
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCold H2–Ar plasma interaction with nickel α-hydroxide as a versatile nanofabrication tool for Ni@Cgr nanoparticles†
Marie-Charlotte
Dragassi
ab,
Sonia
Haj-Khlifa
ab,
Nicolas
Menguy
c,
Michael
Redolfi
*b and
Souad
Ammar
 *a
*a
aUniversité Paris Cité, CNRS UMR-7086, ITODYS, 15 Rue Jean-Antoine de Baïf, Paris 75251, France. E-mail: michael.redolfi@lspm.cnrs.fr; souad.ammar-merah@u-paris.fr
bUniversité Sorbonne Paris Nord, CNRS UPR-3407, LSPM, 99 Avenue Jean-Baptiste Clément, Villetaneuse 93430, France
cSorbonne Université, CNRS UMR-7590, IMPMC, 4 Place Jussieu, Paris 75005, France
First published on 7th June 2024
Abstract
Taking advantage of its open structure and high reactivity, turbostratically disordered, single layered nickel hydroxyacetate was produced by forced hydrolysis in polyol and exposed to cold H2–Ar (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) plasma to study its reactivity toward such reductive conditions. X-ray diffraction and transmission electron microscopy evidenced a quite complex phase transformation kinetics, with the simultaneous production of rhombohedral Ni3C and cubic NiCy (y ≪ 0.25), the former disappearing in favor of the latter, accompanied by a progressive carbon demixing from the cubic nanocrystals, leading to Ni@Cgr core–shell nanoparticles. These results are discussed hereafter, highlighting the role of the organic content of the hydroxide phase, mainly the intercalated acetate ions, which decomposed providing the carbon source for carbon implantation in the in situ formed metal nanoparticles.
:10) plasma to study its reactivity toward such reductive conditions. X-ray diffraction and transmission electron microscopy evidenced a quite complex phase transformation kinetics, with the simultaneous production of rhombohedral Ni3C and cubic NiCy (y ≪ 0.25), the former disappearing in favor of the latter, accompanied by a progressive carbon demixing from the cubic nanocrystals, leading to Ni@Cgr core–shell nanoparticles. These results are discussed hereafter, highlighting the role of the organic content of the hydroxide phase, mainly the intercalated acetate ions, which decomposed providing the carbon source for carbon implantation in the in situ formed metal nanoparticles.
Introduction
The fabrication of tailored functional nanomaterials is at the heart of modern nanoscience and technologies. Various solids are required and different fabrication methods are developed for such a purpose. Among these numerous methods, common gas (H2, CH4, N2, NH3, O2, Ar)-assisted plasma interaction with given nano (in thickness) substrates has attracted increasing research interest. Plasma is a partially ionized gas, consisting of electrons, ions, molecules, free radicals, photons, and excited species, all of which are active species for the preparation and treatment of materials.1 When high energy plasma species come into contact with a material through gas-phase electron-impact or heavy-particle collisional ionization or dissociation of the feedstock gas, physical and chemical changes occur on the exposed surfaces.2 Physical sputtering, chemical etching, reduction and oxidation, among others processes, may take place, leading to topographical and/or chemical transformations. The literature is enriched with such examples.3–5 In all these examples, the material processing control was mainly achieved by adjusting the plasma gas composition and the substrate nature. For instance, applying a microwave H2–Ar plasma on compacted Ni nanoparticles (20–30 nm in diameter)5 or ultrathin Ni film (20 nm in thickness)6 hydrogen-ion implantation may be achieved, leading to Ni2H and/or NiH formation. Microwave discharge oxygen plasma applied to Ti sheets (2 mm in thickness) achieved their oxidation and their surface transformation into the tetragonal rutile phase, the titanium oxidation rate increasing monotonically with the density of oxygen ions.7 Thermal CH4–H2 plasma applied to tungsten hexachloride WCl6 solid allowed the reduction and carburization reactions on the vaporized precursor, synthesizing nanosized WC1−x powders.8 Microwave N2–H2 plasma interaction with TiCl4 and ZrCl4 metal chloride solids allows the production of TiN and ZrN nanopowders.9 For all these reactions, an inert gas was used as a plasma forming and cooling gas while a reactive gas was used as the source of energetic reagents. Also, the flow rate of plasma forming gas, flow rate of carrier gas and microstructure of the raw material appeared as relevant for the whole reaction control.5–9 The reactions may proceed into the solid phase or they may proceed into the gas phase, depending on the plasma energy (cold or hot) and the nature of the precursor.All these examples and others illustrate the power of plasma-assisted functional nanomaterial processing, and focusing on in solid transformations, microwave plasma processing appears as the most relevant. Indeed, microwave plasma offers several advantages. It provides low reaction temperature and uniform temperature field. It also operates at either ambient or low pressures.
From the plasma process point of view, the lower gas temperature and the non-equilibrium conditions limit the densities of the plasma species, which are commonly lower than those of traditional high-temperature plasmas, typically of the order of 1012 to 1015 particles per cubic centimetre. The lower gas temperature and the low plasma density limit the recombination rate and plasma sustainment, providing short lifetimes, typically of the order of microseconds to milliseconds.
From the material science point of view, plasma interaction with inorganic materials leads to ion implantation without “sever damages” (bubbles, blistering…).10–13 It also proceeds only on the extreme surface of the exposed material. For instance, in a Pd foil exposed to 90–10% H2–Ar (0.33 Pa) microwave plasma (1.8 kW and 2.45 GHz), the implantation depth of the main plasma reactive ions was found to be about 201 nm, 115 nm and 24 nm for 40 keV H+, H2+ and Ar+ plasma ions, respectively.14 Thus, it means a total transformation may only proceed on ultrathin films or ultrafine powders, with thicknesses and sizes of some tens of nanometers.
As already mentioned, different solid precursors were tested, but metal hydroxide derivatives were scarcely considered, despite their numerous advantageous. Due to their layered open structure, they are expected to be much more reactive than their oxide or metal counterparts. On these materials, in a classical gas environment, molecules adsorption and/or desorption15–17 as well as oxidation or reduction reactions are easy to achieve.18,19 Focusing on brucite-type MII(OH)2 hydroxides, where MII is a divalent cation, including d transition metal cations like Ni2+, this is particularly true for their α polymorphs. Indeed, most of these hydroxides have two polymorphs, namely, a dense and well-stacked β phase and a less dense one with a more open structure, due to the intercalation of water molecules or anions (compensating the negative charge of the missing hydroxide groups in the inorganic layer), the α polymorph. The interlayer cohesion in the α polymorphs is mainly provided by weak hydrogen bonds between the hydroxyl groups of the inorganic sheets and the intercalated species. As a consequence, their interlayer distance may be varied over a large scale, up to 50 Å,20–23 till the exfoliation limit, whereas the in-plane metal–metal distance remains constant, close to the metal–metal distance in the reference MII(OH)2 β compound, making these allotropes much more reactive than their β counterparts. Moreover, due to their open structure, these α-polymorphs often exhibit stacking faults, like α/β-interstratification and/or turbostratic disorder, thus increasing their reactivity.
In this context, focusing on the nickel system as a case study, a turbostratic disordered Ni(OH)1−x(CH3CO2)x·nH2O α-hydroxide was prepared by forced hydrolysis in polyol.20 The recovered, washed and dried powders were compacted as pellets with 13 mm diameter and 2 mm thickness and then exposed to 90–10% H2–Ar microwave plasma for different times (from 0 to 10 hours) to investigate their reactivity toward such an out-of-equilibrium reductive atmosphere. The kinetics was thus followed thanks to a systematic characterization of the as-produced powders after each exposure time. Interestingly, we evidenced that within our operating conditions, we were able to produce ultrafine cubic Ni crystals (10–20 nm in diameter) coated by a thin graphite layer (2–3 nm in thickness) in the form of core–shell nanoparticles.
Results and discussion
Evidence of nickel-layered hydroxyacetate reduction
A set of characterizations was first achieved on the as-produced hydroxide powder, and its disordered and open structure was confirmed. The unit cell c parameter, inferred from the 00l diffraction line position, was found to be equal to 11.05 Å (see Fig. S1 in the ESI†), significantly larger than that of pure β-Ni(OH)2 (c = 4.63 Å24). The hk0 diffraction lines appeared broadened and asymmetrical, consistent with a turbostratic disorder for a unit cell a parameter of 3.13 Å (Fig. S1, ESI†). Thermogravimetry coupled to infrared spectroscopy allowed to confirm its chemical nature as a hydroxyacetate with a composition close to Ni(OH)1−x(CH3CO2)x·nH2O (x ∼ 0.4 and n ∼ 0.6) (Fig. S2, ESI†). Two set of pellets were then prepared for plasma exposure. A first set was exposed to a 100% H2 atmosphere and the second to a 90–10% H2–Ar atmosphere (1 mbar). For the first atmosphere, no colour change was observed on the pellet, even after 10 h of plasma exposure. The recorded XRD patterns on the exposed pellets only evidence the signature of the starting hydroxide derivative (Fig. 1a). For the second atmosphere, visually, the treated pellets lost their green colour rapidly and became black in colour. The recorded XRD patterns exhibit a rapid and net structural evolution (Fig. 1b).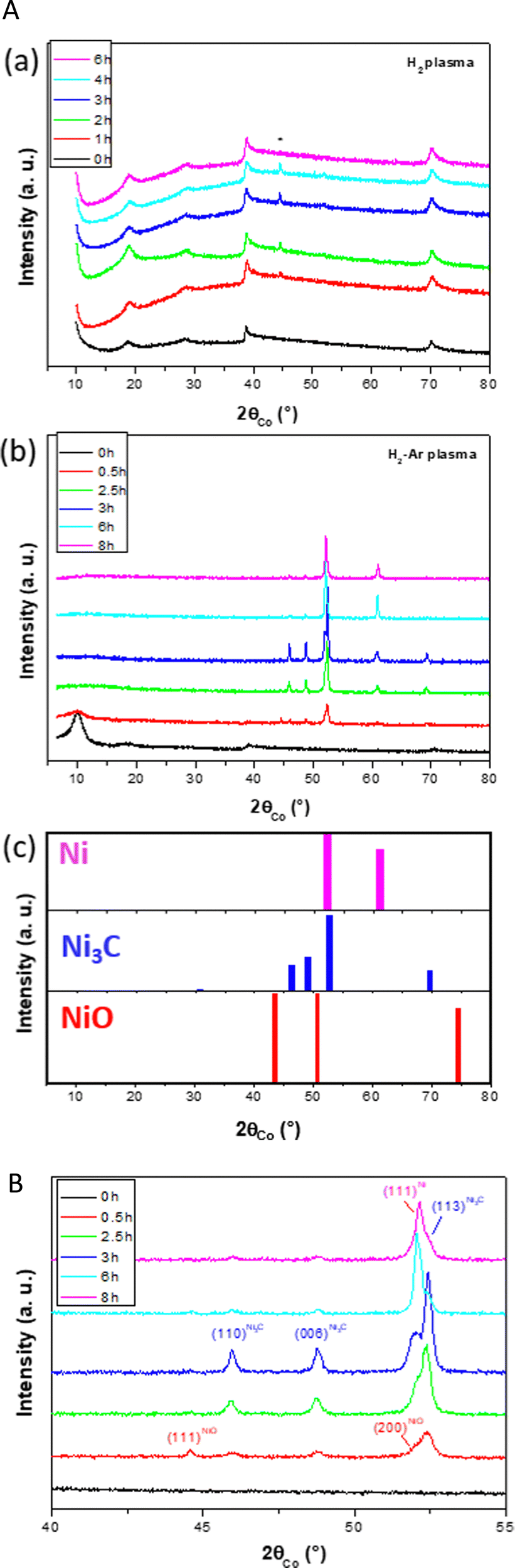 | ||
| Fig. 1 (A) XRD patterns recorded on the turbostratic disordered α-hydroxide powder after different times of (a) H2 (the signature of the silver ink residue, used to attach the pellet to the plasma sampling holder, is highlighted by an apteryx) and (b) H2–Ar plasma exposure. (c) The reference patterns of the evidenced phases Ni (ICDD No. 98-005-2265), Ni3C (ICDD No. 98-001-7005) and NiO (ICDD No. 98-000-9866) are given for information. (B) Zoomed-in XRD patterns recorded in the 40 to 55° 2θ range on the turbostratic disordered α-hydroxide powder after different times of H2–Ar plasma exposure to evidence the main structural changes. | ||
The α-hydroxide signature disappeared after only 1 h of exposure, and it was quickly replaced by the rhombohedral Ni3C carbide (ICDD No. 98-001-7005) and cubic NiO (ICDD No. 98-000-9866) phases. For longer exposure times, the characteristic 111 diffraction line of NiO at 2θ = 43.5° disappears completely in favor of cubic Ni growth (ICDD No. 98-005-2265). This can be followed by the appearance of a new peak matching with the 002 diffraction line of the metal phase at 2θ = 61.1° (Fig. 1c). This means that the hydroxide phase was first dehydroxylated into an oxide, which was in turn rapidly reduced into a metal. Interestingly, the intensity of the metal signature increased while that of the carbide phase decreased. Indeed, the intensity of the peak at 2θ = 52.0°, attributed to the 111 cubic Ni reflection, (ICDD No. 98-005-2265) increased from 1 to 8 h of exposure, while that at 2θ = 52.4, attributed to the 113 rhombohedral Ni3C reflection, decreased till it was almost completely disappeared after more than 6 h of exposure.
Note that nickel carbide and nickel oxide phases seemed to be formed simultaneously at the early stage of plasma/α-hydroxide interaction. This is certainly related to the chemical composition of the precursor. Indeed, it contains hydroxyl and water groups, which might be lost, forming nickel oxide but also structural acetates, whose decomposition may induce carbide formation.
Having all this information, Rietveld refinements using MAUD software25 were performed on the patterns of all the H2–Ar plasma treated samples for exposure times longer than 1 h. The fitting allows a better characterization of the formed Ni and Ni3C phases. Table 1 summarizes the unit cell parameters a (and c), the average crystal size 〈LXRD〉, the average lattice deformation due to micro-strains  , and the weight content of each phase for the related samples.
, and the weight content of each phase for the related samples.
| Time (h) | Phase | a (Å) ± 0.002 | c (Å) ± 0.002 | ΔV (Å) | 〈LXRD〉 (nm) ± 1 |
|
wt% ± 5 |
|---|---|---|---|---|---|---|---|
| 2.5 | Ni | 3.537 | 3.537 | 0.486 | 56 | 0.2 | 33 |
| Ni3C | 4.583 | 13.005 | 4.514 | 96 | <0.1 | 67 | |
| 3 | Ni | 3.533 | 3.533 | 0.336 | 47 | 0.2 | 35 |
| Ni3C | 4.582 | 13.012 | 4.538 | 90 | 0.1 | 65 | |
| 6 | Ni | 3.531 | 3.531 | 0.261 | 45 | <0.1 | 78 |
| Ni3C | 4.582 | 13.010 | 4.501 | 89 | 0.5 | 22 | |
| 8 | Ni | 3.527 | 3.529 | 0.112 | 34 | <0.1 | 84 |
| Ni3C | 4.582 | 13.011 | 4.520 | 90 | 0.2 | 16 | |
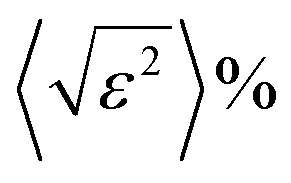
The Rietveld fit quality was confirmed through the perfect superposition of the experimental patterns and the calculated ones for all the analyzed samples, as illustrated in Fig. S3 (ESI†). Clearly, all the in situ formed crystalline phases were nanocrystallized. The average coherence length of the cubic phase appeared to decrease with the increase in the plasma exposure time while that of the rhombohedral phase appeared to be almost constant. Also, the average lattice deformation was found to be not so large for both phases, meaning that even if the crystal lattices contained some defects, the cubic and rhombohedral structures seemed to accommodate them.
Another interesting feature concerned the cell parameter values of the two nickel-based phases. The cell parameter of the cubic phase was found to be larger than that of bulk nickel, with a discrepancy between the two values, which starts increasing for the shortest plasma exposure times and then decreases for the longest ones. Visually, the 111 cubic Ni reflection peak appeared to be less and less shifted toward small 2θ values in the plotted XRD patterns in Fig. 1c, which is reduced by an increase in ΔV (Table 1), which is the difference between the refined metal cell volume and that of bulk nickel (Table 1). Such a dilatation and contraction of the cubic lattice agrees with carbon dissolution and demixing due to supersaturation. In contrast, the a and c cell parameters of the rhombohedral phase were found to be larger than those tabulated for a stoichiometric Ni3C compound, but almost constant, whatever the H2–Ar plasma exposure time. This result suggests a super stoichiometry with an excess of carbon providing a certain instability. But, in the same time, the measured ΔV appears as too large for such exclusive features. Thus, we decided to proceed differently. First, we started by excluding any sample diffraction misalignment and/or goniometer zero offset effects on the collected XRD patterns. To do that, the measurements were repeated while mixing the studied samples with a powdered strain free micrometer-grained CuO reference. The recorded patterns on two representative samples of the studied series mixed with CuO powder are given in the ESI† (Fig. S4). Interestingly, the same features were observed on nickel-based phases, while the peak position of the standard did not change and neither did its refined cell parameters: a = 4.670 ± 0.002 Å; b = 3.430 ± 0.002 Å and c = 5.120 ± 0.002 Å, in agreement with the values reported on bulk CuO (ICDD No. 98-003-1059). Second, a rapid overview of the reported cell parameters for the Ni3C phase allowed us to find values close to those listed in Table 1. For instance, Gaudison et al. obtained by Rietveld XRD refinements a = 4.590 ± 0.005 Å and c = 13.001 ± 0.005 Å for Ni3C nanoparticles.26 Additionally, using first-principles electronic structure methods, Kelling et al., calculated the cell parameter values for rhombohedral Ni3C of a = 4.60 Å and c = 13.00 Å,27 in good agreement with those measured in this work. Last but not least, the ICDD tabulated cell parameters (No. 98-001-7005) of the Ni3C phase were provided from electron diffraction analysis,28 which is known as a less accurate technique compared to X-ray diffraction. Thus, without excluding a real carbon super-stoichiometry, the deviation from stoichiometry must remain modest, significantly smaller than that related to a real unit cell volume variation higher than 4.5 Å3.
To go further in our investigations, transmission electron microscopy (TEM) observations were carried out. The TEM results confirmed the XRD results, and selected area electron diffraction (SAED) measurements showed that Ni3C is the major phase in the sample exposed for 2 h (Fig. 2). Moreover, the micrographs evidenced a large particle size distribution, with diameters ranging between 10 and 100 nm. The particles appear as almost well-faceted and more or less sintered (Fig. 3 and 4), crystallizing in the two rhombohedral Ni3C and cubic Ni structures. Interestingly, the rhombohedral particles appear as large single crystals while cubic ones appear smaller in size (Fig. 3). It should also be noticed that most of the particles are coated with a very thin layer of graphitic carbon (Fig. 3 and 5).
 | ||
| Fig. 2 (a)–(b) TEM bright field image of an assembly of particles recovered after 2 h of H2–Ar plasma exposure of the turbostratic disordered α-hydroxide powder. (c) Corresponding SAED pattern. (d) Radial intensity profile related to the SAED, showing the presence of Ni3C (ICDD No. 98-001-7005) as the major phase and Ni-fcc (ICDD No. 98-005-2265) as the minor phase. | ||
 | ||
| Fig. 3 (a) HRTEM image of an assembly of particles recovered after 2 h of H2–Ar plasma exposure of the turbostratic disordered α-hydroxide powder. The larger one, labelled (b), was identified as an Ni3C crystal using fast Fourier transform (FFT) analysis visible in (b) with d10−2 = 3.39 ± 0.01 Å and d−120 = 2.29 ± 0.01 Å. A smaller one, labelled (c), corresponds to the NiCy particle identified by FFT, as shown in (c) (d020 = d200 = 1.76 ± 0.01 Å). The measured reticular distances fit very well with those calculated using the cell parameters inferred from the XRD analysis (Table S1, ESI†). Note that a thin carbon layer is visible around the particles. Scale bar on (b) and (c) is 0.5 Å−1. | ||
 | ||
| Fig. 4 (a) TEM bright field image of Ni3C and NCy particles attached to each other. (b) HRTEM image of the area labelled (b) in image (a) showing the grain boundary between the Ni3C particles. From the FFT analyses shown in (c) and (d), it can be deduced that the {001} planes of both the crystals are parallel, and d006 was found to be 2.17 ± 0.01 Å, in agreement with the calculated value using the cell parameters inferred from the XRD analysis (Table S1, ESI†). Scale bar on (c) and (d) is 0.5 Å−1. | ||
 | ||
| Fig. 5 (a)–(c) HRTEM images evidencing the presence of graphitic carbon layer on Ni3C and NiCy particles recovered after 2 h of H2–Ar plasma exposure of the turbostratic disordered α-hydroxide powder. (d) and (f) Intensity profiles corresponding to the red lines in (a)–(c); the deduced mean interplanar distances are indicated. | ||
After 6 h of exposure, the recovered powder still exhibits a poly-crystalline character with a bimodal particle size distribution, the smallest particles belonging to the cubic structure while the largest to the rhombohedral one (Fig. 6). The micrographs also evidenced cubic particles with a graphitic coating (Fig. 6), suggesting a carbon demixing, the carbon-enriched NiCy (y < 0.25) transforming itself into Ni@Cgr core–shell particles (Fig. 4). Note that y = 0.25 is the limit of the carbon content in the cubic solid solution, which corresponds to the stoichiometry of the stable Ni3C carbide phase (C/Ni + C = 0.25).29 Altogether, these results suggest that the turbostratic disordered α-hydroxide produces Ni@Cgr core–shell nanoparticles under prolonged H2–Ar plasma exposure time (more than 6 h).
 | ||
| Fig. 6 (a) TEM bright field image of an assembly of particles recovered after 6 h of H2–Ar plasma exposure of the turbostratic disordered α-hydroxide powder, highlighting the bimodal distribution of the Ni-based particles produced. (b) HRTEM image focusing on small particles embedded within their graphitic carbon coating. (c) Image of a large Ni3C particle surrounded by smaller cubic NiCy ones suggesting the production of the former from the latter. The Moiré pattern labelled (e) is due to the superimposition of the Ni3C crystal and the NiCy particle. The Ni3C crystal and the NiCy particles were identified owing to the interplanar distances deduced from the FFT analysis of the areas (d)–(g) labelled in (c). Scale bar for FFTs is 0.5 Å−1 and interplanar distances are respectively: d110(Ni3C) = d2−10(Ni3C) = 2.29 ± 0.01 Å, d202(Ni3C) = 1.89 ± 0.01 Å and d111(Ni) = 2.03 ± 0.01 Å. | ||
Graphite demixing was also confirmed by Raman spectroscopy (Fig. 7), which is usually employed to get insights into the structural properties of the carbon matter. In all the recorded spectra, the two characteristic D and G bands were observed at 1350 and 1590 cm−1, respectively, with an intensity that decreases with the plasma exposure time, without changing their relative intensity ratio significantly. The relative D/G intensity ratio is usually used30 to appreciate the crystalline quality of the produced graphite phase. Indeed, as the intensity of the disorder-induced D-band decreases as the graphite disorder is weak and, reversely, as it increases, the disorder is important, and this is what we observed. For the investigated samples, the total Raman spectral intensity decreases as the plasma exposure time increased, suggesting the thinning of the surrounding carbon layer.
 | ||
| Fig. 7 Raman spectra recorded on the H2–Ar plasma-treated turbostratic disordered α-hydroxide powder for different exposure times, highlighting the presence of almost disordered graphite. | ||
The evaluated carbon shell thickness appeared to be almost constant, ranging between 2 and 3 nm, since carbon demixing should be accompanied by Ar+ sputtering. Thus, a change in the total composition of the analysed powders (the ratio between Ni3C and NiCy particles) and/or an experimental artefact related to the laser focus during the measurement, depending on where the Raman signal is collected, may also induce similar whole spectral intensity decrease. The last hypothesis was minimised by averaging the Raman spectra measured on several points of each sample.
Indeed, the deconvolution of the raw data (Fig. S6, ESI†) shows six main bonds centered at 1080, 1220, 1350, 1450, 1585 and 1690 cm−1 assigned to SL, S, D, V, G, and GL bonds, respectively.31,32 The D band reflects the high regularisation of the carbonaceous material with a combination of benzene-type rings and reveals the possible presence of hetero-atoms like oxygen.32,33 The V absorption band, called the valley, is ascribed to the mixture of two vibrational modes of sp2 carbons, (i) the asymmetric stretch and (ii) the breathing one.31 The S band is attributed to alkyl-type sequences.32 The G band reveals two kinds of vibrations, the quadrant one of the aromatic ring and the Eg2 vibration of graphite.32 The absorption band GL illustrates the presence of the carbonyl functional group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).32 Considering all these contributions at different ratios, it is possible to assess the carbon matter crystallinity by calculating the band area ratios involving the sum of D, V and S on the G absorption bands,32 named here after the D/G ratio. The obtained values are consistent with ratios comprised between 1.1 and 1.8 (Fig. S4, ESI†), allowing us to conclude definitively a disordered graphitic layer.
O).32 Considering all these contributions at different ratios, it is possible to assess the carbon matter crystallinity by calculating the band area ratios involving the sum of D, V and S on the G absorption bands,32 named here after the D/G ratio. The obtained values are consistent with ratios comprised between 1.1 and 1.8 (Fig. S4, ESI†), allowing us to conclude definitively a disordered graphitic layer.
The growth of this disordered graphitic shell around the produced nickel particles acts as a barrier toward any hydrogen ion implantation and diffusion, stopping any eventual hydriding process. This was completely confirmed by the zero amount of released hydrogen during the thermodesorption experiments performed on all the H2–Ar plasma-treated samples (Fig. S6, ESI†).
Interaction mechanism
All the performed material characterizations allowed us to better understand what happened in the plasma reactor. A general scheme can be proposed for the reactivity mechanism as follows.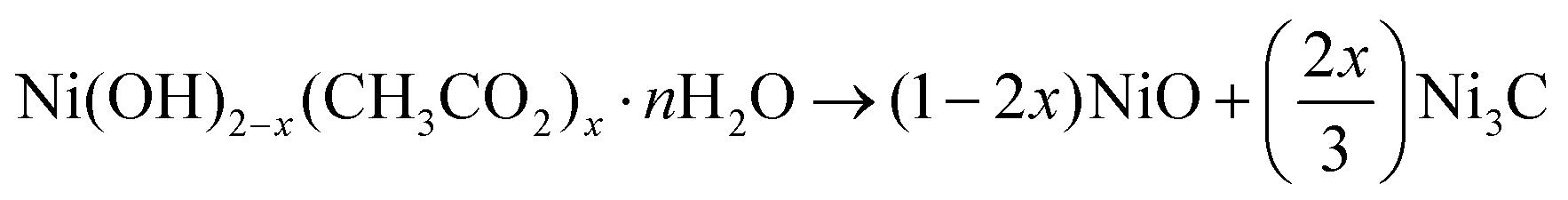 | (1) |
| NiO → Ni → NiCy (y < 0.25) | (2) |
| Ni3C → NiCy (y < 0.25) | (3) |
| NiCy → Ni@Cgr | (4) |
If the first two reaction steps, hydroxyde dehydration into oxide and oxide reduction into metal, followed by carbon dissolution leading to cubic NiCy solid solution, can be understood, the third step involving the transformation of rhombohedral Ni3C into cubic NiCy solid solution remains unclear. The other remaining question is the origin of the carbon atoms in the NiCy crystals and why these crystals are unstable, resulting in the longest plasma exposure time for the production of core–shell Ni@Cgr.
Let us consider the first question, i.e., how Ni3C may transform itself into NiCy. A first hypothesis is the fracturing of the large rhombohedral crystals into smaller cubic crystals due to their super-stoichiometry in carbon. This hypothesis can be supported by the increase in the proportion of very small particles on the recorded TEM micrographs as the plasma exposure time increases. As the weight fraction of the cubic NiCy increases, two crystal populations can be distinguished, the aged cubic NiCy with low carbon content and thick surrounding carbon layer and the fresh cubic NiCy with high carbon content but with a thin surrounding carbon layer. A second scenario consists of the growth of NiCy ultrasmall crystals from the surface of the large super-stoichiometric Ni3C crystals, as suggested by Fig. 6c. Two hypotheses are possible.
Thermodynamically speaking, Ni3C is a metastable phase at room temperature and decomposes above 415 °C (688 K) and 300 °C (673 K) in inert (Ar) and reductive (H2) atmosphere, respectively, into nickel metal,33–35 without excluding non-zero (even small) amount of carbon in the decomposed products.33,35 This carbon content was, for instance, estimated for a nickel cubic phase with a cell parameter of 3.5255 Å, equal to 0.19 ± 0.07 at%.36 In other words, applying the related Vegard law to the NiCy particles produced here, using the cell parameters listed in Table 1, a carbon content varying from 1.65 to 0.38 at% was deduced for cell parameter values varying between 3.537 and 3.527 Å, which remained completely reasonable, far below the 25 at% carbon content of the rhombohedral Ni3C phase.
Last but not least, the phase transformation from rhombohedral Ni3C to cubic Ni and more exactly to cubic NiCy (y ≪ 0.25) proceeds with time,35 which means that as the plasma exposure time is long, the phase transformation would be complete.
Thus, all these features can easily be applied to the 90–10% H2–Ar microwave plasma reactivity conditions with a probable decrease in the transformation temperature down to the temperature usually reached on the surface of microwave plasma-exposed matter (<500 K).37,38
The origin of carbon in the cubic NCy (y ≪ 0.25) crystals has to be also considered with regard to the chemical composition of the starting hydroxide phase. From the beginning of the H2–Ar plasma treatment, pure Ni phase was never stabilised, and only the rhombohedral Ni3C and the cubic NiCy were observed. The decomposition of the organic matter originated from the Ni(OH)1−x(CH3CO2)x·nH2O (x ∼ 0.4 and n ∼ 0.6) precursor, which contributed to the production of Ni3C particles but also provided small hydrocarbon molecules into the plasma chamber, which may themselves interact with the just-formed cubic Ni particles from NiO reduction, leading to the stabilisation of cubic NiCy particles. These particles were then progressively enriched in carbon atoms (accompanied with a cubic unit cell parameter increase) until saturation and carbon demixing as the disordered graphite outer layer (accompanied with a cubic unit cell parameter decrease).
One may assume that acetate decomposes into CH4, CO and/or CO2 species. CO2 was discharged due to the reducing nature of the H2–Ar plasma. Moreover, its presence is incompatible with the carbon graphite layer growth and its persistence along the plasma exposure time. Indeed, CO2 would react with the carbon formed on the Ni crystal surface, cleaning it.39
| CO2 + C(s) → 2CO | (5) |
CH4 is usually used as the gas precursor for carbon nanotube (CNT) growth on Ni nanocatalysts by chemical vapour deposition (CVD).39–45 The observed carbon demixing phenomenon observed in this work is very similar to that reported for CNTs processing. Indeed, it is generally accepted that the carbon precursor (mainly short hydrocarbons) dissociatively adsorbs on the metal surface, releasing hydrogen and forming surface carbon species, which dissolve inside the metal particle. For Ni catalysts, the dissolved carbon atoms occupy the octahedral interstices of the face-centered cubic (fcc) structure. This process continues until the limit of carbon solubility in the metal is reached. At this point, carbon precipitates out of the metal particle and starts to form a CNT. In situ XRD studies demonstrated that in fact, the Ni lattice expands upon carbon dissolution before CNT growth, finally resulting in a cubic nickel–carbon solid solution formation. This unit cell expansion is accompanied by a cell contraction when carbon starts demixing (Fig. 8).45 These carbon-enriched Ni phases possess a face-centred cubic structure with a carbon content that cannot exceed that of the well-known rhombohedral nickel carbide Ni3C.
 | ||
| Fig. 8 Model of carbon diffusion into cubic nickel nanocrystal during H2–Ar plasma exposure starting from CH4 hydrocarbon. Nickel carbide's carbon concentration increases leading, at supersaturation, to its demixing as an outer thin carbon graphitic shell (adapted from ref. 40). | ||
Comparatively, in our plasma reactor, nickel nanoparticles were formed from Ni3C decomposition for long plasma exposure times but also from the plasma hydrogen reduction of NiO particle themselves originated from the progressive dehydration of the hydroxide precursor, with the concomitant decomposition of its organic content, namely, structural acetate, for short plasma exposure times. The temperature on the surface of the thus generated matter stayed less than 500 K,37,38 significantly lower than the temperatures usually employed in the CVD processes for CNT growth (more than 800 K39–45). Also, in our operating conditions, these in situ formed nickel nanoparticles, by NiO reduction, were rapidly exposed to small hydrocarbon derivatives, which originated from the decomposition of acetate organic species. We may thus rightly assume that CH4 molecules are present in the reactor and that they are progressively consumed. By analogy with CNT CVD growth on Ni nanocatalysts, in our reactor, CH4 reacts with the as-formed Ni nanocrystals, providing carbon and hydrogen. The carbon diffuses into the cubic crystal lattice, leading to the stabilisation of the cubic NiCy solid solution. This analogy can be extended to the cubic crystal lattice carbon supersaturation and then carbon demixing, leading to a core–shell Ni@Cgr nanostructures.
To summarize, two types of cubic NiCy nanocrystals are formed in the plasma reactor: (i) carbon-rich solid solutions obtained from NiO reduction and CH4 reaction (major for the short plasma exposure times) and (ii) carbon-deficient solid solutions obtained from Ni3C decomposition (major for the long exposure times). In both cases, carbon demixing proceeds progressively, leading to a carbon content decrease in favour of a coating by a more or less disordered graphite thin layer. Note that this double formation mechanism affects the whole NiCy particle morphology. The NiCy crystals resulting from Ni3C decomposition are very probably small in size (see Fig. 6), certainly smaller than those resulting from NiO reduction, which in proportion explain the observed average coherent length decrease of the cubic phase with the plasma exposure time (see Table 1).
This scenario is supported by the fact that Ni3C is not originated from the carbon supersaturation of Ni nanocrystals but it derives directly from the nickel hydroxyacetate precursor, Ni3C being the most stable nickel carbide phase, according to the Ni–C phase diagram of nanostructures.36
Thus, altogether, these results highlight the richness of microwave plasma reactivity toward nickel hydroxide derivatives, playing with the nature of inorganic matter through dihydroxylation and reduction reactions, organics decomposition and carbon atom implantation. To the best of our knowledge, this feature has never been reported before. It opens serious opportunities for plasma-assisted granular homo- and/or hetero-nanostructure processing.
This processing route also has the advantage, compared to the most common CVD approach, to start from hydroxide derivatives and yet unformed metal nanoparticles, which could be very costly.
Carbon-encapsulated transition metals (Fe, Co, Ni) and their alloys have plenty of technological applications. Among them, Ni@C core–shell NPs appear as one of the most promising functional nanomaterials.
They are seriously explored as electromagnetic absorbers due to the synergic effects of their magnetic and dielectric properties. Optimizing their morphology and composition, in terms of metal core size and carbon shell thickness, allows tuning the lightweight, bandwidth, absorption intensity and reliability. Very interesting results were already reported on the Ni@C core–shell NPs, particularly those exhibiting a metal core size and a carbon shell thickness in the 20–80 nm and 2–4 nm ranges, respectively.46 These nanostructures are usually produced in a two-step process, namely, metal–organic chemical vapour deposition (MOCVD) of Ni nanocores and their subsequent annealing (600 to 800 °C) under vacuum47 or by high temperature (≥400 °C) metal organic framework (MOF) pyrolysis.48 They are also successfully tested as catalysts towards unsaturated hydrocarbon hydrogenation. Thanks to their thin carbon coating and the presence of transition metal in the subsurface layers, they are able to activate H2 dissociative adsorption, which is the key step of hydrogenation reactions. To be as efficient as possible, the metal core size must be as small as possible to increase its specific area, and the thickness of the carbon shell should be thin enough, without exceeding some graphene layers. Such nano-objects are produced by the evaporation of the overheated (2000 °C) liquid drop of metal in the flow of inert gas (Ar) containing a hydrocarbon (butane)49 or by high temperature (≥ 400 °C) MOF pyrolysis,50 among others. Of course, these examples are not at all exhaustive but they clearly illustrate the application potential of such composite nanostructures with the desired morphology, size and shape, and then of their easy-to-achieve plasma processing route.
Experimental
Nickel-layered hydroxyacetate nanopowder synthesis
Forced hydrolysis in polyol was used to produce the desired nanopowders, applying the well-defined protocol of Poul et al.20 In practice, 19.9 g of Ni(CH3CO2)2·4H2O (SIGMA-ALDRICH) was dissolved in 1 L of diethylene glycol O(C2H4OH)2 (ACROS ORGANICS). 14.4 mL of water were then added, and the resulting mixture was heated up to boiling (180 °C) and kept under reflux for 2 h. Once the heating is turned off, the temperature was quickly decreased down to room temperature and a green powder was recovered by centrifugation and washing in ethanol, alternating with sonication and centrifugation. The chemical composition, structure and microstructure of the recovered nanopowders were confirmed by thermogravimetry, FTIR spectroscopy and XRD (see Fig. S1 and S2 in the ESI†).Cold H2–Ar plasma processing
The as-produced nickel-layered hydroxyacetate nanopowders were first compacted using an InfraRed pelletizer (10 t) as discs of 13 mm diameter and 1 mm thickness. The discs were then then introduced in a single-source plasma reactor available at the LSPM laboratory (Villetaneuse, France), equipped with a microwave high frequency (2.45 GHz) source of 180 W. Microwaves are transmitted along a coaxial transmission line to the source. A coaxial line includes a circulator with a water charge adapted to absorb the reflected power (the whole setup is described in Fig. S7, ESI†). At the exit of the source, a plasma is generated5,38 on a 90–10% H2–Ar gas mixture (total pressure of 1 mbar). Argon ions facilitate the ignition of the plasma, its maintenance and its stability.14 Ar+ and ArH+ ions play the role of intermediate ions in the plasma bulk but do not modify the distribution of hydrogen ions in the sheath. Moreover, the argon ions sputtering yield is low in our experimental conditions.38 Last but not least, previous studies performed on cold hydrogen plasma interaction with Ni(OH)238 as well Ni5 ultrafine powders and Ni thin films6 evidenced that the H2–Ar gas ratio did not need further optimization. The current density is stabilized about 6 mA cm−2 after 5 min of plasma ignition. Thanks to a silver ink, the pellet was deposited on a copper sampling holder, which was continuously cooled by an external water circulation and DC polarized at −50 V to attract positively-charged plasma ion species.38 Within these conditions, the incident ionic flux at the surface of the discs was 3.8 × 1016 ions cm−2 s−1.38 The exposition time was then varied from 30 min to 10 h. All the treated pellets were stored under argon to avoid their evolution in air.
Material characterization
The crystalline structure of the compacted powders before and after hydrogen treatment was analyzed by XRD using an X’Pert Pro diffractometer (PANALYTICAL) equipped with a Kα cobalt tube (λ = 0.17889 nm) and operated within θ–θ reflexion geometry. TEM observations were carried out on a JEOL JEM 2100F microscope operated at 200 kV, equipped with a UHR pole piece and a Gatan US4000 CCD camera. Raman spectroscopy was employed to get insights into the structural properties of the carbon matter thanks to a Horiba HR 800 Raman spectrometer, operating with a He–Ne laser beam (633 nm, 63 μW), focusing on carbon–carbon bonding signature in the 800–2000 cm−1 spectral range. Hydrogen quantification was also carried out using a G8 GALILEO H analyzer (Bruker, Palaiseau, France) to check any hydrogen storage in the treated samples. The calibration of the analyzer detector was carried out on a standard sample of 1 g of stainless steel containing 2.7 ppm of hydrogen in weight.Conclusions
Turbostratic disordered acetate-based nickel α-hydroxide was reacted to a cold 90–10% H2–Ar plasma for different times. Thanks to its great reactivity due to its open and disordered structure, it allowed the production of Ni@Cgr core–shell nanoparticles after more than 6 hours of exposure. Experimentally, cubic NiO and carbon super-stoichiometric rhombohedral Ni3C phases are produced at the beginning of the plasma exposure time. Increasing the plasma exposure time led to NiO reduction into cubic Ni nanocrystals, which are rapidly transformed into cubic NiCy (y ≪ 0.25) nanocrystals due to carbon diffusion, carbon being originated from acetate plasma decomposition. Based on Rietveld refinements, the cell parameter of these nanocrystals was found to be larger than that of pure nickel, in agreement with carbon solubilisation into the cubic lattice. It continues increasing before to decrease for a longer exposure time as a consequence of its demixing in a kind of a disordered graphite thin shell (2–3 nm in thickness). This feature is also accompanied by rhombohedral nickel carbide nanoparticle disappearance. An excess of carbon induces its instability and favours the growth of cubic NiCy (y ≪ 0.25) ultrafine nanocrystals, which in turn evolves into Ni@Cgr core–shell nanoparticles. All these features are very similar to those reported for CNT growth by CVD on nickel nanocatalysts using methane gas reaction with solid nickel.Author contributions
MCD and SHK: data curation, formal analysis and writing original draft. NM: TEM data collection and analysis, writing and review. MR and SAM: supervision, project administration, writing and reviewing the submitted draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ANR (Agence Nationale de la Recherche) and CGI (Commissariat à l’Investissement d’Avenir) through the LABEX SEAM (ANR 11 LABX 086) grant. MCD was granted for her PhD from SEGULA technology company, which is largely thanked for its financial support. They also want to thank Sophie Nowak, Stephanie Lau and Alexandre Chevillot (Université Paris Cité) for their assistance during XRD, Raman and FTIR measurements, respectively.Notes and references
- Z. Wang, Y. Zhang, Y. Cornelis Neyts, X. Cao, X. Zhang, B. W. L. Jang and C. Liu, ACS Catal., 2018, 8, 2093 CrossRef CAS 2000, 35, 3523.
- F. Yu, M. Liu, C. Ma, L. Di, B. Dai and L. Zhang, Nanomaterials, 2019, 9, 1436 CrossRef CAS PubMed.
- Y. Ma, Q. Wang, Y. Miao, Y. Lin and R. Li, Appl. Surf. Sci., 2018, 450, 413 CrossRef CAS.
- L. Estevez, D. Reed, Z. Nie, A. M. Schwarz, M. I. Nandasiri, J. P. Kizewski, W. Wang, E. Thomsen, J. Liu, J. G. Zhang, V. Sprenkle and B. Li, ChemSusChem, 2016, 9, 1455 CrossRef CAS PubMed.
- S. Haj-Khlifa, S. Nowak, P. Beaunier, P. De Rango, M. Redolfi and S. Ammar, Nanomaterials, 2020, 10, 136 CrossRef CAS PubMed.
- M. Quaas, H. Wulff, O. Ivanova and C. A. Helm, Z. Kristallogr. Suppl., 2009, 30, 241 CrossRef.
- S. Kakizaka, T. Sakamoto, H. Matsuura and H. Akatsuka, J. Adv. Oxid. Technol., 2007, 10, 253 CAS.
- T. Ryu, H. Y. Sohn, K. S. Hwang and Z. Z. Fang, J. Mater. Sci., 2008, 43, 5185 CrossRef CAS.
- J. L. H. Chau and C. C. Kao, Mater. Lett., 2007, 61, 1583 CrossRef CAS.
- S. Ayadi, Y. Charles, M. Gaspérini, I. Caron Lemaire and T. Da Silva Botelho, Int. J. Hydrogen Energy, 2017, 42, 10555 CrossRef CAS.
- C. Quirós, J. Mougenot, G. Lombardi, M. Redolfi, O. Brinza, Y. Charles, A. Michau and K. Hassouni, Nucl. Mater. Energy, 2017, 12, 1178 CrossRef.
- C. Quiros, J. Mougenot, R. Bisson, M. Redolfi, A. Michau, K. Hassouni and G. Lombardi, Nucl. Mater. Energy, 2019, 20, 100675 CrossRef.
- K. Ouaras, M. Redolfi, D. Vrel, C. Quirós, G. Lombardi, X. Bonnin and K. Hassouni, J. Fusion Energy, 2018, 37, 144 CrossRef CAS.
- S. Tavares, S. Miraglia, D. Fruchart, D. Dos Santos, L. Ortega and A. Lacoste, J. Alloys Compd., 2004, 372, L6 CrossRef CAS.
- T. C. M. Dantas, V. J. F. Junior, A. P. B. dos Santos, F. A. Bezerra, A. S. Araujo and A. P. M. Alves, Adsorpt. Sci. Technol., 2015, 33, 165 CrossRef CAS.
- A. D. Ebner, S. P. Reynolds and J. A. Ritter, Ind. Eng. Chem. Res., 2006, 45, 6387 CrossRef CAS.
- M. K. Ram Reddy, Z. P. Xu, G. Q. Lu and J. C. D. da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504 CrossRef CAS.
- G. Arizaga, K. Satyanarayana and F. Wypych, Solid State Ionics, 2007, 178, 1143 CrossRef CAS.
- E. Hosono, S. Fujihara, T. Kimura and H. Imai, J. Colloid Interface Sci., 2004, 272, 391 CrossRef CAS PubMed.
- L. Poul, N. Jouini and F. Fiévet, Chem. Mater., 2000, 12, 3123 CrossRef CAS.
- M. Taibi, S. Ammar, N. Jouini, F. Fiévet, P. Molinié and M. Drillon, J. Mater. Chem., 2002, 12, 3238 RSC.
- K.-H. Young, L. Wang, S. Yan, X. Liao, T. Meng, H. Shen and W. C. Mays, Batteries, 2017, 3, 6 CrossRef.
- J. Yu, S. Pan, Y. Zhang, Q. Liu and B. Li, Front. Mater., 2019, 6, 124 CrossRef.
- A. J. Tkalych, K. Yu and E. A. Carter, J. Phys. Chem. C, 2015, 119, 24315 CrossRef CAS.
- L. Lutterotti, S. Matthies and H. Wenk, CPD Newsletter, 1999, 21, 14 Search PubMed.
- T. Gaudisson, S. Nowak, Z. Nehme, N. Yaacoub, J.-M. Grenèche and S. Ammar, Front. Mater., 2021, 8, 668994 CrossRef.
- J. Kelling, P. Zahn, J. Schuster and S. Gemming, Elastic and Piezoresistive Properties of Nickel Carbides from First-Principles, arXiv, 2017, preprint, arXiv: 1604.00328v2 [cond-mat.mtrl-sci] DOI:10.48550/arXiv.1604.00328.
- S. Nagakura, J. Phys. Soc. Jpn., 1958, 13, 1005 CrossRef CAS.
- M. Singleton and P. Nash, Bull. Alloy Phase Diagrams, 1989, 10, 121–126 CrossRef CAS.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276 RSC.
- W. Smith, L. Dallmeyer, T. J. Johnson, C. S. Brauer, J.-S. McEwen, J. F. Espinal and M. Garcia-Perez, Carbon, 2016, 100, 678 CrossRef.
- B. Cao, J. Yuan, D. Jiang, S. Wang, B. Barati, Y. Hu, C. Yuan, X. Gong and Q. Wang, Fuel, 2021, 285, 119164 CrossRef CAS.
- Y. Leng, L. Xie, F. Liao, J. Zheng and X. Li, Thermochim. Acta, 2008, 473, 14 CrossRef CAS.
- D. L. Leslie-Pelecky, X. Q. Zhang, S. H. Kim, M. Bonder and R. D. Rieke, Chem. Mater., 1998, 10, 164 CrossRef CAS.
- B. C. Bayer, D. A. Bosworth, F. B. Michaelis, R. Blume, G. Habler, R. Abart, R. S. Weatherup, P. R. Kidambi, J. J. Baumberg, A. Knop-Gericke, R. Schloegl, C. Baehtz, Z. H. Barber, J. C. Meyer and S. Hofmann, J. Phys. Chem. Phys. C, 2016, 120, 22571 CrossRef CAS PubMed.
- M. Singleton and P. Nash, Bull. Alloy Phase Diagrams, 1989, 10, 121 CrossRef CAS.
- L. C. Delacqua, M. Redolfi, K. Ouaras, J. Naël-Redolfi, X. Bonnin, A. Michau, K. Hassouni and G. Lombardi, Phys. Plasmas, 2022, 29, 043508 CrossRef.
- M.-C. Dragassi, S. Haj-Khlifa, N. Menguy, M. Redolfi and S. Ammar, Int. J. Hydrogen Energy, 2024, 50, 1253 CrossRef CAS.
- A. Gili, L. Schlicker, M. F. Bekheet, O. Görke, S. Penner, M. Grünbacher, T. Götsch, P. Littlewood, T. J. Marks, P. C. Stair, R. Schomäcker, A. Doran, S. Selve, U. Simon and A. Gurlo, ACS Catal., 2018, 8, 8739 CrossRef CAS.
- A. Gili, L. Schlicker, M. F. Bekheet, O. Görke, D. Kober, U. Simon, P. Littlewood, R. Schomäcker, A. Doran, D. Gaissmaier, T. Jacob, S. Selve and A. Gurlo, ACS Catal., 2019, 9, 6999 CrossRef CAS.
- N. M. Mubarak, E. C. Abdullah, N. S. Jayakumar and J. N. Sahu, J. Ind. Eng. Chem., 2014, 20, 1186 CrossRef CAS.
- V. K. Portnoi, A. V. Leonov, S. N. Mudretsova and S. A. Fedotov, Phys. Met. Metallogr., 2010, 109, 153 CrossRef.
- V. I. Bogdanov, V. A. Popov, V. K. Portnoi and A. V. Ruban, Phys. Met. Metallogr., 2012, 113, 831 CrossRef.
- M. Diarra, A. Zappelli, H. Amara, F. Ducastelle and C. Bichara, Phys. Rev. Lett., 2012, 109, 185501 CrossRef CAS PubMed.
- A. Rinaldi, J.-P. Tessonnier, M. E. Schuster, R. Blume, F. Girgsdies, Q. Zhang, T. Jacob, S. B. Abd Hamid, D. S. Su and R. Schlögl, Angew. Chem., Int. Ed., 2011, 50, 3313 CrossRef CAS PubMed.
- D. Zhang, Z.-F. Hai, B. Zeng, Y. Qian, Y. Huang and Z. Yang, Chin. Phys. B, 2016, 25, 040201 CrossRef.
- D. Kuang, L. Hou, S. Wang, B. Yu, D. Lianwen, L. Lin, H. Huang, J. He and M. Song, Mater. Res. Express, 2018, 5, 095013 CrossRef.
- X. Wang, Q. Geng, G. Shi, Y. Zhang and D. Li, CrystEngComm, 2020, 22, 6796 RSC.
- A. V. Erokhin, E. S. Lokteva, A. Y. Yermakov, D. W. Boukhvalov, K. I. Maslakov, E. V. Golubina and M. A. Uimin, Carbon, 2014, 74, 291 CrossRef CAS.
- G. Xiaoling, C. Xiao, S. Dangsheng and L. Changhai, Acta Chim. Sin., 2018, 76, 22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00963g |
| This journal is © The Royal Society of Chemistry 2024 |