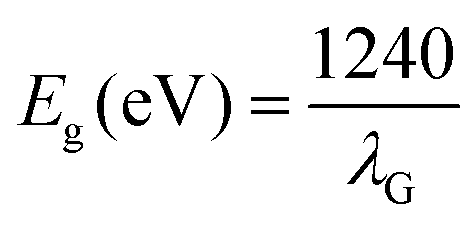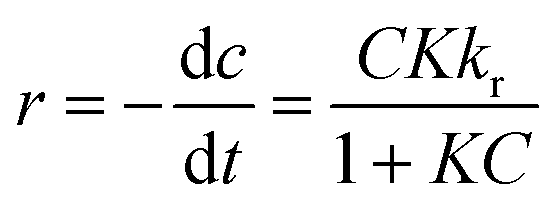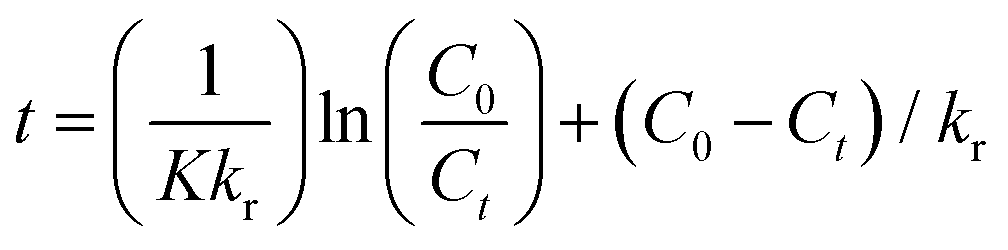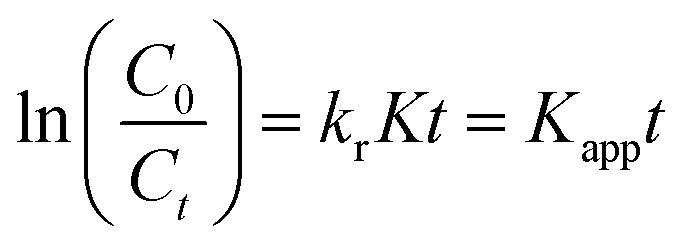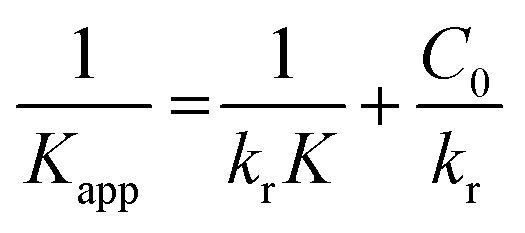DOI:
10.1039/D4MA00431K
(Paper)
Mater. Adv., 2024,
5, 9626-9640
Treatment of chromium-contaminated water using a highly efficient, novel ternary synergistic S–rGO–BiOBr–In2S3 heterojunction
Received
25th April 2024
, Accepted 4th November 2024
First published on 9th November 2024
Abstract
The goal of the current study is to form a Z-scheme heterojunction between narrow band gap In2S3 (indium sulphide) and wide band gap BiOBr (bismuth oxobromide) to minimize the photoinduced charge carrier recombination and increase the visible light harvesting capacity of the nanocomposite for Cr(VI) elimination from polluted water. The exfoliated and corrugated S-doped rGO (sulphur-doped graphene oxide) were used to enhance the surface area and conductivity, encourage nucleation, and act as anchor sites for interfacial contact between BiOBr and In2S3 to facilitate efficient charge transfer. In this work, a BiOBr–In2S3–SrGO nanocomposite was successfully synthesized by a facile hydrothermal method. The optical and physicochemical properties of the synthesized nanomaterials (NMs) were characterized using XRD, FTIR, FE-SEM, EDAX, HRTEM, XPS, UV-DRS, Raman and photoluminescence spectroscopy. The results reveal that almost 96.6% Cr(VI) removal was achieved from an initial Cr(VI) dose of 100 mg L−1 by the nanocomposite within 2 h under the illumination of solar light. On the other hand, the Cr(VI) reduction was limited to 33.4% and 30.9% using the individual NMs of BiOBr and In2S3, respectively. The reduction of Cr(VI) follows pseudo-first-order kinetics. The calculated apparent rate constant (Kapp) of the nanocomposite was 3 times more than the individual NMs of BiOBr and In2S3, with an excellent recycling activity.
1. Introduction
Chromium (Cr(VI)) is highly toxic and bio-accumulates in the food chain. Its presence in natural water and industrial wastewater streams is a significant environmental and public health risk.1 Furthermore, among the hazardous environmental pollutants, Cr(VI) ranked third and is released from effluents of a number of industries; for example: electroplating, textile, dying, tanning nuclear power, and photographic sectors.2 According to recent reports, wastewater typically contains 10–100 mg L−1 of Cr(VI).3 As per the World Health Organization (WHO), the permissible limit of Cr(VI) is less than 0.05 mg L−1 in drinking water.4 In general, Cr(VI) penetrates the cell wall quickly, causing cancer, and damage to the liver, kidneys, and nerve tissue.5 Several techniques have been reported to remove Cr(VI), including ion exchange, chemical precipitation, electrochemical processes, membrane filtration, and adsorption methods. These methods have many drawbacks, including the fouling of membranes, production of carcinogenic by-products, ineffective removal, and high cost.6 One of the most promising methods for the effective removal of Cr(VI) is the reduction of Cr(VI) to Cr(III) because Cr(III) is less toxic, and can be easily precipitated and removed as Cr(OH)3 from contaminated water.7 Compared with other methods, photocatalytic reduction of Cr(VI) is more effective and low-cost, and does not produce any hazardous by-products.8 However, many challenges still exist in the photocatalytic reduction process, such as inefficient solar light harvesting, high rate of charge carrier recombination, and inability to remove the high-strength real Cr(VI) wastewater samples. Thus, the photocatalytic reduction process must be improved to meet the discharge standard.9
BiOBr is an indirect bandgap semiconductor. The unique electronic structure of BiOBr, especially its abundance of Bi 6s orbitals that come from the conduction band (CB) and valence band (VB), produces a tenable electronic state and high charge mobility that promote higher photocatalytic activity.10,11 Nevertheless, the primary issue with BiOBr as a photocatalysts is the high band gap energy of 2.81 eV that prevents them from fully harvesting solar irradiation, especially in the visible spectrum. Furthermore, the low affinity between pollutants and photocatalysts, together with the inadequate specific surface area, inhibit the overall photocatalytic activity.12 Similarly, In2S3 is the ideal choice to enhance the optical characteristics and physical structure of nanomaterials (NMs) for boosting photocatalytic activity. Because of its excellent stability and high photosensitivity, In2S3 with a narrow band gap of 2.0–2.3 eV has been employed extensively as a photocatalyst. However, under visible light excitation, the photogenerated holes produced by In2S3 can react with S2− of the photocatalyst, resulting in catalyst deactivation.13 It is also found that In2S3 has a low photocatalytic nature due to its low mass transfer and high charge carrier recombination rate. These problems have become a bottleneck for effectively developing and applying sulphide-based semiconductors such as In2S3.14 Hence, in this work, a Z-scheme heterojunction has been fabricated between BiOBr and In2S3 to overcome these issues and enhance its photocatalytic efficiency. Similarly, reduced graphene oxide (rGO) is growing in popularity because of its remarkable qualities, which include a high functional group density, an enormous specific surface area (2600 m2 g−1), excellent electron conductive properties (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 V−1 s−1), high chemical and thermal stability, and more. Therefore, it has also been proposed that rGO is an ideal option as a promoter or catalyst carrier. Additionally, the delocalized π electron in sp2 carbon can increase the electron mobility in rGO.15 Furthermore, S-doped rGO was used in the nanocomposite to facilitate efficient charge transfer, which encourages nucleation and acts as an anchor site for interfacial contact between BiOBr and In2S3. In addition, SrGO can significantly increase the structural stability and reaction kinetics and reduce the photo-generated electron–hole recombination ratio, thus boosting the movement of electrons and ions, especially across the interface.16
000 m2 V−1 s−1), high chemical and thermal stability, and more. Therefore, it has also been proposed that rGO is an ideal option as a promoter or catalyst carrier. Additionally, the delocalized π electron in sp2 carbon can increase the electron mobility in rGO.15 Furthermore, S-doped rGO was used in the nanocomposite to facilitate efficient charge transfer, which encourages nucleation and acts as an anchor site for interfacial contact between BiOBr and In2S3. In addition, SrGO can significantly increase the structural stability and reaction kinetics and reduce the photo-generated electron–hole recombination ratio, thus boosting the movement of electrons and ions, especially across the interface.16
The present study synthesized a novel S–rGO–BiOBr–In2S3 Z-scheme heterojunction using a hydrothermal method to remove Cr(III) from contaminated wastewater. The morphological and structural and elemental characterizations of the NMs were performed using XRD, FTIR, FESEM, EDX, HRTEM, XPS, UV-DRS, photoluminescence, and Raman spectroscopy analysis. The kinetics of Cr(III) removal by the nanocomposite was evaluated, and the Cr(VI) reduction mechanism was thoroughly explained. In addition, the reusability capacity of the nanocomposite for Cr(VI) removal was investigated.
2. Materials and methods
2.1. Materials
Analytical grade Bi (NO3)3·5H2O, hydrazine hydrate, H3PO4, and 1,5-diphenylcarbazide (DPC) were purchased from Merck, India. Graphite powder, Na2S·9H2O, was purchased from LOBA chemicals, India, and In2O3·5H2O from Burgoyne India. All other chemicals and reagents were of laboratory and analytical grade.
2.2. Methods
2.2.1. Synthesis of BiOBr.
BiOBr was prepared as reported in the literature.17 Solution A was prepared by dissolving 2.43 g of Bi (NO3)3·5H2O and 0.24 g of citric acid in 20 mL of deionized water. Solution B was prepared by dissolving 0.6 g of KBr in 20 ml of ethanol. Then, solution B was dripped into solution A, which was then stirred for 30 min. The resulting solution was kept at 180 °C for 24 h in a hydrothermal autoclave lined with Teflon. The resulting mixture was centrifuged at 10000 rpm for 10 min. The residue was rinsed with deionised water to remove any potential ionic species, and then dried overnight at 60 °C to obtain the final product.
2.2.2. Synthesis of In2S3.
In2S3 was produced by hydrothermal methods, as reported in the literature.18 Solution A was made by dissolving 0.333 g of In2O3·5H2O in 40 mL distilled water. In another beaker, 0.720 g Na2S·9H2O was dissolved in 40 ml distilled water, named solution B. After being fully dissolved, solution B was added dropwise to solution A. The pH of the resultant reaction mixture was adjusted to 2.8 by adding diluted HNO3. The resulting solution was then kept at 180 °C for 24 h in an autoclave lined with Teflon. Then, the acquired orange precipitate was frequently rinsed with deionized water and ethanol. It was then dried for 12 h at 90 °C in a vacuum oven to obtain the final In2S3 NMs.
2.2.3. Synthesis of SrGO.
Graphite oxide was prepared using a modified Hummers’ process.15 In an ice bath, 80 mL of cold, concentrated H2SO4 was mixed with 2 g of graphite powder. Then 4 g of NaNO3 and 8 g of KMnO4 were added slowly under continuous stirring for 4 h at 10 °C. The mixture temperature was then retained at 35 °C for 4 h. Furthermore, the resultant slurry was diluted with 200 mL of deionized water and stirred for another 1 h. 15 mL of 30% H2O2 solution was added to stop the reaction. The resulting GO mixture was centrifuged at 10000 rpm for 10 min and rinsed a number of times with water and 100% ethanol. The obtained product was then dried in a vacuum oven at 50 °C for 24 h. The SrGO was synthesized according to Tian et al.19 1 g of earlier prepared GO was dissolved in 300 mL distilled water and sonicated for 20 min. After that the solution was purged with nitrogen gas for 10 min. Then, 0.5 mL of hydrazine hydrate was mixed with the solution and sonicated for another 10 min. Then, 3 g of Na2S was added to the solution. The obtained solution was refluxed at 80 °C for 8 h. Then, the acquired residue was repetitively rinsed with deionised water and ethanol and dried at 65 °C in a vacuum oven.
2.2.4. Synthesis of the BiOBr/In2S3/SrGO nanocomposite.
A BiOBr–In2S3–SrGO heterostructure was prepared using the hydrothermal technique. Solution A was prepared by taking 0.333 g In2O3·5H2O in 40 mL of distilled water. Solution B was prepared by dissolving 0.720 g Na2S·9H2O in 40 mL distilled water. After being fully dissolved, solution B was added dropwise to solution A. 330 mg of BiOBr and 500 mg of SrGO were added to the sample. The pH of the solution was retained at 2.8 using diluted nitric acid and then sonicated for 15 min. The resulting solution was then kept at 180 °C for 24 h in an autoclave lined with Teflon. The obtained product was centrifuged at 10000 rpm for 15 min. The residue was collected, repetitively rinsed with deionised water and ethanol, and then dried for 12 h at 90 °C in a vacuum oven to obtain the final nanocomposite.
2.2.5. Experimental set up.
The Cr(VI) removal activity of the synthesised NMs was assessed under the irradiation of solar light. 5 separate 25 mL conical flasks were filled with 25, 50, 100, 150, and 200 mg L−1 Cr(VI) solutions. To eliminate dissolved oxygen, from each flask N2 gas was sparged for 5 min. Then, 1 g L−1, i.e. 25 mg of BiOBr–In2S3–SrGO nanocomposite, was supplemented to each flask containing 25 ml of solution. In another conical flask, a 100 mg L−1 Cr(VI) solution was taken without adding nanocomposite and labelled as a control solution. All the conical flasks containing Cr(VI) solution were kept in dark conditions for 15 min. After that, the photocatalytic experiment was conducted with continuous stirring under solar irradiation. Sample solutions from each flask were collected at regular intervals and centrifuged at 10000 rpm for 15 min. The collected supernatants were employed to measure the remaining Cr(VI) concentration.
2.2.6. Analytical method.
In order to validate the precise synthesis of the NMs, an X-ray powder diffractometer with a CuKα (λ = 1.5405 Å) radiation source was used to perform XRD analysis across a wide range of Bragg's angles (20° ≤ 2θ ≤ 80°) (Model: Rikagu Ultima Japan). The Miller indices, unit cell parameters, and crystal structure of the NMs were all computed employing the X’Pert Highscore Plus software. The molecular vibration and functional groups of the synthesized NMs were investigated by FTIR analysis (Jasco FT/IR-4600 LE, Varian spectrophotometer). The prepared NMs structural configuration and vibrational modes were studied using a Raman microscope equipped with a green 532 nm laser (Renishaw Invia). Using photoluminescence (PL) spectra, the recombination of photo-generated charge carriers was investigated (Edinburgh FLS 1000) at 488 nm, 532 nm, 633 nm, and 785 nm excitation wavelengths. The NM morphology, size, and distribution were examined using FE-SEM Gemini SEM 450 equipment with LaB6 and tungsten as the filament source. The energy dispersive X-ray spectroscopy (EDAX) equipment (FESEM – ZEISS Company, Oxford Instruments) connected to the FESEM was used to carry out the elemental analysis of the synthesized NMs. The investigation was carried out with a 10 kV electron beam in plain view mode. For each nanomaterial, the values of Lα and Kα energies were specified. The morphology of the prepared NMs was studied utilising transmission electron microscopy (TEM) with a Tecnai G2 20 S-TWIN [FEI] electron microscope running at 200 kV. The apparatus has a 0.14 nm line resolution, 0.24 nm point resolution, and 1.0 nm STEM resolution. The surface composition of the nanoparticles was examined using X-ray photoelectron spectroscopy (XPS) using a Thermo VG Scientific MULTILAB instrument with AlKα radiation as a monochromator. Employing Shirley-type background and Lorentzian–Gaussian functions, the XPX peaks 41 software deconvoluted the XPS peaks. For electrochemical Mott–Schottky measurement, 0.1 M Na2SO4 electrolytic solution was employed. The data were recorded by a 3 electrode system employing a Multi Autolab/M204 electrochemical workstation (Netherlands, Metrohm Autolab B.V.), where, Ag/AgCl (3 M KCl), platinum wire, and photocatalyst coated over graphite sheet were applied as the reference, counter, and working electrode, respectively. Before the initiation of the electrochemical experiments, Ar gas was purged for 20 min to eliminate oxygen or any other gas adsorbed on the surface of the sample. For the Mott–Schottky analysis, the samples were scanned varying from −1 V to +1 V (vs. Ag/AgCl and then transformed to potential vs. NHE). The NMs optical absorbance and band gaps were estimated using a UV-visible diffuse reflectance spectrophotometer (UV-Vis DRS; JASCO, Japan). The bandgap energy of the prepared NMs was calculated using Tauc's equation as follows:where,| | 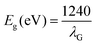 | (2) |
In which A is a constant, Eg represents the band gap energy, v is the photon frequency, and h is the Planck constant. The S factor, equal to 1/2 or 2 for the indirect and direct and transition band gaps, respectively, depends on the kind of electron transfer.20 A colorimetric method was used to assess residual Cr(VI) concentration as depicted by APHA.21
3. Results and discussion
3.1. Nanomaterial characterization
3.1.1. XRD analysis.
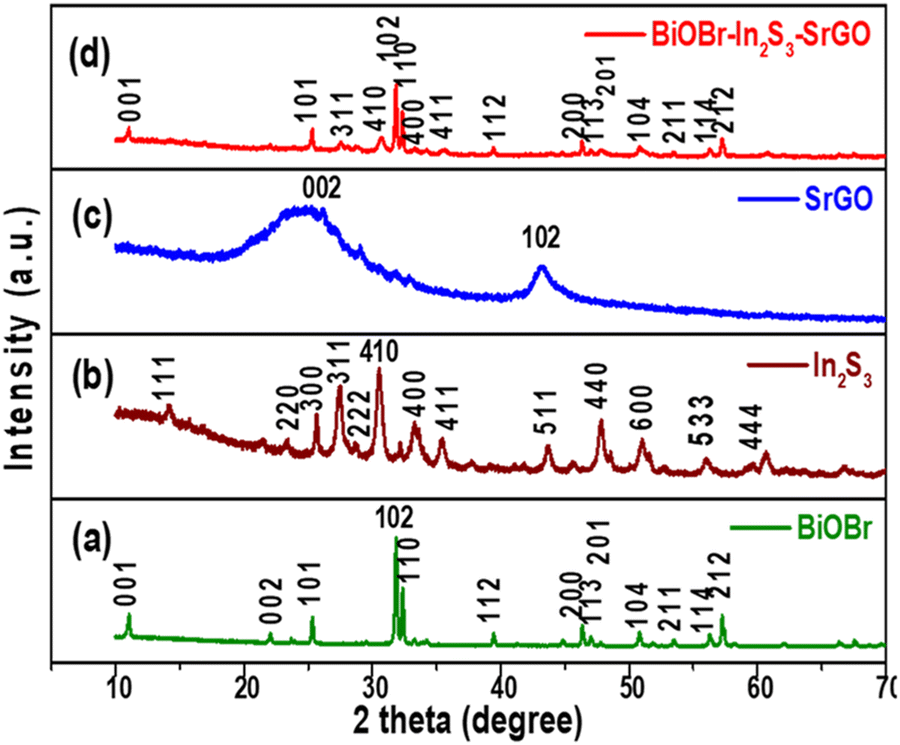
The XRD analysis of the BiOBr, In2S3, SrGO, and BiOBr–In2S3–SrGO nanocomposites is shown in Fig. 1. Fig. 1(a) demonstrates the XRD spectra of BiOBr. The prominent diffraction peaks at 2θ values of 10.9°, 21.9°, 25.1°, 31.6°, 32.2°, 39.3°, 46.2°, 50.6°, and 57.1° were indexed to the (001), (002), (101), (102), (110), (112), (200), (104), and (212) planes, respectively, indicating the tetragonal phase of BiOBr (JCPDS card number 09-0393). Similar observations were also reported in the literature.22,23 Furthermore, the XRD patterns showed an apparent propensity to grow along the (102) and (110) directions, as shown in Fig. 1(a).24 Diffraction peaks at 2θ values of 13.9°, 23.7°, 25.0°, 27.5°, 28.7°, 31.4°, 33.3°, 35.4°, 43.7°, 47.9°, 51.0°, 56.1° and 59.6° in Fig. 1(b) were indexed to the (111), (220), (300), (311), (222), (410), (400), (411), (511), (440), (600), (533) and (444) planes, respectively. This observation matches JCPDS card no. JCPDS-32-0456, 05-0722, which assured the cubic structure of In2S3.25,26Fig. 1(c) shows the XRD spectra of SrGO. The primary diffraction peaks at 2θ values of 26.4° and 43.2° were indexed to the (002) and (102) planes, respectively. The appearance of a maximum intensity peak at a 2θ value of ∼26.4°, which corresponds to the characteristic (002) reflection, reveals the occurrence of parallel graphene layers in SrGO. The literature has reported a similar observation.27 In Fig. 1(d), peaks of both BiOBr and In2S3 phases can be seen, which indicates the successful formation of the BiOBr–In2S3–SrGO nanocomposite. Nevertheless, no clear distinctive peaks of SrGO are seen in the BiOBr–In2S3–SrGO nanocomposite; this could be because the S–rGO sheets are not aggregating to the same extent.28
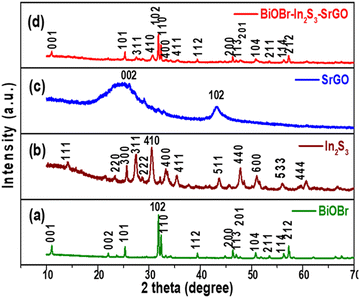 |
| | Fig. 1 X-ray diffraction patterns of (a) BiOBr, (b) In2S3, (c) SrGO, and (d) the BiOBr–In2S3–SrGO nanocomposite. | |
The average crystalline sizes and lattice strains of the synthesized NMs were calculated using the Williamson and Hall (W–H) equation as shown in eqn (3).
| | | βcosθ = 4sinθ + Kλ/D, | (3) |
Here, θ stands for the angle of diffraction, FWHM (full width at half maximum), D stands for average crystal size, K represents the constant with a value of 0.89, and λ denotes wavelength with a value of 0.154 nm. With 4sinθ on the X-axis and cosθ on the Y-axis, a plot is therefore created. The average crystalline size is represented by the Y-intercept, whereas the slope of the curve indicates lattice strain. In this work, the average crystalline size of the BiOBr, In2S3, SrGO, and BiOBr–In2S3–SrGO nanocomposites is 0.02 nm, 0.188 nm, 2.39 nm, and 0.017 nm, respectively.
3.1.2. FTIR Analysis.
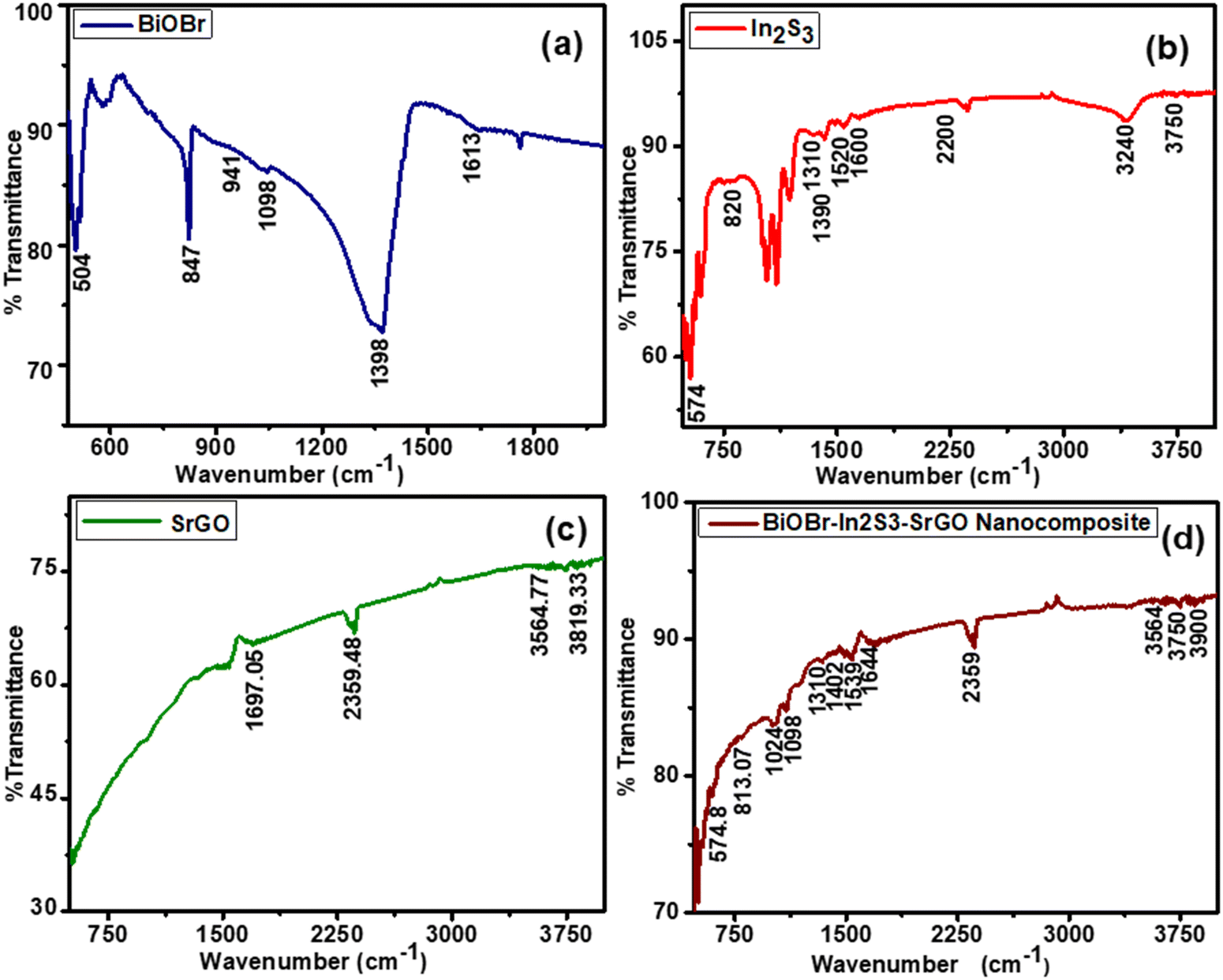
Fig. 2 displays the FTIR spectra of pure BiOBr, In2S3, SrGO, and BiOBr–In2S3–SrGO nanocomposites. As seen in Fig. 2(a), the appearance of a peak below 1000 cm−1 designates the existence of Bi–O bonds, whereas the Bi–Br band appears at absorption peaks between 1000 and 1500 cm−1. The peak centered at 1613 cm−1 represents the flexural vibration mode of O–H in free water molecules.29Fig. 2(b) shows the FTIR spectra of In2S3. The peak at 1600 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the atmospheric CO2 absorbed in the sample.30 The peaks from 3200 to 3750 cm−1 show a vibrational stretching mode of the OH group, which can be attributed to water absorption on the sample.31 Prominent absorption bands at around 1300 to 1600 cm−1 and 574 cm−1 could be attributed to In–S, which is consistent with the XRD patterns.32 FTIR spectra of SrGO are displayed in Fig. 2(c). The OCO starching revealing the occurrence of oxygen in the S–rGO specimen is demonstrated by the strong peak at 2359.48, and the CC stretching vibration is indicated by the peak at 1697.05. The occurrence of the OH group is shown by the peaks at 3564.77 and 3819.33.15,33Fig. 2(d) illustrates the FTIR spectra of the BiOBr–In2S3–SrGO nanocomposites. The absorption peaks at 574.8 and 813.07 cm−1 of the nanocomposite are ascribed to the Bi–O bonds, whereas those between 1000 and 1500 cm−1 are attributed to the Bi–Br band. Similarly, the peaks between 3200–3700 cm−1 and 1644 cm−1 are caused by the O–H stretch vibration of water and the CO stretching vibration of carbon dioxide, absorbed on the nanocomposite, respectively. In addition, a prominent peak at 2359 cm−1 (OCO stretching) is due to the occurrence of oxygen in the SrGO. These results have authenticated that the BiOBr–In2S3–SrGO hetero-structure has been successfully prepared.
O stretching vibration of the atmospheric CO2 absorbed in the sample.30 The peaks from 3200 to 3750 cm−1 show a vibrational stretching mode of the OH group, which can be attributed to water absorption on the sample.31 Prominent absorption bands at around 1300 to 1600 cm−1 and 574 cm−1 could be attributed to In–S, which is consistent with the XRD patterns.32 FTIR spectra of SrGO are displayed in Fig. 2(c). The OCO starching revealing the occurrence of oxygen in the S–rGO specimen is demonstrated by the strong peak at 2359.48, and the CC stretching vibration is indicated by the peak at 1697.05. The occurrence of the OH group is shown by the peaks at 3564.77 and 3819.33.15,33Fig. 2(d) illustrates the FTIR spectra of the BiOBr–In2S3–SrGO nanocomposites. The absorption peaks at 574.8 and 813.07 cm−1 of the nanocomposite are ascribed to the Bi–O bonds, whereas those between 1000 and 1500 cm−1 are attributed to the Bi–Br band. Similarly, the peaks between 3200–3700 cm−1 and 1644 cm−1 are caused by the O–H stretch vibration of water and the CO stretching vibration of carbon dioxide, absorbed on the nanocomposite, respectively. In addition, a prominent peak at 2359 cm−1 (OCO stretching) is due to the occurrence of oxygen in the SrGO. These results have authenticated that the BiOBr–In2S3–SrGO hetero-structure has been successfully prepared.
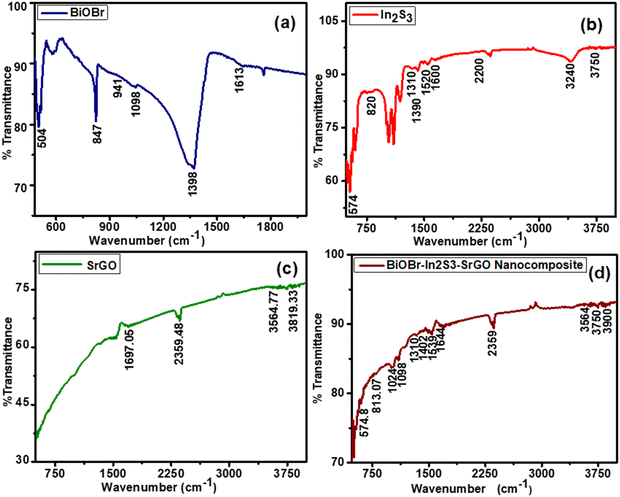 |
| | Fig. 2 FTIR spectra of the synthesized NMs (a) BiOBr, (b) In2S3, (c) SrGO, and (d) the BiOBr–In2S3–SrGO nanocomposite. | |
3.1.3. Raman analysis.
Raman spectroscopy investigation is used to determine the structure and defects of NMs. Fig. 3(a) represents the Raman spectra of BiOBr NMs. There are 2 A1g, B1g, and Eg Raman active modes, as shown in Fig. 3(a). The bands for BiOBr are designated to the A1g internal Bi–Br stretching mode at 57.0, 91.7, and 112.1 cm−1. The E1g Bi–Br stretching mode is attributed to the band at 160.7 cm−1.34 The firm peaks confirm the excellent crystallinity structure of the BiOBr NMs. Fig. 3(b) illustrates the Raman spectrum of In2S3 NMs. From Fig. 3 (b), distinct Raman peaks are observed at about 274, 309, and 369 cm−1. The A1g mode can be attributed to the highly polarized Raman peak at 369 cm−1, the F2g mode at 309 cm−1, and the Eg mode at 274 cm−1.35 These Raman peaks show a strong correlation with the previously reported In2S3 NMs’ active mode in the cubic phase.36,37Fig. 3(c) illustrates the Raman spectra of SrGO. As seen in Fig. 3(c), all of the peaks show a slight red shift (bands appeared at 1351 and 1588 cm−1) and narrowed, which might be due to the reorganization of the structure during the reduction and sulfonation process. When comparing SrGO (2.12) to pure GO, a slight drop in the ID/IG ratio was observed. This suggests that during SrGO's reduction and sulfonation processes, a small number of sp3 defects associated to the oxygenation groups were diminished.38,39Fig. 3(d) shows the Raman spectra of the nanocomposite (BiOBr–In2S3–SrGO). The peaks at 57.02, 69.67, 91.7, 112.1, 160.7, 222.5, and 267.5 cm−1 represent the Raman spectra of the nanocomposite. The bands at 57.02, 69.67, 91.7, and 112.1 cm−1 are designated to the A1g internal Bi–Br stretching mode. The band at 160.7 cm−1 is attributed to the E1g Bi–Br stretching mode. The appearance of a peak at about 267.5 cm−1 represents the in-plane Eg vibration mode of In2S3. The above peaks indicate the cubit structure of the BiOBr–In2S3–SrGO nanocomposite. In summary, the findings of the Raman analysis on the structure of BiOBr, In2S3, and the BiOBr–In2S3–SrGO nanocomposite corroborate with the findings of the XRD study.
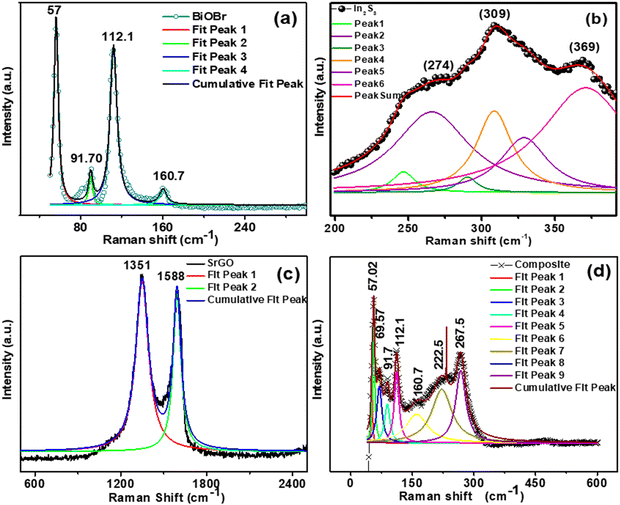 |
| | Fig. 3 Raman shift versus intensity of the synthesized NMs (a) BiOBr, (b) In2S3, (c) SrGO, and (d) the BiOBr–In2S3–SrGO nanocomposite. | |
3.1.4. PL spectra analysis.
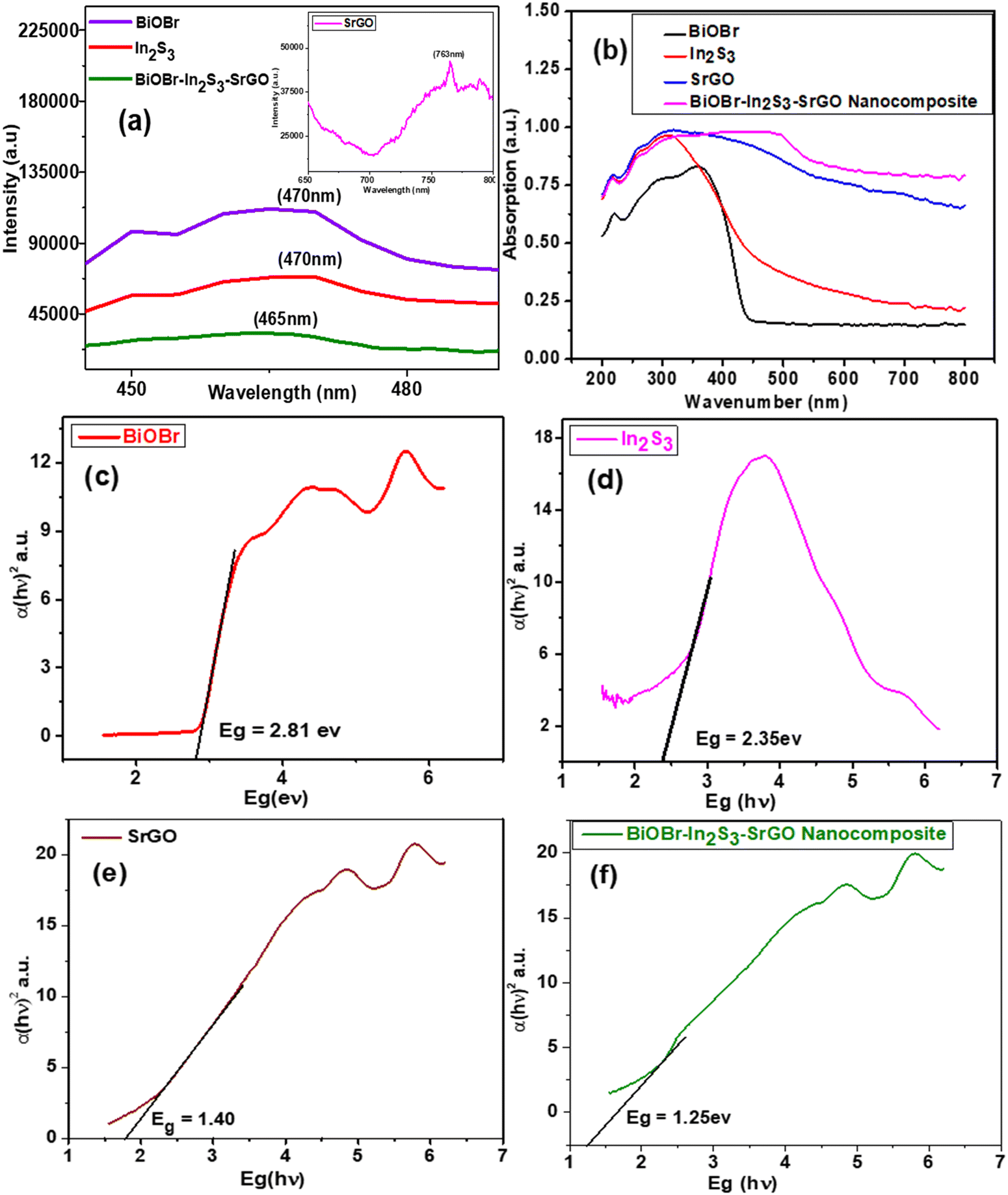
The recombination rate of the photoinduced charge carrier can be predicted using the photoluminescence spectral analysis. Lower photoluminescence intensity denotes a minimum recombination rate of photoexcited electron and hole pairs. Whereas greater photoluminescence intensity indicates a high recombination rate of photogenerated charge.15Fig. 4(a) shows the photoluminescence emission spectra of the BiOBr, In2S3, SrGO, and BiOBr–In2S3–SrGO nanocomposite. The PL spectra of BiOBr, In2S3, and BiOBr–In2S3–SrGO NMs were obtained in the 400–600 nm emission peak range. The bare BiOBr and In2S3 have the highest emission peak, centered at 470 nm.40–42 PL spectra of SrGO were obtained in the emission peak range of 650 to 800 nm, and the emission peak centered at 763 nm, which is in good agreement with the reported literature.43 On the other hand, the composite has the lowest emission peak at 465 nm. It is evident from the figure that the photoluminescence intensity of these NMs diminishes and reaches its lowest value in the synthesized nanocomposite. This phenomenon indicated that the recombination rate of photoinduced charge carriers significantly decreases in the nanocomposite. This observation is likely due to the formation of the Z-scheme heterostructure between the BiOBr and In2S3 that improves the efficiency of photoinduced charge transfer with high redox ability.44
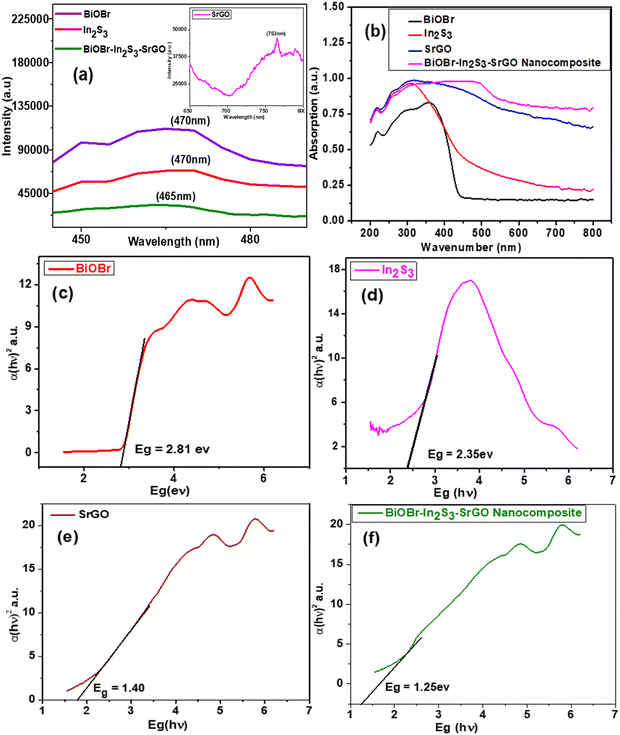 |
| | Fig. 4 (a) Photoluminescence (PL) spectra of BiOBr, In2S3, SrGO and the BiOBr–In2S3–SrGO nanocomposite, (b) UV-DRS spectra of the synthesized NMs, and Willliamson-hall plots of (c) BiOBr, (d) In2S3, (e) SrGO, and (f) the BiOBr–In2S3–SrGO nanocomposite. | |
3.1.5. UV DRS analysis.
In general, the light absorption property heavily relies on the band structure of the semiconductor materials, severely constraining the photocatalytic activity. Thus, UV-VIS DRS was used to compute the light absorption characteristic of NMs.45Fig. 4(b) shows the bathochromic shift of the BiOBr–In2S3–SrGO nanocomposite with respect to the individual NMs. The edges of In2S3, SrGO, BiOBr, and BiOBr–In2S3–SrGO have been redshifted (bathochromic shift) to 311.5 nm, 319.5 nm, 359 nm, and 462 nm, respectively, showing that the formation of the nanocomposite improves the responsiveness to visible light. The band gaps of the produced nanocomposite were determined using Tauc plots, as shown in Fig. 4(c). The computed band gap energy (Eg) of BiOBr is 2.81 eV, which is well supported by many documents in the literature.46,47 The calculated Eg value of In2S3 is 2.35 eV, as shown in Fig. 4(d), which is well correlated by many reports in the literature.48,49Fig. 4(e) shows the Eg value of SrGO, which increased slowly from 0.95 eV to 1.37 eV, implying that the chemical modification of rGO with sulphate groups changed its optical characteristics, almost identical to the reported literature.50 The calculated Eg value of the BiOBr–In2S3–SrGO nanocomposite is 1.25 eV, as shown in Fig. 4(f). As can be seen from Fig. 4(f), BiOBr has a slightly low bandgap energy of 2.81 eV, which suggests that it has moderately visible light absorption efficiency. After forming the nanocomposite, the Eg value was further reduced to 1.25 eV, and thus, the visible light harvesting ability was effectively increased. This shows the unique and productive interaction among the BiOBr, In2S3, and SrGO in the nanocomposite.
3.1.6. FESEM micrographs.
FE-SEM analysis was carried out to observe the morphological features, distribution and size of the prepared NMs. Fig. 5(a) shows a clear, heterogeneous, rectangular 3D nanoplatelet structure of BiOBr with an average scale of 200 nm. The formation of a nanoplatelet structure offers a high surface area and improves the visible light absorption ability. Further formation of the nanoplatelet geometry with a high surface area enhances the contact area with SrGO, facilitating the photogenerated charge transfer efficiency and increasing the photocatalytic properties. A similar sheet-shaped structure with a clear and smooth surface of BiOBr NMs has been documented in the literature.51Fig. 5(b) shows the FE-SEM image of In2S3 NMs. The FE-SEM image reveals a homogeneously distributed spherical and granular structure of In2S3 with an average scale of 300–400 nm.52 It is seen that the particles are dense without pinholes.53 The FESEM image of SrGO is demonstrated in Fig. 5(c), which reveals that the SrGO surface displayed an interconnected 3D porous, corrugated, and exfoliated structure. The conversion of GO to rGO is accountable for this corrugated configuration. Further removal of oxygen-carrying groups from the GO surface while converting to rGO induces the formation of aggregated structures. The π–π stacking phenomena of GO most likely causes the formation of this aggregated structure.15Fig. 5(d) shows the high dispersion and uniformity of the individual constituents of the BiOBr–In2S3–SrGO nanocomposite. It is evident from Fig. 5(d) that a bunch of uniformly distributed rectangular 3D nanoplatelets of BiOBr and dense nanospheres of In2S3 anchored on the corrugated, exfoliated, and 3D porous interconnected configuration of S–rGO.
 |
| | Fig. 5 FE-SEM micrographs of (a) BiOBr, (b) In2S3, (c) SrGO, and (d) the BiOBr–In2S3–SrGO nanocomposite. | |
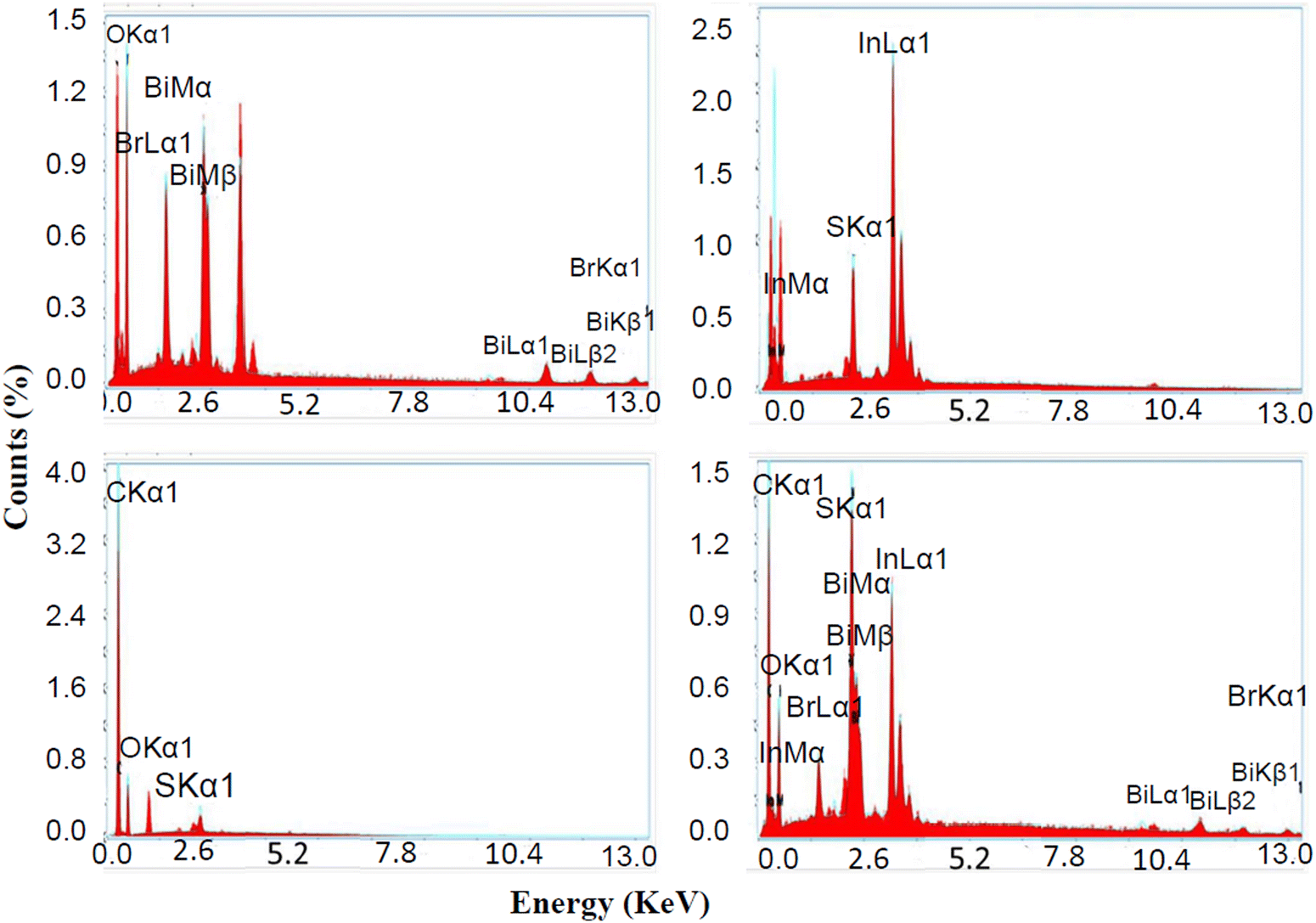
3.1.7. EDAX spectra analysis.
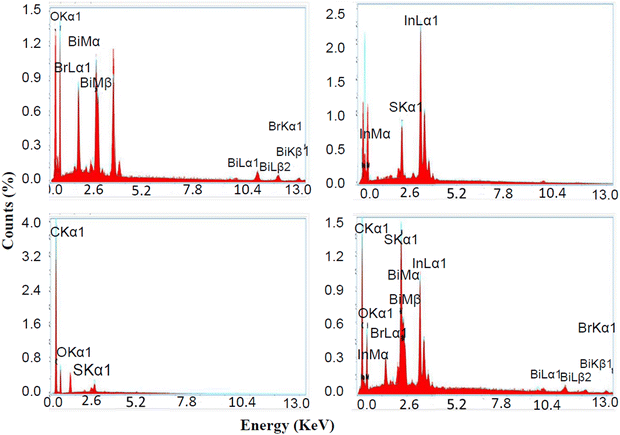
The elemental compositions of BiOBr, In2S3, SrGO, and the BiOBr–In2S3–SrGO nanocomposite were analyzed using EDAX spectroscopy. Fig. 6(a) demonstrates the EDAX spectrum for the BiOBr, which revealed the occurrence of a stoichiometric ratio of Bi, O, and Br in the NMs. Based on the EDAX peaks, the amounts of O2, Bi, and Br present in the BiOBr NMs are 51.8, 30.4, and 17.8%, respectively. Fig. 6(b) displays the EDAX spectra of the synthesized In2S3 NMs. The In2S3 is composed of indium at 90.1 weight%, which is larger than sulfur at 9.9 weight%. The existence of a stoichiometric ratio of In and S elements confirmed that the In2S3 NMs were well synthesized. In Fig. 6(c), the EDAX data of SrGO confirm the occurrence of carbon (C), oxygen (O) and sulphur elements in the NMs. According to the EDAX spectra, the quantity of C in S–rGO is 81.9 weight%, much more than O, which is 17 weight%. As sulfur was applied as a doping agent only, the existence of S in the compound is very small, which is 1.1 weight%. Fig. 6(d) exhibits the EDAX spectra of the synthesized BiOBr–In2S3–SrGO nanocomposite. Fig. 6(d) confirms the presence of Bi, O, Br, In, S, and C in the BiOBr–In2S3–SrGO nanocomposite. The nanocomposite has a larger weight percentage of carbon (46.4), bismuth (18.1), and indium (15) compared to other elements present in it.
 |
| | Fig. 6 Energy dispersive X-rays spectrum (EDX) of (a) BiOBr, (b) In2S3, (c) SrGO, and (d) the BiOBr–In2S3–SrGO nanocomposite. | |
3.1.8. HRTEM micrographs.
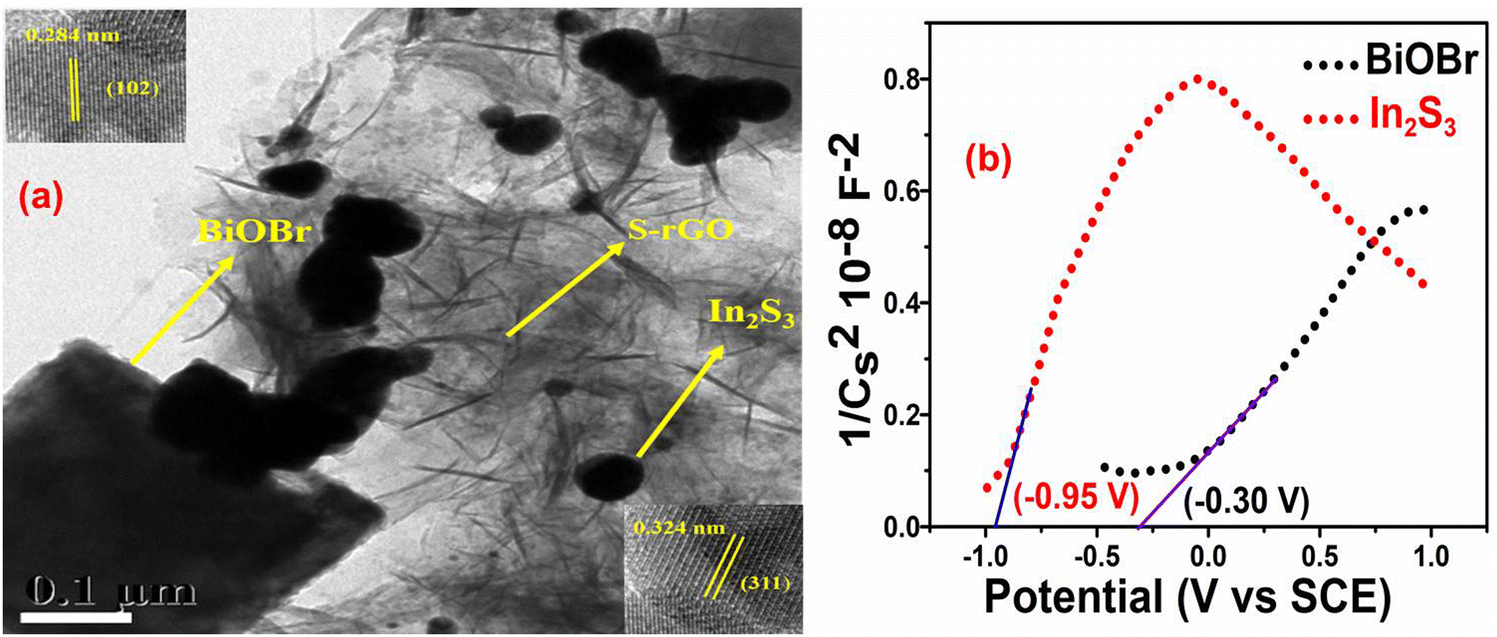
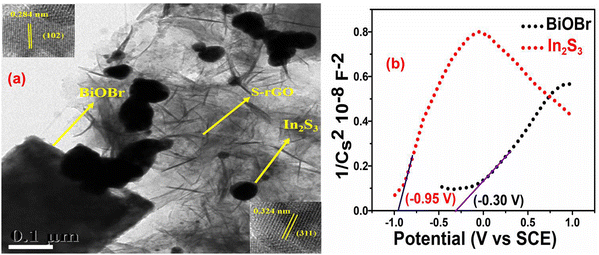
The heterostructure of the BiOBr–In2S3–SrGO ternary nanocomposite was investigated by HRTEM through morphological study. As shown in Fig. 7(a), In2S3 was found to consist of a spherical structure with average sizes of 50 nm, whereas BiOBr was a 3D-nanoplatelet structure with an irregular size and shape of average 400 nm, both of which were decorated on the surface of sheet-like SrGO. The HRTEM image that corresponded to the selected area in the inset of Fig. 7(a) displayed lattice fringes with spacings of 0.324 and 0.284 nm. These fringes could be attributed to the In2S3 crystal plane (311), and the BiOBr crystal plane (102) respectively.54 This aligns well with the XRD results.
 |
| | Fig. 7 (a) TEM image of the BiOBr–In2S3–SrGO ternary nanocomposite (Inset: HRTEM image of BiOBr–In2S3–SrGO ternary nanocomposite). (b) Mott–Schottky plot of BiOBr and In2S3 nanomaterials. | |
3.1.9. Mott–Schottky plot analysis.
In order to ascertain the variations in the electrical characteristics of the BiOBr photocatalyst, Mott–Schottky (MS) measurements were executed by means of the impedance technique.55Fig. 7(b) shows the M-S curves of the n-type characteristics of BiOBr and In2S3, respectively, due to the positive slope.56 The flat potentials of BiOBr and In2S3 are measured to be −0.30 V and −0.95 V (vs. SCE), which are −0.06 V and −0.71 V (vs. NHE). Furthermore, the slope of In2S3 was significantly smaller compared to BiOBr, revealing the generation of more electrons in the In2S3 and therefore comprising a higher charge density for efficient Cr(VI) reduction in the Z-scheme photocatalyst.57 Since the conduction band energy (ECB) position is about 0.10 V above the flat-band potential, in the present study the ECB positions of BiOBr and In2S3 were calculated at about −0.16 V and −0.81 V, respectively, which is well supported with the reported literature.58,59 By deducting Eg (bandgap energy) from the position of the CB minimum, one can determine the VB maximum position60 and thus, in the present study, the calculated VB of BiOBr and In2S3 are 2.65 V and 1.54 V, respectively.
3.1.10. XPS analysis.
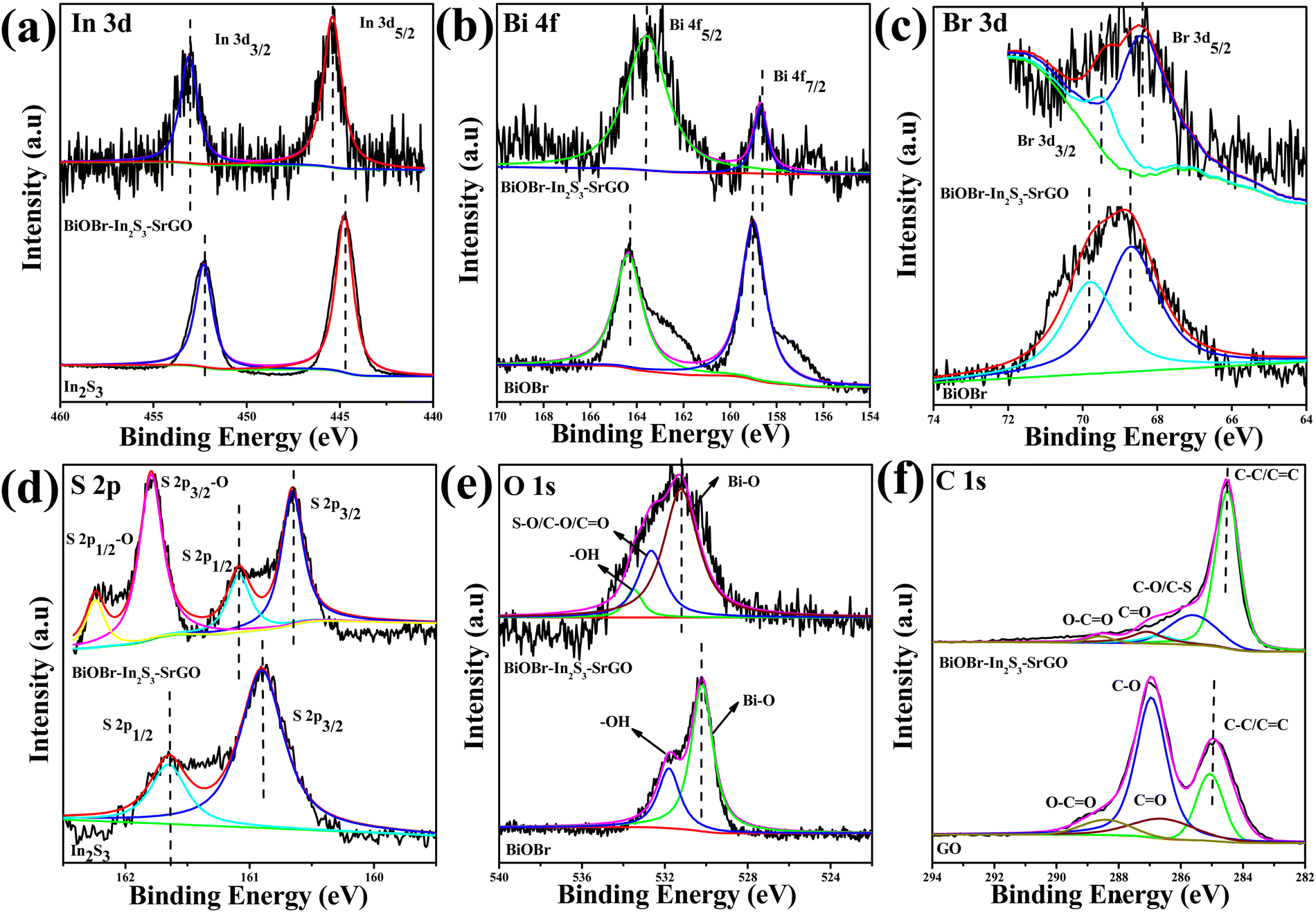
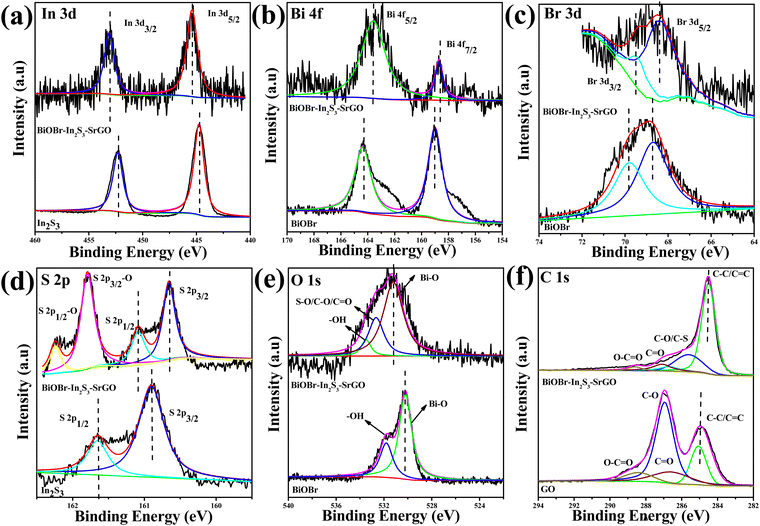
XPS measurements were carried out to examine the valence states and chemical composition of several components in the BiOBr–In2S3–SrGO ternary nanocomposite (Fig. 8). As shown in Fig. 8(a), bare In2S3 shows two distinct peaks located at 445.3 and 453.1 eV that were attributed to In 3d5/2 and 3d3/2 of In3+, respectively.61 Furthermore, the Bi 4f spectrum of bare BiOBr in Fig. 8(b) shows characteristic peaks Bi 4f7/2 and 4f5/2 appearing at a binding energy of 158.6 and 163.5 eV that were assigned to Bi3+, respectively.62 Similarly, the binding energies of 468.6 and 469.8 eV in bare BiOBr in Fig. 8(c) correspond to characteristic peaks of Br 3d (Br 3d5/2 and Br 3d3/2).63 In contrast, when comparing the BiOBr–In2S3–SrGO ternary nanocomposite in Fig. 8(a–c) to bare samples, the In 3d states shifted to higher energies, whereas the Bi 4f and Br 3d states shifted to lower energies. Fig. 8(d) demonstrates the high resolution S 2p spectra of bare In2S3. The two fitted peaks at 162.2 and 164.2 eV were attributed to S(2p3/2) and S (2p1/2) of bare In2S3, respectively.61 On the other hand, the two S 2p peaks of the BiOBr–In2S3–SrGO ternary nanocomposite were shifted to a higher binding energy of 161.4 and 160.2 eV, respectively, and two more doublet peaks were observed at 164.7 and 163.1 eV, respectively. These were ascribed to the S 2p 3/2 and 1/2 peaks of the S–O bonds, which is well supported with the reported literature.64 Two peaks, representing Bi–O (530.2 eV) and –OH (531.8 eV), respectively, were seen in the fitted O 1s spectra of BiOBr in Fig. 8(e).65 However, the BiOBr–In2S3–SrGO ternary nanocomposite shows a new peak at 531.6 eV; however, it may be attributed to the S–O/C–O/CO bonds in S–rGO.66,67 In Fig. 8(f), the deconvoluted C 1s core level spectrum of the BiOBr–In2S3–SrGO ternary nanocomposite reveals four distinct peaks at 284.4, 285.7, 287.1, and 288.6 eV, which are attributed to C–C/CC, C–O/C–S, CO, and O–CO, respectively.68 In comparison with the C 1s core level spectrum of GO, the prominent peaks at CC and the minor peaks at O–CO indicate that the GO was effectively reduced during the synthesis process, confirming the presence of reduced graphene oxide (rGO) in the BiOBr–In2S3–SrGO ternary nanocomposite. Additionally, the peak at 285.7 eV (C–O/C–S) demonstrated that the sulfur ion in the BiOBr–In2S3–SrGO ternary nanocomposite was doped in the carbon lattice to create SrGO rather than physically adsorbed on the graphene sheets. Taking everything into account, it was evident that, as seen in Fig. 8(a–f), each element's binding energy state in the BiOBr–In2S3–SrGO ternary nanocomposite had shifted somewhat when compared to the pure samples. This suggested that photo-excited electrons were moving from BiOBr to In2S3 in the heterostructure through the S-doped rGO.54,69 Because the VB of In2S3 is larger compared to BiOBr and due to the outstanding electrical conductivity and storage ability of S–rGO, the S–rGO can offer interfaces for photoinduced electrons in the CB of BiOBr to combine with the holes of the VB of In2S3 in the composite photocatalysts, which may improve the hole–electron pair separation efficiency. Therefore, a Z-scheme model might describe the charge transfer pattern in this study.
 |
| | Fig. 8 High-resolution XPS spectra of (a) In 3d, (b) Bi 4f, (c) Br 3d, (d) S 2p, (e) O 1s, and (f) C1s from the bare In2S3, BiOBr and BiOBr–In2S3–SrGO ternary nanocomposites. | |
3.2. Cr(VI) removal profile of the BiOBr–In2S3–SrGO nanocomposite
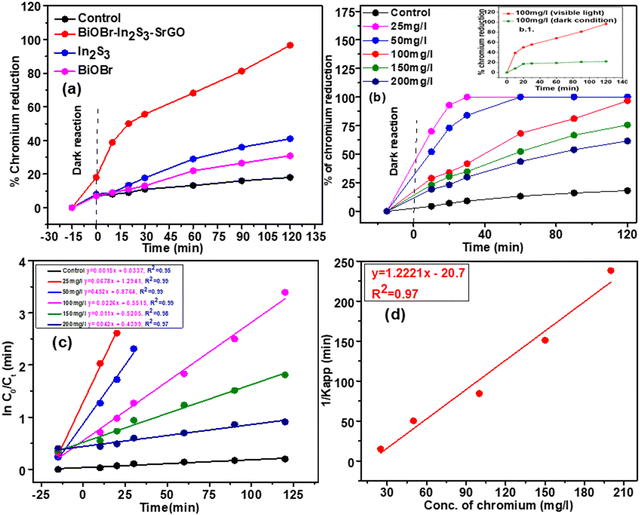
The Cr(VI) removal profile under dark and visible light illumination conditions is shown in Fig. 9(a). From the figure, it can be seen that when the heterojunction was kept under dark conditions for 120 min, it exhibits no noteworthy Cr(VI) removal (22%) at an initial 100 mg L−1 Cr(VI). Upon visible light illumination for 120 min, Cr(VI) removal was increased to 96.6%. When exposed to visible light, the individual NMs of BiOBr and In2S3 had the lowest photo-reduction activity of 30.9 and 33.4%, respectively. On the other hand, the photo-reduction of Cr(VI) is considerably enhanced to 96.6% using the BiOBr–In2S3–SrGO nanocomposite. Table 1 provides a comparative analysis of different photocatalytic systems reported in the literature for Cr(VI) reduction under visible light illumination. Compared to literature reports, the BiOBr–In2S3–SrGO nanocomposite distinctively exhibits superior Cr(VI) reduction activity compared to all other catalysts, as shown in Table 1. The improved photocatalytic activity of the nanocomposite might be due to bathochromic shift and narrowing of the bandgap energy with respect to the individual NMs, as discussed earlier in the UV-DRS analysis shown in Fig. 4(c–f). This is also well supported by the lower photoluminescence intensity of the nanocomposite than individual NMs, as shown in Fig. 4(a). This result suggests that the recombination rate of photogenerated charge carriers is inhibited, which in turn increases the activity of photocatalytic Cr(VI) reduction. The photocatalytic reduction of Cr(VI) was improved further by the addition of S-doped rGO in the nanocomposite because it enhanced the surface area, encouraged nucleation, and provided anchor sites for interfacial contact between BiOBr and In2S3. Thus, the efficiency of electron flow increases, particularly at the interface, which is further accelerated by delocalized π electrons in the sp2 carbon of rGO.15,16 Additionally, S doped r-GO prevents the rate of photo-excited charge carrier recombination. Fig. 9(b) shows the Cr(VI) reduction profile of the BiOBr–In2S3–SrGO nanocomposite at varying initial Cr(VI) doses. The solutions having 25 mg L−1 and 50 mg L−1 initial Cr(VI) doses exhibit 100% reduction within 30 and 60 min, respectively. Meanwhile, 96.6% of the Cr(VI) reduction was achieved by the nanocomposite at 100 mg L−1 of the initial Cr(VI) dose within 120 min. Whereas, Fig. 9b.1 shows the comparison between the reduction efficiency of the BiOBr–In2S3–SrGO nanocomposite under dark and visible light conditions at 100 mg L−1 initial Cr concentration. When the heterojunction was rendered to dark reaction for 120 min, it revealed no substantial Cr(VI) reduction, i.e., about 22% removal at an initial 100 mg L−1 Cr(VI), which was taken as a control. But when irradiated with visible light for 120 min, Cr(VI) removal was boosted, i.e., up to 96.6%.
 |
| | Fig. 9 (a) Demonstration of the Cr(VI) removal profiles by different individual nanomaterials and the nanocomposite at an initial dose of 100 mg L−1. (b) Cr(VI) removal profiles by the nanocomposite at varying initial Cr(VI) doses (b.1) Cr(VI) removal profiles by the nanocomposite under visible light and dark conditions. (c) Pseudo first order Cr(VI) removal kinetics at varying doses of Cr(VI). (d) Variation of the reciprocal of apparent rate constant profile at varying initial Cr(VI) dose. | |
Table 1 Comparative analysis of the BiOBr–In2S3–SrGO nanocomposite's Cr(VI) removal performance with that of other catalysts documented in the literature
| Sl. no. |
Photocatalyst |
Synthesis method |
Experimental condition |
Initial pollutant conc. (mg L−1) |
Cr(VI) reduction efficiency |
Ref. |
| 1 |
MgCr2O4/WO3 |
Sol–gel process |
Visible light |
30 |
100% in 120 min |
70
|
| 2 |
Cu2O/g-C3N4 |
In situ wet chemistry |
Visible light |
20 |
77.7% in 120 min |
71
|
| 3 |
NiFe2O4/MOF-808 |
Mechanochemical |
Visible light |
20 |
100% in 60 min |
72
|
| 4 |
MnFe2O4 and UiO-66-NH2 |
Solvothermal |
Visible light |
10 |
100% in 75 min |
73
|
| 5 |
BiVO4/rGO |
Hydrothermal |
Visible light |
10 |
97.6% in 30 min |
74
|
| 6 |
FeVO4/Bi4O5Br2/BiOBr |
One-step microwave irradiation |
Visible light |
30 |
72.3% in 120 min at PH = 6 |
75
|
| 7 |
BiVO4/FeVO4@rGO |
Hydrothermal |
Visible light |
20 |
90.9% in 90 min |
76
|
| 8 |
rGO-Sm2MoO6-TiO2 |
Hydrothermal |
Visible light |
10 |
96% in 70 min |
77
|
| 9 |
ZnO-Bi2S3 |
Solvothermal |
Visible light |
20 |
95% in 120 min |
78
|
| 10 |
BiOBr–In2S3–SrGO |
Hydrothermal |
Visible light |
100 |
96.6% in 120 min |
This work |
3.3. Reduction profile of chromium at different pH
The Cr(VI) reduction experiment by the nanocomposite was conducted with different pHs of 2, 4, 6, and 8 at 100 mg L−1 initial Cr(VI) concentration and achieved almost 96.6%, 85.3%, 53%, and 47% removal, respectively. In this study, using a BiOBr–In2S3 Z-scheme heterojunction, the Cr(VI) elimination efficacy was 96.6% at pH 2, while at pH 8, it was only 47% under illumination of visible light within 120 min. Typically, Cr2O72− is the primary component under acidic conditions, while CrO42− in alkaline conditions.79 In an acidic environment, the photocatalyst surface was likely positively charged by the H+ ion, which aided in photoelectron migration and enhanced the photocatalytic reduction of Cr2O72− to less hazardous 2Cr(III), as shown in eqn (4).80 Additionally, the conversion of Cr(VI) to Cr(III) may be facilitated by the high redox potential of Cr2O72− at very acidic environments.| | | 14H+ + Cr2O72− + 6e− →2Cr(III) + 7H2O(acid) | (4) |
Moreover, the zeta potential of the NMs is increased at lower pH values, which also lessens the repulsive force of the NMs against Cr2O72− and boosts the Cr(VI) elimination rate. Despite rGO having fewer hydroxyl (OH) and carboxyl (COOH) functional groups than GO at low pH, these functional groups were protonated to be positively charged and captured negatively charged Cr(VI) ions (HCrO4−) via electrostatic attraction, leading to a comparatively high Cr(VI) adsorption.81 Because the Cr(VI) ions adsorbed on GO could readily absorb photoinduced electrons, the photoreduction rate of Cr(VI) increases with the number of Cr(VI) ions adsorbed on GO. Conversely, as the pH increases, the OH and COOH groups on GO deprotonated to exhibit a significant quantity of negative charge, thus causing substantial electrostatic repulsion between Cr(VI) ions (CrO42−) and the rGO, which decreased the Cr(VI) adsorption.81 Nevertheless, Cr(VI) ions occurred in the form of chromate (CrO42−), under alkaline environments, whose reduction products readily converted to Cr(OH)3 and precipitated, as shown in eqn (5).
| | | CrO42− + 4H2O + 3e− → Cr(OH)3 + 5OH−(base) | (5) |
This further weakened the photocatalytic reduction efficiency.80 As a result, the removal efficiency clearly declined at higher pH.
3.4. Kinetics of Cr(VI) removal using the BiOBr–In2S3–SrGO nanocomposite
The photocatalytic process at liquid–solid interfaces can be efficiently assessed by employing the Langmuir–Hinshelwood kinetic model (LR model) as shown below:| | 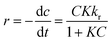 | (6) |
where K stands for the LR adsorption equilibrium constant (l mg−1), and r (−dC/dt) represents the rate of Cr(VI) reduction [mg L−1 min−1]. The t stands for the period of light illumination in min. The photocatalytic reduction rate constant is denoted by kr (mg L−1.min), and C is the dose of Cr(VI) (mg L−1). Eqn (6) can be simplified as follows by integrating it at the Cr(VI) dose (Ct) and light irradiation period t.| | 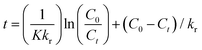 | (7) |
where, the starting and equilibrium dose of Cr(VI) at time t is represented by C0 and Ct, respectively. When the values of C0 are very small, Kc can be ignored, the above eqn (7) can be simplified as follows:| | 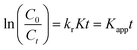 | (8) |
where the apparent first-order rate constant for Cr(VI) reduction is denoted by Kapp (1/min). The pseudo-first-order kinetics curve for the photoreduction of Cr(VI) by the nano catalyst is depicted in Fig. 9(c). The slope of the plot between lnC0/Ctvs. t (time) was used to determine the rate constant (Kapp). The LR kinetic model can be further simplified as shown below.82| | 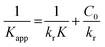 | (9) |
Fig. 9(d) displays the plot of 1/Kapp against the initial Cr(VI) concentration (C0). The higher correlation coefficient value (R2 = 0.97) indicates that the experimental data is well-fitted with this LH model. The intercept and slope of the straight line shown in Fig. 9(c) are used to estimate the values of the photocatalysis rate constant (kr) and Langmuir–Hinshelwood adsorption equilibrium constant (K). In this work, the kr and K values are determined to be 0.818 min−1 and −0.059 mg L−1, respectively. This shows that the rate of adsorption by the nanocomposite is zero, while the rate of Cr(VI) reduction is very high.
3.5. Regeneration and recycling of the BiOBr–In2S3–SrGO nanocomposite
The regeneration and recycling capacities of the BiOBr–In2S3–SrGO nanocomposite were tested, as reported in the literature.83 Following the photocatalytic reaction, the BiOBr–In2S3–SrGO nanocomposite was immersed in a 10% HNO3 solution for 1 h. It was then repeatedly rinsed with deionized water and dried in a vacuum oven. The reborn BiOBr–In2S3–SrGO nanocomposite was re-examined for its Cr(VI) removal ability using a fresh Cr(VI) solution in the identical experimental setup as discussed earlier. The Cr(VI) removal efficiencies in the 1st, 2nd, and 3rd cycles were 96%, 87%, and 81%, respectively, at an initial dose of 100 mg L−1. This finding indicates that recycling the BiOBr–In2S3–SrGO catalyst is both convenient and cost-effective.
3.6. Mechanism of Cr(VI) removal using the BiOBr–In2S3–S–rGO nanocomposite
Upon visible light irradiation, the electrons exited from the VB to their respective CB of both In2S3 and BiOBr, forming a hole in the VB. The work functions of BiOBr, SrGO, and In2S3 are 6.25 eV,84 5.1 eV,85 and 3.95 eV86 respectively. The Fermi level of a semiconductor can be estimated using the following formula: Ef = Evac. − Wf, where, Ef: represents the Fermi level, Evac stands for the vacuum level, and Wf: denotes the work function.84,87,88 Generally, the Fermi level is more negative the greater the work function.89–93 Hence, the Fermi level of BiOBr is more negative compared to SrGO. Similarly, the Fermi level of SrGO is more negative compared to In2S3. So, the photo-induced electrons from the CB of BiOBr can straightforwardly migrate to the S–rGO NMs through the Schottky barrier. In the meantime, the electron from S-doped rGO NMs also transfers to the VB of In2S3, due to the more negative Fermi energy of S–rGO compared to the VB level of In2S3. Furthermore, in the present study, this observation has been validated by Mott–Schottky analysis. Applying Mott–Schottky analysis, the ECB positions of BiOBr and In2S3 were calculated at −0.16 V and −0.81 V, respectively. Similarly, EVB positions of BiOBr and In2S3 were calculated at about 2.65 V and 1.54 V, respectively. This observation clearly indicated that the movement of the e− follows the Z-scheme pattern from the CB of BiOBr (−0.16 V) towards S–rGO (Eg 1.4 V) and then moves to the VB of In2S3 (1.54 V) and finally recombines with the holes of In2S3 as shown in Scheme 1. Thus, VB holes of In2S3 and CB electrons of BiOBr were annihilated efficiently preventing the photoexcited charge carrier's recombination and consequently improving interfacial charge transmission. Moreover, the S–rGO offers a larger surface as well as SP2 hybridization and π–π conjugation, considerably enhancing the mobility of photoinduced charge carriers between BiOBr and In2S3. Finally, the electrons from the CB of In2S3 are used to reduce Cr(VI), as shown in Scheme 1. Furthermore, the ECB of In2S3 (−0.81 eV)13,94 is substantially more negative than the Cr(VI) reduction potential (1.33 eV versus NHE18), thus, the photoinduced electrons at the CB of In2S3 can efficiently reduce Cr(VI) ions to Cr(III) ion. Similarly, the EVB of BiOBr (2.65 eV)59 is more positive than the reduction potential of O2/H2O (1.23 eV against NHE),84 thus, the photoinduced holes at the VB of BiOBr can effectively oxidize 2H2O to O2 and 2H+ simultaneously. The steps of the chemical reaction are presented as follows.| | | In2S3/BiOBr + hv (visible) → In2S3 (eCB−) + BiOBr (hVB+) | (10) |
| | | 2H2O + BiOBr (4hVB+) → O2 + 4H+ | (11) |
| | | Cr2O72− + 14H+ + In2S3 (6eCB−) → 2Cr(III) + 7H2O | (12) |
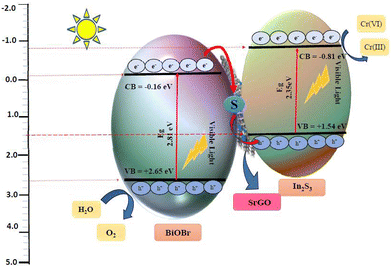 |
| | Scheme 1 Mechanism of Cr(VI) reduction using the BiOBr–In2S3–SrGO nanocomposite. | |
4. Conclusion
A novel BiOBr–In2S3–SrGO Z-scheme photocatalyst was prepared by a facile hydrothermal method. The XRD, FTIR, Raman, UV-Vis DRS, FE-SEM, HRTEM, EDAX, XPS and PL analysis verified that the nanocomposite was well prepared. The results reveal that at an initial 100 mg L−1 Cr(VI), almost 96.6% removal was achieved by the nanocomposite within 2 h under the illumination of solar light. Furthermore, the estimated apparent rate constant (Kapp) of the nanocomposite for Cr(VI) reduction was found to be 3 times more than the individual NMs. This might be due to the efficient inhibition of electron–hole pair recombination, superior charge transfer by formation of a z scheme heterojunction, and application of S-doped rGO. FE-SEM of the nanocomposite proves the uniform distribution of rectangular 3D nanoplatelets of BiOBr and dense nanospheres of In2S3 anchored on the interconnected 3D porous S–rGO. The 3D nanoplatelet structure offers a greater surface area, while the exfoliated and corrugated S–rGO structure facilitates charge transfer and thus enhances photocatalytic activity. Furthermore, the BiOBr–In2S3–SrGO reveals outstanding stability and can be reused for several consecutive cycles. Therefore, this work offers a novel method for creating an effective Z-scheme photocatalyst system for treating Cr(VI)-contaminated wastewater and provides an excellent prospect for application in environmental and aquatic ecosystems.
Author contributions
Satyanjib Sahoo: methodology, conduct experiments and investigation, analysed sample data curation, writing – original draft, writing – review & editing, formal analysis. Naresh Kumar Sahoo: conceptualization, methodology, investigation, visualization, writing – original draft, supervision, review & editing, resources, formal analysis, funding acquisition. Prasanta Kumar Sahoo: conceptualization, data curation, writing – original draft, formal analysis review & editing. Soumya Mishra: methodology, conduct experiments and investigation, analyzed sample, writing – review & editing. Arun Kumar: conduct experiments and investigation, analyzed sample, writing – review & editing. Rundaban Naik: investigation, conceptualization, data curation, writing – original draft, formal analysis review & editing. Prangyan Ranjan Rout: conceptualization, data curation, writing – original draft, formal analysis review & editing.
Data availability
Data will be made available on request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support received from the Department of Biotechnology, Ministry of Science and Technology, Government of India (BT/PR15242/BCE/8/1144/2015 and SAN No. 102/IFD/SAN/3612/2016-2017), for conducting this research work.
References
- Y.-U. Shin, W. Pan, S. K. Patel, J. Lim, L. R. Winter, W. Ma, S. Hong and M. Elimelech, Chem. Eng. J., 2023, 474, 145756 CrossRef CAS.
- S. Singh, A. G. Anil, S. Khasnabis, V. Kumar, B. Nath, V. Adiga, T. S. S. Kumar Naik, S. Subramanian, V. Kumar, J. Singh and P. C. Ramamurthy, Environ. Res., 2022, 203, 111891 CrossRef CAS PubMed.
- M. Abinaya, K. Govindan, M. Kalpana, K. Saravanakumar, S. L. Prabavathi, V. Muthuraj and A. Jang, J. Hazard. Mater., 2020, 397, 122885 CrossRef CAS PubMed.
- G. Yuan, F. Li, K. Li, J. Liu, J. Li, S. Zhang, Q. Jia and H. Zhang, Bull. Chem. Soc. Jpn., 2021, 94, 1142–1155 CrossRef CAS.
- E. T. Anthony and N. A. Oladoja, Environ. Sci. Pollut. Res., 2022, 29, 8026–8053 CrossRef CAS PubMed.
- S. Mitra, A. Sarkar and S. Sen, Nanotechnol. Environ. Eng., 2017, 2, 11 CrossRef.
- N. Doufar, M. Benamira, H. Lahmar, M. Trari, I. Avramova and M. T. Caldes, J. Photochem. Photobiol., A, 2020, 386, 112105 CrossRef CAS.
- C.-C. Wang, X.-D. Du, J. Li, X.-X. Guo, P. Wang and J. Zhang, Appl. Catal., B, 2016, 193, 198–216 CrossRef CAS.
- J. Zeng and Y. Huang, Int. J. Hydrogen Energy, 2024, 51, 423–432 CrossRef CAS.
- L. Meng, Y. Qu and L. Jing, Chin. Chem. Lett., 2021, 32, 3265–3276 CrossRef CAS.
- J. Sun, C. Jiang, Z. Wu, Y. Liu and S. Sun, Chemosphere, 2022, 308, 136107 CrossRef CAS.
- X. Zhong, Y. Liu, S. Wang, Y. Zhu and B. Hu, Sep. Purif. Technol., 2021, 279, 119627 CrossRef CAS.
- Y. Hua, C. Hu, M. Arif, S. Chen, M. Zhang and X. Liu, J. Alloys Compd., 2022, 908, 164488 CrossRef CAS.
- J. Singh and R. K. Soni, Sci. Rep., 2021, 11, 15352 CrossRef CAS PubMed.
- S. Bhattacharya, A. A. Das, G. Chandra Dhal, P. K. Sahoo, A. Tripathi and N. K. Sahoo, J. Environ. Manage., 2022, 302, 114022 CrossRef CAS.
- J. Li, Z. Ding, J. Li, C. Wang, L. Pan and G. Wang, J. Chem. Eng., 2021, 407, 127199 CrossRef CAS.
- Z. Jiang, F. Yang, G. Yang, L. Kong, M. O. Jones, T. Xiao and P. P. Edwards, J. Photochem. Photobiol., A, 2010, 212, 8–13 CrossRef CAS.
- J. Luo, X. Zhou, X. Ning, L. Zhan, L. Ma, X. Xu, Z. Huang and J. Liang, New J. Chem., 2017, 41, 845–856 RSC.
- Z. Tian, J. Li, G. Zhu, J. Lu, Y. Wang, Z. Shi and C. Xu, Phys. Chem. Chem. Phys., 2015, 18, 1125–1130 RSC.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef.
- G. Venkatesan and T. Subramani, Indian J. Mar. Sci., 2019, 48, 528–534 Search PubMed.
- J. Chen, X. Xiao, Y. Wang, M. Lu and X. Zeng, J. Alloys Compd., 2019, 800, 88–98 CrossRef CAS.
- G. D. O. Lima, T. P. Araujo, J. R. D. O. Lima, G. M. P. Prazeres, C. W. A. Paschoal, E. Longo, A. A. Tanaka, A. P. MacIel, D. A. B. Barbosa and M. A. P. Almeida, Bol. Soc. Esp. Ceram. Vidrio, 2021, 60, 2–12 CrossRef CAS.
- Y. Wang, L. Lin, F. Li, L. Chen, D. Chen, C. Yang and M. Huang, Photochem. Photobiol. Sci., 2016, 15, 666–672 CrossRef CAS PubMed.
- M. Kraini, F. W. Aldbea, N. Bouguila and C. Vázquez-Vázquez, J. Appl. Sci., 2018, 17 Search PubMed.
- G. R. Gopinath, R. W. Miles and K. T. Ramakrishna Reddy, Energy Procedia, 2013, 34, 399–406 CrossRef CAS.
- M. Brahmayya, S.-Y. Suen and S. A. Dai, J. Taiwan Inst. Chem. Eng., 2018, 83, 174–183 CrossRef CAS.
- P. Kumar Sahoo, B. Panigrahy and D. Bahadur, RSC Adv., 2014, 4(89), 48563–48571 RSC.
- S. S. Imam, R. Adnan, N. H. Mohd Kaus and M. H. Hussin, J. Mater. Sci.: Mater. Electron., 2019, 30, 6263–6276 CrossRef CAS.
- A. Alsulamei and A. Timoumi, Opt. Mater.: X, 2022, 15, 100176 CAS.
- A. Timoumi, W. Zayoud, A. Sharma, M. Kraini, N. Bouguila, A. Hakamy, N. Revaprasadu and S. Alaya, J. Mater. Sci.: Mater. Electron., 2020, 31, 13636–13645 CrossRef CAS.
- M. Toumi, N. Bouguila, B. Tiss, C. Dias, R. C. Veloso, M. Kraini, J. Ventura and S. Alaya, Solid State Sci., 2022, 133, 107021 CrossRef CAS.
- S. Hassanpoor and E. Tamri, J. Alloys Compd., 2023, 932, 167711 CrossRef CAS.
- J. Wang, Y. Zhang, L. Tian, F. Liu and Q. Xia, J. Nanopart. Res., 2014, 16, 2691 CrossRef.
- T. Sall, A. Nafidi, B. M. Soucase, M. Mollar, B. Hartitti and M. Fahoume, J. Semicond., 2014, 35, 063002 CrossRef.
- O. Surucu, M. Isik, M. Terlemezoglu, N. M. Gasanly and M. Parlak, J. Mater. Sci.: Mater. Electron., 2021, 32, 15851–15856 CrossRef CAS.
- M. Chaudhary, V. Doiphode, P. Shinde, A. Punde, P. Vairale, Y. Hase, A. Waghmare, M. Prasad and S. Jadkar, Mater. Today: Proc., 2019, vol. 39, 1889–1893 Search PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- J. Ji, G. Zhang, H. Chen, S. Wang, G. Zhang, F. Zhang and X. Fan, Chem. Sci., 2011, 2, 484–487 RSC.
- C. Wu, C. Zhou, Y. Chen, Z. Peng, J. Yang and Y. Zhang, J. Nanomater., 2021, 2021, 1–14 Search PubMed.
- M. Jiang, Y. Zhang, J. Chen, Q. Liang, S. Xu, C. Yao, M. Zhou and Z. Li, Cellulose, 2020, 27, 8843–8858 CrossRef CAS.
- L. Wang, B. Zhao, C. Wang, M. Sun, Y. Yu and B. Zhang, J. Mater. Chem. A, 2020, 8, 10175–10179 RSC.
- D. A. Bala, H. Ali, D. Eli and T. Yunana, FUDMA J. Sci., 2019, 3, 226–231 Search PubMed.
- S. S. Imam, R. Adnan and N. H. Mohd Kaus, Appl. Sci., 2019, 1, 845 Search PubMed.
- S. K. Sahoo, A. A. Das, D. Deka, B. Naik and N. Kumar Sahoo, J. Mol. Liq., 2021, 339, 116721 CrossRef CAS.
- A. Han, J. Sun, G. K. Chuah and S. Jaenicke, RSC Adv., 2017, 7, 145–152 RSC.
- Y. Li, H. Jiang, X. Wang, X. Hong and B. Liang, RSC Adv., 2021, 11, 26855–26875 RSC.
- A. R. Warrier, J. Bingi and C. Vijayan, Plasmonics, 2016, 11, 953–961 CrossRef CAS.
- F. Saadallah, N. Jebbari, N. Kammoun and N. Yacoubi, Int. J. Photoenergy, 2011, 2011, 1–4 CrossRef.
- B. D. Ossonon and D. Bélanger, RSC Adv., 2017, 7, 27224–27234 RSC.
- S. Wu, Y. Xie, X. Zhang, Z. Huang, Y. Liu, M. Fang, X. Wu and X. Min, J. Mater. Res., 2019, 34, 3450–3461 CrossRef CAS.
- L. Wang and L. Zan, J. Nanopart. Res., 2020, 22, 325 CrossRef CAS.
- M. Kraini, N. Bouguila, A. Bettaibi, J. Koaib, C. Vázquez-Vázquez, K. Khirouni, M. A. López-Quintela and S. Alaya, J. Mater. Sci.: Mater. Electron., 2016, 27, 11556–11564 CrossRef CAS.
- X. Fu, J. Tao, Z. He, Y. Gao, Y. Xia and Z. Zhao, J. Alloys Compd., 2023, 936, 168202 CrossRef CAS.
- X. Tu, S. Qian, L. Chen and L. Qu, J. Mater. Sci., 2015, 50, 4312–4323 CrossRef CAS.
- J. Shang, Y. Gao, B. Zhao, S. Shen and X. Wang, J. Mater. Sci.: Mater. Electron., 2020, 31, 20858–20867 CrossRef CAS.
- W. Huang, X. Hua, Y. Zhao, K. Li, L. Tang, M. Zhou and Z. Cai, J. Mater. Sci.: Mater. Electron., 2019, 30, 14967–14976 CrossRef CAS.
- Y. Hua, C. Hu, M. Arif, S. Chen, M. Zhang and X. Liu, J. Alloys Compd., 2022, 908, 164488 CrossRef CAS.
- W. Yao, D. Li, S. Wei, X. Liu, X. Liu and W. Wang, ACS Omega, 2022, 7, 36479–36488 CrossRef CAS PubMed.
- A. M. Sadanandan, M. Fawaz, N. P. Dharmarajan, M. Huš, G. Singh, C. Sathish, B. Likozar, Z. Li, A. M. Ruban, C.-H. Jeon, J.-H. Yang, P. Kumar and A. Vinu, Appl. Catal., B, 2024, 124701 Search PubMed.
- J. Xu, B. Luo, W. Gu, Y. Jian, F. Wu, Y. Tang and H. Shen, New J. Chem., 2018, 42, 5052–5058 RSC.
- T. Jia, J. Wu, Y. Xiao, Q. Liu, Q. Wu, Y. Qi and X. Qi, J. Colloid Interface Sci., 2021, 587, 402–416 CrossRef CAS.
- S. Fu, H. Zhu, Q. Huang, X. Liu, X. Zhang and J. Zhou, J. Alloys Compd., 2021, 878, 160372 CrossRef CAS.
- H. Chai, L. Gao, P. Wang, F. Li, G. Hu and J. Jin, Appl. Catal., B, 2022, 305, 121011 CrossRef CAS.
- X. Ren, Y. Sun, H. Xing, K. Wu, W. Wang, J. Yin, S. Yao, X. Zhao and H. Yang, J. Nanopart. Res., 2019, 21, 111 CrossRef.
- J. Park, T. H. Lee, C. Kim, S. A. Lee, M.-J. Choi, H. Kim, J. W. Yang, J. Lim and H. W. Jang, Appl. Catal., B, 2021, 295, 120276 CrossRef CAS.
- H. Y. Hafeez, J. Mohammed, C. E. Ndikilar, A. B. Suleiman, R. S. Sa’id and I. Muhammad, Ceram. Int., 2023, 49, 5269–5278 CrossRef CAS.
- S. Choudhury, S. Pattnayak, U. Sahoo, P. Aparajita and G. Hota, Ind. Eng. Chem. Res., 2023, 62, 8191–8209 CrossRef CAS.
- X.-L. Luo, S.-Y. Yang, Z.-L. Wang and Y.-H. Xu, Sep. Purif. Technol., 2023, 318, 123966 CrossRef CAS.
- S. Habi Ben Hariz, H. Lahmar, G. Rekhila, A. Bouhala, M. Trari and M. Benamira, J. Photochem. Photobiol., A, 2022, 430, 113986 CrossRef CAS.
- B. Dai, Y. Li, J. Xu, C. Sun, S. Li and W. Zhao, Appl. Surf. Sci., 2022, 592, 153309 CrossRef CAS.
- N. Khosroshahi, M. Bakhtian and V. Safarifard, J. Photochem. Photobiol., A, 2022, 431, 114033 CrossRef CAS.
- N. Khosroshahi, M. D. Goudarzi, M. E. Gilvan and V. Safarifard, J. Mol. Struct., 2022, 1263, 132994 CrossRef CAS.
- V. Balakumar, C. Chuaicham, K. Sasaki and K. Sekar, Mater. Today: Proc., 2021, vol. 50, 400–405 CrossRef.
- A. Chachvalvutikul, T. Luangwanta and S. Kaowphong, J. Colloid Interface Sci., 2021, 603, 738–757 CrossRef CAS PubMed.
- R. Yang, Z. Zhu, C. Hu, S. Zhong, L. Zhang, B. Liu and W. Wang, J. Chem. Eng., 2020, 390, 124522 CrossRef CAS.
- A. Raja, P. Rajasekaran, K. Selvakumar, M. Arivanandhan, S. Asath Bahadur and M. Swaminathan, ACS Omega, 2020, 5, 6414–6422 CrossRef CAS PubMed.
- X. Yuan, X. Wu, Z. Feng, W. Jia, X. Zheng and C. Li, Catalysts, 2019, 9, 624 CrossRef CAS.
- G. Wu, Q. Liu, J. Wang, S. Xia, X. Huang, J. Han and W. Xing, Colloids Surf., A, 2022, 653, 130048 CrossRef CAS.
- L. Luo, S. Dong, H. Cui, L. Sun and T. Huang, J. Colloid Interface Sci., 2022, 606, 1299–1310 CrossRef CAS PubMed.
- M. Yu, J. Shang and Y. Kuang, J. Mater. Sci. Technol., 2021, 91, 17–27 CrossRef CAS.
- D. S. Pattanayak, J. Mishra, J. Nanda, P. K. Sahoo, R. Kumar and N. K. Sahoo, J. Environ. Manage., 2021, 297, 113312 CrossRef CAS PubMed.
- S. C. Xu, Y. X. Zhang, S. S. Pan, H. L. Ding and G. H. Li, J. Hazard. Mater., 2011, 196, 29–35 CrossRef CAS.
- Z. Li, B. Liu, X. Zhang, C. Zhang, Y. Bai, J. Liu, Y. Wang, S. Yang, R. Li and C. Fan, Sustainable Energy Fuels, 2024, 8, 262–271 RSC.
- F. Sun, H. Ghosh, J. Wang, Z. Tan and S. Sivoththaman, IEEE Trans. Nanotechnol., 2022, 21, 481–488 CAS.
- J. Zhang, L. Ding, W. Sun, W. Bi, Z. Wu and F. Gao, J. Alloys Compd., 2021, 889, 161770 CrossRef.
- X. Fu, L. Wang, Z. He, Y. Gao, Y. Xia, J. Tao and Z. Zhao, J. Solid State Chem., 2023, 319, 123794 CrossRef CAS.
- Y. Wang, L. Qiu, S. Bao, F. Tian, L. He, W. Yang, Y. Liu and Y. Yu, J. Chem. Eng., 2023, 468, 143768 CrossRef CAS.
- X. Fu, L. Wang, F. Gao, C. Zhu, J. Tao, Z. He, Y. Xia and Z. Zhang, Fuel, 2024, 368, 131700 CrossRef CAS.
- H. Gong, C. Bao, X. Luo, Y. Yu and W. Yang, Microchem. J., 2024, 198, 110109 CrossRef CAS.
- Y. Wang, S. Bao, X. Liu, L. Qiu, J. Sheng, W. Yang and Y. Yu, J. Chem. Eng., 2023, 477, 147050 CrossRef CAS.
- X. Guo, L. Qiu, M. Li, F. Tian, X. Ren, S. Jie, S. Geng, G. Han, Y. Huang, Y. Song, W. Yang and Y. Yu, J. Chem. Eng., 2024, 483, 149264 CrossRef CAS.
- L. Li, F. Tian, L. Qiu, F. Wu, W. Yang and Y. Yu, Catalysts, 2023, 13, 1497 CrossRef CAS.
- X. Fu, J. Tao, Z. He, Y. Gao, Y. Xia and Z. Zhao, J. Alloys Compd., 2023, 936, 168202 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Prasanta Kumar
Sahoo
b,
Soumya
Mishra
a,
Arun
Kumar
c,
Brundabana
Naik
*a,
Prasanta Kumar
Sahoo
b,
Soumya
Mishra
a,
Arun
Kumar
c,
Brundabana
Naik