
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFinely tuning the self-assembled architectures of liquid crystal polymers by molecular engineering: phase transitions derived from terminal group variations
Wenhuan
Yao
*ab,
Yanxia
Wang
a,
Lansheng
Liu
a,
Anzhi
Ma
a,
Jie
Zhao
a,
Zhengrui
Ma
a,
Lanying
Zhang
*c and
Ruochen
Lan
 *d
*d
aKey Laboratory of Eco-Functional Polymer Materials of the Ministry of Education, Key Laboratory of Eco-Environmental Polymer Materials of Gansu Province, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, P. R. China. E-mail: wenhuanyao@163.com
bCollege of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China
cDepartment of Materials Science and Engineering, College of Engineering, Peking University, Beijing 100871, P. R. China. E-mail: zhanglanying@pku.edu.cn
dInstitute of Advanced Materials & Key Lab of Fluorine and Silicon for Energy Materials and Chemistry of Ministry of Education, College of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang 330022, P. R. China. E-mail: lanruochen@pku.edu.cn
First published on 1st March 2024
Abstract
Liquid crystal polymers (LCPs) have gained tremendous scientific attention due to their potentials in fabrication of intelligent soft robots and responsive devices. Modulating the superstructures by molecular engineering via a bottom-up way and gaining insight into the relationships between molecular structures and self-assembled architectures are of significance in understand the self-organization mechanism of mesogens as well as the fabrication of advanced functional materials, given that the properties and stimuli-responsiveness of LCPs are mainly determined by the self-organized superstructures. In this study, a series of innovative homopolymers featuring distinct terminal groups in the side chains were synthesized via thiol–ene click reactions between poly[3-mercaptopropylmethylsiloxane] (PMMS) and a range of monomers with a nearly identical rigid core but varying terminal substituents (4-methylcyclohexyl, p-methylbenzene, and p-biphenyl groups). The thermal behaviour, phase structure, and molecular packing mode were systemically investigated. Experiment results indicate the formation of a smectic E-smectic A-isotropic phase sequence for most polymers. Intriguingly, an additional loosely stacked hexagonal smectic B phase is observed in P-4, which bears biphenyl mesogenic tails, positioned between smectic E and smectic A phases as the temperature increases. This work presents an effective regulatory strategy for achieving diverse phase transition behaviors and hierarchical constructions through subtle tuning of the molecular structure in the side chains of liquid crystalline polysiloxanes.
1. Introduction
Liquid crystalline polymers (LCPs) have attracted extensive attention for decades due to their advantageous mechanical properties, programmable molecular alignment, mechanical anisotropy and stimuli-responsiveness compared to conventional polymers.1–7 Side chain liquid crystalline polymers (SCLCPs), as remarkable models of soft matters with customizable properties, have attracted considerable research attention in multiple fields including polymer chemistry and physics, materials science and soft matters in the past few decades due to their widely tailorable properties, preparation easiness and processing feasibility.8–11SCLCPs consist of a flexible main chain and pendant rigid mesogens. Through finely tuning the chemical structures of these two parts, abundant phase structures of SCLCPs, including smectic,12,13 nematic,14–16 cholesteric,17,18 and blue phases,18,19 have been achieved via the microphase separation between the flexible backbone and rigid mesogens. In this context, the molecular engineering of SCLCPs includes several robust strategies as diverse as changing backbone flexibility/rigidity, altering the length of flexible spacers,13,20 tuning mesogenic content density,21–23 adjusting the molecular weight,24–26 modulating the topology structure,27 size,28–30 polarity,31 and architecture32–34 of the terminal flexible substituents in a mesogenic core, and the linkage groups35–38 between the rigid cores. Revealing relationships between the molecular structure and the superstructure of self-organized SCLCPs generally provides fundamental impetus to the fabrication of functional materials and intelligent devices.39,40
Polysiloxane SCLCPs are one of the earliest reported LCP materials that outperform other LCPs at low glass transition temperatures. In our previously reported works,41,42 we found that there is mysterious influence of subtle molecular structure changes on the phase structure of polysiloxane SCLCPs. The self-assembled structures of SCLCPs are susceptible to the length of a flexible main chain, spacers between rigid mesogens and the main chain, and substitutes of rigid mesogens. Further investigation is required to understand the detailed influence of molecular structures on the self-organized architecture of SCLCPs. In this work, through replacing the p-metoxybenzene terminal group in the side chains with 4-methylcyclohexyl, p-methylbenzene, and p-biphenyl, a series of novel polysiloxane SCLCPs were designed and synthesized by using the same polymer skeleton PMMS. We anticipate that subtle tuning of the molecular structure in the side chains of these liquid crystalline polysiloxanes will yield diverse phase transition behaviours and hierarchical constructions.
2. Experimental section
2.1 Materials
Poly[3-mercaptopropylmethylsiloxane] (PMMS, SMS-992, M.W. 4000–7000, 95 cst, Gelest Inc), ethylparaben (A.R. grade, Sinopharm Chemical Reagent Co. Ltd), cholesterol (A.R. grade, Sinopharm Chemical Reagent Co. Ltd), acetonitrile (A.R. grade, Sinopharm Chemical Reagent Co. Ltd), p-Cresol (99%, Sinopharm Chemical Reagent Co. Ltd), 4-phenylphenol (98%, Sinopharm Chemical Reagent Co. Ltd), 4-methylcyclohexanol (98%, Beijing Dominant Technology Co.), 3-bromopropene (98%, Beijing Dominant Technology Co.), 4-hydroxybenzonitrile (A.R. grade, Beijing Dominant Technology Co.), dimethylaminopyridine (DMAP) (99%, Energy Chemical), N,N′-dicyclohexylcarbodiimide (DCC) (98%, Energy Chemical), potassium hydroxide (A.R. grade, Beijing Chemical Reagents Co.), and anhydrous potassium carbonate (A.R. grade, Beijing Chemical Reagents Co.) were used without any further purification. Toluene (A.R. grade, Beijing Chemical Reagents Co.) was refluxed over sodium and distilled under a nitrogen atmosphere before use. Azodiisobutyronitrile (AIBN) (A.R. grade, Beijing Dominant Technology Co.) was recrystallized with ethanol before use. All other chemical reagents were used as received.2.2 Synthesis
Scheme 1 illustrates the synthetic approach of the precursors, monomers, and target homopolymers. Precursor 4-(allyloxy) benzoic acid and monomers, namely 4-methoxyphenyl 4-(allyloxy) benzoate (M3) and 4-cyanophenyl 4-(allyloxy) benzoate (M5), were synthesized employing the same method as described in our previously published literature. | ||
| Scheme 1 Synthetic route of the monomers and polymers. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) , and C–H), 1711 (CO), 1649 (CC), 1604, 1579, 1508, 1455 (Ar–), 1274 (C–O–C). 1H-NMR (400 MHz, CDCl3, TMS, δ, ppm): 7.99–7.96 (2H, d, Ar−H), 6.93–6.89 (2H, d, Ar−H), 6.09–5.99 (1H, m, CH2CH–CH2–), 5.45–5.39 (1H, dd, one of CH2CH–CH2–), 5.32–5.29 (1H, dd, one of CH2CH–CH2–), 4.91–4.84 (1H, t, –CH2–CH–O–), 4.59–4.57 (2H, d, CH2CH–CH2–), 2.08–2.04 (2H, m, –O–CH–CH2–), 1.79–1.74 (2H, m, another –O–CH–CH2–), 1.52–1.42 (2H, m, –CH2–CH–CH3), 1.40–1.36 (1H, m, –CH2 –CH–CH3), 1.15–1.08 (2H, m, another –CH2–CH–CH3), 0.93–0.91 (3H, d, –CH–CH3).
, and C–H), 1729 (CO), 1658 (CC), 1603, 1509 (Ar–), 1269 (C–O–C). 1H-NMR (400 MHz, CDCl3, TMS, δ, ppm): 8.17–8.13 (2H, d, Ar–H), 7.23–7.20 (2H, d, Ar–H), 7.09–7.06 (2H, d, Ar–H), 7.01–6.97 (2H, d, Ar–H), 6.11–6.04 (1H, m, CH2CH–CH2–), 5.48–5.42 (1H, dd, one of CH2CH–CH2–), 5.36–5.32 (1H, dd, one of CH2CH–CH2–), 4.64–4.62 (2H, d, CH2CH–CH2–), 2.37 (3H, s, –Ar–CH3).
, and C–H), 1729 (CO), 1647 (CC), 1607, 1578, 1511, 1484 (Ar–), 1259 (C–O–C).1H-NMR (400 MHz, CDCl3, TMS, δ, ppm): 8.19–8.16 (2H, d, Ar–H), 7.65–7.62 (2H, d, Ar–H), 7.61–7.59 (2H, d, Ar–H), 7.48–7.43 (2H, dd, Ar–H), 7.37–7.34 (1H, dd, Ar–H), 7.29–7.26 (2H, d, Ar–H), 7.02–7.00 (2H, d, Ar–H), 6.13–6.03 (1H, m, CH2CH–CH2–), 5.48–5.43 (1H, dd, one of CH2CH–CH2–), 5.36–5.33 (1H, dd, one of CH2CH–CH2–), 4.65–4.63 (2H, d, CH2CH–CH2–).
, and C–H), 1711 (CO), 1649 (CC), 1604, 1579, 1508, 1455 (Ar–), 1274 (C–O–C). 1H-NMR (400 MHz, CDCl3, TMS, δ, ppm): 7.99–7.96 (2H, d, Ar−H), 6.93–6.89 (2H, d, Ar−H), 6.09–5.99 (1H, m, CH2CH–CH2–), 5.45–5.39 (1H, dd, one of CH2CH–CH2–), 5.32–5.29 (1H, dd, one of CH2CH–CH2–), 4.91–4.84 (1H, t, –CH2–CH–O–), 4.59–4.57 (2H, d, CH2CH–CH2–), 2.08–2.04 (2H, m, –O–CH–CH2–), 1.79–1.74 (2H, m, another –O–CH–CH2–), 1.52–1.42 (2H, m, –CH2–CH–CH3), 1.40–1.36 (1H, m, –CH2 –CH–CH3), 1.15–1.08 (2H, m, another –CH2–CH–CH3), 0.93–0.91 (3H, d, –CH–CH3).
, and C–H), 1729 (CO), 1658 (CC), 1603, 1509 (Ar–), 1269 (C–O–C). 1H-NMR (400 MHz, CDCl3, TMS, δ, ppm): 8.17–8.13 (2H, d, Ar–H), 7.23–7.20 (2H, d, Ar–H), 7.09–7.06 (2H, d, Ar–H), 7.01–6.97 (2H, d, Ar–H), 6.11–6.04 (1H, m, CH2CH–CH2–), 5.48–5.42 (1H, dd, one of CH2CH–CH2–), 5.36–5.32 (1H, dd, one of CH2CH–CH2–), 4.64–4.62 (2H, d, CH2CH–CH2–), 2.37 (3H, s, –Ar–CH3).
, and C–H), 1729 (CO), 1647 (CC), 1607, 1578, 1511, 1484 (Ar–), 1259 (C–O–C).1H-NMR (400 MHz, CDCl3, TMS, δ, ppm): 8.19–8.16 (2H, d, Ar–H), 7.65–7.62 (2H, d, Ar–H), 7.61–7.59 (2H, d, Ar–H), 7.48–7.43 (2H, dd, Ar–H), 7.37–7.34 (1H, dd, Ar–H), 7.29–7.26 (2H, d, Ar–H), 7.02–7.00 (2H, d, Ar–H), 6.13–6.03 (1H, m, CH2CH–CH2–), 5.48–5.43 (1H, dd, one of CH2CH–CH2–), 5.36–5.33 (1H, dd, one of CH2CH–CH2–), 4.65–4.63 (2H, d, CH2CH–CH2–).
3. Results and discussion
3.1 Synthesis and characterization of monomers and polymers
The precursors and monomers were efficiently and successfully synthesized, beginning with the Williams etherification reaction and concluding with the Steglich esterification reaction, as shown in Scheme 1. All homopolymers were obtained with yields exceeding 86% through the thiol–ene click reaction, employing AIBN as the catalyst. The confirmation of the successful synthesis of precursors, monomers and target homopolymers were achieved through 1H-NMR spectroscopy.As an illustrative example, Fig. 1 exhibits the 1H-NMR spectra of monomer M2, PMMS, and corresponding homopolymer P-2 after the thiol–ene click reaction. The characteristic signals of the vinyl proton at 6.08, 5.45, 5.34 ppm from M2, as well as the mercapto group resonance at 1.37 ppm belonging to PMMS, were completely absent in the spectrum of P-2. Instead, there was a simultaneous appearance of proton peaks corresponding to aromatic rings at 8.15, 7.22, 7.07 and 6.99 ppm, along with the proton peaks of silicomethyl at 0.11 ppm. This observation confirms the success of the reaction and high purity of homopolymer P-2.
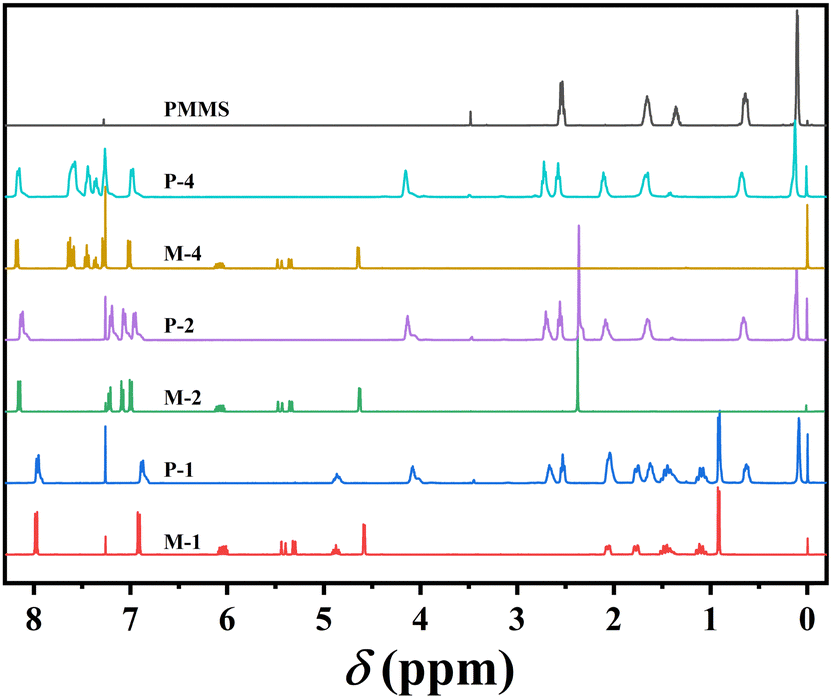 | ||
| Fig. 1 1H-NMR spectra of monomers, PMMS, and polymers in CDCl3. | ||
As listed in Table 1 and Fig. 2a, the trend in the number-average molecular weight (![[M with combining overline]](https://www.rsc.org/images/entities/i_char_004d_0305.gif) n) of these homopolymers, derived from both GPC measurements and calculations based on 1H-NMR results, consistently followed variations in the formula weight of the monomers. Additionally, the broad molecular weight distribution of these homopolymers was inherited from the polymeric starting material PMMS.
n) of these homopolymers, derived from both GPC measurements and calculations based on 1H-NMR results, consistently followed variations in the formula weight of the monomers. Additionally, the broad molecular weight distribution of these homopolymers was inherited from the polymeric starting material PMMS.
| Sample | T g (°C) | T i (°C) |
n
(g mol−1) |
PDIb | T d (°C) | ΔTe (°C) | Yield (%) | |
|---|---|---|---|---|---|---|---|---|
| GPCb | Calculatedc | |||||||
| a Evaluated by DSC during the second heating at a rate of 10 °C min−1. b Determined by GPC in THF using polystyrene standards. c According to the 1H-NMR results. d 5% Weight loss temperature was evaluated by TGA at a rate of 20 °C min−1. e Mesogenic temperature ranges determined by Ti–Tg. f The data of P-3 and P-5 referenced as 41 and 42, respectively, and listed here for comparison only.41,42 | ||||||||
| PMMS | — | — | 800 | 6192 | 2.53 | — | — | — |
| P-1 | −4.5 | — | 1900 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 522 522 |
3.31 | 365 | — | 87.1 |
| P-2 | 5.3 | 85.3 | 2000 | 18252 |
2.07 | 368 | 80.0 | 89.5 |
| P-3f | 17.1 | 95.5 | 2400 | 18972 |
1.45 | 362 | 78.4 | 89.7 |
| P-4 | 21.1 | 133.2 | 2400 | 21042 |
1.67 | 354 | 112.1 | 91.6 |
| P-5f | 2.4 | 92.1 | 2200 | 18747 |
2.84 | 349 | 89.7 | 86.2 |
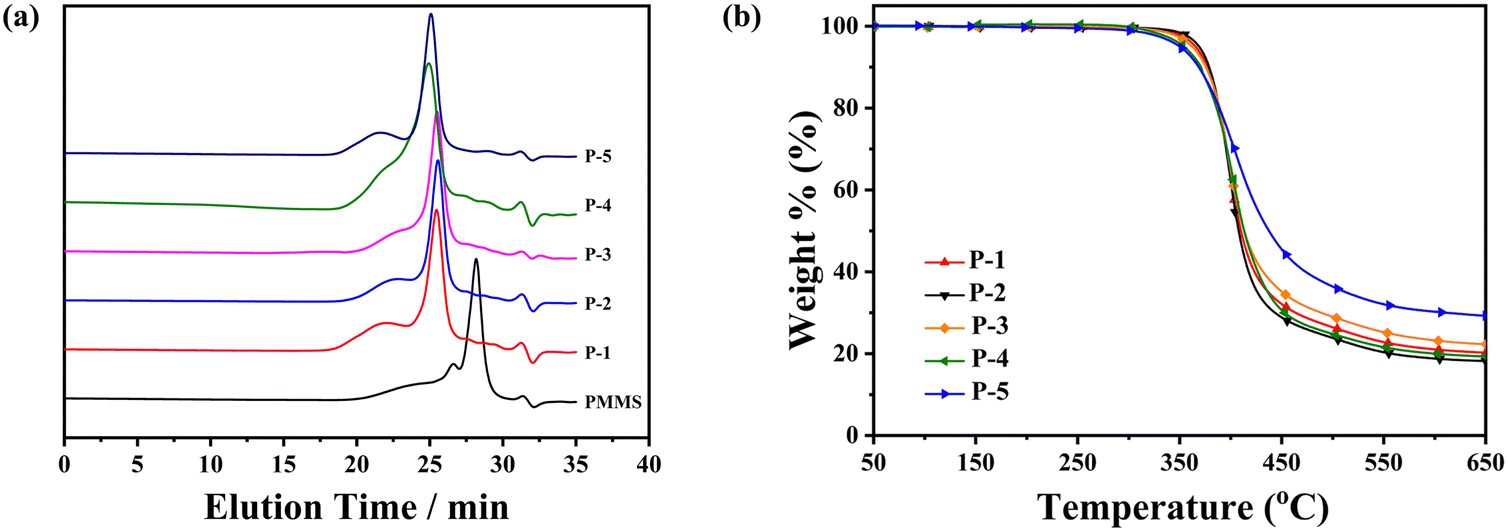 | ||
| Fig. 2 (a) GPC spectra of all the polymers and PMMS and (b) TGA traces of polymers in nitrogen at a rate of 20 °C min−1. | ||
3.2 Thermal and liquid-crystalline properties of homopolymers
The thermal and liquid-crystalline properties of these polymers were investigated through differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and polarized optical microscopy (POM) analyses. As depicted in Table 1 and Fig. 2b, the 5% weight loss temperature of all the homopolymers, assessed from TGA curves under a nitrogen atmosphere, exceeded 349 °C, affirming the excellent thermal stabilities of the polymers.The phase transition behaviours of these homopolymers were investigated through DSC experiments, as shown in Fig. 3. To eliminate the influence of thermal history, the traces during the first cooling and subsequent second heating were recorded at a rate of 10 °C min−1 under a nitrogen atmosphere. Different polymers exhibited diverse liquid-crystalline phases, evident from their distinctive transitions. Homopolymers P-2 and P-4 displayed three or four exothermic/endothermic peaks through their cooling/heating process, while homopolymer P-1 exhibited only one peak. This suggests the potential existence of two or three mesophase structures in the entire mesogenic temperature ranges for samples P-2 and P-4, in contrast with the absence in sample P-1. As anticipated, the glassy transition temperature (Tg) and the clearing point temperature (Ti) of these homopolymers gradually increased, influenced by the rigidity changes in the side chains. Attributing to the collaboration effect of the flexible polysiloxane backbone PMMS and the less rigid side chains, Tg of these homopolymers remained below 22 °C.
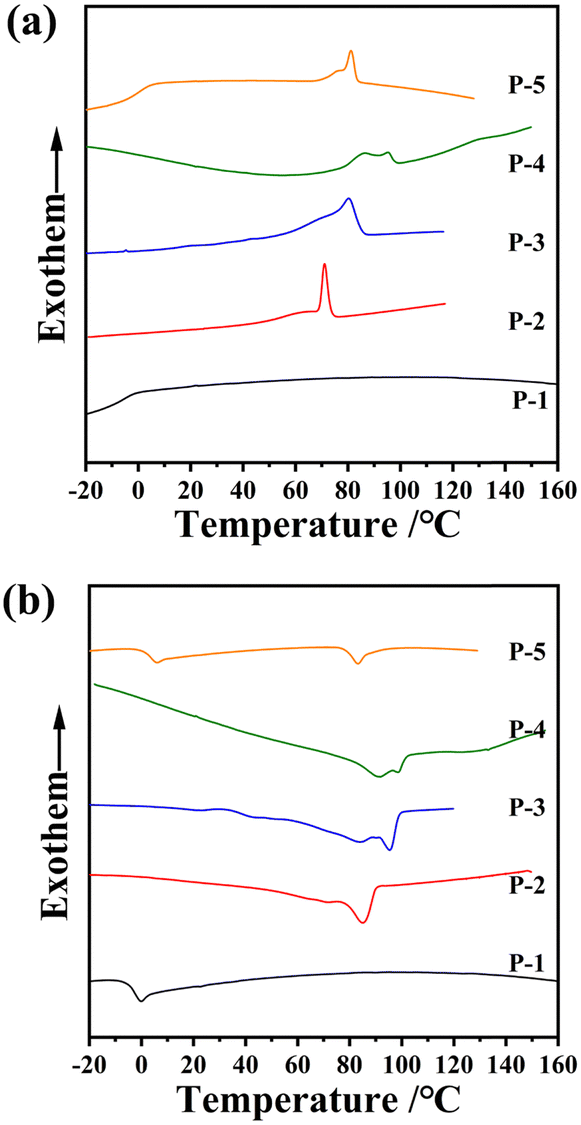 | ||
| Fig. 3 DSC thermograms of the homopolymers during (a) the first cooling and (b) subsequent second heating scans at a rate of 10 °C min−1. | ||
A POM instrument was used to investigate the liquid crystalline birefringence of all powder samples by cooling them from the isotropic state to room temperature at a rate of 1 °C min−1. Some representative POM textures are depicted in Fig. 4. Apparent birefringence was observed for samples P-2 and P-4 during cooling, occurring at 69 °C and 127 °C, respectively. In contrast, sample P-1 exhibited no birefringence throughout the cooling and subsequent heating procedures, indicating its inability to self-assemble into the liquid crystalline phase.
 | ||
| Fig. 4 Representative textures of samples P-4 (a)–(c) and P-2 at (d) and (e) different temperatures cooled from the isotropic state. | ||
For sample P-4, as illustrated in Fig. 4a–c, a sand-shape texture developed as the sample cooled from the isotropic state to 127 °C. Interestingly, upon slow cooling to 125 °C and maintaining that temperature for a while, a fan-shaped texture characteristic of a smectic liquid crystalline phase emerged. This texture remained almost unchanged upon further cooling to room temperature, even with two phase transitions observed in the DSC experiment, except for slight colour changes. This indicates that sample P-4 might possess a series of sub-smectic phases throughout the entire mesomorphic state.
However, homopolymer P-2 exhibited distinct behaviour. As depicted in Fig. 4d and e, a gritty texture was observed as the sample cooled from the isotropic state to 69 °C, and this texture showed no significant difference upon further cooling, even in the presence of a phase transition observed in the DSC experiment.
In comparison to the previously reported homopolymer P-3, which formed a smectic A phase at high temperatures and a smectic E phase at low temperatures (referenced to as PMMS-XChol-0.00 in ref. 41), the only structural disparities among P-2, P-3, and P-4 reside in the termination of their side chains by a methyl group, methoxy group, and phenyl group, respectively. These minor modifications, distinct from the already reported homopolymer P-5, which possessed a smectic A phase throughout the entire mesogenic temperature range due to the dominant polarity interaction arising from the terminal group substitution of the methoxy group with a polar cyano group in the side chains, only result in subtle alterations in molecular rigidity or π–π interaction. Therefore, we boldly speculation that the phase transition behaviour of samples P-2 and P-4 might be similar to that of sample P-3.
3.3 Phase structure identification of homopolymers P-2 and P-4
Given the limited information for phase structure identification based on DSC and POM results, variable-temperature one-dimensional wide-angle X-ray diffraction (1D-WAXD) and small angle X-ray scattering (SAXS) experiments were conducted to discern the phase structures of polymers P-2 and P-4. The structural insights obtained from these experiments offer valuable information regarding self-assembly behaviours influenced by minor molecular structure alterations.Excitingly, in line with our conjecture, the variations in the diffraction patterns of homopolymers P-2 and P-4 in this work appeared similar to the homopolymer P-3, where the side chains were terminated by a methoxy group. The SAXS and 1D-WAXD performance of sample P-2 is presented in Fig. 5a and b during the heating process, respectively. Being consistent with the DSC experiment results, the two phase transition process occurred over the whole temperature range. As shown in Fig. 5a and b, five diffraction peaks with q (q = 4πsinθ/λ) values of 2.46, 4.93, 7.38, 9.87, and 12.32 nm−1 (corresponding to d-spacings of 2.55, 1.27, 0.85, 0.64, and 0.51 nm, respectively) were observed in the low-angle region at low temperatures, with the ratio of scattering vectors q being approximately 1:2:3:4:5, demonstrating a layer packing of an ordered phase. Additionally, four diffraction peaks with q values of 14.27, 14.51, 15.55, and 19.95 nm−1 were observed in the high-angle region at low temperatures (Fig. 5b and c), indicating the existence of molecular packing on the sub-nanometer scale. Through a comprehensive analysis of SAXS and 1D-WAXD results, it is speculated that sample P-2 may develop a lamellar phase with highly ordered molecular packing in the inner layer at low temperatures. Upon heating, the intensity of the diffraction peaks in the high-angle region decreased significantly, the peak at a q value of 14.27 nm−1 became diffused, and the peaks at q values of 14.51, 15.55, and 19.95 nm−1 vanished after heating to 80 °C. Only one sharp diffraction peak with a q value of 2.41 nm−1 remained in the low-angle region, suggesting the loss of molecular packing on the sub-nanometer scale and the emergence of a new phase – a long-range ordered smectic phase (SmA or SmC). Upon further heating to 100 °C, the diffraction peak at the low-angle region disappeared, accompanied by the scattering amorphous broad halo at the high-angle region becoming more diffused and the centre position shifting to a lower angle, indicating the sample entered the isotropic state. The phase transition temperatures (between 70 and 80 °C) and the isotropic temperature (between 80 and 100 °C) for P-2 determined from the SAXS and 1D-WAXD results were consistent with the DSC results. To sum up, considering the similarity of the diffraction peaks at the high-angle region of sample P-2 to that of polymer P-3 (referenced to as PMMS-XChol-0.00 in ref. 41), we concluded that the self-assembly structure of P-2 at low temperatures could be smectic E (SmE), evidenced by the five diffraction peaks in the low-angle region assigned as (001), (002), (003), (004), and (005), as well as the four diffraction peaks in the high-angle region assigned as (110), (111), (200), and (210) diffractions of SmE with a and b values of 0.81 and 0.52 nm,41,43–46 respectively. Furthermore, a simple smectic phase structure of smectic A (SmA) or smectic C (SmC) could be confirmed for P-2 at higher temperatures, as indicated by the diffraction peak at 2.41 nm−1 in the low-angle region and the diffused high-angle amorphous broad halo.
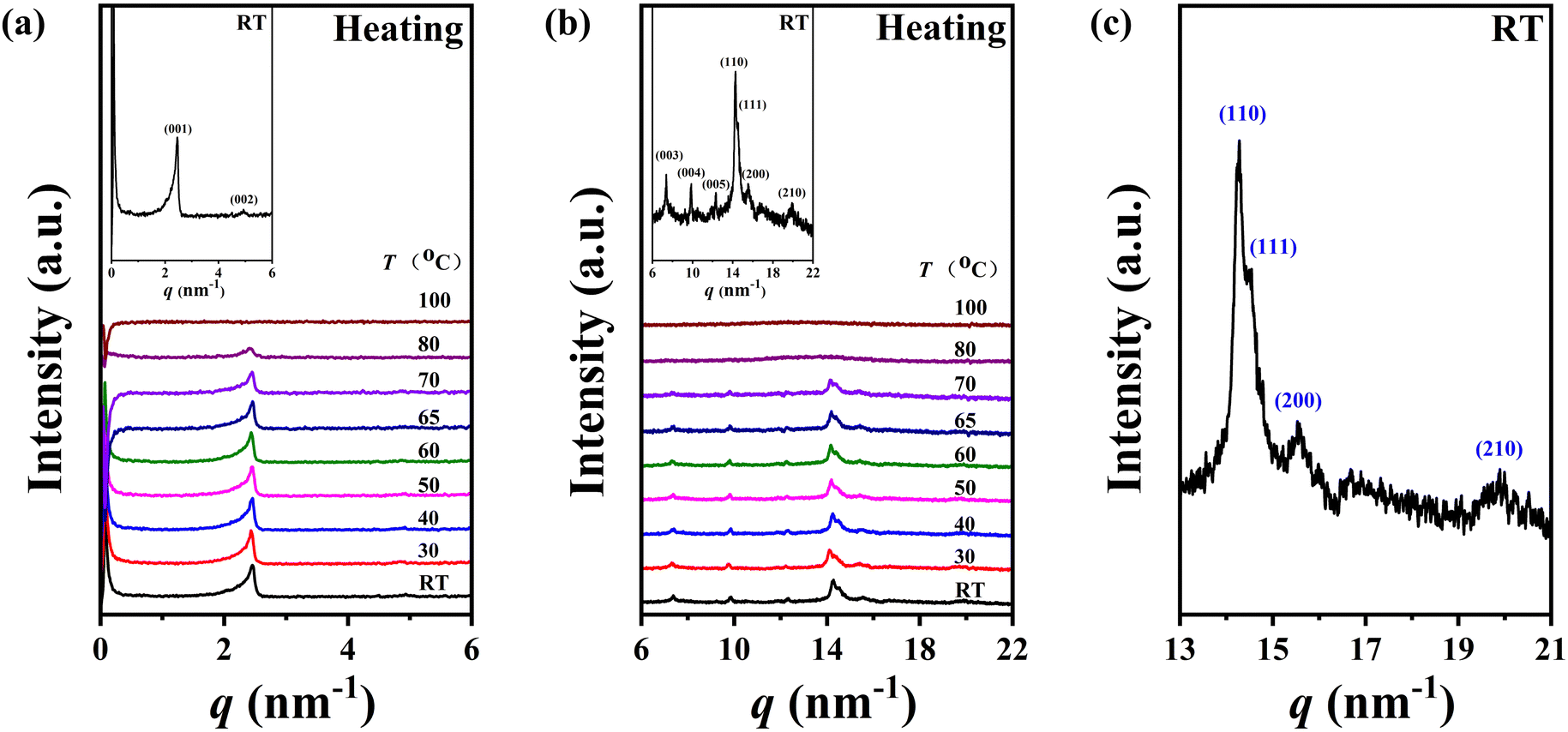 | ||
| Fig. 5 SAXS (the inset shows the enlarged patterns in the low-angle region) (a) and 1D-WAXD (the inset shows the enlarged patterns in the high-angle region) (b) patterns during the heating process, (c) and the enlarged patterns in the high-angle region of 1D-WAXD at room temperature of P-2. | ||
The self-assembly behaviour of homopolymer P-4 exhibited slight differences compared to that of P-2. As shown in Fig. 6a and b, a single sharp diffraction peak with a d-spacing of 2.90 nm in the low-angle region and four diffraction peaks with q values of 14.36, 14.51, 15.53, and 19.97 nm−1 in the high-angle region, assigned as (110), (111), (200), and (210), indicated a characteristic SmE with a and b values of 0.81 and 0.52 nm, below 85 °C (see the inset of Fig. 5b). Upon continuously heating to 93 °C, the intensity of these diffraction peaks significantly decreased, and the peak at a q value of 14.36 nm−1 weakened, shifting to a lower angle (14.16 nm−1, corresponding to a d-spacing of 0.44 nm), while the peaks at q values of 14.51, 15.53, and 19.97 nm−1 vanished. This suggested a reduction in the order of the molecular packing on the sub-nanometer scale, and the development of a new phase (Fig. 6b). After reaching 105 °C, the diffraction peak in the high-angle region transformed into an amorphous broad halo with a blue shift in the centre position, and the intensity of the low-angle region diffraction peak weakened while moving to 2.11 nm−1(corresponding to a d-spacing of 2.97 nm), indicating the loss of molecular packing on the sub-nanometer scale and the formation of a simple smectic phase structure of SmA or SmC. Upon further heating to 145 °C, with the disappearance of the diffraction peak in the low-angle region and the more diffused scatter halo in the high-angle region, sample P-4 entered the isotropic state. For a detailed investigation of the phase transition behaviour of P-4, the values of d-spacing and full width at half-height (FWHH) of the diffraction peak at a q value of 14.36 nm−1 were plotted as a function of temperature variation in Fig. 7. Obviously, the values of d-spacing and FWHM exhibited three jumps between 85 and 93, 96 and 105, 135 and 145 °C, respectively, proving three phase transitions. These findings were in good agreement with the DSC results. In summary, integrating the observed representative fan-shaped texture under POM, and the results from DSC, 1D-WAXD,and SAXS, the self-assembly behaviour of homopolymer P-4 with increasing temperature could be summarized as follows: a SmE phase structure at a low temperature, a smectic phase structure with lower ordered molecular packing on the sub-nanometer scale at a medium temperature, a simple smectic phase structure of SmA or SmC at a high temperature, and the isotropic state at the highest temperature.
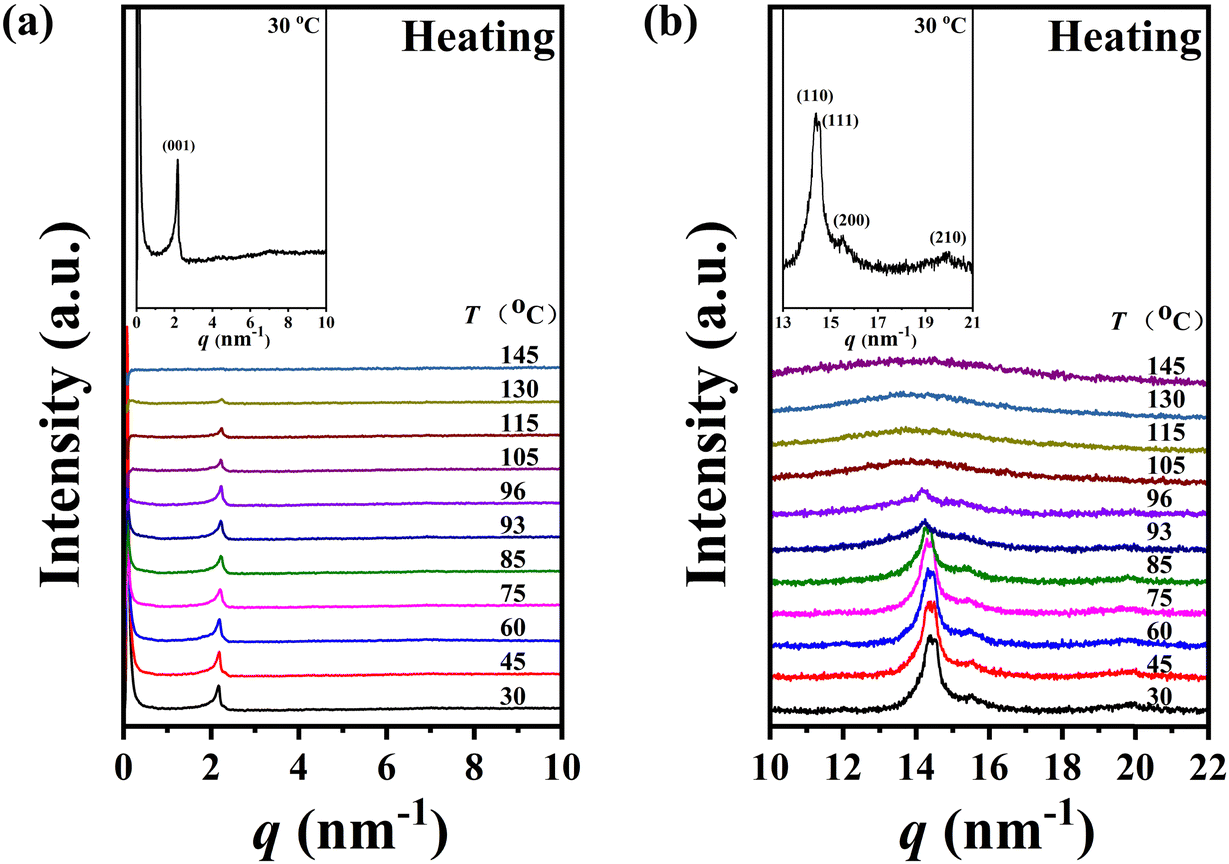 | ||
| Fig. 6 SAXS (the inset shows the enlarged patterns in the low-angle region) (a) and 1D-WAXD (the inset shows the enlarged patterns in the high-angle region) (b) patterns of P-4 during the heating process. | ||
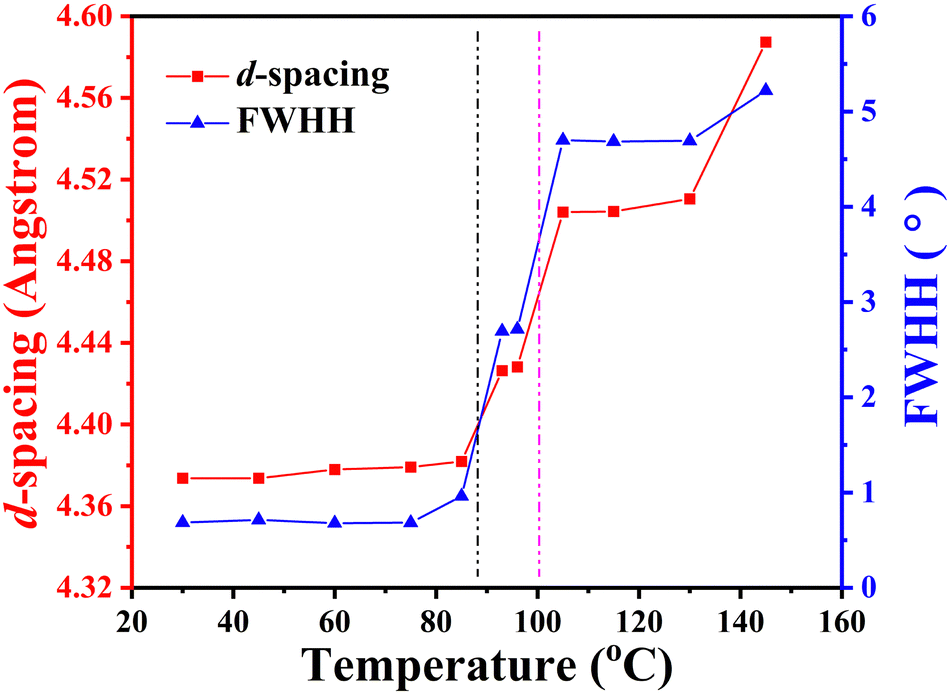 | ||
| Fig. 7 d-spacing and FWHH of the high angle halo as a function of temperature. Below 93 °C, the data correspond to the (110) diffraction of the SmE phase. | ||
The two-dimensional wide-angle X-ray diffraction (2D-WAXD) pattern was intended to complement the lack of dimensionality in the 1D-WAXD and SAXS patterns for a more comprehensive confirmation of the smectic structures of homopolymers P-2 and P-4. Unfortunately, the unexpected brittleness and the continuous phase transition during the cooling process prevented the successful formation of sheared films at low temperatures. Despite attempting various methods, conducting 2D-WAXD experiments on the samples proved challenging.
The calculated lengths of the side chains from the Si atom in the backbone to the last C with the alkyl chains in the all-trans conformation were 2.23 and 2.50 nm for P-2 (Lsc2) and P-4 (Lsc4), respectively. In comparison with the measured smectic layer period (c) of d(001) = 2.55 nm and d(001) = 2.90 nm for P-2 and P-4, where the SmE orthorhombic structure was formed at low temperatures, the measured c values are only 0.32 and 0.40 nm larger, respectively. Consequently, to ensure the parallel closing packing of the mesogen units, the side chains from the two adjacent backbone in the smectic structure are significantly interdigitated. For homopolymer P-4, when the sample formed the lamellar structure with lower ordered molecular stacking on the sub-nanometer scale at medium temperatures, the high-angle diffraction corresponding to a d-spacing of 0.44 nm indicated the separation of every three backbone silicon atoms, to which the mesogens extending on the same side are anchored. This separation is estimated to be 0.52 nm at the maximum under the trans conformation assumption. In other words, the distance between the neighboring mesogen stems is 0.52 nm. Obviously, the value of 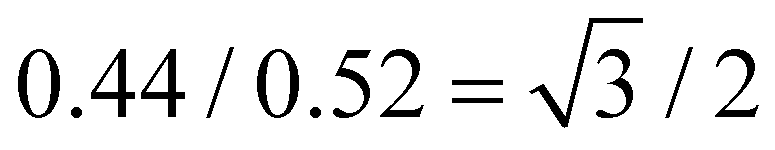 , indicating the existence of hexagonal columnar symmetry between the mesogen units in the side chains within a layer. Consequently, the lamellar structure with lower ordered molecular stacking on the sub-nanometer scale at the medium temperature of P-4 can be identified as a high-ordered smectic B phase.46–48 In essence, upon heating, a loosely staked SmB hexagonal structure emerges from the SmE phase with the orthorhombic structure in homopolymer P-4.
, indicating the existence of hexagonal columnar symmetry between the mesogen units in the side chains within a layer. Consequently, the lamellar structure with lower ordered molecular stacking on the sub-nanometer scale at the medium temperature of P-4 can be identified as a high-ordered smectic B phase.46–48 In essence, upon heating, a loosely staked SmB hexagonal structure emerges from the SmE phase with the orthorhombic structure in homopolymer P-4.
For homopolymer P-3, referenced as PMMS-XChol-0.00 in ref. 41 and exhibiting a liquid crystalline transition sequence of G ↔ SmE ↔SmA ↔ Iso, the orthorhombic molecular stacking structure within a layer degenerated upon heating. Despite this change, the perpendicular relationship between the side chains and the main-chain remained unchanged. Consequently, due to the minor differences in the molecular structure of the mesogen units of P-2 and P-4 compared to that of P-3, as well as the highly similar evolution in the 1D-WAXD and SAX patterns of these samples at high temperatures, the smectic phase structure at high temperatures for samples P-2 and P-4 should also be identified as a SmA phase.
Based on the aforementioned results, we can conclude that the phase transition sequence for sample P-2 is G ↔ SmE ↔ SmA ↔ Iso, while for sample P-4 it is G ↔ SmE ↔ SmB ↔ SmA ↔ Iso. A schematic representation is shown in Fig. 8 which exhibits three self-assembly structures of homopolymer P-4 as it undergoes heating.
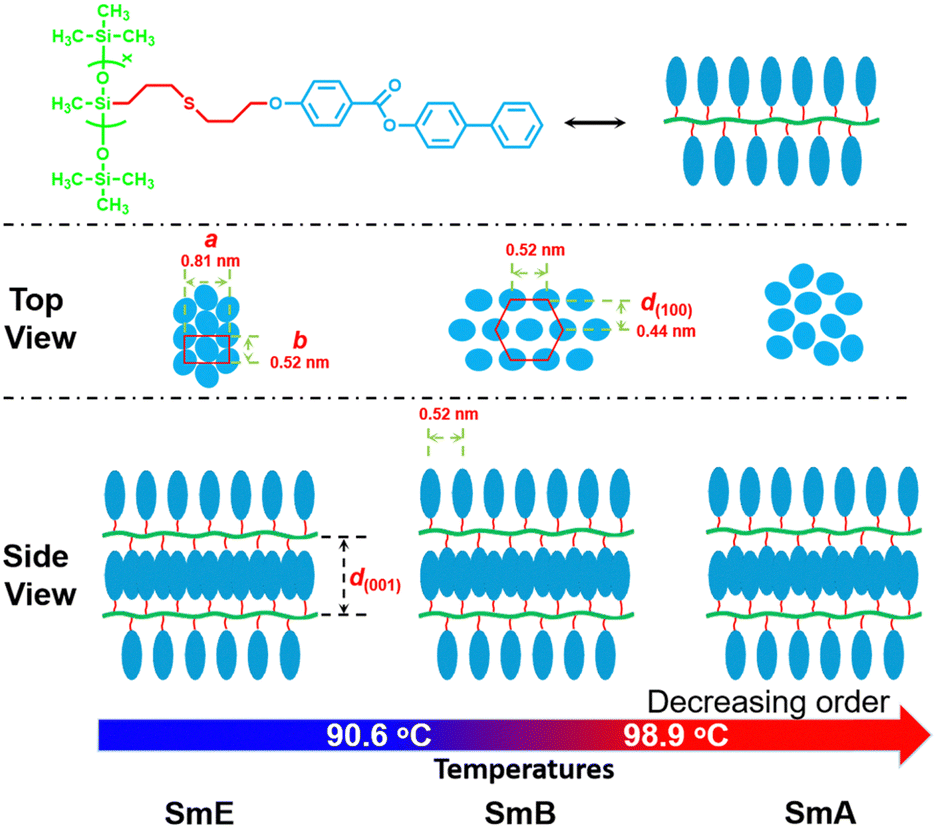 | ||
| Fig. 8 Schematic drawing of the proposed model for the self-assembly of homopolymer P-4 upon heating. | ||
3.4 Dependence of the phase structure on the five terminal groups of the LC building blocks
The phase transitions of all the homopolymers are summarized in Table 2, including the corresponding d-spacing values of the low angle diffraction peaks. Utilizing a semiflexible PMMS backbone in this study, we inferred that the layer spacing is primarily influenced by the side chain length, resembling the calculated molecular length when fully extended and closely packed. This inference aligns well with the experimental confirmation. For sample P-1, where the LC building blocks are terminated by 4-methylcyclohexanol, the insufficient rigidity (corresponding to a weak π–π interaction) of the side chains prevents the polymer from forming a liquid crystalline phase. However, with increased rigidity achieved by replacing the cyclohexanol structure with an aromatic ring (corresponding to an increase in π–π interaction), sample P-2 exhibits a clear LC transition sequence of G ↔ SmE ↔ SmA ↔ Iso. The alteration of the terminal group from methyl (P-2) to methoxy (P-3) does not induce changes in the phase transitions but rather weakens the intensity of the high-angle diffraction peaks and eliminates some detailed patterns like (111) diffraction. This could be attributed to the electron-absorbing effect of methoxy, which results in a reduction of π–π interaction. Remarkably, introducing an additional highly ordered SmB phase between the SmE and SmA phases becomes evident when changing the terminal group from methoxy (P-3) to aromatic (P-4, corresponding to a stronger π–π interaction). Amazingly, substituting the terminal aromatic group with cyano, possessing greater polar, results in only the SmA phase occupying the entire mesogenic temperature range due to the decisive role of polarity interaction.42 Thus, we can conclude that tiny variations of the molecular structure lead to significant self-assembly construction evolution. This finding unveils the delicate role of molecular details in guiding hierarchical structures.| Sample | d (001) (nm) | Phase transitionsb (°C) | Lattice parametera (nm) | c (nm) |
|---|---|---|---|---|
| Heating | ||||
| Cooling | ||||
| a Calculated at room temperature for all the polymers and according to the XRD results. b According to the DSC results, G: glass state, SmAs: the smectic A phase with a single layer, SmB: the smectic B phase, and Iso: isotropic state. c Minimized layer spacing of the periodic arrangement. d The data of P-3 and P-5 referenced as 41 and 42, respectively, and listed here for comparison only.41,42 | ||||
| P-1 | — | — | — | — |
| P-2 | 2.55 | G5.33SmE70.5SmAs85.3 Iso | a = 0.81, b = 0.52 | 2.55 |
| Iso71.1SmAs61.1SmE3.8G | ||||
| P-3 d | 2.62 | G17.1SmE83.4SmAs95.5I | a = 0.81, b = 0.52 | 2.62 |
| I80.5SmAs68.6SmE15.2G | ||||
| P-4 | 2.90 | G21.1SmE90.6SmB98.9SmAs133.3Iso | a = 0.81, b = 0.52 | 2.90 |
| Iso129.6SmAs95.5SmB85.8SmE19.1G | ||||
| P-5d | 3.20 | G2.4SmAs92.1Iso | — | 3.20 |
| Iso81.3SmAs-1.4G | ||||
4. Conclusions
In summary, the synergistic interaction between the flexible polysiloxane backbone and rigid mesogenic side chains facilitated the successful design and synthesis of a series of smectic homopolymers characterized by high thermal stability. Systematic investigations revealed that minor variations in the molecular structure, achieved by altering the terminal groups of the mesogenic side chains, led to distinct self-assembly behaviours and phase transitions. When the terminal substituents of the side chain mesogens were methyl or methoxyl groups, a typical smectic E to smectic A phase transition order was observed in response to temperature changes, in contrast to phenyl group substitution. Intriguingly, the introduction of a phenyl group as the terminal substituent resulted in an additional high-ordered smectic B phase between the smectic E and smectic A phases with the increasing temperature. This study underscores the significance of the rational molecular design, specifically the modulation of terminal groups in building blocks, as an efficient approach for achieving diverse phase transition behaviours and adjusting phase structures. Ongoing research in our group aims to further regulate the self-assembly nanostructures through hierarchical architecture construction, utilizing these homopolymers as fundamental building blocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation (Grant No. 52263033), the China Postdoctoral Science Foundation Funded Project (Grant No. 2021M690954), the Natural Science Foundation of Gansu Province (Grant No. 20JR10RA105), the Natural Science Foundation of Hunan Province (Grant No. 2022JJ30152), and the graduate research funding project of Northwest Normal University (Grant No. 2023KYZZ-S157) are gratefully acknowledged.Notes and references
- T. J. White and D. J. Broer, Nat. Mater., 2015, 14, 1087–1098 CrossRef CAS PubMed.
- M. Pilz da Cunha, M. G. Debije and A. P. H. J. Schenning, Chem. Soc. Rev., 2020, 49, 6568–6578 RSC.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2022, 122, 4887–4926 CrossRef CAS PubMed.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 34 Search PubMed.
- J. Liu, Z.-P. Song, L.-Y. Sun, B.-X. Li, Y.-Q. Lu and Q. Li, Responsive Mater., 2023, 1, e20230005 CrossRef.
- W. Kang, Y. Tang, X. Meng, S. Lin, X. Zhang, J. Guo and Q. Li, Angew. Chem., Int. Ed., 2023, 62, e202311486 CrossRef CAS.
- H. Wang, Y. Tang, H. Krishna Bisoyi and Q. Li, Angew. Chem., Int. Ed., 2023, 62, e202216600 CrossRef CAS PubMed.
- V. P. Shibaev and A. Y. Bobrovsky, Russ. Chem. Rev., 2017, 86, 1024–1072 CrossRef.
- V. P. Shibaev, Polym. Sci., Ser. A, 2014, 56, 727–762 CrossRef CAS.
- C. S. Hsu, Prog. Polym. Sci., 1997, 22, 829–871 CrossRef CAS.
- G. Siva Mohan Reddy, J. Jayaramudu, S. S. Ray, K. Varaprasad and E. Rotimi Sadiku, in Liquid Crystalline Polymers: Volume 2–Processing and Applications, ed. V. K. Thakur and M. R. Kessler, Springer International Publishing, Cham, 2015, pp. 389–415 DOI:10.1007/978-3-319-20270-9_16.
- Y. D. Chen, P. Lu, Z. Y. Li, Y. J. Yuan and H. L. Zhang, Polym. Chem., 2021, 12, 2572–2579 RSC.
- D. Shi, W. Y. Chang, X. K. Ren, S. Yang and E. Q. Chen, Polym. Chem., 2020, 11, 4749–4759 RSC.
- S. M. Alauddin, A. R. Ibrahim, N. F. K. Aripin, T. S. Velayutham, O. K. Abou-Zied and A. Martinez-Felipe, Eur. Polym. J., 2021, 146, 110246 CrossRef.
- X. L. Zheng, Y. J. Zhan, Y. C. Liu, M. P. Lu, E. X. Jiao, H. Z. Zhang, J. Shi, M. G. Lu and K. Wu, Polym. Chem., 2022, 13, 3915–3929 RSC.
- H. Yang, M. X. Liu, Y. W. Yao, P. Y. Tao, B. P. Lin, P. Keller, X. Q. Zhang, Y. Sun and L. X. Guo, Macromolecules, 2013, 46, 3406–3416 CrossRef CAS.
- S. Gao, Z. P. Liu, Y. H. Cong, X. Z. He, B. Y. Zhang, F. B. Meng and Y. G. Jia, Dyes Pigm., 2020, 181 Search PubMed.
- Z. H. Cheng, H. Cao, D. Y. Zhao, W. Hu, W. L. He, X. T. Yuan, J. M. Xiao, H. Q. Zhang and H. Yang, Liq. Cryst., 2011, 38, 9–15 CrossRef CAS.
- J. M. Gilli, M. Kamaye and P. Sixou, J. Phys., 1989, 50, 2911–2918 CrossRef CAS.
- E. Mehravar, A. Iturrospe, A. Arbe, J. M. Asua and J. R. Leiza, Polym. Chem., 2016, 7, 4736–4750 RSC.
- K. Mukai, M. Hara, S. Nagano and T. Seki, Angew. Chem., Int. Ed., 2016, 55, 14028–14032 CrossRef CAS PubMed.
- W. Y. Chang, D. Shi, X. Q. Jiang, J. D. Jiang, Y. Zhao, X. K. Ren, S. Yang and E. Q. Chen, Polym. Chem., 2020, 11, 1454–1461 RSC.
- L. Han, H. W. Ma, Y. Li, S. Q. Zhu, L. C. Yang, R. Tan, P. B. Liu, H. Y. Shen, W. Huang and X. C. Gong, Macromolecules, 2016, 49, 5350–5365 CrossRef CAS.
- T. Michinobu, N. Fujii, M. Tokita, J. Watanabe and K. Shigehara, Macromol. Rapid Commun., 2008, 29, 1593–1597 CrossRef CAS.
- S. Chen, H. b Luo, H. L. Xie and H. L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 924–935 CrossRef CAS.
- C. Ye, H. L. Zhang, Y. Huang, E. Q. Chen, Y. L. Lu, D. Y. Shen, X. H. Wan, Z. H. Shen, S. Z. D. Cheng and Q. F. Zhou, Macromolecules, 2004, 37, 7188–7196 CrossRef CAS.
- D. Y. Kim, D. G. Kang, S. Shin, T. L. Choi and K. U. Jeong, Polym. Chem., 2016, 7, 5304–5311 RSC.
- Y. F. Zhu, Z. Y. Zhang, Q. K. Zhang, P. P. Hou, D. Z. Hao, Y. Y. Qiao, Z. H. Shen, X. H. Fan and Q. F. Zhou, Macromolecules, 2014, 47, 2803–2810 CrossRef CAS.
- Y. H. Cheng, W. P. Chen, Z. H. Shen, X. H. Fan, M. F. Zhu and Q. F. Zhou, Macromolecules, 2011, 44, 1429–1437 CrossRef CAS.
- S. Chen, L. Y. Zhang, L. C. Gao, X. F. Chen, X. H. Fan, Z. H. Shen and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 505–514 CrossRef CAS.
- S. Q. Dai, J. X. Li, J. C. Kougo, H. Y. Lei, S. Aya and M. J. Huang, Macromolecules, 2021, 54, 6045–6051 CrossRef CAS.
- X. Mei, J. Zhang, Z. H. Shen and X. H. Wan, Polym. Chem., 2012, 3, 2857–2866 RSC.
- X. Mei, Y. Chu, J. Cui, Z. Shen and X. Wan, Macromolecules, 2010, 43, 8942–8949 CrossRef CAS.
- Y. Guan, X. Chen, Z. Shen, X. Wan and Q. Zhou, Polymer, 2009, 50, 936–944 CrossRef CAS.
- Q. F. Zhou, X. L. Zhu and Z. Q. Wen, Macromolecules, 1989, 22, 491–493 CrossRef CAS.
- D. Zhang, Y. Liu, X. Wan and Q. F. Zhou, Macromolecules, 1999, 32, 4494–4496 CrossRef CAS.
- Z. G. Zhu, J. G. Zhi, A. H. Liu, J. X. Cui, H. Tang, W. Q. Qiao, X. H. Wan and Q. F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 830–847 CrossRef CAS.
- D. Zhang, Y. X. Liu, X. H. Wan and Q. F. Zhou, Macromolecules, 1999, 32, 5183–5185 CrossRef CAS.
- Y. Zhang, Y. Xue, L. Gao, R. Liao, F. Wang and F. Wang, Chin. Chem. Lett., 2023, 109217 CrossRef.
- Y. Xue, S. Jiang, H. Zhong, Z. Chen and F. Wang, Angew. Chem., Int. Ed., 2022, 61, e202110766 CrossRef CAS.
- W. H. Yao, Y. Z. Gao, X. Yuan, B. F. He, H. F. Yu, L. Y. Zhang, Z. H. Shen, W. L. He, Z. Yang, H. Yang and D. K. Yang, J. Mater. Chem. C, 2016, 4, 1425–1440 RSC.
- W. H. Yao, Y. Z. Gao, C. H. Zhang, C. Y. Li, F. S. Li, Z. Yang and L. Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1765–1772 CrossRef CAS.
- T. Miyazawa, Y. Yamamura, M. Hishida, S. Nagatomo, M. Massalska-Arodz and K. Saito, J. Phys. Chem. B, 2013, 117, 8293–8299 CrossRef CAS PubMed.
- H. L. Xie, C. K. Jie, Z. Q. Yu, X. B. Liu, H. L. Zhang, Z. Shen, E. Q. Chen and Q. F. Zhou, J. Am. Chem. Soc., 2010, 132, 8071–8080 CrossRef CAS PubMed.
- N. Hida, T. Nakajima, M. Hara, T. Seki and S. Nagano, Macromol. Rapid Commun., 2023, 44, 2200761 CrossRef CAS PubMed.
- R. Imanishi, Y. Nagashima, K. Takishima, M. Hara, S. Nagano and T. Seki, Macromolecules, 2020, 53, 1942–1949 CrossRef CAS.
- J. M. Seddon, Handbook of Liquid Crystals Set, 1998, pp. 635–679 DOI:10.1002/9783527619276.ch8ca.
- H. Kuroda, H. Goto, K. Akagi and A. Kawaguchi, Macromolecules, 2002, 35, 1307–1313 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |