
Evaluation of two methods allowing the full preparation in a single day of silicate rocks in view of radiogenic isotope (Nd, Sr, and Pb) analyses
Christian
Pin
a and
Abdelmouhcine
Gannoun
 *b
*b
aLaboratoire de Géochimie Isotopique Environnementale (GIS), Université de Nîmes, 150 Rue Georges Besse, 30035 Nimes CEDEX 1, France
bCNRS, IRD, OPGC, Laboratoire Magmas et Volcans, UMR 6524, Université Clermont Auvergne, F-63000, Clermont-Ferrand, France. E-mail: mouhcine.gannoun@uca.fr
First published on 15th November 2023
Abstract
In this paper, two approaches are described aiming to circumvent the bottle-neck associated with sample decomposition of silicate samples and subsequent isolation of three elements (Nd, Sr, and Pb) of major interest for their radiogenic isotope compositions. These new methods alleviate the need for any evaporation and repeated dissolution steps and make it possible to achieve in a single day the complete preparation of a batch of 10 samples. Specifically, samples are decomposed either by alkaline fusion with a LiBO2 flux followed by quenching in nitric acid, or by quick dissolution in an HF–HNO3 mixture immediately followed by reaction with an aqueous solution of boric acid to neutralize excess HF and dissolve sparingly soluble fluorides. In both cases, the resulting sample solutions are directly loaded onto small chromatographic columns filled with RE and Sr resins, respectively, used in tandem, in order to achieve the concomitant separation of the LREE, Sr, and Pb from matrix elements. While Sr and Pb fractions suitable for isotopic analyses are directly stripped from the Sr resin, an additional column of DGA resin is used, in tandem with the RE resin column, to obtain a Nd fraction isolated from the other LREE. The potential of these most straightforward approaches is demonstrated by repeated analyses of eleven geological reference materials (RMs) spanning a wide range of major element concentrations.
1. Introduction
The variations of radiogenic isotopes of Sr, Pb, and Nd, caused by radioactive decay of 87Rb, 235,238U + 232Th, and 147Sm, respectively, provide a powerful means for tracing the origin and evolution of natural systems. Accordingly, these three isotopic systems, used separately or preferably in combination, make up part of the standard tool box of scientists involved in environmental, geological, and planetary sciences. By virtue of the direct sample introduction at atmospheric pressure inherent to this technique, MC-ICP-MS has greatly simplified the isotope ratio measurement step, increased the overall analytical throughput and, in the case of lead, made it possible to correct instrumental mass fractionation more accurately. However, wherever direct solid sampling by laser ablation is not possible, that is, in all cases but a few accessory minerals containing the elements of interest at high concentrations (e.g., Sr in apatite or some plagioclase and Nd in phosphate phases), it is necessary to greatly concentrate and purify the element to be analyzed from the sample matrix. This is generally achieved by using liquid chromatography techniques, which in turn require decomposing the sample and then putting it in the form of a solution appropriate for the separation process. These steps involve lengthy dissolution and evaporation steps typically requiring several days, much longer than the chromatographic separation proper.In this paper, we describe two new procedures, differing by their sample decomposition steps (viz., alkaline fusion with LiBO2vs. a modified version of acid dissolution with HF), but using the same separation scheme, based on the adaptation of well-established extraction chromatographic techniques. Because they do not involve any evaporation or repeated acid treatments and use miniaturized columns filled with fairly selective extraction chromatographic materials, these protocols make it possible to prepare in a single day a set of 10 samples for measuring 143Nd/144Nd, 87Sr/86Sr, and 208,207,206Pb/204Pb ratios.
2. Experimental
2.1 Chemicals
Water was first deionized with conventional ion-exchange resins and then further purified to a resistivity of 18.2 MΩ cm with an Aqua System Distribution (Bondoufle, France) device. Reagent grade hydrofluoric, nitric and hydrochloric acids (Fluka, Seelze, Germany) were purified by subboiling distillation in PFA DST-100 systems (Savillex, Eden Prairie, MN, USA). Orthoboric acid (Merck, Suprapur, 99.9999%) and ascorbic acid (Sigma, analytical grade, >99%) were used without further purification. The LiBO2 flux used for sample decomposition by fusion was prepared in-house by sintering at 550 °C a mixture in stoichiometric proportions of high-purity Li2CO3 and H3BO3, both of Suprapur grade (supplied by Merck). Eleven geological materials were investigated during the course of this work; eight were provided by the United States Geological Survey (USGS), namely AGV-2 (andesite), BCR-2, BHVO-2 and BIR-1a (basalts), G-2 (granite), GSP-2 (granodiorite), RGM-1 (rhyolite), and STM-1 (nepheline syenite). We also analysed two RMs from the Geological Survey of Japan, namely JA-1 (andesite) and JB-3 (basalt), and finally one basalt (BE-N) from the “Centre de Recherches Pétrographiques et Géochimiques” (CRPG, Nancy, France).2.2 Extraction chromatography materials
Three extraction chromatography (EXC) materials obtained from Triskem International (Bruz, France) were used in this work, respectively, the RE resin, based on octyl(phenyl)-N,N-diisobutylcarbamoylmethylphosphine oxide (CMPO) dissolved in tributyl phosphate,1,2 the DGA resin, based on tetra(n-octyl)diglycolamide,3 and the Sr resin, based on the crown ether di-tert-butylcyclohexano-18 crown-6 (DtBuCH18C6) in 1-octanol.4 In every case, these ligands were sorbed onto Amberchrom CG-71 acrylic ester beads of 50–100 μm particle size.2.3 Instrumentation
Except for the alkaline fusion stage, all chemical handlings were made under class 10 vertical laminar flow hoods in a laboratory supplied with an overpressure of filtered air, at a temperature of 20 °C.A 2.4 kW EasyHeat induction heater (Ambrell Co., Rochester, NY, USA) was used for sample decomposition by fusion with a LiBO2 flux, in home-made, graphite crucibles with a rounded bottom machined from 25 mm diameter high-purity graphite rods (Ringsdorff, Bonn, Germany) covered with a flat graphite disk during RF heating.
Screw cap 15 ml PFA vessels (Savillex, Eden Prairie, MN, USA) were used for sample dissolution. The separated fractions of Sr, Pb, and Nd were collected in 5 ml screw cap PFA vials with a conical bottom (Savillex) and evaporated to dryness.
Three different columns, fitted with polyethylene frits at the bottom and at the top of the resin bed, were involved. The first short column, ca. 5 mm i.d., is prepared from a “fine tip, large bulb”, model 234 (Samco Scientific) obtained from Thermo Scientific. It contains 100 mg of RE resin, slurry-packed in dilute HNO3, making a ca. 10 mm high resin bed. The second short column, ca. 5 mm i.d., is made of silica glass and contains 83 mg of Sr resin.
The third, longer column is made of a ca. 4 mm i.d. transfer pipette (model 235 (Samco Scientific)) filled with a slurry containing 500 mg of DGA resin, corresponding to a resin height of ca. 80 mm. These are depicted in Fig. 1.
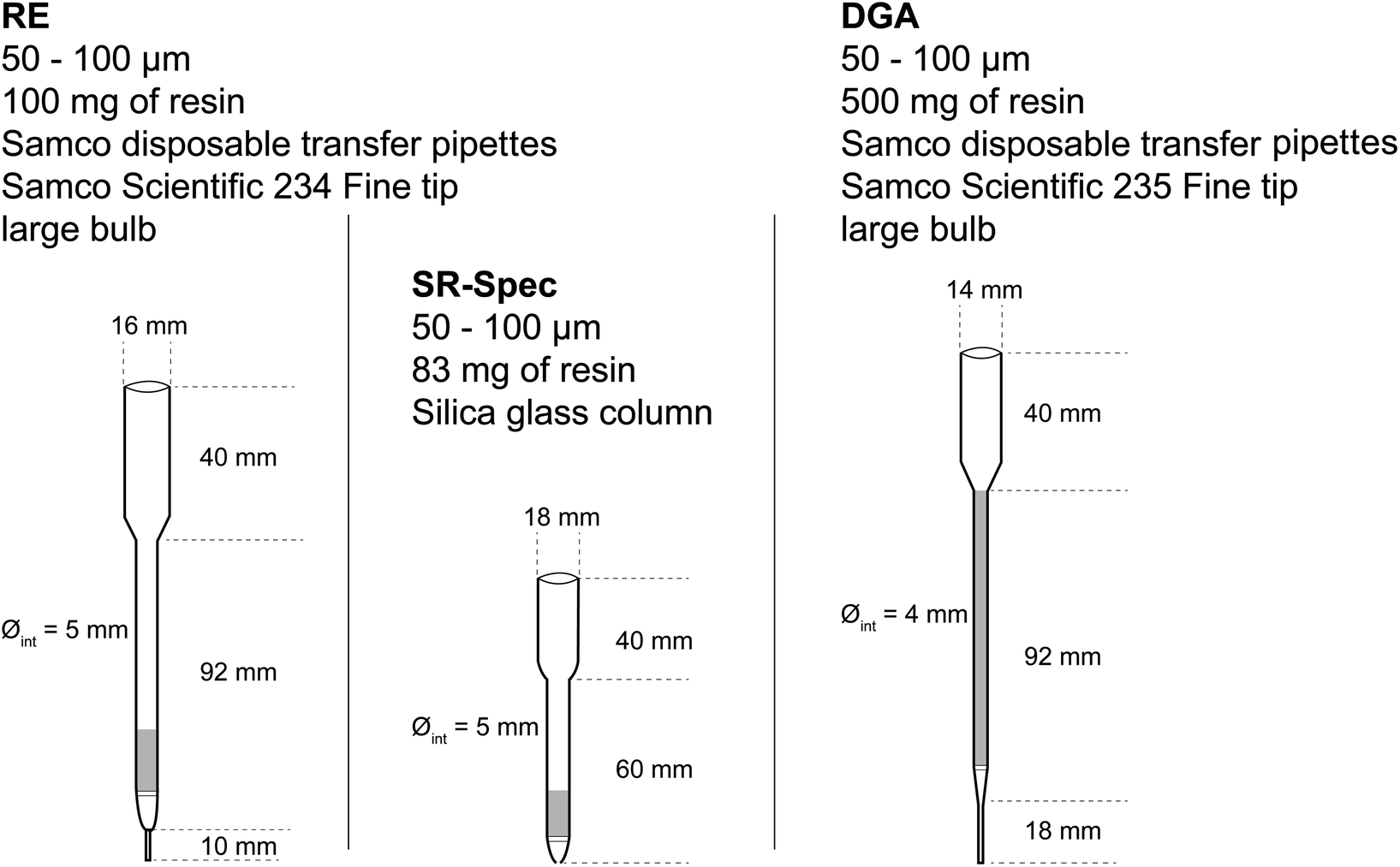 | ||
| Fig. 1 Summary of the geometry and resins of the three extraction chromatography columns used in this work. | ||
Two quadrupole ICP-MS (Agilent 7500 and 8900, Agilent Technologies, Les Ulis, France) instruments were used for setting up the separation method and further characterization of the protocol, respectively. All isotope ratio measurements were made with a Neptune Plus MC-ICP-MS (Thermo Scientific) with a conventional sample introduction system, and operating parameters are given in Table 1.
| a Optimized daily for maximum Sr, Nd or Pb sensitivity. | |
|---|---|
| Analysis | Sr, Nd and Pb isotopes |
| Laboratory | Magmas et Volcans |
| MC-ICP-MS model | Thermo Neptune Plus |
| Plasma condition | Wet (cyclonic spray chamber) |
| RF power | 1200 W |
| Resistors | 1011 ohms |
| Resolution | Low (M/ΔM ∼ 400) |
| Cool gas flow (Ar)a | 15–16 L min−1 |
| Auxiliary gas (Ar)a | 0.7–0.8 L min−1 |
| Sample gas (Ar)a | 0.9–1.1 L min−1 |
| Sample uptake | 100 μl min−1 |
| Sample cone | Standard |
| Skimmer cone | H |
| Sensitivity (V per μg per g) | ∼60 |
| Scanning mode | Static multi-collection |
| Integration time | 8.4 s |
| Number of cycles | 60 |
| Number of blocks | 1 |
2.4 Sample digestion
2.5 Chemical separation
Whatever the digestion technique used, the ca. 6 ml sample solution in 2 mol per L HNO3 containing ascorbic acid is loaded onto the RE column placed over the Sr column (both preconditioned in 2 mol per L HNO3). The loading is made in 6 portions of 1 ml each, in order to keep to a minimum the surface of the column reservoir in contact with the solution. After complete draining, the two columns, still in tandem, are rinsed twice with 0.5 ml of 2 mol per L HNO3–0.145 mol per L HF. Then, the RE and Sr columns are decoupled. The RE column is further rinsed with 0.5 ml of 1 mol per L HNO3. Following this, the rest of the unwanted matrix elements and the heavy REE are stripped by using a suite of small fractions of nitric acid of decreasing strength, specifically, 2 × 0.25 ml of 0.5 mol per L HNO3, 2 × 0.25 ml of 0.25 mol per L HNO3, 2 × 0.25 ml of 0.125 mol per L HNO3, and finally 0.15 ml of 0.05 mol per L HNO3. At this stage, the DGA column (preconditioned with 3 mol per L HCl) is placed below the RE column, and the LREEs are eluted from the RE resin and simultaneously loaded onto the DGA column by means of 2 ml of 3 mol per L HCl. Then, the columns are decoupled and the lightest lanthanides (La–Ce–Pr) are eluted from the DGA column with 3.55 ml of 2 mol per L HCl. Finally, the Nd fraction is recovered with 2.75 ml of 1 mol per L HCl.After decoupling from the RE column, the Sr column is rinsed with 0.5 ml of 2 mol per L HNO3 and then 2 ml of 7 mol per L HNO3 to get rid of matrix elements left in the free column volume and Ba, respectively. A small, intermediate fraction of 0.5 ml of 2 mol per L HNO3 precedes the elution of the Sr fraction with 2 ml of 0.05 mol per L HNO3. Finally, Pb is stripped with 2 ml of 6 mol per L HCl. The whole procedure of chemical separation is outlined in Table 2.
| Reagent | Volume (ml) | Step Durationc (min) | |
|---|---|---|---|
| a The column pre-cleaning is performed the day before processing the samples. The duration of sample preparation and digestion was ∼120 min. b The duration time reported here is for the whole step after decoupling Sr-spec and RE columns, since they were processed in parallel on separate column supports. c The duration time given here is for a set of 10 columns. | |||
| Column pre-cleaning and preconditioning | |||
| Sr-spec column (83 mg of Sr-spec resin) | 6 mol L per HCl | 5 | |
| 0.05 mol L per HNO3 | 5 | ||
| Preconditioning | 2 mol L per HNO3 | 0.5 | |
| 3 mol L per HCl | 4 | ||
| RE column (100 mg of RE resin) | 0.05 mol L per HNO3 | 4 | |
| Preconditioning | 2 mol L per HNO3 | 0.5 | |
| DGA column (500 mg of DGA resin) | 0.1 mol L per HCl–0.29 mol L per HF | 4 | |
| 0.05 mol L per HCl | 4 | ||
| Preconditioning | 3 mol L per HCl | 0.5 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| RE and Sr-spec columns in tandem | ∼90 | ||
| Sample loading | 2 mol L per HNO3–C6H8O6 | 6 × 1 | |
| Reservoir and column rinsing | 2 mol L per HNO3–0.145 mol L per HF | 2 × 0.5 | |
|
|||
| Columns decoupling and further elutionb | ∼120 | ||
| Sr Spec column | |||
| Elution of Ba | 7 mol L per HNO3 | 2 | |
| Rinse | 2 mol L per HNO3 | 0.5 | |
| Sr stripping | 0.05 mol L per HNO3 | 2 | |
| Transition | 3 mol L per HCl | 4 | |
| Pb stripping | 6 mol L per HCl | 2 | |
| RE column | |||
| Stripping of unwanted elements (residual major and minor elements, HREE) | 1 mol L per HNO3 | 0.5 | |
| 0.5 mol L per HNO3 | 0.5 | ||
| 0.25 mol L per HNO3 | 0.5 | ||
| 0.125 mol L per HNO3 | 0.5 | ||
| 0.05 mol L per HNO3 | 0.15 | ||
|
|||
| RE and DGA columns in tandem | |||
| Elution/loading of the LREE | 3 mol L per HCl | 2 | ∼60 |
|
|||
| Columns decoupling and further elution | ∼240 | ||
| DGA column | |||
| Reservoir and column rinsing | 3 mol L per HCl | 0.5 | |
| Pre-Nd fraction (La–Ce–Pr) | 2 mol L per HCl | 3.55 | |
| Nd stripping | 1 mol L per HCl | 2.75 | |
| Sm stripping | 0.5 mol L per HCl | 1.5 | |
The total duration of the whole procedure, for a batch of 10 samples, including weighings is about 12 hours.
3. Results
The elution profiles of the elements of interest on the RE and DGA columns used in tandem are compared in Fig. 2 in order to evaluate the potential effect of the alternative sample digestion modes on chromatographic separation. Although a subtly earlier breakthrough of La can be noticed in the fusion-digested profile, there is no significant shift of the LREE peaks, in spite of the fact that the HFSEs, including titanium, display contrasting behaviours in F-bearing and F-free contexts. Indeed, if the RE resin does not extract HFSEs when these elements are complexed by fluorine – as is the case when the sample has been dissolved with HF, it has a distinct affinity for these elements in the absence of fluoride (DZr > 400 and DTica. 25 in 2 mol per L HNO3).2 As a result, the effective column capacity for the LREE might have been somewhat reduced when processing samples digested by fusion. However, the similarity of the elution profiles shows that the combined RE-DGA EXC approach is robust enough to make such effects negligible and can handle sample solutions prepared by contrasting methods, even when micro-columns are used.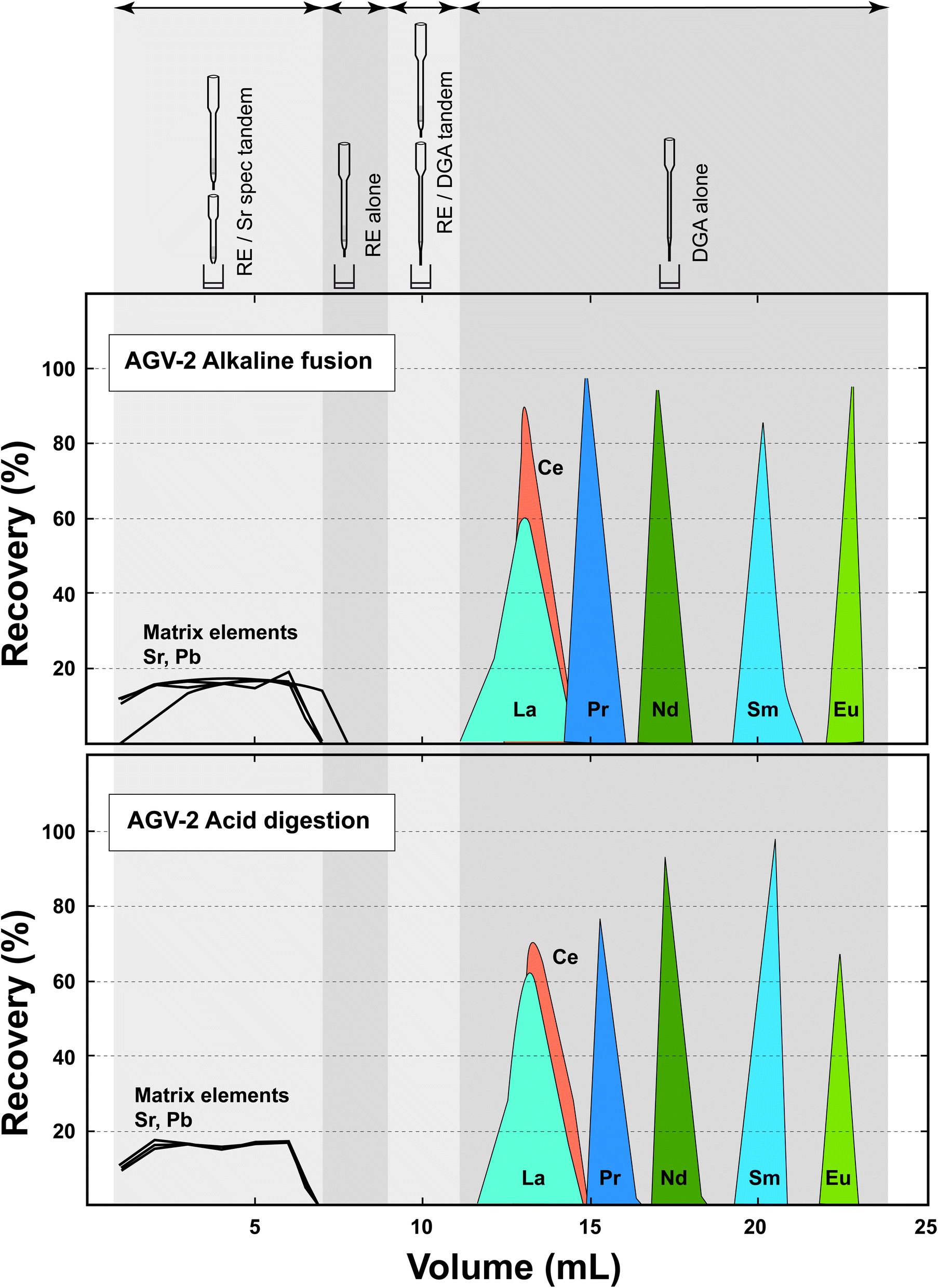 | ||
| Fig. 2 Elution profiles on the RE and DGA columns for the RM AGV-2 digested by LiBO2 fusion and HF dissolution. The detailed protocol including the volumes and types of acid used is shown in Table 2. | ||
The ranges of chemical recoveries measured for the three elements are listed in Table 3 for both sample digestion methods. While essentially identical values around 90% were obtained for Nd, it can be observed that slightly lower recoveries (in the 80–90% range) were achieved for Sr and Pb when the samples were digested by fusion. The cause of this difference is not clear at present. However, because the LiBO2 flux is likely to contain non-negligible amounts of potassium, the only element (besides Ba, Sr and Pb) to be significantly extracted using the Sr resin,4 it is tentatively suggested that this element might have reduced the capacity of the column and caused a partial loss of Sr and Pb during sample loading. If this explanation proves to be true, this problem could be circumvented by using a slightly greater amount of resin, or obtaining a flux cleaner in K. Nevertheless, it is noteworthy that good chemical recoveries can be obtained from both expeditious silicate sample digestion procedures investigated.
| Yields (%) | Alkaline fusion method | HF flash method | ||
|---|---|---|---|---|
| Min | Max | Min | Max | |
| Sr | 80 | 88 | 85 | 93 |
| Pb | 79 | 94 | 95 | 100 |
| Nd | 85 | 95 | 82 | 93 |
The analytical contamination associated with the reagents and columns used in this work can be evaluated from the data given in Table 4, together with the total procedural blanks measured for the two HF-based and LiBO2-based methods. Not surprisingly, total procedural blanks achieved for the procedure based on sample digestion by fusion are significantly larger than those obtained with the acid dissolution. However, the results do not differ by a factor greater than three for Sr, and even two for Pb and Nd, thus indicating that – contrary to a widespread opinion – the sample digestion approach by fusion does not suffer from an intrinsic, irremediable flaw. Indeed, from a practical point of view, such levels of contamination would not be excessive for processing a wide range of common samples, in which a 50 mg sample portion typically contains several μg of Sr and several tens of ng of Pb and Nd.
| 7 mol per L HNO3 (pg ml−1) | 6 mol per L HCl (pg ml−1) | Column blank (pg) | Total blank (fusion) (pg) | Total blank (HF flash) (pg) | |
|---|---|---|---|---|---|
| Sr | 5.1 ± 1.0 | 4.3 ± 1.2 | 14.1 ± 3.2 | 125 ± 37 | 45 ± 17 |
| Pb | 3.1 ± 0.7 | 1.5 ± 0.4 | 7.8 ± 2.1 | 95 ± 28 | 42 ± 15 |
| Nd | 0.9 ± 0.2 | 1.1 ± 0.3 | 6.3 ± 1.9 | 21 ± 6 | 13 ± 5 |
| (n = 4) | (n = 4) | (n = 6) | (n = 3) | (n = 3) |
In order to demonstrate the potential of these two single day preparation methods prior to radiogenic isotope measurements, eleven reference materials have been processed by using both protocols, and the Nd, Sr, and Pb fractions isolated in this way were analyzed by MC-ICP-MS for 143Nd/144Nd, 87Sr/86Sr, and 208,207,206Pb/204Pb ratios. The results for Nd are listed in Table 5, along with the average value of the isotopic standard JNdi-1, and the net values measured for the ion beams at m/z 140 and 147, which are used to monitor Ce and Sm, the two elements having isobaric interferences with Nd at mass 142 and 144, respectively. These values document that Nd was very well isolated from these two troublesome elements, although the significantly higher signal for 147Sm+ than for 140Ce+ (73 ± 48 (SD) μV against 9 ± 25 μV, respectively) suggests that, unless a very minor polyatomic interference occurred at m/z 147, the volume of 1 mol per L HCl used to recover the Nd fraction could be slightly reduced. The overall accuracy of our results can be judged from a comparison with the ranges of values obtained worldwide for these geostandards, as compiled by GeoReM.5 It can be seen that the 143Nd/144Nd ratios measured by both modes of digestion agree very well within analytical precision. The only possibly significant difference is observed for GSP-2, a ca. 1.4 Ga old granodiorite, for which the two subsamples digested by fusion are very slightly less radiogenic than that dissolved with HF. This might suggest that a refractory phase with an even less radiogenic signature was not entirely opened during the HF attack. However, all three data points are well within the range of values of the GeoReM compilation and the isotopic dispersion might equally well be interpreted in terms of subtle heterogeneity of that RM, prepared from a coarse-grained igneous rock prone to the “nugget effect” for trace elements such as Nd. It is concluded from these data that swift HF dissolution promptly followed by treatment with boric acid does not introduce any perceptible bias, as far as Nd isotopes are concerned, in part because the highly refractory phase zircon generally does not play a major role in the mass balance of that element.
| Geostandards | Method | 140Ce (V) | 144Nd (V) | 147Sm (V) | 143Nd/144Nda (±2 s.e.) | 143Nd/144Nd | |
|---|---|---|---|---|---|---|---|
| Min. GEOREM | Max. GEOREM | ||||||
| a The mean 143Nd/144Nd of the JNdi-1 standard is 0.512116 ± 0.000008 (2 SD; N = 7). | |||||||
| JA-1 | Fusion | 4.0 × 10−5 | 3.52 | 2.8 × 10−5 | 0.513095 ± 0.000006 | 0.513047 | 0.513112 |
| JA-1 | Flash HF | −1.3 × 10−5 | 2.99 | 4.8 × 10−5 | 0.513096 ± 0.000007 | ||
| BHVO-2 | Fusion | 1.5 × 10−5 | 6.01 | 8.2 × 10−5 | 0.512990 ± 0.000005 | 0.512115 | 0.513013 |
| BHVO-2 | Flash HF | −2.9 × 10−5 | 2.95 | 3.5 × 10−5 | 0.512990 ± 0.000007 | ||
| BIR-1a | Fusion | −2.2 × 10−5 | 0.78 | 3.0 × 10−5 | 0.513086 ± 0.000013 | 0.513088 | 0.513106 |
| BIR-1a | Flash HF | −1.8 × 10−5 | 0.47 | 7.7 × 10−6 | 0.513092 ± 0.000018 | ||
| JB-3 | Fusion | 2.1 × 10−5 | 3.34 | 3.5 × 10−5 | 0.513063 ± 0.000006 | 0.513024 | 0.513091 |
| JB-3 | Flash HF | 1.8 × 10−6 | 2.49 | 4.4 × 10−5 | 0.513066 ± 0.000007 | ||
| BE-N | Fusion | 2.8 × 10−6 | 7.01 | 7.7 × 10−5 | 0.512882 ± 0.000005 | 0.512862 | 0.512895 |
| BE-N | Flash HF | 3.1 × 10−5 | 5.24 | 6.1 × 10−5 | 0.512883 ± 0.000006 | ||
| BCR-2 | Fusion | −9.8 × 10−6 | 5.14 | 9.5 × 10−5 | 0.512642 ± 0.000006 | 0.5126 | 0.5129 |
| BCR-2 | Flash HF | 3.8 × 10−5 | 3.11 | 3.5 × 10−5 | 0.512638 ± 0.000007 | ||
| BCR-2 | Flash HF | 3.9 × 10−6 | 0.29 | 6.5 × 10−5 | 0.512649 ± 0.000010 | ||
| STM-1 | Fusion | 1.5 × 10−6 | 8.41 | 1.1 × 10−4 | 0.512916 ± 0.000005 | 0.512908 | 0.512939 |
| STM-1 | Flash HF | 9.9 × 10−6 | 4.38 | 6.7 × 10−5 | 0.512919 ± 0.000005 | ||
| RGM-1 | Fusion | −1.1 × 10−5 | 4.10 | 4.3 × 10−5 | 0.512800 ± 0.000006 | 0.512784 | 0.512816 |
| RGM-1 | Fusion | 1.6 × 10−5 | 4.39 | 5.0 × 10−5 | 0.512797 ± 0.000006 | ||
| RGM-1 | Flash HF | 5.3 × 10−6 | 1.91 | 3.9 × 10−5 | 0.512812 ± 0.000009 | ||
| RGM-1 | Flash HF | −2.3 × 10−5 | 2.19 | 3.6 × 10−5 | 0.512807 ± 0.000009 | ||
| GSP-2 | Fusion | 5.1 × 10−5 | 6.65 | 1.4 × 10−4 | 0.511362 ± 0.000004 | 0.511348 | 0.511389 |
| GSP-2 | Fusion | 3.9 × 10−5 | 11.09 | 2.5 × 10−4 | 0.511354 ± 0.000003 | ||
| GSP-2 | Flash HF | 6.3 × 10−5 | 3.86 | 1.1 × 10−4 | 0.511373 ± 0.000005 | ||
| AGV-2 | Fusion | −2.0 × 10−5 | 7.44 | 8.6 × 10−5 | 0.512788 ± 0.000004 | 0.512755 | 0.512802 |
| AGV-2 | Flash HF | 1.6 × 10−5 | 2.66 | 6.1 × 10−5 | 0.512778 ± 0.000007 | ||
| G-2 | Flash HF | 1.5 × 10−5 | 5.66 | 1.2 × 10−4 | 0.512232 ± 0.000006 | 0.512215 | 0.512258 |
Lead isotope data do not show clear discrepancy between those samples decomposed by fusion and those dissolved with HF (Table 6). Only the results for GSP-2 digested by fusion display slightly more radiogenic 206Pb/204Pb and 207Pb/204Pb and less radiogenic 208Pb/204Pb ratios, consistent with a more complete opening of an ancient, accessory mineral with a high U/Th ratio, presumably zircon, as commonly found in crustal plutonic rocks. The fusion data for JA-1 display more radiogenic signatures of both uranogenic and thorogenic ratios than those of the acid dissolution ones, but an interpretation in terms of an ancient component rich in U and Th is not very attractive in this volcanic sample. More work would be required to get further insights into this difference. The agreement of our data with the 206Pb/204Pb ratios compiled by GeoReM documents the overall accuracy of the method. Based on the similar ion beam sizes obtained for both digestion modes, no evidence was found for any significant loss of lead as a volatile species during fusion, in contrast with the observation of Totland et al.6 who used much longer (20 min.) fusions in a muffle furnace.
| Geostandards | Method | 208Pb (V) | 206Pb/204Pba | 207Pb/204Pba | 208Pb/204Pba | 206Pb/204Pb | 88Sr (V) | 87Sr/86Srb | 87Sr/86Sr | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| This study | This study | This study | Min. GEOREM | Max. GEOREM | This study | Min. GEOREM | Max. GEOREM | ||||
| a The Pb instrumental mass fractionation was corrected during measurement by doping the Pb solutions with the Tl NBS SRM 997 standard. The NBS 981 standard was measured between every two samples to correct Pb ratios for instrumental drift and the slight difference in mass fractionation between Pb and Tl. Data were normalized to the NBS981 standard using the values of Galer et al.7 (206Pb/204Pb = 16.9405, 207Pb/204Pb = 15.4963, and 208Pb/204Pb = 36.7219). Repeated analysis of the NBS SRM 981 standard (N = 25) every two samples yielded an average intra-session 2SD reproducibility of 119 ppm for both the 206Pb/204Pb and 207Pb/204Pb ratios and 128 ppm for the 208Pb/204Pb ratio. b Strontium isotope compositions were corrected for mass fractionation using 88Sr/86Sr = 8.375209. Repeated measurement of the NBS SRM 987 standard (N = 12) during the course of the study yielded 87Sr/86Sr = 0.710167 ± 25 (2SD). Sample data were renormalized to the accepted value of 0.710245. | |||||||||||
| JA-1 | Fusion | 10.7 | 18.3259 ± 0.0003 | 15.5654 ± 0.0003 | 38.3481 ± 0.0009 | 18.267 | 18.320 | 6.9 | 0.703592 ± 0.000010 | 0.70350 | 0.07036 |
| JA-1 | Flash HF | 8.6 | 18.3189 ± 0.0006 | 15.5416 ± 0.0005 | 38.2675 ± 0.0013 | 8.1 | 0.703522 ± 0.000009 | ||||
| BHVO-2 | Fusion | 10.3 | 18.6412 ± 0.0004 | 15.5517 ± 0.0004 | 38.2966 ± 0.0011 | 18.620 | 18.662 | 9.3 | 0.703503 ± 0.000005 | 0.70349 | 0.70502 |
| BHVO-2 | Flash HF | 10.4 | 18.6400 ± 0.0004 | 15.5457 ± 0.0004 | 38.2721 ± 0.0012 | 6.6 | 0.703492 ± 0.000006 | ||||
| BIR-1a | Fusion | 11.1 | 18.8378 ± 0.0004 | 15.6263 ± 0.0005 | 38.3613 ± 0.0013 | 18.674 | 18.853 | 3.0 | 0.703120 ± 0.000013 | 0.70307 | 0.70311 |
| BIR-1a | Flash HF | 10.8 | 18.8529 ± 0.0004 | 15.6456 ± 0.0004 | 38.4494 ± 0.0011 | 2.1 | 0.703108 ± 0.000012 | ||||
| JB-3 | Fusion | 9.0 | 18.2838 ± 0.0003 | 15.5310 ± 0.0003 | 38.2226 ± 0.0010 | 18.287 | 18.296 | 8.1 | 0.703486 ± 0.000008 | 0.70338 | 0.70367 |
| JB-3 | Flash HF | 7.6 | 18.2868 ± 0.0012 | 15.5307 ± 0.0010 | 38.2331 ± 0.0026 | 9.3 | 0.703445 ± 0.000008 | ||||
| BE-N | Fusion | 10.9 | 19.1857 ± 0.0005 | 15.6001 ± 0.0004 | 38.9198 ± 0.0011 | 19.172 | 19.249 | 10.5 | 0.703791 ± 0.000006 | 0.70328 | 0.70403 |
| BE-N | Flash HF | 10.3 | 19.1849 ± 0.0004 | 15.6046 ± 0.0005 | 38.9110 ± 0.0014 | 9.5 | 0.703770 ± 0.000005 | ||||
| BCR-2 | Fusion | 5.0 | 18.7584 ± 0.0008 | 15.6243 ± 0.0007 | 38.7300 ± 0.0018 | 18.740 | 18.790 | 9.4 | 0.705167 ± 0.000008 | 0.70350 | 0.70517 |
| BCR-2 | Flash HF | 6.0 | 18.7539 ± 0.0007 | 15.6218 ± 0.0007 | 38.7123 ± 0.0019 | 9.8 | 0.705160 ± 0.000009 | ||||
| STM-1 | Fusion | 4.9 | 19.5158 ± 0.0010 | 15.6298 ± 0.0009 | 39.2506 ± 0.0025 | 19.515 | 19.519 | 7.0 | 0.703818 ± 0.000006 | 0.70374 | 0.70382 |
| STM-1 | Flash HF | 6.0 | 19.5134 ± 0.0007 | 15.6295 ± 0.0006 | 39.1793 ± 0.0018 | 9.3 | 0.703792 ± 0.000005 | ||||
| RGM-1 | Fusion | 6.1 | 19.0032 ± 0.0007 | 15.6321 ± 0.0007 | 38.7028 ± 0.0018 | 18.988 | 19.005 | 6.8 | 0.704166 ± 0.000010 | 0.70419 | 0.70426 |
| RGM-1 | Fusion | 6.0 | 18.9952 ± 0.0007 | 15.6298 ± 0.0007 | 38.6480 ± 0.0020 | 5.8 | 0.704183 ± 0.000010 | ||||
| RGM-1 | Flash HF | 6.3 | 19.0012 ± 0.0007 | 15.6293 ± 0.0007 | 38.6932 ± 0.0018 | 5.7 | 0.704208 ± 0.000012 | ||||
| RGM-1 | Flash HF | 6.1 | 18.9965 ± 0.0006 | 15.6289 ± 0.0006 | 38.6868 ± 0.0016 | 4.3 | 0.704177 ± 0.000015 | ||||
| GSP-2 | Fusion | 6.1 | 17.5966 ± 0.0007 | 15.5257 ± 0.0007 | 50.1153 ± 0.0025 | 17.527 | 17.615 | 9.3 | 0.765136 ± 0.000009 | 0.76500 | 0.76522 |
| GSP-2 | Flash HF | 7.3 | 17.5728 ± 0.0007 | 15.5147 ± 0.0007 | 50.3088 ± 0.0022 | 8.5 | 0.765062 ± 0.000008 | ||||
| AGV-2 | Flash HF | 6.0 | 18.8612 ± 0.0007 | 15.6104 ± 0.0006 | 38.5662 ± 0.0017 | 18.851 | 18.907 | 10.5 | 0.703942 ± 0.000006 | 0.70393 | 0.70408 |
| G-2 | Flash HF | 6.2 | 18.3777 ± 0.0005 | 15.6314 ± 0.0005 | 38.8778 ± 0.0015 | 18.370 | 18.422 | 9.9 | 0.709727 ± 0.000005 | 0.70830 | 0.70990 |
Although the results for Sr (Table 6) do not show a large bias between the values obtained after fusion and HF dissolution, respectively, it can be observed that, in most cases, slightly higher 87Sr/86Sr ratios were measured for those samples digested by fusion. The largest offset of 8 × 10−5 is observed for the most radiogenic ratios measured in GSP-2, a very ancient rock, which developed large isotopic differences among its rock-forming minerals, and is therefore not likely to be perfectly homogeneous at the 50 mg subsample scale, as far as radiogenic isotopes are concerned. In the other cases, the higher 87Sr/86Sr might be interpreted to reflect the effect of the higher procedural blank inherent to the fusion method. However, this explanation would not account for the conspicuous absence of negative correlation between the offset of 87Sr/86Sr ratios and the amount of sample Sr processed; indeed, samples with the highest Sr mass fraction (STM-1: 700 μg per g Sr and BE-N: 1370 μg per g Sr)8 show the same positive bias (3 × 10−5 and 2 × 10−5, respectively) than the much poorer BIR-1 basalt (110 μg per g Sr, 2 × 10−5), while RGM-1 (110 μg per g Sr) has a negative bias of the same amplitude. It is therefore suspected that the observed differences might be due to a limited accuracy, not adequately reflected by the quoted within-run precision of 87Sr/86Sr ratios measured by MC-ICP-MS. It is tentatively suggested that residual amounts of Li left in the Sr fraction, quite likely to occur after single pass separation, might have subtle effects on instrumental mass bias which is the Achilles' heel of that technique. Further work involving measurements by TIMS would be required to get clearer insights into this question.
Whatever the reasons for these small discrepancies, it is emphasized that our Sr and Pb isotope data are encompassed by those compiled by GeoReM, thereby documenting an overall satisfactory accuracy. This is noteworthy because the fusion method is generally deemed “dirty”, not only because of the use of the alkaline flux, but also because it involves sample handling in a HT environment that is not easy to keep under very clean conditions.
4. Discussion
4.1 Sample size
A nominal sample size of 50 mg was chosen because, in most cases, this provides more than enough of the three target elements to measure radiogenic isotope ratios. Besides this, processing as small a sample mass as possible facilitates the digestion process and keeps to a minimum the amount of expensive and/or hazardous high-purity reagents involved in the subsequent steps. Certainly, such a mass is on the lower end of the acceptable range of sample quantity to keep powder in-homogeneity issues at a low level. These are particularly acute in the case of granitic (s.l.) samples, where many trace elements (REE, HFSE, and radiogenic Pb) are mostly hosted by accessory minerals, which are not evenly distributed. The size of the final powder portion taken for analysis can be significantly reduced by adding a further step to the standard powder preparation procedure. Specifically, a subsample of ca. 1 gram of the powder obtained following the conventional grinding is taken, poured in a small (ca. 5 ml) agate-lined jar together with a few small agate balls, placed in a “mixer mill” apparatus, and subjected to vigorous shaking for half an hour or so. This treatment produces a final powder that is both very finely ground and well homogenized, thereby allowing the analyst to use sample aliquots as small as a few tens of mg.4.2 Sample digestion
The two methods used in this work share a crucial common feature, namely, enabling the analyst to decompose silicate rocks and obtain a solution suitable for chemical separation in a few tens of minutes. This compares very favourably to the conventional HF dissolution procedure which requires several days, starting from digestion proper – often made during tens of hours – to final dissolution in a medium adapted to the isolation of the element(s) of interest, through repeated evaporation and conversion steps to remove un-reacted HF and destroy sparingly soluble fluorides formed during attack.In order to obviate the need for prohibitively expensive fusion vessels made of platinum, relatively low-cost crucibles machined in-house from high-purity graphite were used throughout this study.10,14,15 Cost-effective digestions with adequate blank levels are achieved in this way, at the price of the introduction in the quenching solution of inert graphite particles which need to be removed by filtration and/or centrifugation before chromatographic separation.
| % in precipitate | Ti | Rb | Sr | Ba | Cs | Zr | Nb | Y |
|---|---|---|---|---|---|---|---|---|
| BE-N | 15 | 16 | 27 | 9 | 7 | 19 | 25 | 23 |
| BHVO-2 | 1 | 49 | 11 | 13 | 82 | 5 | 6 | 14 |
| BHVO-2 | 5 | 68 | 34 | 20 | 76 | 16 | 46 |
| % in precipitate | La | Ce | Nd | Sm | Eu | Dy | Yb | Lu |
|---|---|---|---|---|---|---|---|---|
| BE-N | 12 | 12 | 11 | 12 | 14 | 17 | 22 | 24 |
| BHVO-2 | 5 | 5 | 6 | 6 | 12 | 13 | ||
| BHVO-2 | 26 | 24 | 29 | 36 | 45 | 48 | 55 |
| % in precipitate | Hf | Ta | W | Pb | Th | U |
|---|---|---|---|---|---|---|
| BE-N | 21 | 36 | 1 | 28 | 42 | 4 |
| BHVO-2 | 6 | 13 | 20 | 3 | ||
| BHVO-2 | 18 | 19 | 43 | 7 |
In order to circumvent the problems related to digestion proper, and to the subsequent solution handlings, two radical measures have been investigated in this work.
Firstly, a short dissolution time of ca. 15 minutes (hereafter referred to as “flash attack”) was used. Admittedly, highly refractory minerals such as zircon are not fully digested under these conditions, particularly if the rock powder is not very finely ground. Accordingly, those samples containing zircon in a significant amount – as far as the LREE and uranogenic Pb budgets are concerned – would require either a further step of isolation of the undissolved residue and its treatment in a steel jacketed high pressure PTFE bomb,33 or alternatively, to be decomposed by alkaline fusion, as described above, thereby preserving the advantage of a single stage, rapid process.
Secondly, the evaporation steps were purely and simply eliminated. Following the digestion step, the sample solution is immediately treated with a slight excess of a 4 wt% boric acid solution in order to neutralize any un-reacted HF and dissolve the “young” fluorides formed during the attack. This approach avoids the heating and evaporation steps involved in the removal of HF and the difficult conversion of fluorides to soluble species. It was developed in the 1960s and 1970s to prepare solutions in view of major – including silica – and trace element determinations by AAS or ICP-AES and widely used34–41 but, to our knowledge, it was not yet applied prior to separation chemistry in view of radiogenic isotope work. In the scope of isotopic analyses, its outstanding advantage is to produce in a very straightforward way sample solutions ready – at least for the three target elements of this study – for separation by EXC, thereby obviating the need for the time-consuming evaporation and conversion steps which put the strongest limitation on the overall analytical throughput.
Provided that the digestion process starts the evening before the rest of the procedure, a variant of HF-based digestion could be used, in which ammonium acid bifluoride (NH4HF2, a compound with melting and boiling/decomposition temperatures of 125 °C and 240 °C, respectively42) is employed in the molten state instead of the usual aqueous solution of HF.43,44 Because it involves a reagent with a much higher boiling point than 48% HF, this method does not require high pressure devices, but can be used in simple screw cap PFA vessels, and it is claimed to better dissolve zircon by virtue of working temperatures around 220–230 °C. In this case, the digestion would be performed overnight. After cooling to room temperature, the solid residue produced by this mode of attack is taken up with HNO3 and immediately diluted with boric acid. This method was not used to produce the results given in this study, but preliminary experiments suggest that solutions prepared in such a manner are compatible with subsequent separations based on our protocol.
It is emphasized that these modes of digestion are not appropriate for a concomitant determination of Sm/Nd ratios by isotope dilution, which would require some further steps. In the case of fusion with LiBO2, a total spiking approach with a mixed 149Sm–150Nd tracer could be used by adding the tracer before fusion, as described elsewhere.15 By virtue of the very high temperatures reached during the melting process, this method is likely to ensure a perfect isotopic equilibration between the sample and tracer and it would allow the determination of both the Nd concentration and 143Nd/144Nd ratio to be made through a single mass spectrometric run. In all the other cases, a precisely weighed, small aliquot portion of the solution left after fusion and quenching, or boric acid treatment of the sample dissolved in HF–HNO3, should be taken, mixed with a weighed amount of isotopic tracer, e.g. a 149Sm–145Nd mixed spike. Then, the sample-tracer blend should be evaporated to dryness (after addition of HF to get rid of Si as volatile fluoride, in the case of fusion) and submitted to a thorough treatment with HCl and/or HNO3 and preferably HClO4 to promote isotopic equilibration between the sample and tracer. Finally, the dry residue left after this treatment could be taken up in 1 mol per L HNO3 and passed through a small column of TRU or RE resin to get rid of matrix elements and obtain a LREE fraction suitable for the simultaneous measurement of 145Nd/146Nd and 149Sm/147Sm isotope ratios by MC-ICP-MS. Bearing in mind the small sample quantities and volumes of acids involved, it is suggested that these steps might be made in parallel with the processing of the main solution used for unspiked, isotope composition measurements.
4.3 Separation chemistry
The separation stages are based on well-established EXC methods which were found, after very minor adaptations, to be fully compatible with the two swift sample digestion methods used in this study. The first stage achieves the on-line separation of LREEs, Sr and Pb from matrix elements. Following earlier work,45 two EXC columns in tandem configuration are used to isolate, in a single step, the analytes from matrix elements; specifically, a first column filled with a CMPO-based resin coupled to a second column of Sr resin, similar to those used previously46,47 for the concomitant extraction of the LREE, Sr and Pb. Here, the RE resin was used instead of the TRU resin because it features higher weight distribution ratios of the LREE in 2–3 mol per L HNO3, specifically, DW of Nd ca. 230 (ref. 48) vs. ca. 150 for TRU,2 thereby permitting the use of significantly larger volumes during the column loading step – as required by the two methods of attack used – without a risk of incipient breakthrough of Nd. The sample solution is loaded onto the RE column and Sr column in tandem, thereby achieving the extraction of the LREE using the upper column and of Sr and Pb (plus Ba) using the lower one.After rinsing, the two columns are decoupled and further processed separately. The Sr column is treated as described previously,47 that is, further rinsed with 7 mol per L HNO3 to get rid of Ba, followed by the sequential elution of Sr and Pb with very dilute (0.05 mol L−1) nitric acid and strong (6 mol L−1) HCl, respectively.
The second stage is required to isolate Nd from adjacent REEs. As described in an earlier contribution,49 the LREE fraction is stripped from the RE column with 3 mol per L HCl and directly loaded onto an on-line column of DGA resin. This lower column allows the lanthanides to be sequentially separated from the lighter ones to the heavier ones by using a gradient of decreasing HCl molarity. In this way, Nd fractions are easily obtained that are essentially free of Ce and Sm which would produce isobaric interferences at m/z 142 and 144, respectively.
On a wider scale, this study highlights the great flexibility and robustness of extraction chromatography, which proves to be able to accommodate solutions prepared by a variety of methods. Indeed, unlike the small volumes of final solutions obtained after repeated evaporation and conversion stages of conventional digestion methods, larger volumes were involved, consisting either of pure nitric acid with a high charge of salts, or a more complex mixture of fluoboric and nitric acids. In either case, the extraction of the nitrate species of the three target elements was not significantly impeded, thereby documenting the efficiency and high selectivity of the CMPO and crown ether ligands used to impregnate an inert support with good chromatographic characteristics to produce the RE and Sr resins, respectively.
5. Conclusions
The alternative protocols investigated in this work of fast digestion of silicate rocks and further prompt treatment of the sample solution without any evaporation and conversion step are compatible with a straightforward separation of the LREE, Sr, and Pb from matrix elements by miniaturized extraction chromatography. Good chemical recoveries of the three target elements and blank levels adequate for routine work on common rocks are achieved, and the isotope ratios measured on several geological RMs spanning a range of bulk compositions agree well with published values obtained by using conventional methods, thus documenting the overall validity of these new, single day approaches. With a minor adaptation of the final DGA column, these protocols could include Ce in view of isotope analyses. Last but not least, the solutions obtained from both sample digestion methods would allow, at the price of a precise splitting of the overall starting solution into appropriate aliquots, the concomitant determination of major (including silica) and many trace element concentrations to be made on one and the same sample. This would require that ICP-AES and ICP-QMS instruments are available in the laboratory and that a well-functioning organization of the technical staff involved is achieved. In this case, rather complete chemical and radiogenic isotope characterization of silicate samples could be obtained in a very short amount of time, with minimal cost and analyst time and effort.Author contributions
Both authors designed the analytical procedure, carried out the chemical separation, performed the measurements on mass spectrometers and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Chantal Bosq for preparing purified acids, Mhammed Benbakkar and Eric Brut for building an easy-to-use induction furnace from the EasyHeat RF generator, Krzysztof Suchorski for sharing his experience on digestion with NH4HF2, Jean-Luc Piro for making the Agilent 7500 ICP-QMS easily available for early development work, and Mathis Neimard for further analyses with the Agilent 8900 triple quad MS. We thank Urs Klötzli and one anonymous referee for their perceptive reviews which led to significant improvements in the manuscript and Derya Kara Fisher for editorial handling. This is contribution no. 620 of the ClerVolc program of the International Research Center for Disaster Sciences and Sustainable Development of the University of Clermont Auvergne. CP dedicates this article to the director of LMV who, 3 years ago, kicked him out (“l'a mis à Laporte”) by refusing him the possibility of applying for the status of emeritus researcher when he was retiring from the CNRS after 40 years of work in this laboratory.References
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond and D. M. Nelson, Anal. Chim. Acta, 1993, 281, 361–372 CrossRef CAS.
- E. A. Huff and D. R. Huff, Presented at 34th ORNL/DOE Conference on Analytical Chemistry in Energy Technology, Gatlinburg, Tennessee, October, 1993 Search PubMed.
- E. P. Horwitz, D. R. McAlister, A. H. Bond and R. E. Barrans Jr., Solvent Extr. Ion Exch., 2005, 23, 319–344 CrossRef CAS.
- E. P. Horwitz, R. Chiarizia and M. Dietz, Solvent Extr. Ion Exch., 1992, 10, 313–336 CrossRef CAS.
- GeoReM Database, https://georem.mpch-mainz.gwdg.de, accessed July 2023 Search PubMed.
- M. Totland, I. Jarvis and K. E. Jarvis, Chem. Geol., 1992, 95, 35–62 CrossRef CAS.
- S. J. G. Galer and W. Abouchami, Mineral. Mag., 1998, A62, 491–492 CrossRef.
- K. Govindaraju, Geostand. Newsl., 1994, XVIII, 158 Search PubMed.
- C. O. Ingamells, Talanta, 1964, 11, 665–666 CrossRef.
- C. O. Ingamells, Anal. Chim. Acta, 1970, 52, 323–334 CrossRef.
- M. Cremer and J. Schlocker, Am. Mineral., 1976, 61, 318–321 Search PubMed.
- C. Feldman, Anal. Chem., 1983, 55, 2451–2453 CrossRef.
- S. Shirey and G. N. Hanson, Geochim. Cosmochim. Acta, 1986, 50, 2631–2651 CrossRef.
- B. Le Fèvre and C. Pin, Anal. Chem., 2001, 73, 2453–2460 CrossRef.
- B. Le Fèvre and C. Pin, Anal. Chim. Acta, 2005, 543, 209–221 CrossRef.
- P. J. Potts, A Handbook of Silicate Rock Analysis, Blackie, Glasgow & London, 1987 Search PubMed.
- R. B Vocke, G. N. Hanson and M. Grünenfelder, Contrib. Mineral. Petrol., 1987, 95, 145–154 CrossRef.
- A. Pourmand, N. Dauphas and T. J. Ireland, Chem. Geol., 2012, 291, 38–54 CrossRef.
- N. H. Suhr and C. O. Ingamells, Anal. Chem., 1966, 38, 730–734 CrossRef.
- J. Carignan, CRPG-CNRS, Nancy (France), Pers. Comm.
- R. H. Boer, G. J. Beukes, F. M. Meyer and C. B. Smith, Chem. Geol., 1993, 104, 93–98 CrossRef.
- K. E. Jarvis, Chem. Geol., 1990, 83, 89–103 CrossRef.
- R. Bock, A Handbook of Decomposition Methods in Analytical Chemistry, International Textbook Co., 1979 Search PubMed.
- Z. Sulcek and P. Povondra, Methods of Decomposition in Inorganic Analysis, CRC Press, Boca Raton, Florida, 1989 Search PubMed.
- I. W. Croudace, Chem. Geol., 1980, 31, 153–155 CrossRef.
- F. J. Langmyhr and K. Kringstad, Anal. Chim. Acta, 1966, 35, 135–137 CrossRef.
- T. Yokoyama, A. Makishima and E. Nakamura, Chem. Geol., 1999, 157, 175–187 CrossRef.
- R. Tanaka, A. Makishima, H. Kitagawa and E. Nakamura, J. Anal. At. Spectrom., 2003, 18, 1458–1463 RSC.
- F. J. Langmyhr, Anal. Chim. Acta, 1967, 39, 516–518 CrossRef.
- R. S. Cohen, P. Hemmes and J. H. Puffer, Chem. Geol., 1975, 16, 307–309 CrossRef.
- C. O. Ingamells and F. F. Pitard, Applied Geochemical Analysis, Wiley Interscience, New York, 1986 Search PubMed.
- D. M. Unruh, P. Stille, P. J. Patchett and M. Tatsumoto, 1984, Lunar and Planetary Science Conference, 14th, Houston, TX, J. Geophys. Res., 1984, 89, B459–B477 CrossRef.
- C. Pin and J. F. Santos Zalduegui, Anal. Chim. Acta, 1997, 339, 79–89 CrossRef.
- B. Bernas, Anal. Chem., 1968, 40, 1682–1686 CrossRef.
- D. E. Buckley and R. E. Cranston, Chem. Geol., 1971, 7, 273–284 CrossRef.
- N. T. Bailey and S. J. Wood, Anal. Chim. Acta, 1974, 69, 19–25 CrossRef.
- H. Agemian and A. S. Y. Chau, Anal. Chim. Acta, 1975, 80, 61–66 CrossRef PubMed.
- W. J. Price and P. J. Whiteside, Analyst, 1977, 102, 664–671 RSC.
- H. Uchida, T. Uchida and C. Iida, Anal. Chim. Acta, 1979, 108, 87–92 CrossRef.
- D. Silberman and G. Fisher, Anal. Chim. Acta, 1979, 106, 299–307 CrossRef.
- C. R. M Rao and G. S. Reddi, Anal. Chim. Acta, 1990, 237, 251–252 CrossRef.
- National Library of Medicine Online, https://pubchem.ncbi.nlm.nih.gov/compound/Ammonium-bifluoride, accessed June 2023.
- W. Zhang, Z. Hu, Y. Liu, H. Chen, S. Gao and R. M. Gashnig, Anal. Chem., 2012, 84, 10686–10693 CrossRef PubMed.
- G. Újvári, U. Klötzli, M. Horschinegg, W. Wegner, D. Hippler, G. I. Kiss and L. Palcsu, Rapid Commun. Mass Spectrom., 2021, 35, 9081 CrossRef.
- C. Pin, D. Briot, C. Bassin and F. Poitrasson, Anal. Chim. Acta, 1994, 298, 209–217 CrossRef.
- C. Deniel and C. Pin, Anal. Chim. Acta, 2001, 426, 95–103 CrossRef.
- C. Pin, A. Gannoun and A. Dupont, J. Anal. At. Spectrom., 2014, 29, 1858–1870 RSC.
- B. Le Fèvre, PhD thesis, Blaise Pascal University, Clermont-Ferrand, 2002.
- C. Pin and A. Gannoun, J. Anal. At. Spectrom., 2019, 34, 2136–2146 RSC.
| This journal is © The Royal Society of Chemistry 2024 |