
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface engineering assisted CoNiP nanosheet arrays for electrochemical overall water splitting†
Linfei
Zhang‡
 *a,
Ruilong
Wei‡
b,
Chenyi
Liu
a,
Ningfa
He
a,
Xinru
He
a,
Yuhuan
Jiang
a,
Changshuai
Guo
a,
Yong
Hu
a and
Shengliang
Zhong
*b
*a,
Ruilong
Wei‡
b,
Chenyi
Liu
a,
Ningfa
He
a,
Xinru
He
a,
Yuhuan
Jiang
a,
Changshuai
Guo
a,
Yong
Hu
a and
Shengliang
Zhong
*b
aSchool of Automotive Engineering, Guangdong Polytechnic of Science and Technology, ZhuHai 519090, China. E-mail: dafeiuli@163.com
bCollege of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang 330022, China
First published on 24th May 2024
Abstract
Electrochemical overall water splitting is a green, environmentally friendly, and efficient hydrogen production method that involves two half-reactions: the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER). Due to the slow reaction kinetics of the OER and HER, electrochemical overall water splitting often requires a higher potential than the theoretical value, resulting in significant energy loss. Here, we report that a hybrid catalyst synthesized using cobalt oxyhydroxide (CoOOH) nanoparticles and 2D metallic CoNiP nanosheets on a conductive carbon fiber paper (CoNiP/CoOOH-CFP) shows significantly enhanced overall water splitting activity in alkaline electrolytes. The high activity of CoNiP/CoOOH-CFP may be due to the dual-function mechanism used in this hybrid catalyst, in which CoOOH promotes the adsorption and dissociation of water, thereby providing protons for the subsequent hydrogen evolution reaction on CoNiP and reducing the free energy of the oxygen evolution reaction. It is worth noting that our self-made alkaline electrolytic cell assembled with CoNiP/CoOOH-CFP as a dual-function catalyst can achieve a water splitting current density of 20 mA cm−2 at a low battery voltage of 1.732 V. When a bias voltage of 1.73 V was applied, the performance of the hybrid catalyst remained unchanged after a 50 hour working cycle, indicating its good stability.
1. Introduction
The synthesis of high purity and environmentally friendly hydrogen using low energy consuming water splitting is of great significance.1–3 However, the electrolytic water splitting reaction is hampered by the slow kinetics of its two half-reactions, namely the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER).4 Although precious metal Pt and RuO2/Ir-based materials are considered to be the most advanced electrocatalysts for the HER and OER in industry, the high cost and element scarcity of these precious metal based electrocatalysts greatly limit their widespread application.5,6 To solve these problems, it is necessary to design cost-effective and earth-abundant catalysts for water splitting. Transition metal-based electrocatalysts are favored by researchers due to the high earth abundance, diversity, potential stability and theoretical catalytic activity of transition metals, such as transition metal carbides,7 sulfides,8 phosphates9 and selenides.10 However, most reported catalysts are specialized to catalyze either the HER or OER, respectively, and the best operating conditions reported often do not match. When a water splitting device uses different catalyst materials as the cathode and anode, the incompatibility of the two electrodes can lead to reduced efficiency.11 Therefore, it is necessary to develop bifunctional catalysts with high activity for both the HER and OER in the same medium, so as to improve the overall efficiency of water splitting.Cobalt phosphide (CoP) has great potential as a bifunctional catalyst for integrated water splitting. Theoretical studies have proved that the metal atoms Co and non-metal P atoms in CoP have cationic and anionic states, respectively, and the P atoms with anionic states act as active sites to promote charge transfer in alkaline media for the water splitting process.12 In order to overcome the shortcomings of low HER electrocatalytic performance and OER instability of pure CoP in an alkaline medium, hybridization of CoP-based catalysts with other catalytic active materials is a promising method,13 and it has also been shown that the interaction of heterogeneous metals with two components can improve the water splitting performance of such polymetallic hybrid structures.14 Recently, Yang et al. prepared a single crystal (FexNi1−x)2P catalyst with excellent electrocatalytic activity, which shows that doped metal atoms can change the charge distribution near the P active site, thus making P more active and able to reduce the kinetic energy barrier.15 In addition, Zatovsky's research showed that a symmetrical water electrolyzer constructed from a graded NiCoP nanocone array requires a voltage of 1.64 V to achieve a current density of 20 mA cm−2.16 However, most reported CoP hybridization structures have in common foreign cations or anions being introduced to produce homologous ligand compositions (e.g. NiCo2Px,17 3D-NiCoP,18 Co2−xNixP,19etc.), so the space for further improving its catalytic activity and stability is limited.
The modification of CoP itself without the introduction of an external catalyst for efficient charge transfer and activation of H2O/H+ is an interesting and challenging task. Recently, it has been reported that organic functionalization of the CoP surface is a simple and effective strategy to promote catalytic activity. Li et al. prepared hydroxylated porous carbon nitride using a hydrothermal method in the presence of ammonium hydroxide,20 and the modified carbon nitride improved the specific surface area and hydrogen evolution efficiency of porous carbon nitride. For the lithium storage performance of highly flexible two-dimensional transition metal carbides reported by Zhang RuiFeng et al., it is shown that the five thermogenically preferred Ti2CTn terminated by O− and F− co-functional groups21 all show excellent mechanical flexibility and strength, and show an increasing trend in activity with a decrease of the F/O ratio. Therefore, compared with other surface engineering methods to construct electrocatalysts, such as defect, interface and doping, functional group modification is a new strategy.22–25 Through the development of controllable modification strategies, it is shown that surface functional groups are important for the improvement of electrocatalytic performance, especially H+ activation. More importantly, it is crucial for understanding the nature of surface functional group modification, which is of great help for the intelligent design of high-performance electrocatalysts.
In this work, we report a surface modification method that increases the hydrophilicity of CoNiP nanosheets by covering them with a thin layer of CoOOH, thereby enhancing the overall water splitting activity of metallic CoNiP nanosheets under alkaline conditions. By adjusting the oxidation time, the CoNiP/CoOOH-CFP hybrid catalyst can achieve a cathode current density of 20 mA cm−2 in the 1.0 M KOH electrolyte with a HER overpotential of about 148 mV, which greatly reduces the original CoP overpotential (175 mV). Similarly, the surface modified catalyst used in the anodic OER also showed good performance. The current density of the CoNiP/CoOOH-CFP catalyst reached 20 mA cm−2 under an applied bias voltage of 1.499 V. In addition, when a bias voltage of 1.73 V was applied for 50 h, the overall water splitting performance maintained basic stability. System experiments have shown that the CoOOH thin layer on the surface of the CoNiP catalyst can reduce the activation energy of the system, making the surface of CoNiP more prone to water absorption and desorption, greatly improving the water splitting performance.
2. Experimental section
2.1 Synthesis of the CoNi(OH)2-CFP precursor
Before electrodeposition, each carbon fiber paper (CFP, 3 mΩ cm2) was cut into a 1 × 2.0 (cm2) rectangle. 10 exposed CFPs were randomly weighed with an average weight of 18.28 mg. During electrodeposition, 1 × 1.5 (cm2) CFP was immersed in the electrolyte. Electrodeposition was carried out in a standard three-electrode glass electrolytic cell with a clean CFP as the working electrode, a platinum plate as the counter electrode and Ag/AgCl (in a saturated KCl aqueous solution) as the reference electrode in an aqueous electrolyte solution of 0.08 M Co(NO3)2·6H2O and 0.02 M Ni(NO3)2·6H2O at room temperature. CoNi(OH)2 double hydroxide nanosheets were electrodeposited on the CFP. The electrodeposition potential was −1.0 V (vs. Ag/AgCl). After 8 minutes of electrodeposition, the color of the CFP changed from black to pink, indicating the successful formation of hydroxide on the CFP. With the assistance of ultrasonic treatment, the pink CFP was carefully rinsed several times with deionized water and anhydrous ethanol, and finally dried in air. For the synthesis of Co(OH)2/CFP, all conditions are the same, except that the zero Ni content during electrodeposition can be changed accordingly.2.2 Synthesis of CoNiP-CFP intermediates
In a typical synthesis, CoNi(OH)2-CFP and NaH2PO2·H2O are placed in two separate porcelain boats, and the porcelain boat containing NaH2PO2·H2O is placed upstream of the furnace tube. The mole ratio of Co to P is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. Subsequently, the sample is heated to 300 °C at a rate of 5 °C min−1 and then annealed at 300 °C for 2 h. Finally, they are naturally cooled to room temperature to obtain a CoNiP-CFP intermediate sample. The whole process is carried out under constant Ar air flow (100 sccm). For the synthesis of CoP-CFP, all other conditions are the same, except that the precursor is changed from CoNi(OH)2-CFP to Co(OH)2-CFP.
:10. Subsequently, the sample is heated to 300 °C at a rate of 5 °C min−1 and then annealed at 300 °C for 2 h. Finally, they are naturally cooled to room temperature to obtain a CoNiP-CFP intermediate sample. The whole process is carried out under constant Ar air flow (100 sccm). For the synthesis of CoP-CFP, all other conditions are the same, except that the precursor is changed from CoNi(OH)2-CFP to Co(OH)2-CFP.
2.3 Synthesis of the CoNiP/CoOOH-CFP electrocatalyst
The newly synthesized CoNiP-CFP was soaked in ethanol solution (40 mL) containing 0.2 M CoCl2 and NH4HCO3 (1 mmol). After 5 minutes of ultrasonic treatment, the suspension was stirred in an ambient atmosphere at 400 rpm magnetic force for 8 h. The sample was then removed with tweezers, alternately washed twice with deionized water and ethanol, then dried in a vacuum and stored in a vacuum. At the end of the process, 10 CFPs were weighed again, with an average weight of 20.12 mg. Therefore, the loading capacity of CoNiP/CoOOH on the CFP was determined to be 0.61 mg cm−2. Specifically, similar samples were prepared for performance and structural comparison.2.4 Characterization
The samples were characterized by field emission scanning electron microscopy (FESEM, ZEISS-Merlin SEM), and transmission electron microscopy (TEM) images and energy dispersive X-ray spectroscopy (EDS) data were obtained using a TEM-HT7700 transmission electron microscope operated at 80 kV with a drop of hydrosol placed on a 400-mesh carbon-coated copper grid. High-resolution TEM (HRTEM) characterization was performed using an FEI Tecnai G2 F30 Super-Twin TEM system operated at 300 kV. X-Ray powder diffraction (XRD) was carried out on a Bruker D8 X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) and a scintillation counter, and X-ray photoelectron spectroscopy (XPS) measurement was performed on a PerkinElmer Model PHI 5600 XPS system with a resolution of 0.3–0.5 eV and a monochromatic aluminum anode X-ray source.2.5 Electrochemical measurements
All electrochemical measurements were performed with a conventional three-electrode cell using a CHI 760E, where the CoNiP-CFP electrode or CoNiP/CoOOH-CFP (1 × 1.5 cm2) and a standard Hg/HgO electrode were used as the working electrode and reference electrode, respectively. A graphite rod was used as the counter electrode in the HER and OER experiments, respectively. A solution of 1.0 M KOH was used as the electrolyte (pH = 13.60). The electrochemical impedance spectra (EIS) of the HER and OER were recorded in the frequency range of 100 kHz to 0.01 Hz using an alternating current (AC) amplitude of 10 mV and applying various overpotentials (vs. a reversible hydrogen electrode (RHE)). The EIS results were simulated using Z-view software.Linear sweep voltammetry (LSV) was conducted at a scan rate of 10 mV s−1. For comparison, CoP/CoOOH@CFP was used as the working electrode, which was dispersed in a solution of isopropyl alcohol with Nafion (5 wt%, Sigma Aldrich) and then drop-casted onto the CFP with a load of about 0.61 mg cm−2. All overpotentials presented were corrected for iR losses, the impedance spectra of CoP/CoOOH-CFP were recorded at −0.2 V versus a RHE in 1.0 M KOH using a three-electrode setup, and the solution resistance was measured to be ∼1.0 Ω. All overpotential values measured were normalized with reference to the RHE using the following equation:
| ERHE = EHg/HgO + 0.098 + 0.0592pH. |
The overpotentials of the OER were calculated using the equation η = ERHE − 1.23 V. The Tafel plots were extracted from the LSV data based on the Tafel equation η = a + blog(j), where η is the overpotential, j is the current density, and b is the Tafel slope.
3. Results and discussion
The porous CoNiP nanosheets were readily fabricated through topotactic transformation from the presynthesized two dimensional cobalt nickel double hydroxide (2D CoNi(OH)2) nanosheets using a vapor phosphorization strategy, as schematically illustrated in Fig. 1. The precursor of CoNi(OH)2 nanosheets was synthesized through electrochemical deposition modification containing several steps.26 On the cathode (CFP, the SEM image of Fig. S1 (ESI†) displays a smooth surface of the bare CFP with a diameter of about 10 μm before the electrochemical deposition), water was electrolyzed into H2 and OH−, and then CoNi(OH)2 was formed by the combination of Co2+ or Ni2+ ions and the generated OH−. Meanwhile, NO3− was reduced to ammonia and OH− ions near the cathode. On the anode (Pt electrode), some OH− ions were directly oxidized into O2. We then annealed the CFP-supported CoNi(OH)2 nanosheets with NaH2PO2·H2O under an argon atmosphere to form porous CoNiP nanosheets. Subsequently, the porous CoNiP-CFP was placed in a weakly alkaline ethanol solution containing CoCl2 to modify an ultrathin layer of amorphous cobalt oxyhydroxide (CoOOH).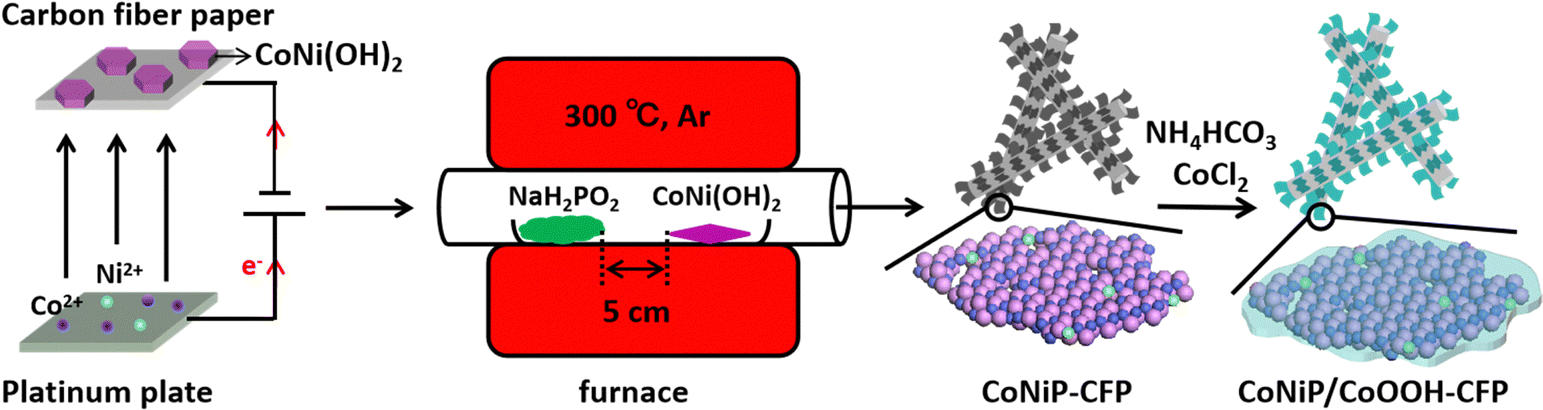 | ||
| Fig. 1 Schematic diagram of the synthesis route of CoNiP/CoOOH CFP, including the first step of electrochemical deposition, followed by a solid phosphating precursor, and finally the sol–gel synthesis of CoOOH. | ||
To investigate how the chemical composition affects the morphology of the as-prepared sample, the role of CFP and nickel species is preliminarily explored as shown in Fig. S2–S5 (ESI†). It is evident from the SEM images that the product can be more evenly dispersed and grown on the CFP carrier. Similarly, the introduction of nickel species can make the nanosheets thinner, as the product is a well-known layered double hydroxide (CoNi-LDH).27 FESEM images of samples prepared in different stages are provided in Fig. 2. In Fig. 2(A)–(C), it can be seen that after electrochemical deposition, the bare carbon fiber surface is covered with a large number of 3D structured nanosheets, which are interconnected and interwoven like fishing nets. The thickness of these nanosheets is about 20 nm and the surface is smooth. It is worth noting that the basic 3D structured nanosheet remains unchanged with the implementation of the phosphating process, as shown in Fig. 2(D)–(F). The magnified observation of the nanosheets revealed a highly porous structure, which is formed due to the different diffusion rates of OH− and P during the phosphorylation process after heating.26 Under high temperature conditions, the sheet-like precursor collides with PH3, stimulating the absorbed energy of PH3, which meets the energy conditions required for the growth of phosphide crystals. During the reaction process, phosphide crystals grow into a sheet-like structure through structural rearrangement, and their size and morphology depend on the reaction conditions and the decomposition rate of NaH2PO2. Therefore, the sheet-like structure likely undergoes subtle changes, such as thickening or enlargement of nanosheets.28,29 The TEM image shows that the sheet-like crystals with a size of approximately 200 nm come from peeled off individual nanosheets (Fig. S6, ESI†). Fig. S7 (ESI†) shows that ultra-thin CoNiP/CoOOH nanosheets are interlaced to form a hierarchical structure, and an amorphous layer is also observed. From Fig. 2(G)–(I), it can be seen that the pores in the 2D structured porous nanosheets are almost completely filled, and the thickness increases relatively, which indicates that the surface of the porous nanosheets is covered with a pre-designed CoOOH layer.
 | ||
| Fig. 2 FESEM images of the as-prepared products at different stages of the catalyst preparation process: (A)–(C) CoNi(OH)2-CFP, (D)–(F) CoNiP-CFP, and (G)–(I) CoNiP/CoOOH-CFP. | ||
The crystallinity and phase purity of the as-prepared catalysts are confirmed by XRD analysis (Fig. 3(A)). The diffraction peaks at 31.4, 36.2, 46.3, and 48.2° are attributed to the (011), (111), (112), and (211) lattice planes of cubic CoP (JCPDS no. 29-0497), which is consistent with the XRD patterns of CoNiP-CFP and CoNiP/CoOOH-CFP. The peak located at 41.2° belongs to NiP. It is obvious that no diffraction peak of CoOOH was observed, indicating that CoOOH is likely amorphous. The HRTEM images in Fig. 3(B) and (C) confirm the formation of a thin amorphous layer of CoOOH. Furthermore, the HRTEM image of CoNiP/CoOOH depicts distinct lattice fringes with d-spacings of about 0.283 nm and 0.189 nm (Fig. 3(C)), corresponding to the CoP (011) and (211) crystalline planes, respectively. These results indicate that Ni exists in a doped state within the CoP lattice. X-ray photoelectron spectroscopy (XPS) measurements were further carried out for the CoP/CoOOH sample to examine the electronic states of the Co and P elements and the high-resolution spectra of Co 2p and P 2p are presented in Fig. S8 (ESI†). High-resolution Co 2p and P 2p XPS reveal the characteristic peaks of Co and P, and that Co and P have +3 and −3 valence states. At the same time, the P–O bond peaks are obvious, indicating that oxidation occurs on the CoP surface (Fig. S9, ESI†).
 | ||
| Fig. 3 (A) XRD patterns of CoNiP-CFP and CoNiP/CoOOH-CFP. (B) TEM images of the as-prepared CoNiP/CoOOH-CFP. (C) HRTEM image taken from the region indicated within the box in (B). | ||
Fig. 4 compares the XPS of CoNiP/CoOOH with that of pure CoP/CoOOH, and it was found that the corresponding binding energy remained basically unchanged, indicating that there was no obvious electronic interaction between CoNiP and CoOOH. Concretely, the two main peaks located at 778.8 eV and 797.6 eV are ascribed to the Co–P bonds and the fitted peaks located at 781.6 eV and 797.9 eV are the Co 2p3/2 and Co 2p1/2 peaks ascribed to the Co–O bond, caused by spin–orbit effects, respectively (Fig. 4(A)). It is noteworthy that both the Co–P and Co–O bond peaks are slightly positively shifted due to the nickel incorporation in the host lattice.30 The Ni 2p3/2 spectrum is shown in Fig. 4(B), and the main peak located at 856.6 eV along with its shakeup satellite peak correspond to Ni2+ arising from superficial oxidation of Ni–O.31 It should be noted that the weak peak located at 853.0 eV can be assigned to the formation of a Ni–P bonding structure. The P 2p spectrum shown in Fig. 4(C) is deconvoluted into three peaks: the doublet peaks located at 129.5 eV and 130.4 eV, which can be attributed to the metal–phosphorus bonds in CoP, and the third peak at 133.7 eV, which is attributed to the oxidized surface of CoNiP nanosheets. The results from surface-structural and compositional analysis using XRD, FESEM, TEM, and XPS have confirmed the successful formation of porous Ni–CoP nanosheets. As shown in Fig. 4(D), the O 1s spectrum of CoNiP/CoOOH is deconvoluted into three peaks: a peak at 529.5 eV is attributed to the oxygen bound to metal atoms, revealing the metal–oxyhydroxide (M–OOH) formation,32 a prominent peak at 531.7 eV is ascribed to the OH− groups arising from the presence of metal hydroxide and a peak at 533.4 eV is is attributed to the physical adsorption of water molecules on the surface. The XRD, HRTEM, and XPS results indicate that the generated product is Ni doped CoP nanosheets coated with an amorphous CoOOH layer. A N 1s peak was also detected for the surface of the sample, indicating that a small amount of NH4+ was adsorbed on the surface, which came from NH4HCO3 (Fig. S8, ESI†).
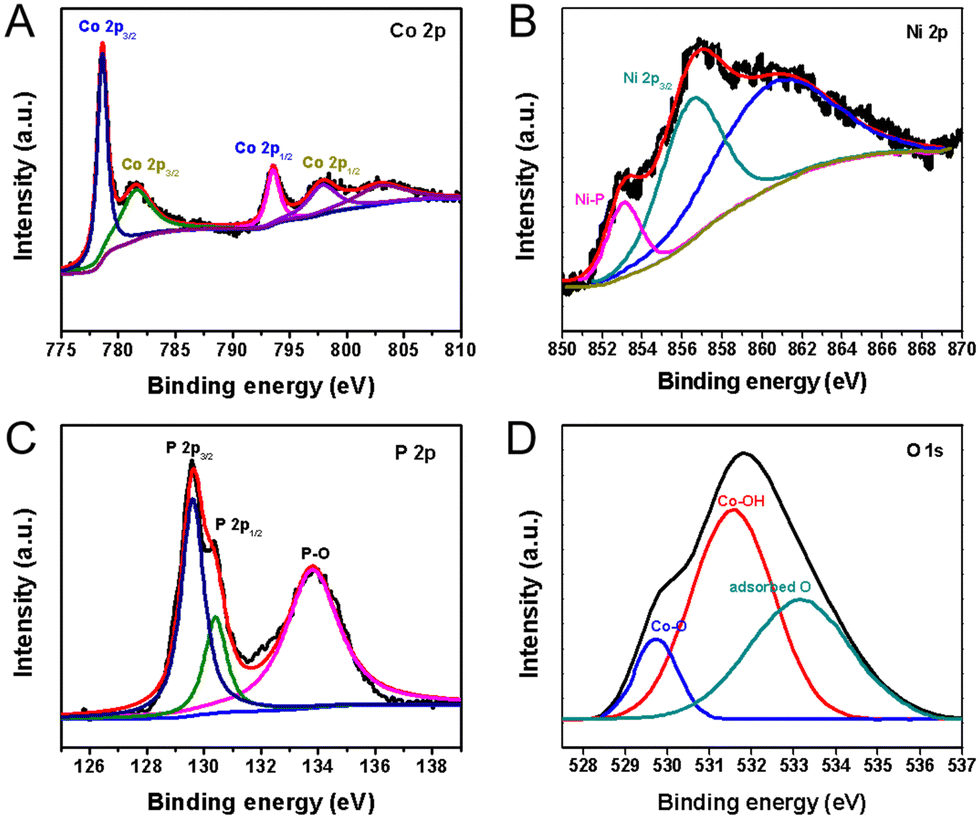 | ||
| Fig. 4 High-resolution XPS spectra of the as-prepared CoNiP/CoOOH-CFP hybrids catalysts. (A) Co 2p spectrum, (B) Ni 2p3/2 spectrum, (C) P 2p spectrum, and (D) O 1s spectrum. | ||
4. Overall water splitting
Electrochemical hydrogen evolution and oxygen evolution were carried out using a Shanghai Huachen CHI 760E electrochemical workstation. The standard three-electrode test was conducted with Hg/HgO as the reference electrode and a graphite rod as the counter electrode. The HER and OER activities of NiCoP/CoOOH nanosheet arrays supported by the carbon fiber paper were studied in KOH aqueous solution with a pH of 13.6. Prior to the test, the electrolyte in the cell was injected with high purity nitrogen and bubbled for 30 minutes, and other studied catalysts (e.g., CoP and NiCoP) were used as references. The polarization curve in Fig. 5(A) showed that for a NiCoP/CoOOH catalyst, the HER starts only at an initial potential of −58 mV relative to a reversible hydrogen electrode (RHE). At 20 mA cm−2, the recorded overpotential of NiCoP/CoOOH was 0.148 V, while those of CoP, CoP/CoOOH and CoNiP were 0.175 V, 0.144 V and 0.150 V, respectively. It can be seen that due to the coverage of pores and the reduction of contact sites, the HER performance of CoNiP/CoOOH is indeed weaker than that of CoNiP. Additionally, nitrogen adsorption and desorption isotherm measurements also showed that the specific surface area of CoNiP/CoOOH was the smallest (21.3 m2 g−1), but similar to that of the porous CoNiP nanosheets, and the CoOOH layer did not completely encapsulate the porous CoNiP nanosheets, as shown in Fig. S10 (ESI†). The Tafel slope of the CoNiP/CoOOH catalyst is 76 mV dec−1, which is smaller than those of other control samples but comparable to that of CoNiP (Fig. S11, ESI†). This indicates two points: (i) Ni doping changes the electronic structure of CoP, which is the main factor for improving the HER performance, while the CoOOH layer has little effect on the improvement of the HER performance and (ii) the HER conducted on CoNiP/CoOOH catalysts may follow the Volmer–Heyrovsky mechanism.3 Considering the small overpotential and Tafel slope, the HER performance of the as-prepared CoNiP/CoOOH catalyst in an alkaline electrolyte outperformed that of many previously reported Co-based HER catalysts, as briefly summarized in Table S1 (ESI†). At the same time, hydrogen evolution performance under acidic conditions was also investigated, and it was found that all catalysts had a similar performance, which also confirmed the presence of CoOOH on the surface, which completely dissolved under acidic conditions without improving the performance (Fig. S12, ESI†). In addition to the OER performance, CoNiP/CoOOH porous nanosheets also showed catalytic effects for electrolytic water oxidation (Fig. 5(B)). The initial potential of the composite catalyst is 180 mV, and the overpotential of η20 is 1.499 V, which is much better than those of CoP, CoP/CoOOH and CoNiP. The OER kinetics process was further assessed using the Tafel plot shown in Fig. S13 (ESI†). CoNiP/CoOOH exhibited a relatively low Tafel slope (45 mV dec−1) under alkaline conditions, which is lower than those of CoP (71 mV dec−1), CoP/CoOOH (50.5 mV dec−1) and CoNiP (65 mV dec−1), demonstrating the kinetic advantage of the CoNiP/CoOOH nanosheets for water oxidation. The OER performance of the CoNiP/CoOOH nanosheets also exceeds that of most of the reported Co-based nonprecious OER electrocatalysts in alkaline media (Table S2, ESI†). Although the Ni content in CoNiP/CoOOH is low, according to the XPS study, it can play a role as the active site for the OER. Thus, the enhanced OER activity of CoNiP/CoOOH can be attributed to higher Ni valence states resulting from strong electron transfer. Based on the performance for two half-reactions, CoNiP/CoOOH was ultimately chosen as a typical catalyst. The stability of CoNiP/CoOOH hybrids was examined by continuous measurement for 2000 cycles in 25 h and a chronoamperometry test carried out in 1.0 M KOH. For both the HER and OER, CoNiP/CoOOH exhibits a small growth of overpotential to maintain a current density of 20 mA cm−2 (Fig. 5(C) and (D)). This excellent durability is expected to facilitate its commercial application in catalysis.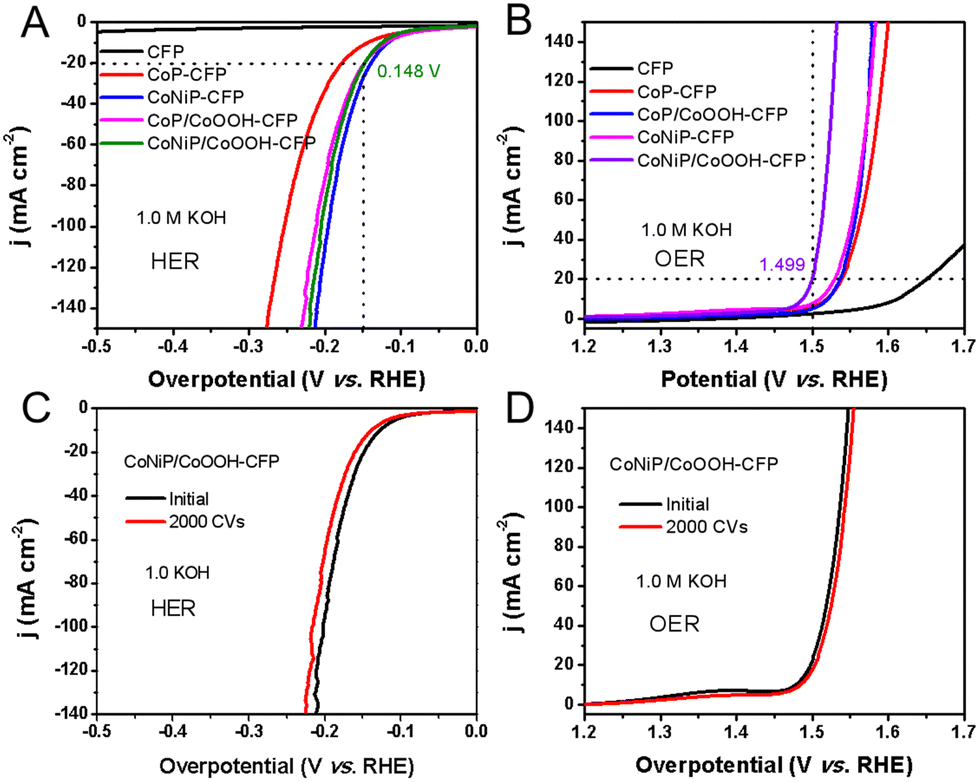 | ||
| Fig. 5 (A) and (B) Polarization curves of the HER and OER on the bare carbon fiber paper, CoP, NiCoP, CoP/CoOOH and NiCoP/CoOOH. (C) and (D) HER and OER polarization curves for evaluating the accelerated durability of CoNiP/CoOOH. | ||
Regulation of the electronic structure through heteroatom doping represents one of the most powerful strategies to boost the electrocatalytic performance of the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). Experimental results manifest that the appropriate Ni incorporation into CoP can dramatically modulate the electronic structure of CoP and alter the adsorption free energies of reaction intermediates, thus promoting the HER and OER activities. Specifically, the thermodynamic equilibrium potential of the OER is 1.23 V, and the OER under alkaline conditions typically has a higher energy barrier, requiring a higher overpotential to overcome the energy barrier.33 Previous reports suggest that solving the energy relationship between OHads and OOHads is the key to achieving ideal conditions for low overpotential.32 Therefore, the artificial rational design of an intermediate CoOOH layer and the synergistic effect of heteroatom doping may be effective in reducing the reaction adsorption free energy, as it can directly provide intermediate groups (M–OOH) and change the electronic structure of CoP.
Then, at room temperature, a self-built alkaline electrolytic cell was assembled using NiCoP/CoOOH supported by the carbon fiber paper as the anode and cathode for overall water splitting in N2 saturated 1.0 M KOH aqueous solution (Fig. S14, ESI†). By applying a battery voltage of 1.73 V to the constructed device, water splitting current densities of 20 mA cm−2 can be achieved. CoNiP/CoOOH performs extremely well as a dual catalyst for overall water splitting, outperforming CFP or other reported dual-function water splitting catalysts (Fig. 6(A)). Due to the formation of a new interface between CoNiP/CoOOH and oxides, the catalytic performance is still better than that of pure CoP.34 Distinct H2 and O2 bubbles can be observed on the surface of both electrodes. The long-term stability of the water splitting reaction was assessed by continuous operation at 1.73 V for 50 hours (Fig. 6(B)). After a long term stability test, CoNiP/CoOOH nanosheets were analysed using FESEM, XRD and XPS to evaluate the surface chemical composition, valence state modulation and changes in the morphology. The sheet-like structure of the catalyst is partially etched and forms oxides (CoO or NiO) on the surface (Fig. S15A, ESI†), resulting in a slight increase in overpotential.35 Despite a slight current increase at the onset of the chronoamperometry measurement, the anodic current density of CoNiP/CoOOH exhibits no significant degradation over a duration of 50 h. The XRD pattern (Fig. S15B, ESI†) also shows a diffraction peak of the oxide, which is located at 42.5° and belongs to the CoO (200) lattice plane (JCPDS no. 48-1719). The main peak of the Co 2p XPS spectrum at 781.6 eV is enhanced, confirming an increase in the oxide content on the catalyst surface (Fig. S15C, ESI†). At the beginning of the first hour, the degradation is small, and then the activity remains stable or even increases, which proves its excellent chemical stability and its great potential for application in practical electrolytic systems.
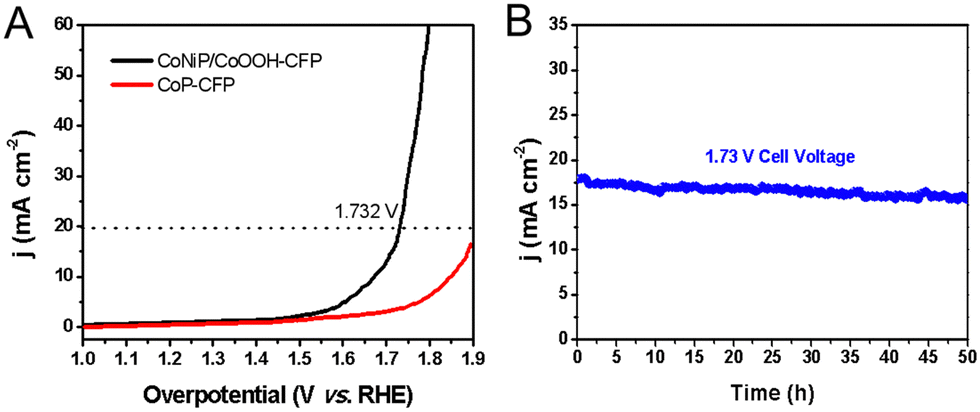 | ||
| Fig. 6 (A) LSV curve for overall water splitting in a two-electrode configuration. (B) Stability of water splitting for 50 hours at an applied bias pressure of 1.73 V. | ||
5. Conclusions
In summary, we proposed a refined Ni doping and surface engineering strategy to enhance the electrocatalytic performance of CoNiP/CoOOH porous nanosheets for overall water splitting in an alkaline medium. A catalyst was prepared through a simple three-step route, with mainly the third step being the surface modification process for the successful growth of an intermediate CoOOH layer. The hybrid nanosheets can be used as bi-functional electrocatalysts, which can effectively drive the reduction and oxidation reactions of water in an alkaline pH electrolyte. Consequently, in our self-made water electrolysis cell, these NiCoP/CoOOH porous nanosheets as electrode materials also showed good catalytic ability for large-scale water electrolysis, and have good practical application prospects. This novel hybrid structure with highly porous surfaces enhances catalytic performance, which can be further improved by adjusting the metal and its ratio in the compound. We anticipate that this oxidation surface modification method will contribute to the development of electrocatalysts for alkaline electrolysis cells.Author contributions
L. F. Zhang and S. L. Zhong designed the research and interpreted the experimental results. R. L. Wei collected important experimental data. C. Y. Liu, C. S. Guo, and Y. Hu performed the experiments. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Projects in Guangzhou (grant no. 202201010817) and the Guangdong Provincial Department of Education's Characteristic Innovation Project for Ordinary Higher Education Institutions (grant no. 2021KTSCX215 and 2021KTSCX216).References
- J. Li, Z. Peng and E. Wang, J. Am. Chem. Soc., 2018, 140, 10629–10638 CrossRef CAS PubMed.
- K. Kawashima, R. A. Márquez, L. A. Smith, R. R. Vaidyula, O. A. Carrasco-Jaim, Z. Q. Wang, Y. J. Son, C. L. Cao and C. B. Mullins, Chem. Rev., 2023, 123, 12795–13208 CrossRef CAS PubMed.
- Y. M. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- A. B. Laursen, S. Kegnæs, S. Dahl and L. B. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577–5591 RSC.
- C. F. Chen, A. P. Wu, H. J. Yan, Y. L. Xiao, C. G. Tian and H. G. Fu, Chem. Sci., 2018, 9, 4746–4755 RSC.
- H. Yin, F. Rong and Y. B. Xie, Int. J. Hydrogen Energy, 2024, 52B, 350–375 CrossRef.
- S. P. Wang, J. Wang, M. L. Zhu, X. B. Bao, B. Y. Xiao, D. F. Su, H. R. Li and Y. Wang, J. Am. Chem. Soc., 2015, 137, 15753–15759 CrossRef CAS PubMed.
- C. Zhang, Y. M. Shi, Y. F. Yu, Y. H. Du and B. Zhang, ACS Catal., 2018, 8, 8077–8083 CrossRef CAS.
- Y. Q. Wu, X. Tao, Y. Qing, H. Xu, F. Yang, S. Luo, C. H. Tian, M. Liu and X. H. Lu, Adv. Mater., 2019, 31, 1900178 CrossRef PubMed.
- Y. P. Yang, W. B. Zhang, Y. L. Xiao, Z. P. Shi, X. M. Cao, Y. Tang and Q. S. Gao, Appl. Catal., B, 2019, 242, 132–139 CrossRef CAS.
- J. Zhang, Q. Y. Zhang and X. L. Feng, Adv. Mater., 2019, 31, 1808167 CrossRef PubMed.
- J. Yang, D. H. Guo, S. L. Zhao, Y. Lin, R. Yang, D. D. Xu, N. E. Shi, X. S. Zhang, L. Z. Lu, Y. Q. Lan, J. C. Bao and M. Han, Small, 2019, 15, 1804546 CrossRef PubMed.
- J. W. Joo, T. Y. Kim, J. Y. Lee, S. L. Choi and K. Lee, Adv. Mater., 2019, 31, 1806682 CrossRef PubMed.
- L. L. Chai, Z. Y. Hu, X. Wang, Y. W. Xu, L. J. Zhang, T. T. Li, Y. Hu, J. J. Qian and S. M. Huang, Adv. Sci., 2020, 7, 1903195 CrossRef CAS PubMed.
- W. Zhang, Y. H. Zou, H. L. Liu, S. Chen, X. Wang, H. W. Zhang, X. L. She and D. J. Yang, Nano Energy, 2019, 56, 813–822 CrossRef CAS.
- J. Z. Li, G. D. Wei, Y. K. Zhu, Y. L. Xi, X. X. Pan, Y. Ji, I. V. Zatovsky and W. Han, J. Mater. Chem. A, 2017, 5, 14828–14837 RSC.
- R. Zhang, X. X. Wang, S. J. Yu, T. Wen, X. W. Zhu, F. X. Yang, X. N. Sun, X. K. Wang and W. P. Hu, Adv. Mater., 2017, 29, 1605502 CrossRef PubMed.
- B. Ma, Z. C. Yang, Y. T. Chen and Z. H. Yuan, Nano Res., 2019, 12, 375–380 CrossRef CAS.
- Y. Pan, Y. J. Chen, Y. Lin, P. X. Cui, K. A. Sun, Y. Q. Liu and C. G. Liu, J. Mater. Chem. A, 2016, 4, 14675–14686 RSC.
- X. J. Wang, X. Tian, F. T. Li, Y. P. Li, J. Zhao, Y. J. Hao and Y. Liu, Int. J. Hydrogen Energy, 2016, 41, 3888–3895 CrossRef CAS.
- S. Chen, Z. H. Fu, H. Zhang, D. Legut, T. C. Germann, Q. F. Zhang, S. Y. Du, J. S. Francisco and R. F. Zhang, Adv. Funct. Mater., 2018, 28, 1804867 CrossRef.
- G. Huang, Z. H. Xiao, R. Chen and S. Y. Wang, ACS Sustainable Chem. Eng., 2018, 6, 15954–15969 CrossRef CAS.
- J. H. Lin, P. C. Wang, H. H. Wang, C. Li, X. Q. Si, J. L. Qi, J. Cao, Z. X. Zhong, W. D. Fei and J. C. Feng, Adv. Sci., 2019, 6, 1900246 CrossRef PubMed.
- L. F. Zhang, J. T. Zhu, Z. Wang and W. J. Zhang, Int. J. Hydrogen Energy, 2020, 45, 19246–19256 CrossRef CAS.
- B. Zhang, F. Yang, X. D. Liu, N. Wu, S. Che and Y. F. Li, Appl. Catal., B, 2021, 298, 120494 CrossRef CAS.
- R. Wu, B. Xiao, Q. Gao, Y. L. Zheng, X. S. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., Int. Ed., 2018, 57, 15445–15449 CrossRef CAS PubMed.
- W. Qiao, B. W. Jin, W. F. Xie, M. F. Shao and M. Wei, J. Energy Chem., 2022, 69, 9–15 CrossRef CAS.
- Y. Zhao, S. D. Wang, N. Li, Q. Zhang, G. X. Li and J. Y. Shen, Catal. Commun., 2019, 124, 67–70 CrossRef CAS.
- R. A. Acedera, A. T. Dumlao, D. D. Matienzo, M. Divinagracia, J. A. Paraggua, P. A. Chuang and J. Ocon, J. Energy Chem., 2024, 89, 646–669 CrossRef.
- H. W. Huang, C. Yu, C. T. Zhao, X. T. Han, J. Yang, Z. B. Liu, S. F. Li, M. D. Zhang and J. S. Qiu, Nano Energy, 2017, 34, 472–480 CrossRef CAS.
- W. G. Xi, G. Yan, Z. L. Lang, Y. Y. Ma, H. Q. Tan, H. T. Zhu, Y. H. Wang and Y. G. Li, Small, 2018, 14, 1802204 CrossRef PubMed.
- C. Wang, P. L. Zhai, M. Y. Xia, W. Liu, J. F. Gao, L. C. Sun and J. G. Hou, Adv. Mater., 2023, 35, 2209307 CrossRef CAS PubMed.
- C. S. Wang, W. B. Chen, D. Yuan, S. S. Qian, D. D. Cai, J. T. Jiang and S. Q. Zhang, Nano Energy, 2020, 69, 104453 CrossRef CAS.
- X. W. Zhong, L. F. Zhang, J. Tang, J. W. Chai, J. C. Xu, L. J. Cao, M. Y. Yang, M. Yang, W. G. Kong, S. J. Wang, H. Cheng, Z. G. Lu, C. Cheng, B. M. Xu and H. Pan, J. Mater. Chem. A, 2017, 5, 17954–17962 RSC.
- J. F. Liu, Z. X. Wang, J. David, J. Llorca, J. S. Li, X. T. Yu, A. Shavel, J. Arbiol, M. Meyns and A. Cabot, J. Mater. Chem. A, 2018, 6, 11453–11462 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00114a |
| ‡ L. F. Zhang and R. L. Wei contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |