially HER, as well as the high cost caused by long synthetic routes and low yields still hinder the practical industrial application. Therefore, the development of new conjugated polymer materials and a synthesis strategy is crucial for the continuous improvement of their photocatalytic water-splitting hydrogen production performance.
000 μmol h−1 g−1 (>420 nm without cocatalyst), while F8T2 dots did not show any consistent reactivity to light-driven proton reduction under the same conditions.20 Although some remarkable results have been achieved based on these CP materials, their overall photo-catalytical performance, especially HER, as well as the high cost caused by long synthetic routes and low yields still hinder the practical industrial application. Therefore, the development of new conjugated polymer materials and a synthesis strategy is crucial for the continuous improvement of their photocatalytic water-splitting hydrogen production performance.
For organic conjugated polymer-based photocatalysts, good planarity and skeleton rigidity are beneficial to enable π-electron delocalization, narrow the bandgap, suppress interannular rotation, and enhance intrinsic charge carrier mobility, thus leading to high photocatalytic water splitting activity.16 In this way, the electron-deficient thieno[3,4-c]pyrrole-4,6-dione (TPD) with symmetric coplanar structure is introduced into the backbone as an “A” unit to increase the conjugation and planarity of polymer and enhance interchain π–π stacking through intramolecular noncovalent interactions between the oxygen and the sulfur (or hydrogen) atoms, resulting in high charge carrier mobility and efficient charge transport of the obtained materials.26 In addition, as revealed by previous studies, the introduction of functional groups with great electronegative atoms, such as O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO,16,18 CO,27,28 F,29 and CN,30 can modify the surface chemistry and the energy band structure of organic polymer photocatalysts, thus significantly affecting the photocatalytic performance. For example, Lin et al. developed a new conjugated small molecule photocatalyst (Y6CO) by introducing one carbonyl group in the fused-ring core as a “claw” to efficiently anchor Pt cocatalyst through σ–π coordination, achieving state-of-the-art HER performance.27 Therefore, it could be envisioned that TPD may offer a good affinity for metallic cocatalysts and water and lower the HOMO energy level of the polymer, thereby achieving improved photo-catalytical performance.
SO,16,18 CO,27,28 F,29 and CN,30 can modify the surface chemistry and the energy band structure of organic polymer photocatalysts, thus significantly affecting the photocatalytic performance. For example, Lin et al. developed a new conjugated small molecule photocatalyst (Y6CO) by introducing one carbonyl group in the fused-ring core as a “claw” to efficiently anchor Pt cocatalyst through σ–π coordination, achieving state-of-the-art HER performance.27 Therefore, it could be envisioned that TPD may offer a good affinity for metallic cocatalysts and water and lower the HOMO energy level of the polymer, thereby achieving improved photo-catalytical performance.
However, CPs applied as photocatalysts for hydrogen production are usually synthesized using Suzuki and Stille coupling polymerization methods, which suffer from multistep synthesis and unsustainable environmental issues despite their high reaction efficiency and broad universality. Recently developed direct C–H/C–H coupling polycondensation, where C–C bond formation occurs directly from C–H bonds of the non-functionalized monomers, has enabled the greener and more sustainable synthesis of these polymers through the reduction of waste, elimination of toxic and harmful by-products, and through the reduction of overall synthetic steps.31–38 Unfortunately, there are no examples of conjugated polymers prepared using such a polymerization method for photocatalytic hydrogen evolution so far.
In this context, we combined electron-deficient TPD units with widely used electron donor units, including 2,2′-bithiophene (2T), cyclopenta[2,1-b:3,4-b]dithiophene (TT), and benzo [1,2-b:4,5-b′] dithiophene (BDT), to synthesize three new D–A typed conjugated polymers, namely, TPD2T, TPDTT and TPDBDT via the direct C–H/C–H coupling polycondensation (Scheme 1) and carefully evaluate their HER under visible light. Based on the experimental results and DFT simulations, the mechanism of photo-catalytical water splitting in the presence of these polymers was also discussed.
 | ||
| Scheme 1 Synthetic routes for the D–A conjugated polymers. | ||
Results and discussion
Synthesis of polymer
The synthetic routes of polymers are shown in Scheme 1. To screen the reaction conditions, the effect of catalyst, additive, solvents, and reaction temperature on the C–H/C–H cross-coupling polymerization was systematically evaluated by taking the reaction of 5-(2-octyl-1-dodecyl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (TPD) with 3,3′-dioctyl-2,2′-bithiophene (2T) as a model system, and the results are summarized in Table S1.† The reaction was first carried out under the same conditions as that for efficient homo-polymerization of TPD, as reported in our previous work39 (entry 1, Table S1†), yielding polymer TPD2T with a low number molecular weight (Mn) of 3.8 kDa (PDI 1.87) and a moderate yield of 58.7%. After that, various additives, including PCy3, P(2-MeOC6H4)3, PCy3–HBF4, and P(t-Bu)2MeHBF4, were used to promote the polymerization of TPD with 2T (entries 2–5, Table S1†). The Mn of TPD2T was boosted to 10.7 kDa with an increased yield of 70.4% in the presence of P(t-Bu)2MeHBF4 (entry 5, Table S1†). Further, different palladium catalysts were used to explore their influence on the C–H/C–H cross-coupling polymerization (entries 6–8, Table S1†). Among the various tested catalysts, Pd(PPh3)4 gave the highest Mn of 12.5 kDa (PDI 1.21) and a tolerable yield (63.4%). Besides, the effect of solvent on polymerization was assessed (entries 9–12, Table S1†). In the mixed solvent system, as the xylene content increased from 20:1 to 1:1, Mn of TPD2T increased up to 18 kDa with a moderate yield of 57.5% (entry 12, Table S1†). Overall, under optimized conditions, direct C–H/C–H cross-coupling polymerization of TPD with 2T using 0.1 equiv. of Pd(PPh3)4 as catalyst, DMAc/xylene(v/v = 1:1) as solvent at 80 °C can produce TPD2T with the highest Mn of up to 18.1 kDa with a yield of 57.5%.
TPDTT and TPDBDT were also synthesized using 5-(2-octyl-1-dodecyl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (TPD) and 4,4-dioctyl-cyclopenta[2,1-b:3,4-b']dithiophene (TT) and 4,8-bis((2-ethylhexyl)oxy)benzo[1,2-b:4,5-b']dithiophene (BDT) as A and D units under the above optimal reaction conditions for TPD2T (Scheme 1). In contrast to TPD2T, TPDTT and TPDBDT have higher molecular weights (Mn = 25.3 kDa for TPDTT, and Mn = 27.5 kDa for TPDBDT) with moderate yields (Table 1). Three polymers exhibit good solubility in common organic solvents, such as tetrahydrofuran, chloroform, and chlorobenzene. From the thermal gravimetric analysis (TGA) curve shown in Fig. S1,† it can be observed that the polymers exhibit excellent thermal stability under N2 atmosphere with thermal decomposition temperatures at 418, 385, and 350 °C, respectively, at 5 wt% weight loss.
| Sample |
M
na (kDa) |
PDI | Tdb (°C) | UV-visc | Photoluminescence (PL)c | Oxidation (versus SCE) | Reduction (versus SCE) | E g CV,f | Residual Pdg (wt%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ solmax | λ NPsmax | E optg,d | λ solmax | λ NPsmax |
Φ
fe (%) |
E onset (V) | E HOMO (eV) | E onset (V) | E LUMO (eV) | ||||||
| a Estimated by GPC measurements (eluent: THF, standard: polystyrene). b Decomposition temperature taken as the temperature corresponding to a 5% weight loss in the thermogravimetric run (10 °C min−1, N2). c Optical data are given in CHCl3 (sol) and nanoparticles (NPs). d E optg is an optical bandgap obtained from the onset wavelength of polymer nanoparticles. e Fluorescence quantum efficiency of polymers. f E CVg = EHOMO − ELUMO. g Residual Pd contents were determined by ICP-OES. | |||||||||||||||
| TPD2T | 18.1 | 1.26 | 418 | 490 | 547 | 1.76 | 564/609 | 745 | 14.73 | 1.51 | −6.07 | −0.95 | −3.61 | 2.46 | 0.0054 |
| TPDTT | 25.3 | 1.62 | 385 | 525 | 549 | 1.87 | 669 | 733 | 9.18 | 1.67 | −6.23 | −0.82 | −3.74 | 2.49 | 0.0032 |
| TPDBDT | 27.5 | 1.52 | 350 | 505 | 548 | 1.85 | 635 | 743 | 0.07 | 1.73 | −6.29 | −0.70 | −3.86 | 2.43 | 0.0012 |
Structural analyses of polymers
The chemical structure of the polymer was confirmed by 1H and 13C NMR spectroscopy (Fig. S2–S7†). As a representative, the NMR spectra of TPD2T are shown in Fig. 1. Peak (a) at 3.56 ppm in its 1H NMR spectrum could be assigned to the hydrogen on α-carbon directly connected to an amide of TPD, while peak (b) at 2.55–2.28 ppm corresponds to the hydrogen of the α-carbon of the thiophene side chain (Fig. 1a and S2†). The integral ratio of Ha and Hb was estimated to be 1:1, suggesting that the number of TPD and 2T units in the polymer was almost equal. Further, the edited heteronuclear single quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC) NMR spectroscopy of TPD2T were recorded to affirm the coupling sites of TPD and 2T in the polymer TPD2T (Fig. 1b and c). As shown in Fig. 1b, the strong signals at positions (2.54, 30.21 ppm), (7.05, 132.09 ppm), and (3.64, 43.13 ppm) reflect the C–H bond connectivity of C1 and Hb, C2 and Hc, and C3 and Ha, respectively. Moreover, the cross points at position (7.05, 143.33 ppm) in the HMBC spectrum (Fig. 1c) imply the connectivity between C4 and Hc. These NMR results suggest that polymer TPD2T has a D–A alternating structure. Similarly, the 1H and 13C NMR spectra of TPDTT and TPDBDT were also collected, as illustrated in Fig. S4–S7.† All signals of hydrogen and carbon atoms of monomers can be observed in their 1H and 13C NMR spectra of the corresponding polymers, indicating the successful polymerization of TPD and the corresponding “D” molecules.
 | ||
| Fig. 1 (a) 1H NMR (b) HSQC and (c) HMBC spectroscopy of TPD2T. | ||
DFT simulations
Density functional theory (DFT) calculations were performed with the Gaussian 09 W software package and Gaussian 09 package at the B3LYP3/6-31G (d) level to study the backbone structure and the electronic states of polymers. Their geometries were optimized without any restriction at the theoretical level of B3LYP/6-31G (d), which is followed by frequency calculations to confirm that the located geometries are real minimums. In this study, four repeated units were adopted, and all side chains were replaced with methyl groups to simplify the calculations. The HOMO and LUMO level values were obtained using MO topologies based on the optimized geometry.Fig. S8† shows the optimized molecular geometries of TPD2T, TPDTT, and TPDBDT. From Fig. S8,† it can be clearly observed that TPDTT and TPDBDT tend to adopt planar conformation along polymeric main chains with small dihedral angles of about 0.4° and 1.5° between TPD and TT (or BDT). The TPD2T backbone became twisted with a large dihedral angle (33.9°) between the two thiophene rings. Thus, the introduction of fused ring donors, such as cyclopentadithiophene (TT) and benzodithiophene (BDT), caused a more rigid skeleton and more planar conformation of the resultant TPDTT and TPDBDT.
Additionally, the HOMOs are mainly distributed in the main chain of the polymer, including the TPD (A) units and D segments, and the HOMO energy levels are approximately −5.03, −4.78, and −5.12 eV for TPD2T, TPDTT, and TPDBDT, respectively. The LUMOs are dominantly localized on the sulfur atoms and D units, and the LUMO energy levels were estimated to be −2.62, −2.78, and −2.80 eV, respectively. It is also noted that the HOMO electron clouds of TPDBDT are mainly rhomboidal distribution unlike those of TPD2T and TPDTT, which have a mainly trapezoidal distribution (Fig. S9†) beneficial to charge transport.
Fabrication and characterization of nanoparticles
The surface wettability of the three polymers against water was first assessed by contact angle measurements (Fig. 2). The water contact angles of the polymer films TPD2T, TPDTT, and TPDBDT were 97.1°, 96.3°, and 90°, respectively. Among them, the TPDBDT film shows the highest hydrophilic property with water contact angles of 90°, which should be attributed to the hydrophilic side chains on the BDT segments. This indicates better interface contact between TPDBDT and water, as well as shorter paths for separated charges emigrating to the polymer–water surface, which is highly desired for hydrogen evolution.40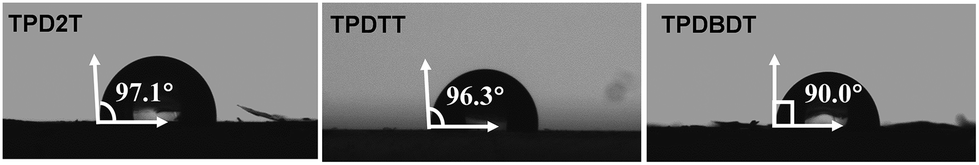 | ||
| Fig. 2 Water contact angles of TPD2T, TPDTT, and TPDBDT. | ||
To obtain the small and stable polymer nanoparticles that can provide a short exciton diffusion length and reduce the exciton recombination within the organic photocatalysts, three polymers were separately encapsulated into an amphiphilic block copolymer (PEG–PBLA20) through a nanoprecipitation process,41,42 yielding the corresponding composite nanoparticles (NPs). Transmission electron microscopy (TEM) images of NPs reveal that they have nearly spherical shapes with diameters of 194, 208, and 102 nm for TPD2T NPs, TPDTT NPs, and TPDBDT NPs, respectively (Fig. 3a–c). The corresponding mean hydrodynamic diameters of the three NPs were about 260, 236, and 168 nm, respectively, as measured by dynamic light scattering (DLS) (Fig. 3d–f).
 | ||
| Fig. 3 (a–c) TEM images and (d–f) hydrodynamic diameter spectra of TPD2T NPs, TPDTT NPs, and TPDBDT NPs. | ||
Optical properties
The optical properties of the three polymers (TPD2T, TPDTT, and TPDBDT) were evaluated using UV-vis absorption spectroscopy and photoluminescence (PL) spectroscopy. Fig. 4a shows the UV-vis absorption spectra of three polymers in dilute chloroform solution (1 × 10−5 M) and their nanoparticles dispersed in deionized water, and the corresponding data are summarized in Table 1. These polymers exhibited broad absorption in the range of 350–700 nm with absorption peaks at 490, 525, and 505 nm (ε = 8.7 × 104, 2.4 × 104 and 6.4 × 104 mol−1 cm−1), respectively. The strong absorption of three polymers in the visible region is favourable for sunlight harvesting and charge transfer between frontier molecular orbitals (FMOs)43 and thus enhances the ability of photocatalysis. Compared with the solution of polymers, their corresponding NPs exhibited significantly red-shifted absorption extended to the near-infrared region (Fig. 4a and Table 1). The optical band gaps (Eoptg) of the three polymers were estimated to be about 1.8 eV from the absorption edge of their nanoparticles. The narrow band gap could enhance their visible light-harvesting ability.44 | ||
| Fig. 4 (a) UV-vis absorption of TPD2T, TPDTT, TPDBDT solution and their corresponding nanoparticles; (b) energy levels of TPD2T, TPDTT, and TPDBDT. | ||
All TPD-based conjugated polymers in dilute CHCl3 solution (1 × 10−5 M) exhibit broad emission in the range of 500–800 nm with fluorescence quantum efficiency of 14.73% for TPD2T, 9.18% for TPDTT and 0.04% for TPDBDT, respectively (Fig. S10† and Table 1). The characteristic two emission peaks located in the high and low energy regions should be attributed to the energy transfer of π*–π of conjugated backbone and the excited state of donors to TPD units within D–A copolymers,45 respectively. When the polymers form nanoparticles, their fluorescence is substantially quenched because of the well-known aggregation-caused fluorescence quenching (ACQ) effect, which indicates that the D–A architecture can effectively inhibit radiative recombination upon aggregation, thus probably leading to high activity of photocatalysis.
Electrochemical property
Cyclic voltammetry (CV) measurements were conducted to assess the electrochemical properties of the polymers using ferrocene/ferrocenium (Fc/Fc+) as internal standards with an absolute energy of −4.8 eV (Fig. S11†). All polymer samples show both oxidation (p-doping) and reduction (n-doping) peaks in their CV curves. The energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were calculated based on the following formulas:| HOMO = −e(Eoxonset + 4.8) (eV) |
| LUMO = −e(Eredonset + 4.8) (eV) |
The electrochemical data for the three polymers are summarized in Table 1. The onset oxidation potentials are found to be at 1.51, 1.67, and 1.73 V for TPD2T, TPDTT, and TPDBDT, respectively, versus Fc/Fc+, corresponding to the HOMO energy levels of −6.07, −6.23, and −6.29 eV, respectively. Additionally, the potential onsets of the reduction waves of the polymers are calculated to be −0.95, −0.82, and −0.70 V, respectively. Therefore, the LUMO levels of TPD2T, TPDTT, and TPDBDT are −3.61, −3.74, and −3.86 eV, respectively. The LUMO positions of the polymers are much higher than the water reduction potential (−5.43 eV), suggesting that the thermodynamical force can drive the water-splitting half reaction.46 The electrochemical band gaps (ECVg) are 2.46, 2.49, and 2.43 eV for TPD2T, TPDTT, and TPDBDT, respectively, matching the requirements of the band gap (1.23 eV) for photocatalytic hydrogen production. It was noted that the ECVg of the three polymers was significantly higher than that of their Eoptg, which should be related to the measurement conditions and the exciton binding energy of conjugated polymers (0.4–1.0 eV).47,48
Photocatalytic hydrogen production
Benefitting from the strong light absorption capability of three polymers, suitable HOMO and LUMO levels as well as good dispersion stability in water, their photocatalytic hydrogen evolution (PHE) activity was first evaluated in the presence of ascorbic acid (AA) as a sacrificial electron donor owing to the matching narrow bandgap.44 The pH value of the photocatalytic system was carefully regulated with sodium hydroxide (NaOH) aqueous solution (0.1 M) to ensure that it is close to the pKa of AA (pKa = 4.04). In this case, the proton source of AA can be eliminated.Fig. 5a depicts the hydrogen generation activity of TPD2T and TPDTT NPs at different concentrations ranging from 50 to 250 μg mL−1. It was clearly observed, from Fig. 5a, that the hydrogen evolution rates (HER) of TPD2T NPs gradually decreased, while TPDTT NPs increased first and then decreased with the increase in NP concentrations, and the rates reached the maxima at different concentrations, that is, 378.7 μmol h−1 g−1 at 50 μg mL−1 for TPD2T NPs and 812.2 μmol h−1 g−1 at 150 μg mL−1 for TPDTT NPs. These results should be tentatively explained as follows: the increase in concentrations of photocatalysts could improve the absorption ability of solar light and thus enhance hydrogen production activity. Moreover, aggregation of NPs could happen at higher concentrations, which reduces the photocatalytic efficiency owing to the longer distance of photogenerated charges to the surface of the photocatalyst and increased probability of electron and hole recombination.49
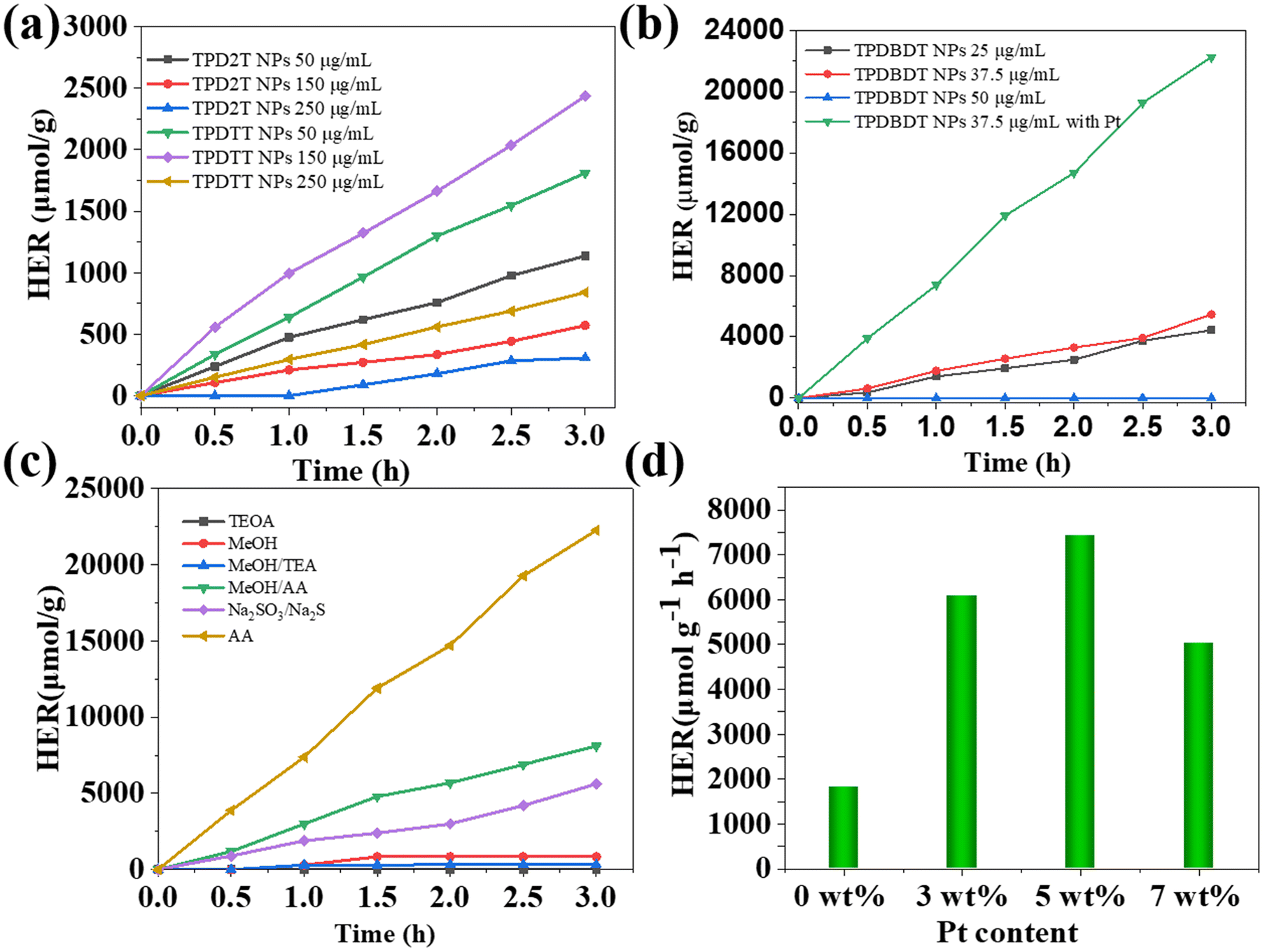 | ||
| Fig. 5 (a) Hydrogen evolution rate of TPD2T NPs and TPDTT NPs and (b) hydrogen evolution rate of TPDBDT NPs dispersed in varied concentrations under visible light. (c) HERs of TPDBDT NPs in the presence of various sacrificial agents. (d) Average hydrogen evolution rates of TPDBDT NPs under different amounts of Pt (0.2 M AA). | ||
In addition, it was noted that the HER of TPDTT NPs was slightly faster than that of TPD2T NPs at the same concentrations (Fig. 5a). This is related to lower LUMO levels and a more planar structure of TPDTT as revealed by DFT calculations (Fig. S8†), because the good co-planarity of polymer is believed to increase the amount of reaction sites required for surface photocatalysis, thus enhancing H2 production activity.16 Very interestingly, TPDBDT NPs at 37.5 μg mL−1 rendered a predominant hydrogen generation rate of 1819.4 μmol h−1 g−1 without any cocatalyst, which is about 2.2 times faster than that of TPDTT NPs at 150 μg mL−1. This should be attributed to the following reasons: the rigid fused ring structure of BDT makes TPDBDT more coplanarity, leading to more reaction sites; the presence of oxygen atoms of BDT increases the hydrophilicity of TPDBDT. Once the concentration of TPDBDT NPs was further increased to 50 μg mL−1, the inferior photocatalytic properties were obtained, which may be caused by its poor dispersibility in H2O/AA mixed solution.
Numerous studies have shown that the type of sacrificial agent has an important effect on the performance of photocatalytic systems. Accordingly, choosing an effective and suitable sacrificial agent is very important for the enhancement of hydrogen evolution activity.18,50 In this study, we selected the five widely used sacrificial agents, including methanol, methanol/triethylamine (TEA), triethanolamine (TEOA), AA and Na2SO3 (Fig. 5c). In the case of TEOA, methanol/TEA addition, TPDBDT NPs exhibit poor photocatalytic activity mainly because of their alkaline environment and low LUMO energy levels. When methanol is used as a sacrificial agent, the hydrogen production rate is 840 μmol g−1 in the test period of 3 h. The hydrogen production rate was increased to 8098 μmol g−1 in the presence of methanol/AA, but the hydrogen production rate was as high as 22253 μmol g−1 with AA. This may be because methanol affects the hydrophilic groups on the surface of the nanoparticles. The addition of methanol results in visible solid precipitation during the catalytic process. Inorganic sacrificial agents, such as Na2SO3/Na2S, are also used to detect the photocatalytic activity of TPDBDT NPs, and the hydrogen production rate is only 1800 μmol g−1 h−1. Therefore, AA should be an appropriate sacrificial agent for our systems.
Further, the effect of the amount of co-catalyst Pt on the PHE activity of the polymers was assessed using TPDBDT NPs as an example. As shown in Fig. 5d, with the increase in the amount of co-catalyst Pt from 0 to 7 wt%, the photocatalytic hydrogen evolution (PHE) activity of the nano-system increased first and then decreased. The PHE activity reaches its maximum value in the presence of 5 wt% Pt, and the excessive loading of Pt over 5 wt% restricts the PHE performance owing to the light-shield effect.51 Further, hydrogen production rates of TPDBDT NPs are positively correlated to their concentrations and irradiation time (Fig. 5b). When 5 wt% Pt was added as a co-catalyst, the HER of the TPDBDT NPs remarkably increased up to 7420.0 μmol h−1 g−1, maintaining the other experimental condition fixed (Fig. 5b). However, the addition of Pt to the TPD2T or TPDTT system hardly increased HER. Notably, trace amounts of palladium could remain in the polymers owing to the use of a Pd catalyst in their preparation process, which may result in an enhanced performance of photocatalytic hydrogen evolution. To clarify this issue, the residual Pd contents of the three polymers were measured to be approximately 0.0054, 0.0032, and 0.0012 wt% for TPD2T, TPDTT, and TPDBDT (Table 1), respectively, using an inductively coupled plasma optical emission spectrometer (ICP-OES). It can be clearly observed that TPDBDT NPs demonstrated the highest photocatalytic activity compared with TPD2T and TPDTT NPs although they had the least amount of Pd in their nanoparticles. The results indicate that the effect of residual Pd in polymer NPs on photocatalytic activity is not significant for our system, which is consistent with the results reported in the literature.52
To understand the differences in hydrogen evolution performances of the three polymer-based NPs, their charge separation and transfer properties were carefully assessed by photoluminescence lifetimes (Fig. 6a), electrochemical impedance spectroscopy (EIS) (Fig. 6b) and photocurrent measurements (Fig. 6c). The photoluminescence lifetimes of the three polymers were measured at their most intense emission maxima of the polymers (Fig. 6a and Table S2†). The average lifetimes (τav) of TPD2T, TPDTT, and TPDBDT were calculated to be 2.04, 1.27, and 0.95 ns, respectively, by fitting the curves to biexponential decays. The short photoluminescence lifetimes of the three polymers indicate a faster electron transfer rate to the surface, which should be favourable for the HER.28,49,53–55 As shown in Fig. 6b, TPDBDT NPs exhibited the smallest Nyquist radius, indicating that they had the highest quality of the interface among the three TPD-based polymers, which is in favour of charge transport at the NP interface. In addition, transient photocurrent measurements showed that TPDTT NPs and TPDBDT NPs have similar photocurrent densities but are much higher than those of TPD2T NPs under visible irradiation (Fig. 6c), suggesting their good photo response ability. Besides, the highest HER of TPDBDT NPs among the three TPD-based materials should be related to the fact that the alkoxy side chains of TPDBDT can significantly promote charge transport on the interface between photocatalyst and aqueous solution owing to their hydrophilicity, leading to effective photo-excited charge separation.21,43
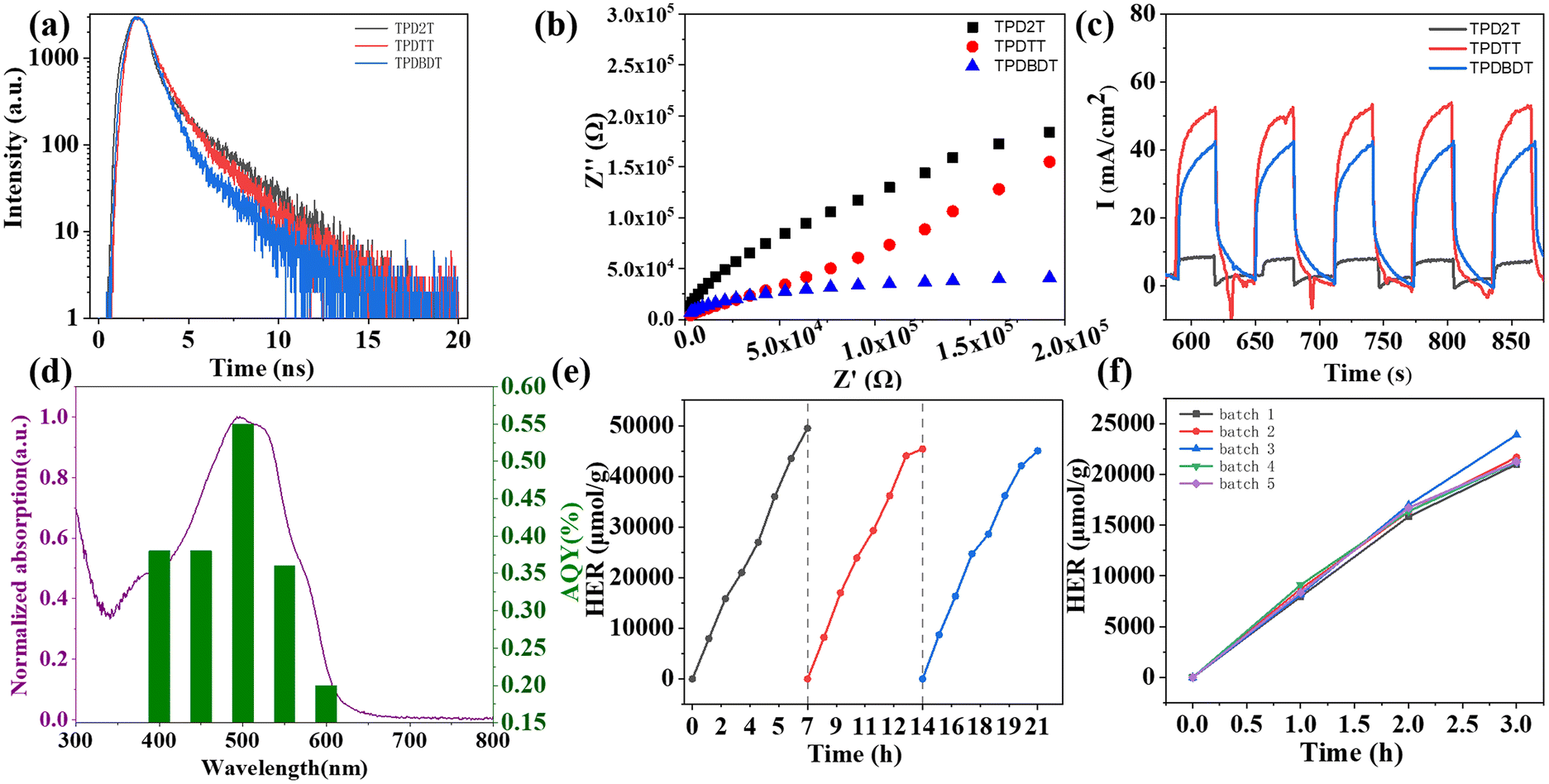 | ||
| Fig. 6 (a) Time-resolved photoluminescence decay curve of TPD2T, TPDTT, and TPDBDT. (b) EIS spectra of TPD2T, TPDTT, and TPDBDT in 0.2 M Na2SO4 aqueous solution under visible light. (c) Transient photocurrent of TPD2T, TPDTT and TPDBDT measured in 0.2 M Na2SO4 aqueous solution. (d) Normalized absorption and apparent quantum yield of the TPDBDT NPs at different wavelengths. (e) Stability test and (f) HER of different batches of TPDBDT NPs. | ||
To measure the spectral contributions to the H2 evolution based on the polymer photocatalysts, the apparent quantum yield (AQY) values of the TPDBDT NPs are calculated under the optimized conditions at 400, 450, 500, 550, and 600 nm using monochromatic light (Fig. 6d). The maximum AQY value of 0.55% occurs at 550 nm, which is consistent with its absorption spectrum (Fig. 4).
Stability test
In this study, TPDBDT NPs were chosen for the photocatalytic stability test owing to their high visible-light activity, surpassing TPD2T and TPDTT NPs. When three consecutive photocatalytic cycles of 21 h (7 h each) under visible-light irradiation were applied to TPDBDT NPs, it was observed that the TPDBDT NPs maintained stable photo-catalytic activity in the cycling measurement with average HERs of over 6.4 ± 0.4 mmol h−1 g−1 after continuous photo-catalytic reaction for 21 h (Fig. 6e). After the long-term reaction, 1H NMR and UV–vis spectrum (Fig. S12, ESI†) of TPDBDT NPs after H2 production shows no obvious change compared with that of the pristine polymer TPDBDT, indicating that the chemical structure of polymer did not change essentially before and after the reaction. Different batches of TPDBDT NPs were performed under the same conditions to investigate the reproducibility of the photocatalytic performance. As shown in Fig. 6f, the photo-catalytic activity did not show obvious changes for different batches of the polymer under visible irradiation, implying excellent photo-catalytic reproducibility.Proposed mechanism of hydrogen production
Heteroatoms in conjugated polymers are regarded as active sites that can interact with protons and catalyze hydrogen evolution reaction.20,21,56 To theoretically investigate the effect of electronic properties on their activities, the Mulliken atomic charges of the fragmental structures were employed. As shown in Fig. 7a, the oxygen atom in the imide group of TPD units possesses the largest concentrated negative Mulliken charge in all cases, suggesting a strong ability in photogenerated electron concentrating, which possesses a large driving force for the hydrogen evolution reaction. This implies that the electron-concentrating ability accounts for the impressive activity obtained in the three polymers. In other words, the atomic charge screening in TPD2T, TPDTT, and TPDBDT demonstrates that the oxygen atom in the imide group of TPD units is a reactive site (Fig. 7a). To further elucidate the role of the O atom in the catalytic activity of the hydrogen evolution reaction, we first employed density functional theory-based calculations to assess the hydrogen binding free energy, ΔGH, which has been recognized as a good descriptor for such a reaction.57,58 Here, hydrogen should be adsorbed neither too weakly (to facilitate the protonation process) nor too strongly (to avoid high barriers for H2 formation and release), which is achieved at an optimal value of ΔGH = 0 eV.57,58 As an example, the metallic catalyst Pt exhibits a good value of ΔGH = −0.1 eV, approximately.59–61 The oligomeric structures have been used to model the properties of the polymers, and ΔGH was computed for hydrogen adsorbed on O sites (see the method section for more computational details). The results are shown in Fig. 7b. As can be observed in Fig. 7b, there is a distinct difference in ΔGH when different sites are hydrogenated. The hydrogen binding at the oxygen site on imides is 1.48, 1.52, and 1.03 eV for TPD2T, TPDTT, and TPDBDT, respectively. It was observed that TPDBDT exhibited the lowest ΔGH of the oxygen site in the imide groups. The oxygen atom in the imide groups is used as the active site for hydrogen production, as mentioned above. The oxygen with lone–pair electrons can bond with an empty orbital of H to become activated H and subsequently react with another H to generate hydrogen, which in turn favours the energetics toward H2 formation. As shown in Fig. 7a, the oxygen atom on the BDT also has a certain concentrated negative Mulliken charge. The hydrogen binding free energy at the oxygen site on alkoxy groups is also calculated at about 1.99 eV, which is a high overpotential for HER. Therefore, the oxygen atom on the alkoxy group may not have become an active site but rather act more like an H+ transport channel, enabling closer contact between H+ and the conjugated backbones. Additionally, as described in the literature,21 the oxygen atom on the alkoxy group adsorbed by H+ can significantly lower the energy levels of conjugated polymers. Therefore, the oxygen atom of BDT units provides many more adsorption sites for H+ loading, which could potentially accelerate the photocatalytic progress. These results suggest that the outstanding photocatalytic activity of TPDBDT is derived from the lowest ΔGH* in the three polymers owing to the suitable electron configuration, which avoids the big barrier for hydrogen formation and release.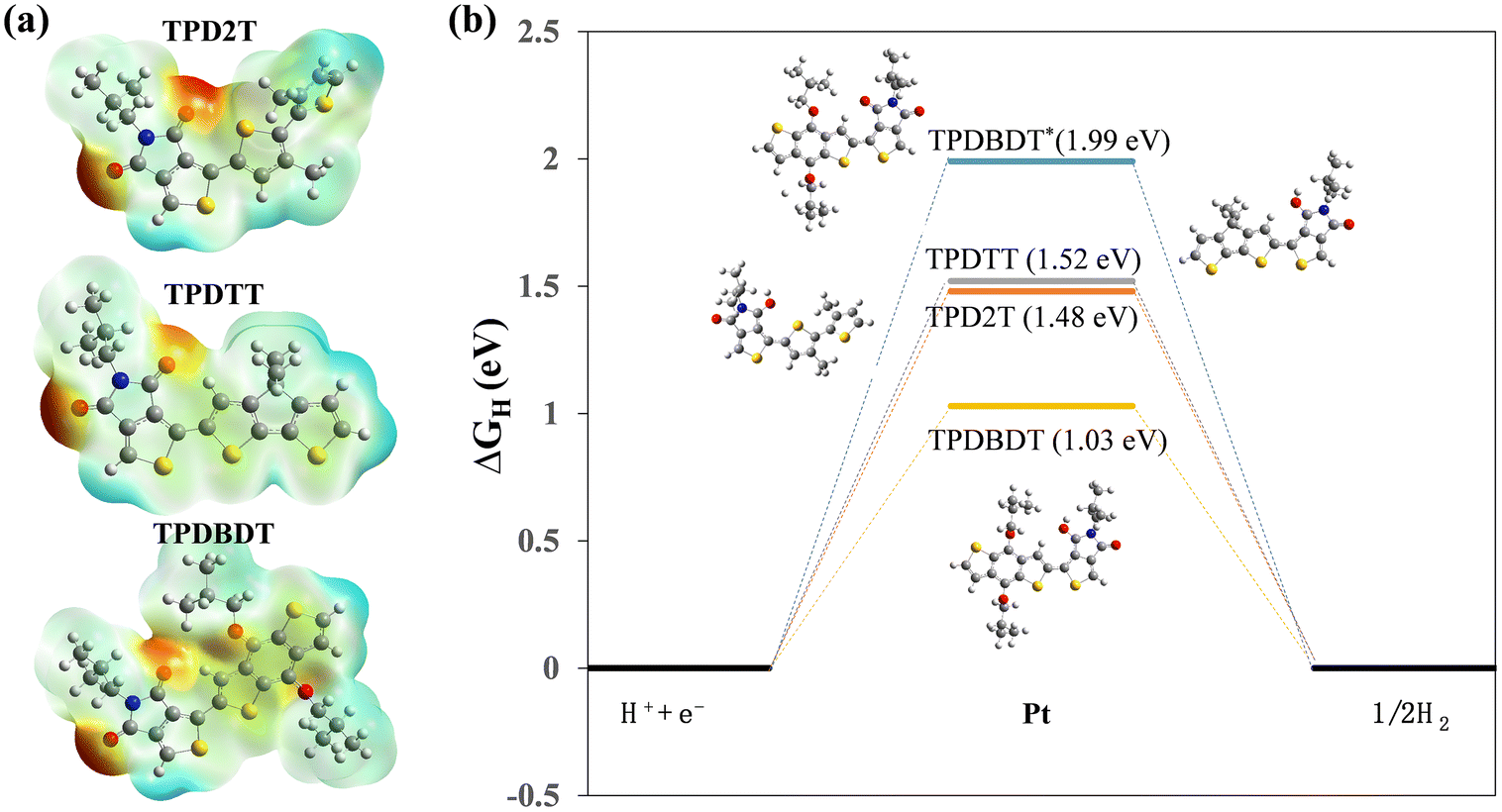 | ||
| Fig. 7 (a) Simulated Mulliken atomic charges of the repeat units of the polymers. (b) Calculated hydrogen binding free energy, ΔGH, at oxygen sites: the case of one hydrogen atom. The insets indicate the structures of the transition states. | ||
Based on the above results, the proposed photocatalytic reaction mechanism is shown in Fig. 8 by taking TPDBDT NPs as an example. As shown in Fig. 8, TPDBDT has a typical donor–acceptor character and separates HOMO and LUMO orbits. During photoexcitation, the photogenerated electrons migrate from the HOMO orbits to the LUMO orbits of the TPDBDT. The strong pull–push interaction between D and A units might accelerate photogenerated electron transfer from BDT to TPD segments.18 When the TPDBDT was encapsulated into an amphiphilic block polymer (PEG–PBLA20) to form nanoparticles, TPDBDT in NPs aggregated together through interchain π–π stacking, significantly shortening the charge diffusion path. Thus, some excited electrons (e−) quickly and efficiently transfer from the LUMO orbits to the NP interface to facilitate a proton reduction reaction (Fig. 8). When platinum was photo-deposited onto the materials, some excited electrons were further transferred from the LUMO orbits to Pt nanoparticles because of the positive Schottky barrier at TPDBDT/Pt interface, which enhanced the electron transfer more efficiently to facilitate proton reduction reaction (Fig. 8). Meanwhile, the photogenerated holes (h+) in the HOMO orbits were consumed by sacrificial reagent AA, which reduced the recombination of the electron–hole pairs and promoted a photocatalytic water-splitting reaction.
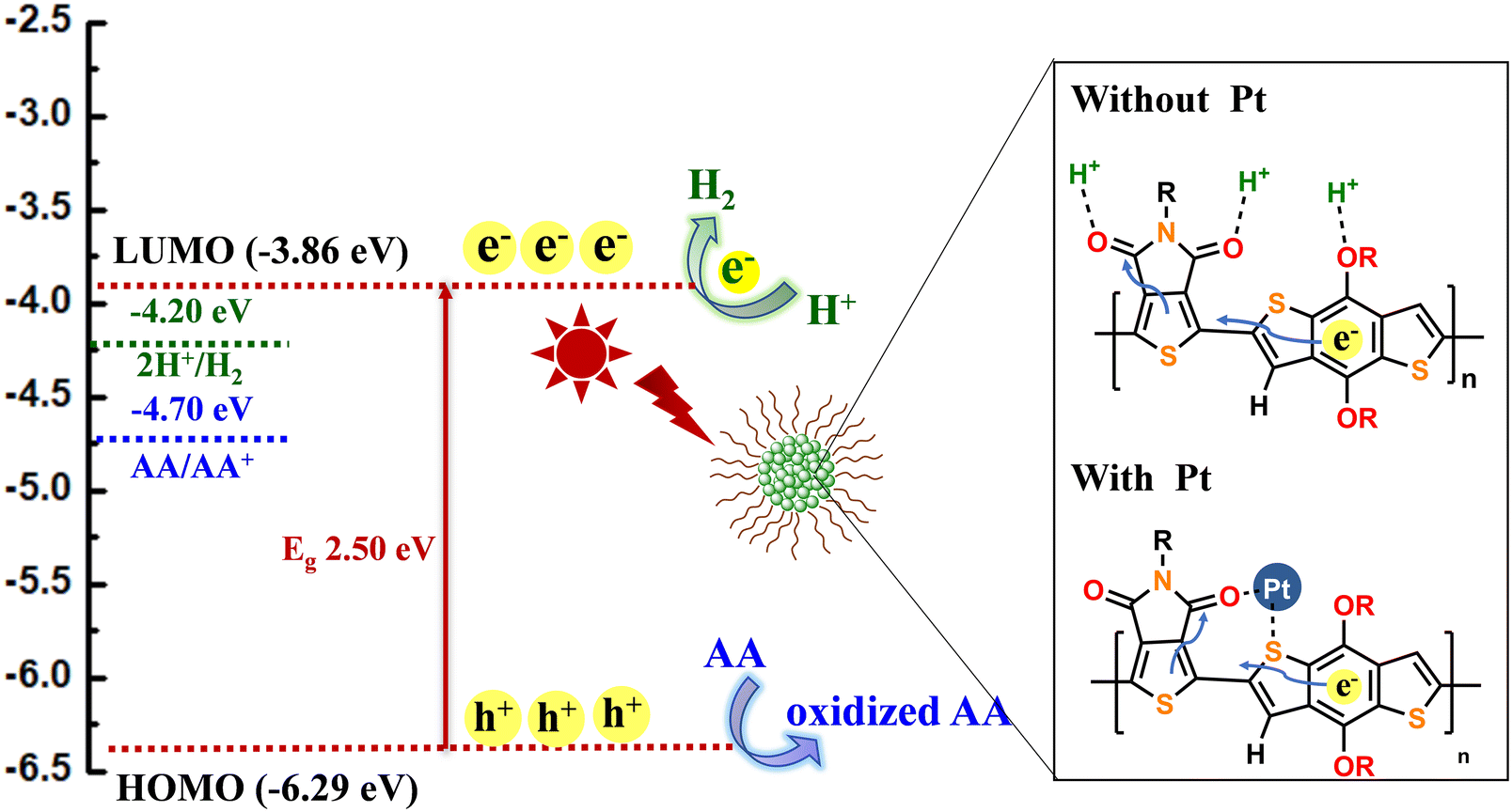 | ||
| Fig. 8 The proposed photocatalytic reaction mechanism of TPDBDT NPs. | ||
Additionally, the HER performance of TPDBDT NPs is significantly enhanced (about 4.1 times) using a Pt co-catalyst. This is also attributed to the fact that multiple chelating Pt sites on TPDBDT can anchor more Pt, as shown in Fig. 8. Based on DFT simulations, the carbonyl group of TPD and sulfur atom of BDT as well as the oxygen atom on the BDT side chains and hydrogen atom on the thiophene ring of BDT could bind Pt effectively. Furthermore, the effect of NP size on HER performance may not be ignored, as previously reported systems.62 Compared with TPD2T and TPDTT NPs, TPDBDT NPs have the smallest hydration diameter (about 102 nm), as revealed in the DLS (Fig. 3), which may increase their total available surface area for proton reduction. In the follow-up work, further research on the relationship between the size of NPs and HER performance may be needed to understand thoroughly the process of producing hydrogen based on these TPD-containing polymers.
Experimental
Synthesis of polymer
5-(2-Octyl-1-dodecyl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (0.10 mmol), thiophene derivatives (0.10 mmol), palladium catalyst (10% mol), additive (20% mol), Ag2CO3 (0.4 mmol) and KOAc (0.4 mmol) were put in a 15 mL round bottom flask. A mixture of DMAc and xylene (v/v = 1:1, 1 mL) was added, and the reaction was heated using an oil bath at various temperatures for 48 h. The mixture was cooled to room temperature and poured into 100 mL of cold methanol. The precipitate was filtered, followed by sequential Soxhlet extractions with methanol, hexanes, and chloroform. After that, the chloroform phase was concentrated to 2 mL and poured into cold methanol (100 mL) to precipitate again. The precipitate was filtered, isolated and dried under vacuum at 40 °C for 12 hours to yield pure polymers. TPD2T: a dark purple solid, 0.047 g, yield: 57.5%. 1H NMR (400 MHz, CDCl3) δ 7.18–6.87 (m, 2H), 3.61 (d, 2H), 2.79–2.31 (m, 4H), 1.94 (m, 1H), 1.32–1.21 (m, 58H), 0.86 (d, 12H). 13C NMR (101 MHz, CDCl3) δ 162.00, 161.83, 143.86, 133.97, 133.76, 133.68, 133.33, 133.27, 133.22, 132.88, 132.76, 128.67, 128.60, 128.48, 127.01, 43.31, 36.76, 31.88, 31.83, 31.76, 31.72, 31.30, 30.51, 30.11, 29.88, 29.76, 29.67, 29.52, 29.50, 29.43, 29.35, 29.22, 29.17, 29.07, 28.75, 26.17, 22.60, 22.53, 13.95. TPDTT: a dark purple solid, 0.055 g, yield: 66.17%. 1H NMR (400 MHz, CDCl3) δ 7.01 (br, 1H), 3.63 (br, 2H), 2.09 (s, 1H), 1.95 (br, 4H), 1.25 (br, 46H), 0.92 (br, 12H). 13C NMR (101 MHz, CDCl3) δ 162.77, 162.60, 143.81, 135.50, 134.77, 134.74, 134.53, 134.06, 133.53, 133.46, 132.83, 128.14, 127.11, 54.57, 50.51, 44.08, 37.54, 35.75, 32.65, 32.60, 32.54, 32.08, 30.88, 30.66, 30.54, 30.44, 30.29, 30.21, 30.12, 29.99, 29.94, 29.17, 27.90, 26.95, 23.38, 23.30, 14.73, 11.34, 11.16. TPDBDT: a dark purple solid, 0.048 g, yield: 54.82%. 1H NMR (400 MHz, CDCl3) δ 8.28–7.29 (m, 2H), 3.89 (br, 4H), 3.66–3.51 (br, 2H), 2.09 (s, 1H), 1.95 (s, 2H), 1.26 (br, 45H), 0.86 (br, 19H). 13C NMR (101 MHz, CDCl3) δ 161.15, 160.97, 146.35, 133.11, 132.91, 132.41, 131.90, 124.32, 122.04, 76.31, 76.20, 76.00, 75.68, 64.31, 40.94, 36.07, 31.73, 31.03, 30.98, 30.91, 30.54, 30.45, 29.25, 29.03, 28.91, 28.82, 28.68, 28.54, 28.50, 28.37, 28.31, 28.09, 26.07, 25.39, 22.31, 22.07, 21.75, 21.68, 18.71, 13.10, 10.09.
Preparation of nanoparticles
Polymer nanoparticles (NPs) were prepared using the nanoprecipitation method.41,42 Briefly, 5 mg of polymer and 1 mg of amphiphilic polyethylene glycol-poly aspartic acid benzyl ester (PEG–PBLA20) were added to 5 mL of tetrahydrofuran (THF). The resultant mixture was sonicated for 15 min and then slowly dropped into 50 mL of ultrapure water under sonication, followed by sonication for 60 min to ensure complete dispersion. Subsequently, the organic phase (THF) was completely removed by bubbling nitrogen gas and polymer NPs were obtained (100 μg mL−1) after constant volume.Photocatalysis test
Aqueous dispersion of polymer NPs (5 mL) and aqueous solution of ascorbic acid (AA) with a concentration of 0.2 M was added to a glass photoreactor (10 mL). The pH of the mixed solution was adjusted with NaOH solution (0.1 M) and sonicated for 15 min. After that, the resultant mixture was degassed by argon bubbling before illumination using a xenon lamp (300 W, λ > 300 nm) as the light source. The temperature of the sample was maintained at 25 °C using flowing cooling water. The generated hydrogen was collected using a gas-tight syringe and injected into a GC-7900 gas chromatograph with argon as the carrier gas. The hydrogen was detected with a thermal conductivity detector, referencing a standard hydrogen of known concentration. The increase in pressure owing to evolved hydrogen was ignored in the calculations.63 The Pt cocatalyst was photo-deposited in situ on the surface of polymers from H2PtCl6 (3 wt%) for 1 h.Conclusions
In summary, we systematically evaluated for the first time the photo-catalytical property of the conjugated polymers synthesized through a greener C–H/C–H coupling polycondensation strategy than the classical Stille and Suzuki coupling methods. Because photocatalytic hydrogen production is also recognized as a green chemical technology, the combination of the two significantly advances the development of clean energy production more sustainably. In this work, three new TPD-containing conjugated polymers, namely TPD2T, TPDTT, and TPDBDT, were efficiently synthesized via C–H/C–H cross-coupling polymerization. Among the three polymers, the TPDBDT-based NPs exhibited the highest HER performance of 1819.4 μmol h−1 g−1 and 7420.0 μmol h−1 g−1 in the absence and presence of the Pt co-catalyst, respectively. More Pt chelating sites on TPDBDT may be responsible for its higher HER performances than those of TPD2T and TPDTT. This work indicates that the electron donor units in D–A conjugated polymers play a crucial role in determining their photocatalytic activity and provides a novel design guideline for organic photocatalysts with high-performance hydrogen evolution.Author contributions
M. Chang and X. Zhang: Investigation, data curation, formal analysis, writing – original draft. L. Wang: Software, data curation, formal analysis. D. Wang: Validation, formal analysis. Q. Zhang: Conceptualization, supervision, writing – original draft, review & editing, funding acquisition. Y. Lu: Supervision, project administration, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully appreciate the financial assistances from the NSFC (No. 21604063 and 52173010), the Project funded by China Postdoctoral Science Foundation (No. 2016M591392), and the Program for Youth Reserve Talent of Tianjin Educational Committee for financial support. We also would like to greatly thank Professor Tongbu Lu and Dr Wen Zhang for help with photocatalytic hydrogen production test.References
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- S. Luo, Z. Zeng, H. Wang, W. Xiong, B. Song, C. Zhou, A. Duan, X. Tan, Q. He, G. Zeng, Z. Liu and R. Xiao, Prog. Polym. Sci., 2021, 115, 101374 CrossRef CAS.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- J. L. Young, M. A. Steiner, H. Döscher, R. M. France, J. A. Turner and T. G. Deutsch, Nat. Energy, 2017, 2, 17028 CrossRef CAS.
- L. Fan, F. Li and S. Ramkumar, Particuology, 2008, 6, 131–142 CrossRef CAS.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1907296 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS.
- M. Liras, M. Barawi and V. A. de la Pena O'Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- M. Mansha, T. Ahmad, N. Ullah, S. Akram Khan, M. Ashraf, S. Ali, B. Tan and I. Khan, Chem. Rec., 2022, 22, e202100336 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- C. Han, S. Xiang, S. Jin, L.-W. Luo, C. Zhang, C. Yan and J.-X. Jiang, J. Mater. Chem. A, 2022, 10, 5255–5261 RSC.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef.
- Y. Liu, J. Wu and F. Wang, Appl. Catal., B, 2022, 307, 121144 CrossRef.
- A. M. Elewa, J. Jayakumar, Y.-W. Huang, M. H. Elsayed, C.-L. Chang, L.-Y. Ting, W.-C. Lin, C.-C. Chueh and H.-H. Chou, J. Environ. Chem. Eng., 2022, 10, 106927 CrossRef.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef PubMed.
- J.-L. Wang, G. Ouyang, D. Wang, J. Li, J. Yao, W.-S. Li and H. Li, Macromolecules, 2021, 54, 2661–2666 CrossRef.
- W.-J. Xiao, Y. Wang, W.-R. Wang, J. Li, J. Wang, Z.-W. Xu, J. Li, J. Yao and W.-S. Li, Macromolecules, 2020, 53, 2454–2463 CrossRef.
- J. Yu, X. Sun, X. Xu, C. Zhang and X. He, Appl. Catal., B, 2019, 257, 117935 CrossRef.
- J. Wang, W. Zhu, Y. Zhang, X. Yang, G. Bai, Q. Zhang, Y. Chen and X. Lan, Macromolecules, 2022, 55, 10842–10853 CrossRef.
- S.-Z. Geng, W.-T. Yang, J. Gao, S.-X. Li, M.-M. Shi, T.-K. Lau, X.-H. Lu, C.-Z. Li and H.-Z. Chen, Chin. J. Polym. Sci., 2019, 37, 1005–1014 CrossRef.
- Y. Liang, T. Li, Y. Lee, Z. Zhang, Y. Li, W. Si, Z. Liu, C. Zhang, Y. Qiao, S. Bai and Y. Lin, Angew. Chem., Int. Ed., 2023, 62, e202217989 CrossRef.
- L. Hao, R. Shen, S. Chen, W. Bi, L. Wang, G. Liang, P. Zhang and X. Li, J. Mater. Chem. A, 2022, 10, 24064–24072 RSC.
- C. Cheng, X. Wang, Y. Lin, L. He, J.-X. Jiang, Y. Xu and F. Wang, Polym. Chem., 2018, 9, 4468–4475 RSC.
- D.-N. Ye, Y.-J. Zhang, Z.-R. Tan, Y.-Q. Xing, Z. Chen, J.-B. Qiu and S.-Y. Liu, Chem. Commun., 2022, 58, 12680–12683 RSC.
- T. Doba, L. Ilies, W. Sato, R. Shang and E. Nakamura, Nat. Catal., 2021, 4, 631–638 CrossRef.
- Q. Zhang, M. Chang, Y. Lu, Y. Sun, C. Li, X. Yang, M. Zhang and Y. Chen, Macromolecules, 2018, 51, 379–388 CrossRef.
- Y. Yang, M. Nishiura, H. Wang and Z. Hou, Coord. Chem. Rev., 2018, 376, 506–532 CrossRef.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef.
- B. Chakraborty and C. K. Luscombe, Angew. Chem., Int. Ed., 2023, 62, e202301247 CrossRef PubMed.
- Q. Zhang, Y. Li, L. Deng, L. Zhao, C. Li and Y. Lu, Chin. J. Polym. Sci., 2018, 36, 1019–1026 CrossRef.
- G. Albano, A. Punzi, M. A. M. Capozzi and G. M. Farinola, Green Chem., 2022, 24, 1809–1894 RSC.
- T. Tian, Z. Li and C.-J. Li, Green Chem., 2021, 23, 6789–6862 RSC.
- Q. Zhang, X. Wan, Y. Lu, Y. Li, Y. Li, C. Li, H. Wu and Y. Chen, Chem. Commun., 2014, 50, 12497–12499 RSC.
- Y. Hu, Y. Liu, J. Wu, Y. Li, J. Jiang and F. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 42753–42762 CrossRef.
- Z. He, L. Zhao, Q. Zhang, M. Chang, C. Li, H. Zhang, Y. Lu and Y. Chen, Adv. Funct. Mater., 2020, 30, 1910301 CrossRef.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef.
- Z.-R. Tan, Y.-Q. Xing, J.-Z. Cheng, G. Zhang, Z.-Q. Shen, Y.-J. Zhang, G. Liao, L. Chen and S.-Y. Liu, Chem. Sci., 2022, 13, 1725–1733 RSC.
- W.-C. Lin, J. Jayakumar, C.-L. Chang, L.-Y. Ting, M. H. Elsayed, M. Abdellah, K. Zheng, A. M. Elewa, Y.-T. Lin, J.-J. Liu, W.-S. Wang, C.-Y. Lu and H.-H. Chou, Appl. Catal., B, 2021, 298, 120577 CrossRef CAS.
- Z.-W. He, Q. Zhang, C.-X. Li, H.-T. Han and Y. Lu, Chin. J. Polym. Sci., 2022, 40, 138–146 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- Y. Zhu, R. D. Champion and S. A. Jenekhe, Macromolecules, 2006, 39, 8712–8719 CrossRef CAS.
- X.-J. Li and Y.-F. Li, Acta Polym. Sin., 2022, 53, 995–1004 CAS.
- P. Zhao, L. Wang, Y. Wu, T. Yang, Y. Ding, H. G. Yang and A. Hu, Macromolecules, 2019, 52, 4376–4384 CrossRef CAS.
- Y. Liu, B. Li and Z. Xiang, Small, 2021, 17, 2007576 CrossRef CAS PubMed.
- R. Shen, Y. Ding, S. Li, P. Zhang, Q. Xiang, Y. H. Ng and X. Li, Chin. J. Catal., 2021, 42, 25–36 CrossRef CAS.
- R. Chen, P. Hu, Y. Xian, X. Hu and G. Zhang, Macromol. Rapid Commun., 2022, 43, 2100872 CrossRef CAS.
- W.-C. Lin, M. H. Elsayed, J. Jayakumar, L.-Y. Ting, C.-L. Chang, A. M. Elewa, W.-S. Wang, C.-C. Chung, C.-Y. Lu and H.-H. Chou, Int. J. Hydrogen Energy, 2020, 45, 32072–32081 CrossRef CAS.
- J. Yang, Y. A. Liu, M.-M. Zhai, J.-J. Qin, W.-B. Hu, H. Yang and K. Wen, Polym. Chem., 2023, 14, 1507–1513 RSC.
- X. Xu, H. Zhong, W. Huang, Y. Sui, R. Sa, W. Chen, G. Zhou, X. Li, D. Li, M. Wen and B. Jiang, Chem. Eng. J., 2023, 454, 139929 CrossRef CAS.
- Z. Pei, J. Gu, Y. Wang, Z. Tang, Z. Liu, Y. Huang, Y. Huang, J. Zhao, Z. Chen and C. Zhi, ACS Nano, 2017, 11, 6004–6014 CrossRef CAS.
- C. Ling, L. Shi, Y. Ouyang and J. Wang, Chem. Mater., 2016, 28, 9026–9032 CrossRef CAS.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- W.-R. Wang, J. Li, Q. Li, Z.-W. Xu, L.-N. Liu, X.-Q. Chen, W.-J. Xiao, J. Yao, F. Zhang and W.-S. Li, J. Mater. Chem. A, 2021, 9, 8782–8791 RSC.
- C. Li, H. Wu, D. Zhu, T. Zhou, M. Yan, G. Chen, J. Sun, G. Dai, F. Ge and H. Dong, Appl. Catal., B, 2021, 297, 120433 CrossRef CAS.
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712–757 CrossRef CAS.
- M. H. Elsayed, J. Jayakumar, M. Abdellah, T. H. Mansoure, K. Zheng, A. M. Elewa, C.-L. Chang, L.-Y. Ting, W.-C. Lin, H.-H. Yu, W.-H. Wang, C.-C. Chung and H.-H. Chou, Appl. Catal., B, 2021, 283, 119659 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04350a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |