
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrating hydroxyapatite and bovine bone mineral into cellulose–collagen matrices for enhanced osteogenesis†
Tudor
Pinteala
ab,
Paul-Dan
Sirbu
 ab,
Narcis
Anghel
*c,
Irina
Rosca
c,
Geanina
Voicu
d,
Manuela
Calin
d and
Iuliana
Spiridon
c
ab,
Narcis
Anghel
*c,
Irina
Rosca
c,
Geanina
Voicu
d,
Manuela
Calin
d and
Iuliana
Spiridon
c
aDepartment of Orthopedics and Traumatology, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, 700115 Iasi, Romania
bDepartment Orthopedics and Traumatology, Clinical Rehabilitation Hospital, 700661 Iasi, Romania
c“Petru Poni” Institute of Macromolecular Chemistry, Grigore Ghica–Vodă 41, 700487 Iasi, Romania. E-mail: anghel.narcis@icmpp.ro
d“Medical and Pharmaceutical Bionanotechnologies” Laboratory, Institute of Cellular Biology and Pathology “Nicolae Simionescu” of the Romanian Academy, B.P. Hasdeu 8, 050568 Bucharest, Romania
First published on 7th November 2024
Abstract
This study investigates novel biomaterials developed for bone regeneration, using cellulose and collagen type I matrices enhanced with hydroxyapatite or InterOss. These materials demonstrate significantly improved mechanical properties, notably the compressive modulus, indicating their potential for effective structural support in bone regeneration. Incorporating hydroxyapatite into these matrices markedly improves their physical properties, increasing the Brunauer–Emmett–Teller area and monolayer capacity, thereby facilitating superior cell adhesion and proliferation. This enhancement promotes more effective osteoblast activity and viability over extended periods compared to matrices containing InterOss. Furthermore, the scaffolds comprising cellulose modified with (3-amino-4-methylphenyl) boronic acid exhibit significantly enhanced antibacterial properties, effectively inhibiting both Gram-positive and Gram-negative bacteria, which is crucial for preventing post-surgical infections. Materials that incorporate hydroxyapatite (HA) have displayed a rougher and more intricate surface compared to those that include InterOss® particles, suggesting that HA promotes the development of an enhanced mineralized skeleton within the composites. Cytocompatibility studies revealed that the scaffold containing cellulose, collagen, and hydroxyapatite provided the most favorable environment for sustaining cell viability, with significant improvements noted from day 7 onwards. Despite initial cytotoxicity challenges, long-term exposure showed improved cell viability, suggesting degradation of cytotoxic products over time. This research underscores the clinical potential of these biomaterials in bone regeneration, highlighting their ability to enhance structural integrity, support osteogenic activity, and prevent bacterial infections, thus promising to improve patient outcomes in bone-related therapies.
1. Introduction
Bone fractures are a major public health issue due to their high morbidity rates and substantial economic burden on healthcare systems. The problem is becoming more urgent as global populations age and bone-related diseases like cancer, osteoporosis, and dental anomalies become more prevalent. Notably, proximal tibial plateau fractures in the elderly are on the rise,1 with 95% of patients over 70 years of age experiencing some degree of osteoporosis, which complicates both the condition and its treatment.2,3The growing need for advanced bone regeneration techniques is underscored by the critical role that bone plays in maintaining essential physiological functions, such as regulating blood calcium levels. In response to these challenges, there has been a surge of interest in bone substitute materials,4 which include both inorganic and organic components that enhance structural integrity, stiffness, and mineral homeostasis.5,6
Natural and synthetic polymers have become focal points in research on bone repair and regeneration due to their biocompatibility, mechanical strength, and biodegradability.7 Natural polymers, such as collagen, chitosan, and hyaluronic acid, provide excellent biocompatibility and osteoconductivity, facilitating bone cell attachment and minimal immunogenic responses. However, their mechanical stability and inconsistent degradation rates often limit their long-term application in bone tissue engineering. Synthetic polymers, in contrast, can be engineered to provide controlled degradation rates and improved cellular attachment by incorporating negatively charged chemical groups, which makes them more adaptable to therapeutic requirements.8
Collagen derived from marine environments, in particular, has shown promise in facilitating cellular migration and adhesion, two key factors in effective tissue integration and regeneration. Marine-derived collagen stands out for its biocompatibility, low immunogenicity, and biodegradability, along with its ability to attract migrating cells – qualities that make it ideal for tissue engineering applications.9–11 Scientific research has shown that marine-derived collagen can promote osteogenic differentiation. For instance, Liu and Sun12 demonstrated that fish-derived collagen scaffolds enhance the osteogenic differentiation of human mesenchymal stem cells, highlighting the material's relevance in bone regeneration.
Novel biomaterials were developed using cellulose and collagen type I derived from bovine Achilles tendon as the matrices for bone regeneration. These materials combine the beneficial structural and functional properties of both components, which are critical for effective bone tissue engineering. Cellulose, a polysaccharide composed of β-D-glucose units, is known for its biodegradability,13 biocompatibility,14 and strong mechanical properties.15 These characteristics have made it suitable for various applications, particularly in dentistry.16,17 However, cellulose's limited bioactivity poses challenges in bone regeneration, so osteogenic bioactive agents are frequently added to enhance its performance in these applications.18
Hydroxyapatite (HA), chemically represented as Ca10(PO4)6(OH)2, is another critical component in bone regeneration. It closely mimics the natural composition of human bone, making it highly biocompatible and osteoconductive, promoting new bone growth and cell attachment.19 The material is bioactive and supports the proliferation of bone cells, facilitating osseointegration – the process of bonding implants with bone tissue. Studies by Cool et al.20 emphasize the role of HA in facilitating this critical process. HA's osteoconductivity allows it to serve as a scaffold for new bone growth, integrating seamlessly with surrounding bone tissue. Moreover, HA can be engineered into various forms, such as powders, granules, and porous structures, making it versatile for different types of bone defects and applications. Its mechanical properties can also be tailored to match those of bone, reducing the risk of implant failure.21–23
InterOss®, a natural bone substitute material derived from bovine bones, is widely used in dental and orthopedic applications for bone grafting and regeneration. InterOss® consists primarily of hydroxyapatite and undergoes rigorous processing to remove organic components, which prevents immunogenic reactions and ensures biocompatibility. Its highly porous structure facilitates vascularization and promotes rapid host tissue integration, making it a preferred choice for bone regeneration in dental implantology and orthopedic surgeries. Studies have shown that InterOss® is effectively maintaining bone volume and density post-grafting, which is critical for the long-term success of implants and other orthopedic devices.24–27
In addition to advancements in bone substitute materials, the role of antioxidants in bone regeneration has garnered increasing attention, particularly in managing osteoporosis and bone inflammatory conditions. Oxidative stress, induced by reactive oxygen species (ROS), has a significant impact on bone metabolism, contributing to bone degradation and inflammatory diseases. Antioxidants are now being incorporated into bone regeneration materials to combat ROS-induced oxidative stress and promote bone health.28,29
Trolox, a water-soluble analog of vitamin E, is widely recognized for its potent antioxidant properties. It acts as a scavenger of ROS, neutralizing free radicals that cause cellular damage. Trolox has been extensively studied for its role in enhancing cell viability and protecting tissues from oxidative stress, which is particularly beneficial in tissue engineering and regenerative medicine.30
In the field of bone regeneration, Trolox has shown the potential to improve the viability and functionality of cells under oxidative stress conditions. Studies have demonstrated that it enhances the resilience of osteoblasts, the cells responsible for bone formation, in harsh environments, thereby supporting tissue regeneration and healing. Morabito et al.31 assessed the impact of Trolox on osteoblastic cells, showing that it significantly improved cell viability under oxidative conditions. These findings highlight the potential of incorporating antioxidants like Trolox into bone regeneration materials to improve their efficacy.
The study aims to evaluate and develop innovative biomaterials for bone regeneration, specifically integrating hydroxyapatite and InterOss® into a matrix consisting of cellulose or cellulose modified with (3-amino-4-methylphenyl) boronic acid, and bovine collagen. Mechanical properties, cell adhesion, and antibacterial performance of these novel scaffolds in comparison to existing materials like InterOss® alone were assessed. The goal is to enhance the structural and biological efficacy of bone regeneration materials, offering improved solutions for clinical applications. Results showed that the collagen–cellulose matrix not only promoted superior cell adhesion but also supported the prolonged viability of MG-63 osteoblast-like cells, particularly in materials that incorporated hydroxyapatite.32
The study's findings suggest that these HA-enhanced collagen–cellulose matrices outperformed InterOss® in key areas such as mechanical strength and cellular interaction. This performance underscores the potential of these novel biomaterials to improve bone regeneration outcomes. As the global population continues to age and the prevalence of bone pathologies increases, ongoing research and investment in bone health technologies are critical to addressing the challenges of bone repair and regeneration. By leveraging cutting-edge materials and technologies, the medical community is better equipped to meet the complex demands of bone repair and regeneration, ultimately improving patient outcomes and addressing this global health challenge.
2. Experimental section
2.1. Materials
Cellulose (cotton linters, ∼20 micrometers, 240 kDa), Collagen hydrolysate, a polypeptide made by further hydrolysis of denatured collagen type I from bovine Achilles's tendon (molecular weight of 96 kDa), 1-allyl-3-methylimidazolium chloride, trolox ((±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid) and hydroxyapatite (HA) were purchased from Sigma-Aldrich and used without further purification. InterOss® from SigmaGraft Inc (Fullerton, CA, USA) was used as received.2.2. Synthesis of dialdehyde cellulose by periodate oxidation
The cellulose was oxidized according to the method described by Toshikj et al.,33 with slight modifications. 0.2 g of KIO4 were added to 1.5 g of cellulose in 50 mL of acetate buffer (pH 4) at room temperature, in dark, upon continuous stirring, for 6 hours. The product (dialdehyde cellulose – DAC) was separated from the mixture through filtration, washed successively with distilled water and ethanol, and dried at room temperature. Yield 1.435 g.2.3. Reductive amination of DAC with (3-amino-4-methylphenyl) boronic acid
Reductive amination of DAC was done according to method described by Simon et al.34 A mixture of 1 g of DAC and 0.1 g of (3-amino-4-methylphenyl) boronic acid in 30 mL of ethanol was stirred at 80 °C for 12 hours. The product was filtered, washed with ethanol, and resuspended in 30 mL of ethanol. 0.2 g of sodium borohydride is added under stirring. After 60 minutes, the product was filtered, washed with water and ethanol, successively and dried at room temperature. Yield 0.877 g. The synthesis of the reaction product, aminomethylphenylboronic cellulose (CellD), was confirmed through FTIR and EDX analysis.2.4. Development of biomaterials
Cellulose or cellulose derivative (1 g) and collagen (0.25 g) were dissolved in 10 g of 1-allyll-3-methylimidazolium chloride at 100 °C, under stirring for 6 hours. Hydroxyapatite or InterOss® (0.025 g), and Trolox (0.005 g) were added into Cel–Coll matrix and various formulations were obtained (Table 1).| Sample code | Cellulose, g | Cellulose derivative, g | Collagen, g | InterOss®, g | Hydroxyapatite, g | Trolox, g |
|---|---|---|---|---|---|---|
| Cell-Coll-InterOss® | 1 | 0.25 | 0.025 | 0.005 | ||
| Cell-Coll-HA | 1 | 0.25 | 0.025 | 0.005 | ||
| CellD-Coll-InterOss® | 1 | 0.25 | 0.025 | 0.005 | ||
| CellD-Coll-HA | 1 | 0.25 | 0.025 | 0.005 |
2.5. Fourier transform infrared spectroscopy (FTIR)
Brüker FTIR Spectrophotometer, Vertex 70 (Billerica, MA, USA) equipped with an attenuated total reflection (ATR) device was used to evidence the interactions between composites’ components. All samples were acquired using a diamond crystal with ZnSe focusing element at room temperature. The analysis was performed in the spectral region of 4000–600 cm−1 with a resolution of 4 cm−1 and an average of 64 scans, at a 45° angle of incidence.2.6. Mechanical testing
All samples were immersed in phosphate buffer saline (PBS) for 24 h to reach saturation prior their testing in wet state. An initial compressive force of 0.1 N was applied in order to ensure a complete contact between the surface of the plate and that of the sample. The compression experiments were conducted at room temperature, using a Shimadzu Testing Machine (EZ-LX/EZ-SX Series, Kyoto, Japan) equipped with a 500 N load cell, at a 1 mm min−1 crosshead speed downwards at 25 °C.2.7. Dynamic vapor sorption (DVS)
An IGAsorp fully automated gravimetric analyzer (Hiden Analytical, Warrington – UK), was used to determine the dynamic water vapor sorption capacity of the studied materials. Measurements were made at 25 °C, in an RH range of 0–90% with 10% increments. The sorption capacity (% d.b.) was measured and the specific surface area was calculated by means of the Brunauer–Emmett–Teller (BET) equation.2.8. Scanning electron microscopy (SEM)
The SEM imaging was performed using a Verios G4 UC Scanning electron microscope (Thermo Scientific, Czech Republic) equipped with energy dispersive X-ray (EDX) spectroscopy analyzer (Octane Elect Super SDD detector, USA). SEM investigations were performed in high vacuum mode at accelerating voltage of 5 kV.2.9. Antimicrobial activity
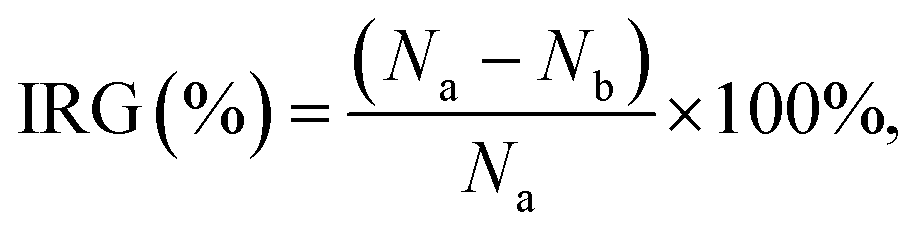 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 according to supplier recommendation.
:4 according to supplier recommendation.
At every experimental time point, the cells grown in monolayer or scaffolds were washed with PBS, fixed with 4% PFA in 0.1 M phosphate buffer (pH 7.4) for 60 minutes at room temperature (RT), and permeabilized with 0.5% Triton X-100 for 15 minutes. Then, the cells were stained with 200 ng mL−1 phalloidin-FITC (catalog no. RD-5782, R&D Systems, Minneapolis, MN, USA) for 1 hour and with 2 μg mL−1 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific, Waltham, Massachusetts, USA, catalog no. 62247) for 15 minutes at RT, protected from light. The samples were examined with the 20× objective through an Inverted Microscope Olympus IX81 (Olympus Corporation Shinjuku, Tokyo, Japan) using the green (FITC) and blue (DAPI) detection filters. For scaffolds, consecutive Z-stacks were acquired with a step size of 7 μm. The images were reconstructed in ImageJ version 1.8.0 software developed at the National Institutes of Health (NIH), USA.
2.10. Cytotoxicity assessment of scaffolds
2.11. Cell toxicity of scaffold leachates (XTT assay)
The four scaffolds were incubated at 37 °C in a complete cell culture medium to obtain the leachates. The scaffold extracts were collected at 3, 7, 14, and 21 days and replaced with fresh medium. The MG-63 cells were incubated with the leachates withdrawn at different intervals and the cytotoxicity was determined using XTT assay. Before incubation with the leachates, MG-63 cells were seeded at a density of 0.1 × 104 cells per well in a 96-well plate for 24 hours. After 24, 48, and 72 hours of cells’ incubation with the leachates, the medium was replaced with 0.2 mg mL−1 XTT solution that contains 5 μM PMS (phenazine methosulfate) in colorless DMEM (1 hour at 37 °C). In the XTT assay, the tetrazolium salt of XTT [(2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)]-2H-tetrazolium-5-carboxyanilide salt) is reduced by the mitochondrial enzymes in healthy cells. It is formed orange-colored water-soluble formazan that can be spectrophotometrically detected. The optical absorbance of the medium was measured at 450 nm using an Infinite® M200 PRO microplate reader spectrophotometer (Tecan, Männedorf, Switzerland). The experiments were performed in duplicate and the results were expressed as percentages relative to control cells maintained in a complete medium.2.12. Cell viability assay (MTS assay)
Human osteoblast-like MG-63 cell line (CLS Cell Lines Service GmbH, Eppelheim, Germany) were seeded in 96-well tissue culture-treated plates at 105 cells per mL in alpha-MEM medium supplemented with 10% fetal bovine serum (both from PAN-Biotech GmbH, Aidenbach, Germany) and 1% penicillin–streptomycin-amphotericin B mixture (Gibco, Thermo Fisher Scientific, Waltham, MA USA). The next day, cells were incubated with leachate material samples (10% in complete cell culture medium) or control medium (10% PBS in complete cell culture medium). After 24 h incubation, biocompatibility assessment was performed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI USA). Absorbance readings at 490 nm were done with a FLUOstar® Omega microplate reader (BMG LABTECH, Ortenberg, Germany) and viability of treated cells was calculated as percentage of the viability of control cells (means ± standard deviation).2.13. Quantification of alkaline phosphatase activity in osteoblasts incubated with the scaffold leachates
To investigate the osteogenic differentiation of MG-63 osteoblasts after incubation with the leachates collected from the collagen-based scaffolds, we determined the alkaline phosphatase (ALP) activity by a colorimetric reaction that uses para-nitrophenyl phosphate (pNPP) as substrate. ALP is a glycoprotein located on the cellular surface and represents an early marker of osteogenic differentiation. The enzymatic activity of ALP was determined after 7 days of MG-63 osteoblasts incubation with the leachates collected at different intervals (7, 14, and 21 days). As controls, osteoblasts cultivated in monolayer and maintained either in DMEM complete medium or osteogenic medium37 (OM, DMEM with 4.5 g L−1 glucose, 50 μg mL−1 ascorbic acid, 10 mM β-glycerophosphate, 10 nmole dexamethasone) were used. After 7 days, the cells were processed for ALP assay using SensoLyte pNPP Alkaline Phosphatase Assay Kit (AnaSpec, Inc. Fremont, CA, USA), according to the manufacturer's instructions.38 The absorbance of the samples was determined at 405 nm with a spectrophotometer TECAN Infinite® M200 PRO and the results were calculated using a standard curve.2.14. Degradation and swelling properties
The scaffolds were cut and immersed in PBS solution (pH = 7.4) at 37 °C. Their weights were recorded before and after immersion on days 2, 4, and 6. The percentage of degradation was determined using eqn (2):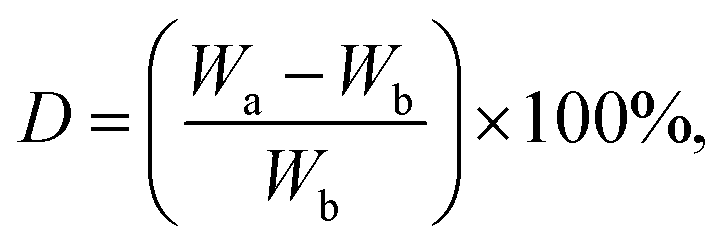 | (2) |
The swelling behavior of the scaffolds was evaluated by immersing them in PBS at 37 °C. The PBS solution was prepared by dissolving 8 g of NaCl, 200 mg of KCl, 1.44 g of Na2HPO4, and 245 mg of KH2PO4 in 800 mL of distilled water, followed by pH adjustment to 7.4 and dilution to a final volume of 1 L. The scaffolds were cut and immersed in the PBS solution for various time intervals: 15, 30, 60, 120, 240, and 480 minutes. Weights were measured before and after immersion at each time point. The swelling percentage was calculated using eqn (3):
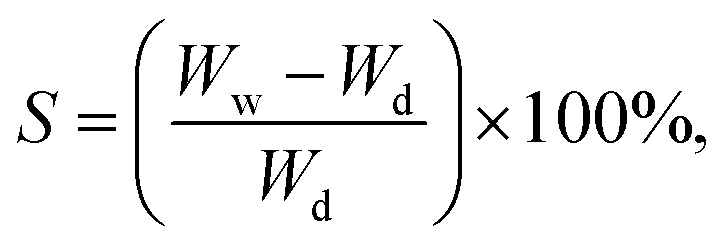 | (3) |
3. Results
3.1. FTIR analysis of biomaterials
The FTIR spectrum of cellulose (Fig. S1a, ESI†) presents a broad absorption peak at 3346 cm−1 corresponding to the stretching of O–H bond, due to the presence of hydroxyl groups in cellulose backbone.Shifts of this band toward the lower frequencies was recorded in cellulose derivative, namely aminomethylphenylboronic cellulose (Fig. S1b, ESI†), due to superimposed absorptions of the both hydroxyl and secondary amine groups.
The absorption band at around 3411 cm−1 corresponds to N–H bending, while the bands allocated to aromatic ring vibrations appear in the spectral range of 1100–1000cm−.39 The new peak at 800.4cm−1 is ascribed to the deformation out-of-plane vibrations -B–O andtorsional deformation vibration of O–H bound.40 The hydroxyl index (HI – attributed to the OH groups stretching) calculated as A3400/A2916 absorbance ratio presents a decrease from 0.517 (cellulose) to 0.4018 (after treatment with 3-amino-4-methylphenylboronic acid) and confirms that reaction was done.
EDX mapping (Fig. S2, ESI†) confirmed the presence of N and B elements in cellulose derivative.
FTIR spectra of the obtained materials are shown in Fig. S3 (ESI†).
Materials comprising cellulose derivative present a peak at 1726 cm−1 ascribed to ester carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). Collagen presence is confirmed by peaks at 1643 cm−1 (amide I) and at ∼1568 cm−1 (amide II).41 The orthophosphates (PO43−) were observed in the range25 960–1120 cm−1 and 623 cm−1.
O). Collagen presence is confirmed by peaks at 1643 cm−1 (amide I) and at ∼1568 cm−1 (amide II).41 The orthophosphates (PO43−) were observed in the range25 960–1120 cm−1 and 623 cm−1.
3.2. Mechanical properties
Materials engineered for bone regeneration should possess mechanical properties that mirror those of natural bone. Liu et al.42 documented that a hybrid material composed of collagen and oxidized cellulose exhibits superior mechanical strength. This enhancement is primarily due to the hydrogen bonds and electrostatic interactions among the constituents. Fig. 1 illustrates that the compressive strength of the synthesized materials varies between 0.14 and 0.24 MPa, while the typical range observed in human trabecular bone is in the range 0.70–15 MPa.43 Importantly, the compressive modulus of these materials surpasses that found in the pericellular matrix of human cartilages and organs (0.144–0.954 MPa),44 highlighting their potential efficacy in bone regeneration applications, including those utilizing InterOss®.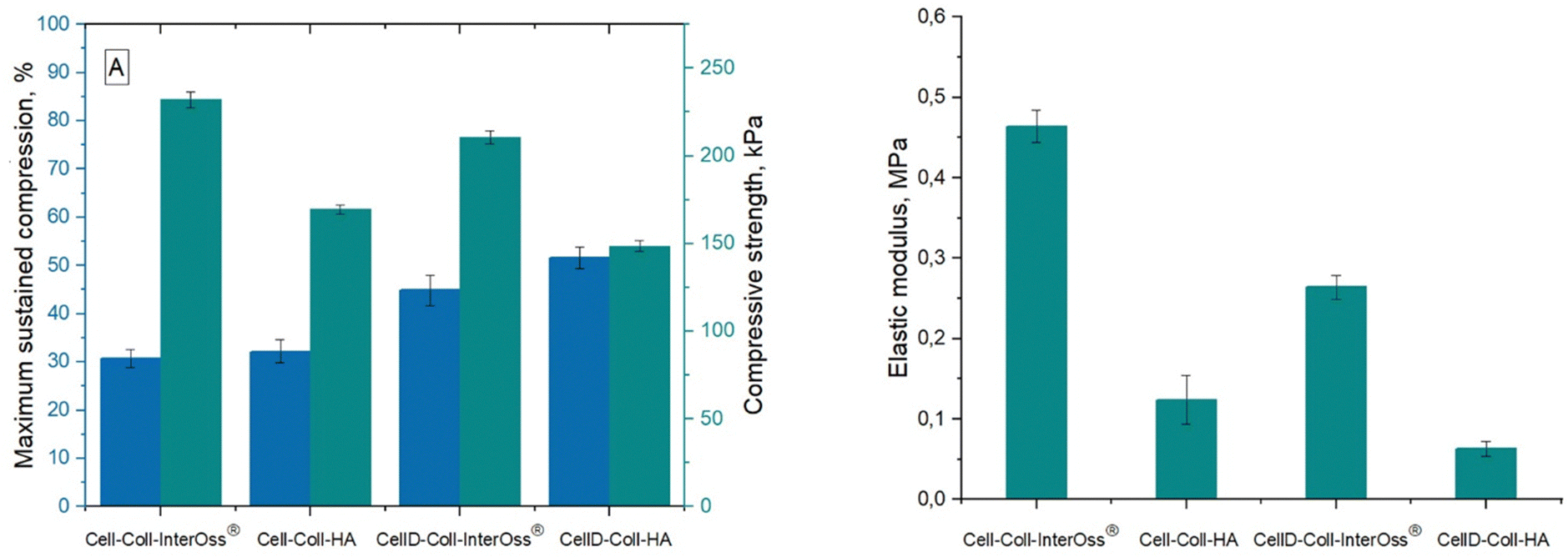 | ||
| Fig. 1 (A) Maximum sustained compression (blue columns) and compressive strength (green columns); (B) the values of compressive elastic modulus are determined from the linear dependence of stress–strain profiles. | ||
Materials incorporating the cellulose derivative demonstrated lower compression strength and elastic modulus, yet they endured significantly higher stress levels. This characteristic can be attributed to the derivative's diminished ability to form robust hydrogen bonds among matrix macromolecules compared to native cellulose. The Hydrogen Bond Index (HBI) values were as follows: 3.5281 (CellD-Coll-InterOss®) > 3.5252 (Cell-Coll-InterOss®) > 3.2585 (Cell-Coll-HA) > 4.077 (CellD-Coll-HA).
Furthermore, the elastic modulus of all tested materials remained below that of human cortical bone, which spans 7 to 30 GPa.45 Notably, materials incorporating InterOss® exhibited higher (with about 43–49%) elastic modulus values, suggesting enhanced mechanical strength attributable to the InterOss® particles. This reinforcement is likely due to the strong interfacial adhesion between the matrix and InterOss® particles, as documented in scanning electron microscopy (SEM) images.
3.3. Dynamic vapor sorption (DVS)
The water absorption capacity of biomaterials is crucial for their effectiveness in absorbing exudate from tissue flaws and their overall compatibility within the tissue environment. It has been noted that the specific surface area plays a vital role in bone tissue engineering, enhancing the uniform distribution, attachment, and the migration, proliferation, and differentiation of biological cells.46 The porous structure of the surface also facilitates vascularization and bone ingrowth. To model the sorption isotherms, the Brunauer–Emmett–Teller (BET) and Guggenheim–Anderson–de Boer (GAB) equations were employed. The experimental results indicated a better fit with the BET model compared to the GAB model.47Results from Fig. 2 and Table 2 indicate that incorporating InterOss® particles into the cellulose–collagen matrix significantly enhances the Brunauer–Emmett–Teller (BET) area and monolayer capacity. Conversely, in systems utilizing a cellulose derivative, an adverse effect was observed on these parameters. Notably, the material identified as CellD-Coll-HA, which exhibited the highest BET value (with about 60.2% compared to the rest of the tested stuffs), demonstrated superior cell adhesion capabilities compared to materials containing hydroxyapatite (HA) alone. This suggests that the specific architectural features of the CellD-Coll-HA matrix may more effectively promote cell attachment and subsequent tissue integration.
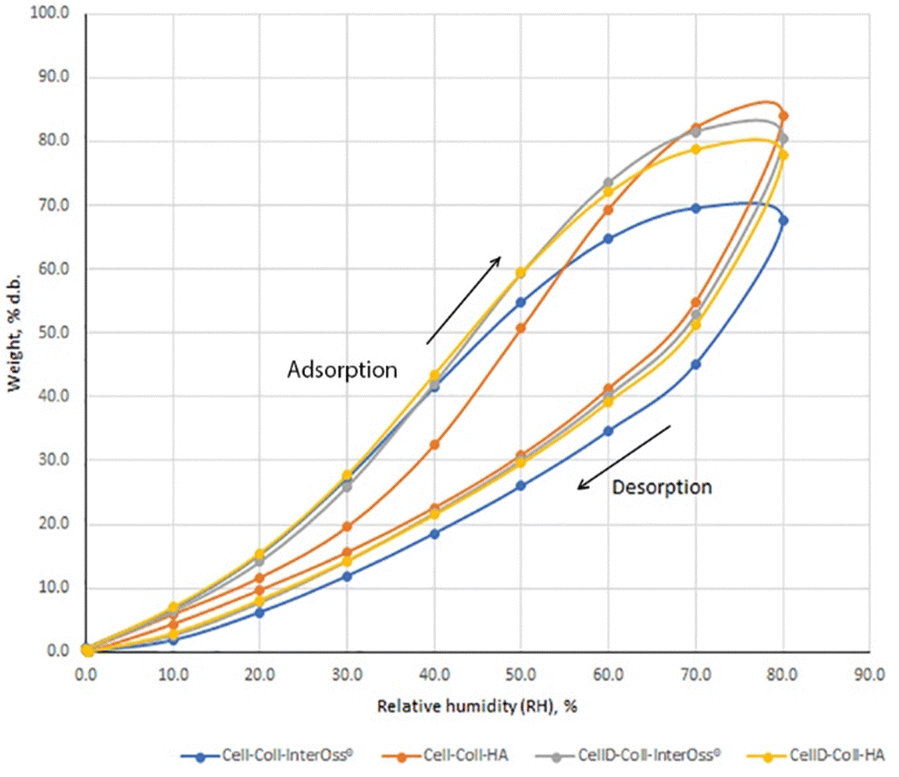 | ||
| Fig. 2 Dynamic vapor sorption isotherms of biomaterials. | ||
| Sample code | Sorption capacity, % | BET area, m2 g−1 | Monolayer, g g−1 |
|---|---|---|---|
| Cell-Coll-InterOss® | 67.62 | 1387.854 ± 872.498 | 0.395291 |
| Cell-Coll-HA | 84.02 | 944.020 ± 79.280 | 0.268878 |
| CellD-Coll- InterOss® | 80.43 | 941.313 ± 164.116 | 0.268107 |
| CellD-Coll-HA | 77.74 | 2375.867 ± 244.708 | 0.276700 |
3.4. Scanning electron microscopy (SEM)
The surface morphology of the materials, as illustrated in Fig. 3, plays a crucial role in influencing cellular behaviors, including adhesion, proliferation, and migration. Scanning electron microscopy analysis reveals that mineral particles are evenly distributed throughout the polymer matrix. However, the morphology of materials that incorporate a cellulose derivative shows a higher incidence of microcracks, indicating that the added fillers are not tightly embedded within the polymeric matrix. These microcracks suggest that the interfacial bonding between the components is insufficient for effective energy dissipation.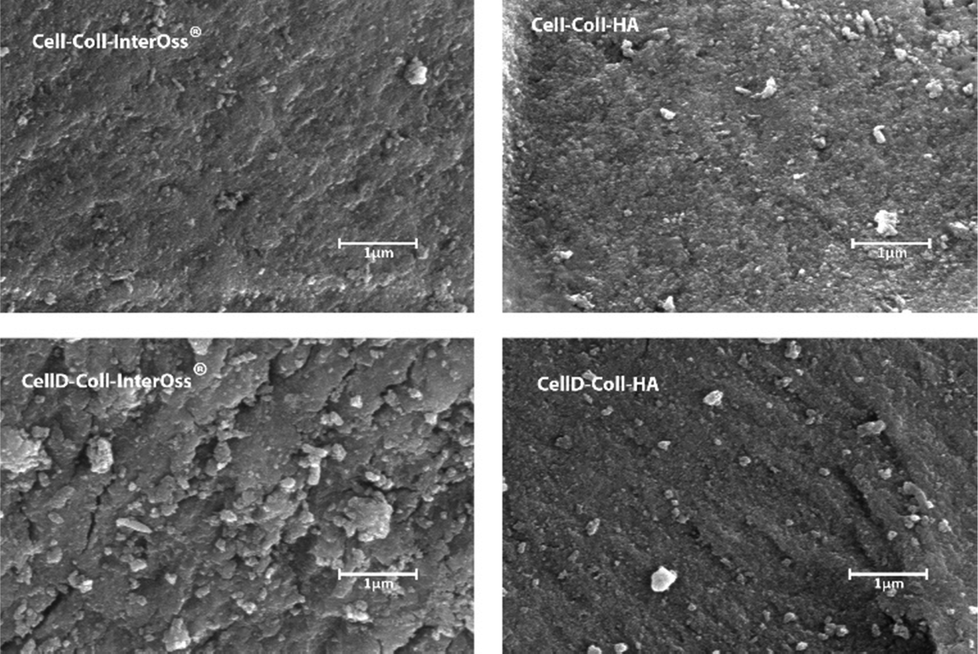 | ||
| Fig. 3 SEM images of biomaterials. | ||
Consequently, materials that include cellulose derivatives in their matrix demonstrate lesser fracture resistance compared to those that incorporate unmodified cellulose. Materials that incorporate hydroxyapatite (HA) have displayed a rougher and more intricate surface compared to those that include InterOss® particles, suggesting that HA promotes the development of an enhanced mineralized skeleton within the composites. The rough surface texture and greater surface area associated with HA composites are undoubtedly more conducive to cell attachment and proliferation. On the other hand, materials containing InterOss® particles exhibit a more compact structure, which correlates well with their improved mechanical properties. This distinction in surface morphology between the two types of composites highlights their different functional potentials in tissue engineering applications.
EDX spectrum of all materials (Fig. S2, ESI†) have similar elements, including chlorine, carbon, oxygen, and nitrogen, derived from the collagen and cellulose dissolved in ionic liquid, while calcium and phosphorus are derived from the HA or InterOss® particles. The presence of B in materials comprising cellulose derivative was evidenced.
3.5. Antimicrobial activity of biomaterials
The antibacterial efficacy of the materials was evaluated, as illustrated in diagrams presented in Fig. 4(a) and (b), which depict the inhibition of bacterial colony growth on the samples tested. The materials exhibited varying degrees of antibacterial activity against Staphylococcus aureus and Escherichia coli. Specifically, against the Gram-positive bacterial strain S. aureus, there was a significant reduction in cell viability, dropping to 1–5% after just 4 hours of incubation. Subsequently, no colonies were detected on the plates up to 72 hours of incubation, as shown in Fig. 5(c). This indicates a potent antibacterial effect of the materials against S. aureus, highlighting their potential for preventing bacterial infections in clinical applications. In the case of the Gram-negative bacterial colonies, represented by Escherichia coli, the antibacterial efficacy of the materials was notably. After just 2 hours of incubation, cell viability was reduced to less than 1%, as depicted in Fig. 4(b). Furthermore, the development of bacterial cells was completely halted after 4 hours, with this effect sustained for up to 72 hours, as shown in Fig. 5(d).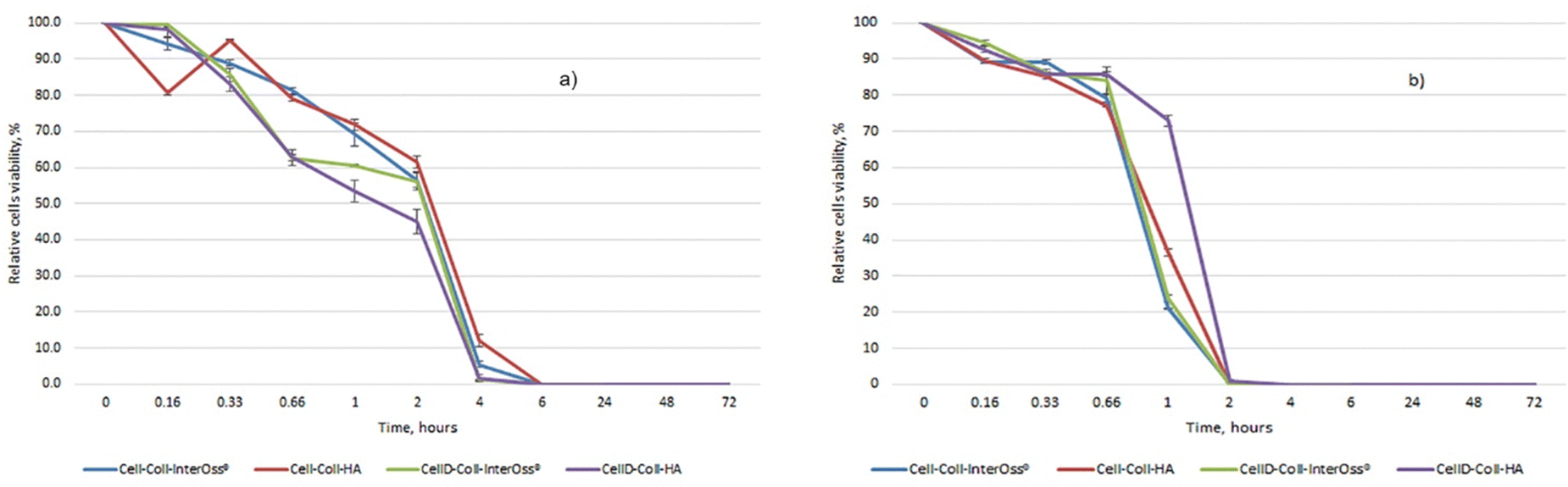 | ||
| Fig. 4 Relative cells viability: (a) S. aureus; (b) E. coli. | ||
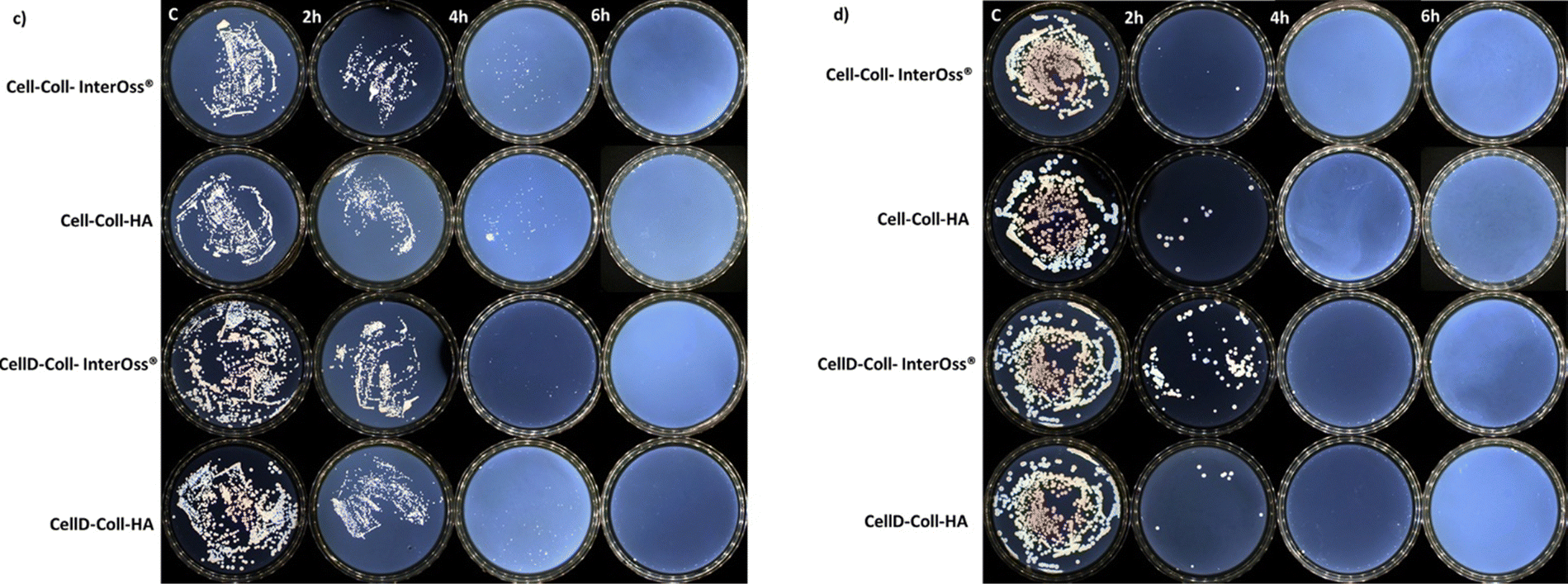 | ||
| Fig. 5 Images of plates cultured with bacteria over time: (c) S. aureus; (d) E. coli. | ||
Intriguingly, all materials demonstrated consistent antibacterial activity regardless of their composition. They reached peak antibacterial effectiveness after 6 hours of incubation for Staphylococcus aureus and just 2 hours for E. coli. This indicates a rapid and sustained antibacterial action that is effective against both Gram-positive and Gram-negative bacteria, underscoring the potential of these materials in clinical settings where infection prevention is critical. The enhanced antibacterial effectiveness against Escherichia coli might be attributed to the presence of a lipopolysaccharide component in the outer membrane of Gram-negative bacteria, which can hinder the proper trans-membrane transport of bioactive substances present in the tested materials.48,49 The incorporation of cellulose modified with (3-amino-4-methylphenyl) boronic acid as a component of the matrix significantly improves the antibacterial capabilities against both Gram-positive and Gram-negative bacteria. This modification likely enhances the interaction of the matrix with bacterial cell membranes, thereby increasing the material's efficacy in inhibiting bacterial growth and viability.
3.6. Swelling and degradation behavior
The ability of a scaffold to swell directly correlates with its capacity to absorb fluids, which is critical for nutrient and oxygen diffusion within the scaffold (Fig. 6). Higher swelling capacity can lead to better cell viability, especially for osteoblast cultures, as it allows for an improved supply of essential nutrients. From the swelling experimental data, CellD-Coll-HA shows the highest swelling capacity (39.48% at 240 minutes), which likely enhances the biological function of osteoblasts by supporting an environment rich in nutrients.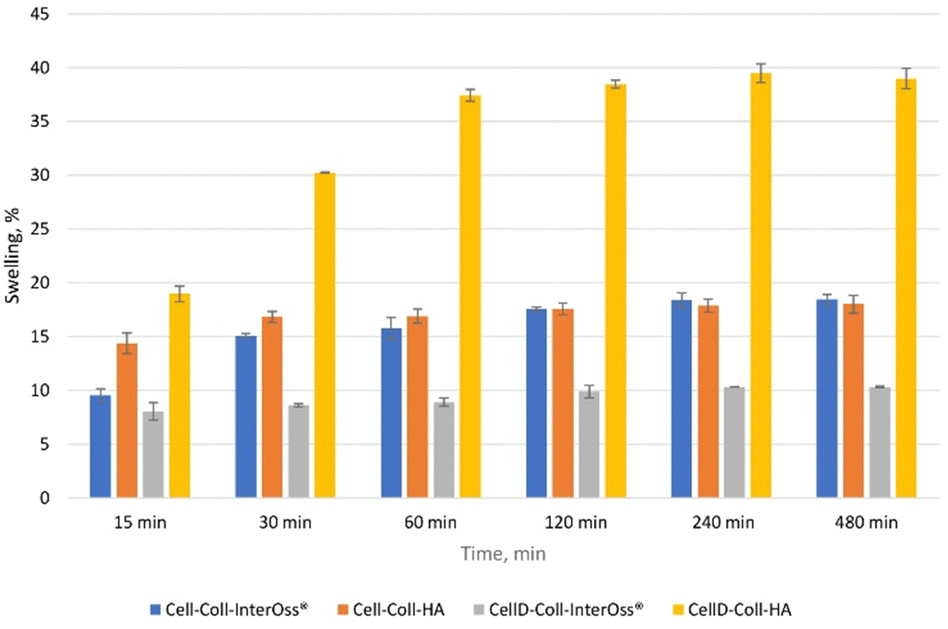 | ||
| Fig. 6 Swelling capacity of scaffolds. | ||
Hydroxyapatite (HA) promotes better osteoblast activity. This can be tied to its high swelling capacity, which allows scaffolds to retain more fluid and therefore create a conducive environment for cell proliferation. In materials such as CellD-Coll-HA, which exhibited superior swelling behavior, the enhanced swelling likely provides better support for osteoblast proliferation and adhesion. This scaffold also exhibited superior biocompatibility and cell viability, as shown in longer-term studies.
Swelling also affects the porosity of the scaffold, which is critical for osteoblast migration and integration into the matrix. A more porous structure (corelated with a higher swelling percentages) allows for better vascularization and integration of bone tissue. The correlation between swelling and BET surface area suggests that higher swelling leads to better cell distribution and proliferation within the scaffold, especially in CellD-Coll-HA matrices.
The swelling capacity of scaffolds influences key biological parameters, particularly cell adhesion, proliferation, and osteoblast differentiation. Materials like CellD-Coll-HA, with higher swelling percentages, offer improved environments for osteoblast cultures, supporting nutrient absorption, cell integration, and overall osteogenic activity. However, this needs to be balanced with the scaffold's mechanical properties to ensure structural integrity during bone regeneration.
Although high swelling capacity is generally beneficial for cell proliferation and nutrient transport, it may compromise the mechanical integrity of the scaffold if the material becomes too soft or loses its structural support. Materials like Cell-Coll-InterOss®, which exhibit moderate swelling, might offer a balance between sufficient hydration and mechanical support. This makes it suitable for applications where mechanical stability is as important as biological compatibility.
Hydroxyapatite supports osteogenic differentiation by mimicking natural bone composition. The swelling capacity of HA-based scaffolds could facilitate the transport of osteogenic factors, such as calcium and phosphate ions, to osteoblasts, enhancing their differentiation and bone-forming capacity. The scaffold's ability to sustain long-term cell viability and promote ALP activity (a marker of osteogenic differentiation) could be tied to its swelling behavior, allowing osteoblasts to thrive in a nutrient-rich and structurally supportive environment.
The degradation behavior of scaffolds (Fig. 7), combined with their swelling capacity, directly impacts the scaffold's ability to support osteoblast proliferation, differentiation, and long-term tissue integration. CellD-Coll-HA, with its faster degradation rate, promotes quicker scaffold resorption, facilitating early bone formation and osteoblast activity, while Cell-Coll-InterOss®, with slower degradation, provides prolonged structural support. Achieving the right balance between degradation, swelling, and mechanical properties is crucial for optimizing the scaffold for bone regeneration applications.
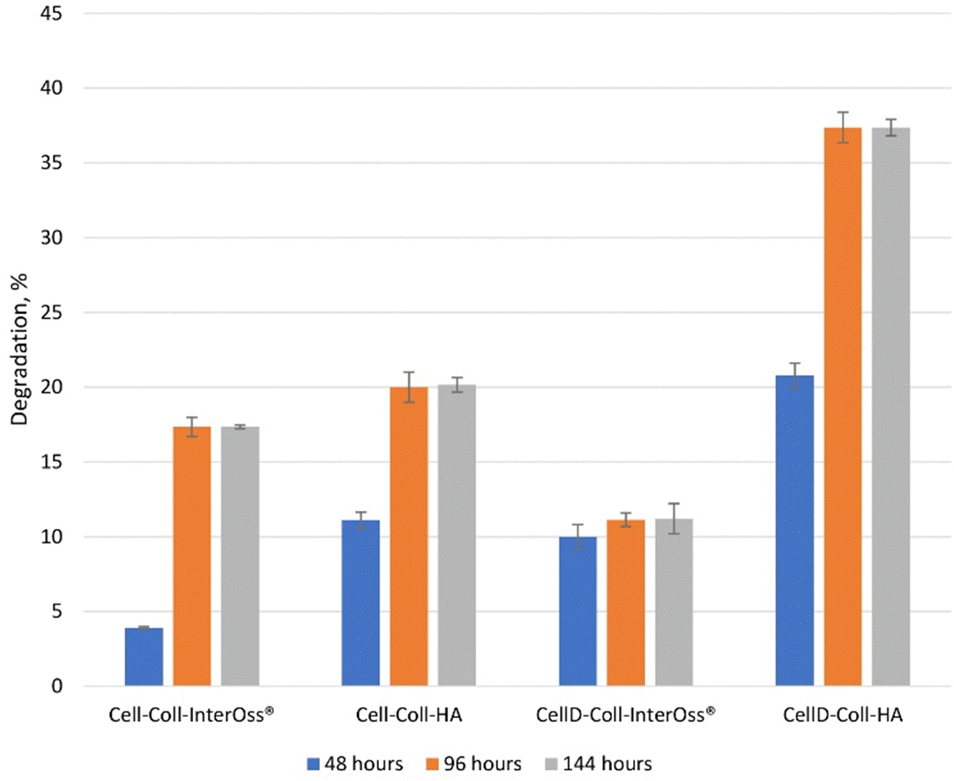 | ||
| Fig. 7 Time-dependent material degradation. | ||
3.7. Cell viability
The in vitro biocompatibility of the samples was evaluated after 24 hours using the MTS assay. Overall, the samples demonstrated biocompatibility, with cell viability ranging from 91% to 104% (Fig. 8). The viability exceeding 100% can be attributed to the treated cells exhibiting higher proliferation rates compared to the control cells, resulting in increased absorbance at 490 nm. Consequently, cell viability values above 100% are not uncommon and may indicate enhanced cell proliferation in response to the treatment.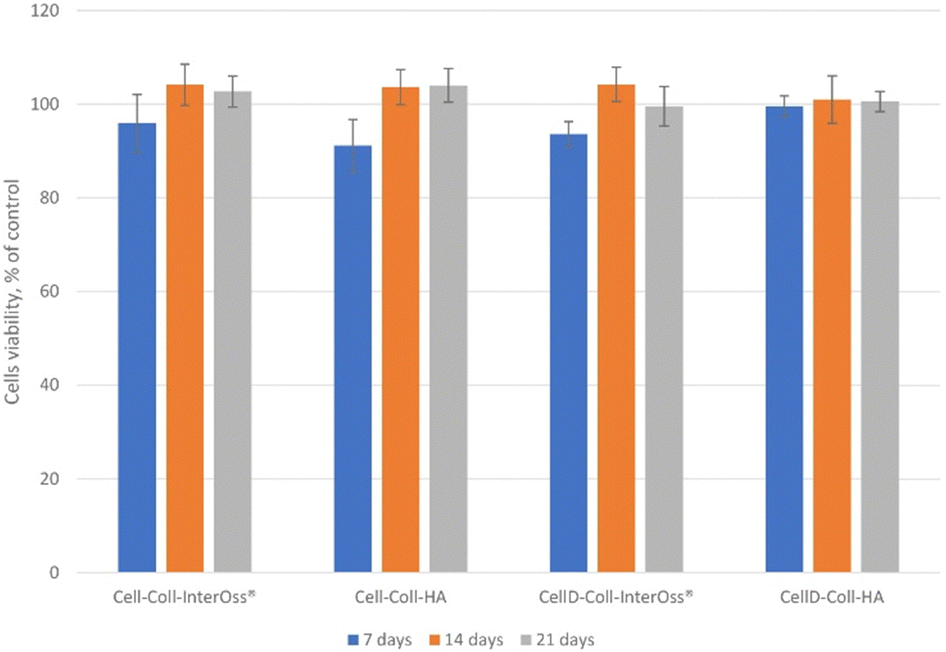 | ||
| Fig. 8 Cell viability under leachate exposure over time. | ||
3.8. Cytocompatibility of the scaffolds
The cytocompatibility of various collagen-based scaffolds was assessed using the human osteosarcoma-derived osteoblast cell line MG-63, which was seeded into four different types of scaffolds and cultured over periods of 3, 7, 14, and 21 days. Cellular F-actin fibers were stained with Phalloidin-FITC to monitor cell viability and morphology, as depicted in Fig. 9. | ||
| Fig. 9 Fluorescence images showing the morphology of MG-63 osteoblasts seeded for different intervals in a 2D culture model or scaffolds by staining the F-actin fibers with Phalloidin-FITC (green) and nuclei with DAPI (blue). Scale bar: 100 μm. | ||
For the Cell-Coll-InterOss® scaffold, no viable cells were observed on or within the scaffold throughout the study period, likely due to its high degree of compaction, which may impede cell infiltration and survival. In contrast, the Cell-Coll-HA scaffold supported cell presence; cells were observed proliferating within the scaffold throughout the 21-day culture period. However, these cells tended to grow in isolation without spreading extensively within the scaffold.
The CellD-Coll-InterOss® scaffold showed enhanced cell proliferation starting from day 14, indicating a delayed but effective adaptation of the cells to the scaffold environment. The CellD-Coll-HA scaffold exhibited the most favorable environment for sustaining cell viability, with significant improvements noted from day 7 onwards. Fluorescent imaging revealed a broad distribution of osteoblasts within this scaffold, displaying a morphology akin to that seen in traditional 2D cell cultures by day 21. Over time, from 14 to 21 days, there was a noticeable increase in the density of viable osteoblasts, which actively infiltrated deeper into the scaffold, suggesting excellent integration and growth potential within this scaffold composition. The analysis of adenylate kinase (AK) release into the culture medium, a marker of cell damage, supports findings from fluorescence microscopy involving live/dead staining. Fig. 11 illustrates that the AK levels detected in the medium after 3 days of culturing MG-63 cells in scaffolds were notably low, which correlates with the minimal cell survival observed in the scaffolds, as shown in Fig. 9 and 10.
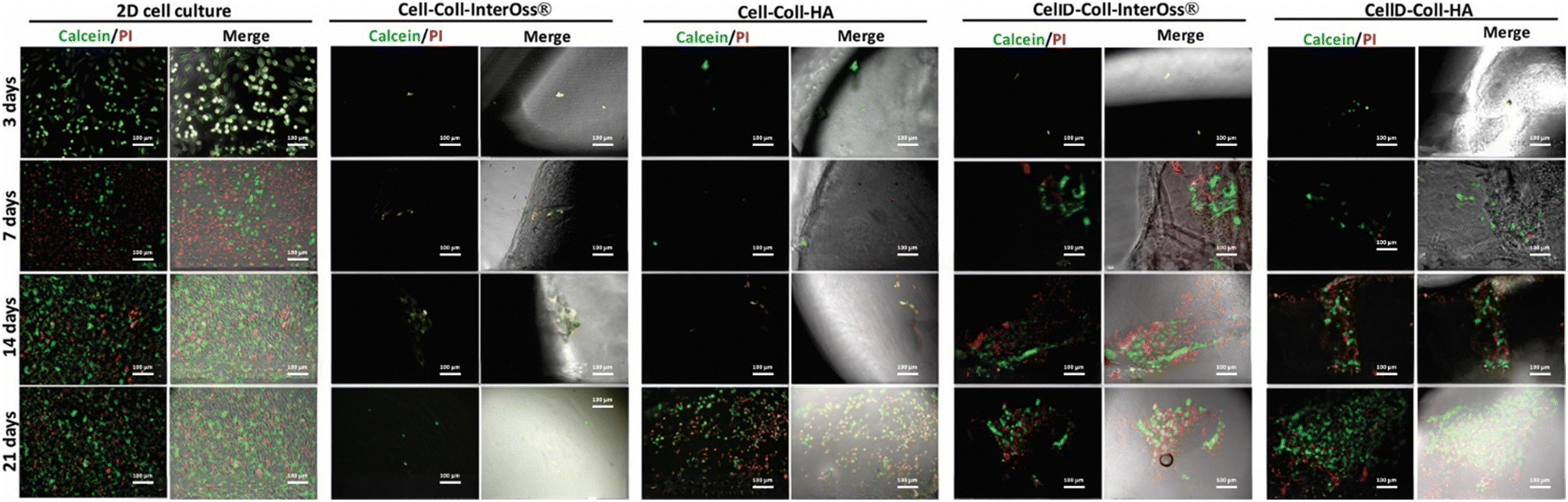 | ||
| Fig. 10 Live (green)/dead (red) staining on MG-63 osteoblasts seeded in a 2D cell culture system or scaffolds at different time intervals. Scale bar: 100 μm. | ||
Throughout the 21-day culture period, the release of AK was lowest in the Cell-Coll-InterOss® scaffold, indicating minimal cell activity and survival. In contrast, scaffolds such as Cell-Coll-HA, CellD-Coll-InterOss®, and CellD-Coll-HA exhibited higher AK release, suggesting increased cell proliferation over extended periods within these scaffolds. Despite this proliferation, cell mortality remained high, as evidenced not only by live/dead staining results shown in Fig. 11 but also by the elevated AK release levels. This pattern underscores a dynamic environment within the scaffolds where cell growth is accompanied by significant cell death, indicating challenges in maintaining long-term cell viability within these structures.
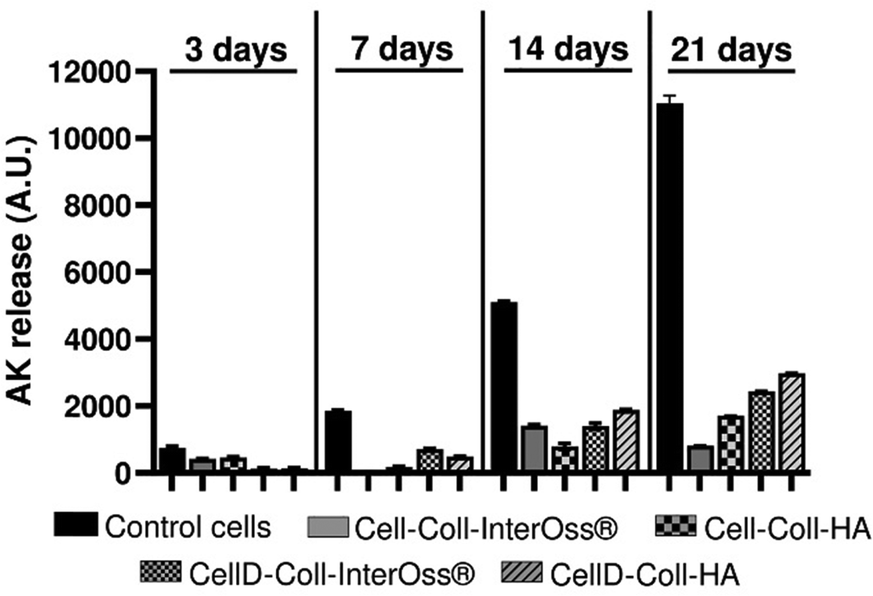 | ||
| Fig. 11 Adenylate kinase release from MG-63 osteoblasts seeded in a 2D cell culture system or scaffolds, at different time intervals. Data are presented as arbitrary units from experiments performed in duplicate (n = 2). | ||
3.9. The cytotoxicity of scaffold leachates
The effects of leachates collected from various scaffolds on the viability of MG-63 osteoblasts were extensively studied, and findings are detailed in Fig. 12. Initial exposure of the osteoblasts to leachates collected on the third day of incubation did not cause any significant change in cell viability compared to control cells, which were incubated with standard cell culture medium (Fig. 12(A)). This suggests that short-term exposure to these leachates does not have an immediate cytotoxic effect.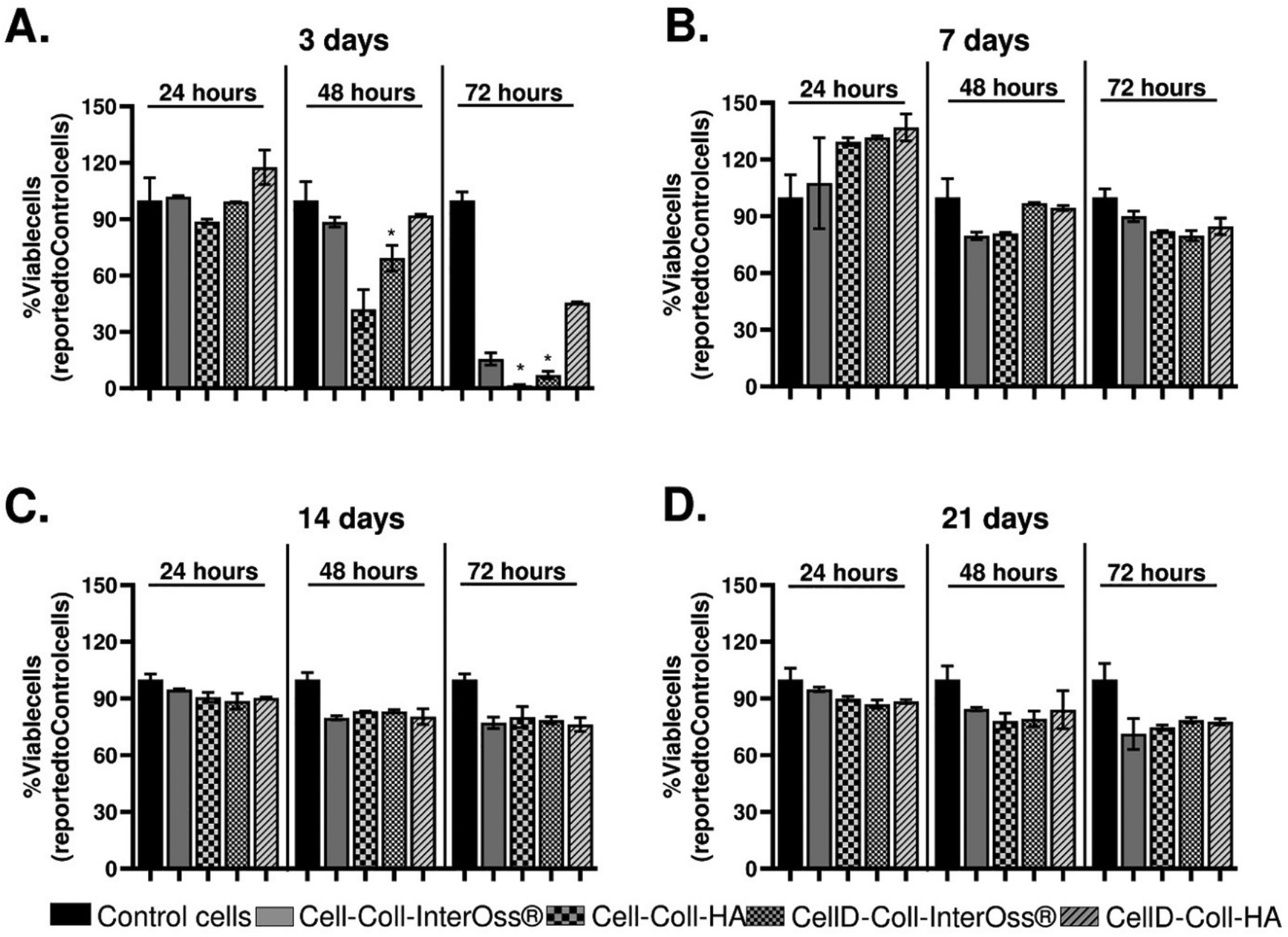 | ||
| Fig. 12 The viability of MG-63 osteoblasts seeded in a 2D cell culture system and incubated for 24, 48, and 72 hours with the leachates collected from the scaffolds after three (A), seven (B), fourteen (C), and twenty-one (D) days. Data are presented as mean ± S.D. of one experiment performed in duplicate (n = 2), *p < 0.05 versus control. | ||
However, as the exposure time increased, a notable decline in cell viability was observed. Specifically, after 48 hours of exposure, osteoblast viability decreased by approximately 10% with leachates from the Cell-Coll-InterOss® and CellD-Coll-HA scaffolds, by about 60% with the Cell-Coll-HA scaffold, and by 30% with the CellD-Coll-InterOss® scaffold. After 72 hours, the decline in viability became more pronounced: approximately 75% lower viability for cells exposed to leachates from Cell-Coll-InterOss®, more than 90% reduction for those incubated with leachates from Cell-Coll-HA and CellD-Coll-InterOss®, and around 55% reduction for CellD-Coll-HA scaffold leachates. These observations suggest a higher initial cytotoxicity in the leachates, aligning with results from direct cell seeding experiments on scaffolds. A possible explanation for this initial cytotoxicity could be the presence of toxic degradation products in the scaffolds, which are most prominent in the conditioned medium collected on day 3.
Interestingly, leachates collected on the 7th day showed no significant effect on cell viability over 24, 48, and 72 hours of exposure, and even appeared to induce slight cell proliferation at the 24-hour mark in leachates from the Cell-Coll-HA, CellD-Coll-InterOss®, and CellD-Coll-HA scaffolds (Fig. 12(B)). Additionally, no significant decrease in viability was noted with leachates collected on the 14th and 21st days (Fig. 12(C) and (D)), regardless of exposure time. This improvement over time suggests that the initial cytotoxic components may degrade or dissipate, reducing their impact on cell viability in longer-term cultures.
4. Conclusions
This study demonstrated that scaffold materials incorporating hydroxyapatite (HA) or InterOss® into a cellulose–collagen matrix significantly enhance mechanical properties, with a compressive modulus surpassing that typically associated with the pericellular matrix of human cartilages and organs. Specifically, these materials show a compressive modulus that indicates their potential efficacy for structural support in bone regeneration applications.Notably, the introduction of HA into these matrices greatly improves their physical and functional properties. It increases the specific surface area, which is crucial for promoting better cell adhesion and proliferation. This attribute is particularly beneficial in the context of bone regeneration, where enhanced cell attachment can accelerate healing processes. Our results demonstrate that scaffolds containing HA support the viability and functionality of MG-63 osteoblasts more effectively and for longer periods than those modified with InterOss®, indicating a significant improvement in biocompatibility and cellular response.
The incorporation of cellulose modified with (3-amino-4-methylphenyl) boronic acid is another innovative aspect, which notably enhances their antibacterial properties. This modification not only prevents bacterial growth but does so effectively against a broad spectrum of bacterial strains, both Gram-positive and Gram-negative. Such antibacterial efficacy is critical in preventing post-surgical infections and is a desirable property for any material used in invasive procedures.
Among the materials tested, the CellD-Coll-HA scaffold stands out. It not only supports cell adhesion and proliferation but also shows a notable increase in the density of viable osteoblasts over time. This scaffold's properties are particularly aligned with the needs of bone tissue engineering, where long-term cell viability and integration into the host tissue are crucial for successful regeneration.
The addition of HA into the cellulose–collagen matrix notably improved the Brunauer–Emmett–Teller (BET) area and monolayer capacity, achieving a BET area as high as 2375 m2 g−1 for the CellD-Coll-HA scaffold. This enhancement is crucial as it indicates a superior capacity for cell adhesion compared to scaffolds incorporating InterOss®, which is essential for initiating and sustaining cellular activities necessary for tissue integration and healing.
Cytocompatibility studies revealed that the CellD-Coll-HA scaffold provided the most favorable environment for sustaining cell viability, with significant improvements observed from day 7 onwards. Fluorescent imaging confirmed a broad distribution of osteoblasts within this scaffold, showing a morphology akin to cells maintained in a traditional 2D culture by day 21. This suggests not only compatibility but also the promotion of natural cellular behaviors crucial for effective tissue engineering.
However, cytotoxicity assessments indicated some initial challenges. The leachates from the scaffolds tested showed varying degrees of cytotoxicity over time. Notably, after 72 hours of exposure, the viability of MG-63 osteoblasts decreased significantly, with reductions of about 75% for Cell-Coll-InterOss® and more than 90% for Cell-Coll-HA and CellD-Coll-InterOss® scaffolds. Interestingly, leachates collected after 7 days did not significantly affect cell viability, suggesting that any cytotoxic degradation products present initially may degrade or dissipate over time, highlighting the importance of temporal dynamics in scaffold behavior.
Furthermore, the activity of alkaline phosphatase (ALP), a key marker of osteoblast differentiation, was consistently maintained at levels comparable to controls across all scaffold types when exposed to leachates collected at 7, 14, and 21 days. This consistent ALP activity, regardless of exposure duration to scaffold leachates, underscores the potential of these scaffolds to support osteogenic differentiation without adverse effects on this critical differentiation pathway.
In conclusion, the findings from this study underscore the potential of HA-enhanced cellulose–collagen scaffolds, particularly those modified with (3-amino-4-methylphenyl) boronic acid, in promoting superior mechanical properties, enhanced cell adhesion, and sustained osteoblast viability—key attributes for successful bone regeneration. The favorable cytocompatibility and controlled cytotoxicity further support their clinical potential. However, the initial cytotoxicity observed necessitates further investigation into the long-term biological interactions and the functional longevity of these scaffolds in vivo, to fully harness their capabilities for bone tissue engineering applications.
The integration of these advanced biomaterials into clinical practice promises to transform the management of bone fractures and disorders. Tailoring material properties to suit individual patient needs supports the principles of precision medicine, enabling more effective and personalized treatment strategies. This approach not only aims to improve patient outcomes but also strives to alleviate the economic burden on healthcare systems by enhancing treatment efficiencies and reducing the incidence of complications.
Looking forward, the field of bone regeneration is prone for further innovation. Emerging technologies such as 3D bioprinting and the integration of bioactive molecules into scaffold designs are paving the way for next-generation bone repair solutions. These advancements are set to redefine the standards of patient care in orthopedics, contributing to more sustainable healthcare practices and better quality of life for patients suffering from bone-related conditions.
Author contributions
Conceptualization, methodology, I. S.; writing original draft preparation, I. S., M. C., T. P., N. A.; writing and review, I. S., N. A., P.-D. S; editing I. S. N. A.; methodology, investigation, writing original draft preparation I. S.; N. A., M. C.; G. V.; T. P. All authors have read and agreed to the published version of the manuscript.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.References
- U. Bengner, O. Johnell and I. Redlund-Johnell, Acta Orthop. Scand., 1986, 57, 334–336, DOI:10.3109/17453678608994405.
- E. R. Bogoch, V. Elliot-Gibson, D. E. Beaton, S. A. Jamal, R. G. Josse and T. M. Murray, J. Bone Joint Surg., 2006, 88, 25–34, DOI:10.2106/JBJS.E.00198.
- A. Malviya, M. R. Reed and P. F. Partington, Injury, 2011, 42, 1368–1371, DOI:10.1016/j.injury.2011.06.198.
- J. Lee, H. Byun, S. K. Madhurakkat Perikamana, S. Lee and H. Shin, Adv. Healthcare Mater., 2019, 8, e1801106, DOI:10.1002/adhm.201801106.
- F. S. Kaplan, Bone biology, American Academy of Orthopedic Surgeons, Columbus, USA, 1994, p. 127 Search PubMed.
- A. L. Boskey, Elements, 2007, 3, 385–391, DOI:10.2113/gselements.3.6.385.
- Q. Wei, N. N. Deng, J. Guo and J. Deng, Int. J. Biomater., 2018, 7158621, DOI:10.1155/2018/7158621.
- L. Roseti, V. Parisi, M. Petretta, C. Cavallo, G. Desando, I. Bartolotti and B. Grigolo, Mater. Sci. Eng., C, 2017, 78, 1246–1262, DOI:10.1016/j.msec.2017.05.017.
- M. Abedi, M. Shafiee, F. Afshari, H. Mohammadi and Y. Ghasemi, Appl. Biochem. Biotechnol., 2024, 196, 5563–5603, DOI:10.1007/s12010-023-04793-3.
- E. E. Antoine, P. P. Vlachos and M. N. Rylander, Tissue Eng., Part B, 2014, 20, 683–696, DOI:10.1089/ten.TEB.2014.0086.
- S. S. Mathew-Steiner, S. Roy and C. K. Sen, Bioengineering, 2021, 8, 63, DOI:10.3390/bioengineering8050063.
- C. Liu and J. Sun, ACS Omega, 2020, 5, 28360–28368, DOI:10.1021/acsomega.0c04360.
- R. Brunsek, D. Kopitar, I. Schwarz and P. Marasovic, Polymers, 2023, 15, 3532, DOI:10.3390/polym15173532.
- H. Seddiqi, E. Oliaei, H. Honarkar, J. Jin, L. C. Geonzon, R. G. Bacabac and J. Klein-Nulend, Cellulose, 2021, 28, 1893–1931, DOI:10.1007/s10570-020-03674-w.
- N. Fatema, R. M. Ceballos and C. Fan, Front. Bioeng. Biotechnol., 2022, 10, 993711, DOI:10.3389/fbioe.2022.993711.
- S. Ye, S. He, C. Su, L. Jiang, Y. Wen, Z. Zhu and W. Shao, Molecules, 2018, 23 DOI:10.3390/molecules23082082.
- L. Zheng, S. Li, J. Luo and X. Wang, Front. Bioeng. Biotechnol., 2020, 8, 593768, DOI:10.3389/fbioe.2020.593768.
- J. Shi, W. Dai, A. Gupta, B. Zhang, Z. Wu, Y. Zhang, L. Pan and L. Wang, Materials, 2022, 15, 8474, DOI:10.3390/ma15238475.
- Y. Li, C. Liao and S. C. Tjong, Nanomaterials, 2019, 9, 590, DOI:10.3390/nano9040590.
- S. M. Cool, B. Kenny, A. Wu, V. Nurcombe, M. Trau, A. I. Cassady and L. Grondahl, J. Biomed. Mater. Res., Part A, 2007, 82, 599–610, DOI:10.1002/jbm.a.31174.
- Z. Sheikh, C. Sima and M. Glogauer, Materials, 2015, 8, 2953–2993, DOI:10.3390/ma8062953.
- Z. Wang, T. Han, H. Zhu, J. Tang, Y. Guo, Y. Jin, Y. Wang, G. Chen, N. Gu and C. Wang, Nanoscale Res. Lett., 2021, 16, 67, DOI:10.1186/s11671-021-03522-1.
- S. V. Dorozhkin, Biomatter, 2011, 1, 3–56, DOI:10.4161/biom.1.1.16782.
- InterOss® – SigmaGraft biomaterials, https://sigmagraft.com/inteross/ (accessed January 2024).
- D. S. H. Lee, Y. Pai and S. Chang, Mater. Chem. Phys., 2014, 146, 99–104, DOI:10.1016/j.matchemphys.2014.03.004.
- F. C. Cheng, C. Y. Wang and C. P. Chiang, J. Dent. Sci., 2023, 18, 1444–1446, DOI:10.1016/j.jds.2023.02.014.
- Y. J. Kim, C. E. T. Saiki, K. Silva, C. K. M. Massuda, A. P. de Souza Faloni, P. H. Braz-Silva, D. Pallos and W. R. Sendyk, Int. J. Dent., 2020, 2494128, DOI:10.1155/2020/2494128.
- V. Domazetovic, G. Marcucci, T. Iantomasi, M. L. Brandi and M. T. Vincenzini, Clin. Cases. Miner. Bone Metab., 2017, 14(2), 209–216, DOI:10.11138/ccmbm/2017.14.1.209.
- T. Xia, M. Kovochich, M. Liong, L. Mädler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134, DOI:10.1021/nn800511k.
- J. H. Lee, B. Kim, W. J. Jin, J. W. Kim, H. H. Kim, H. Ha and Z. H. Lee, Biochem. Pharmacol., 2014, 91, 51–60, DOI:10.1016/j.bcp.2014.06.005.
- B. E. Minter, D. A. Lowes, N. R. Webster and H. F. Galley, Antioxidants, 2020, 9, 195, DOI:10.3390/antiox9030195.
- C. Morabito, S. Guarnieri, A. Cucina, M. Bizzarri and M. A. Mariggio, Int. J. Mol. Sci., 2020, 21, 3638, DOI:10.3390/ijms21103638.
- E. Toshikj, A. Tarbuk, K. Grgić, B. Mangovska and I. Jordanov, Cellulose, 2018, 26, 777–794, DOI:10.1007/s10570-018-2133-4.
- J. Simon, L. Fliri, J. Sapkota, M. Ristolainen, S. A. Miller, M. Hummel, T. Rosenau and A. Potthast, Biomacromolecules, 2023, 24, 166–177, DOI:10.1021/acs.biomac.2c01022.
- Z. Zhou, D. Yan, X. Cheng, M. Kong, Y. Liu, C. Feng and X. Chen, Int. J. Biol. Macromol., 2016, 89, 471–476, DOI:10.1016/j.ijbiomac.2016.02.036.
- M. Turtoi, M. Anghelache, S. M. Bucatariu, M. Deleanu, G. Voicu, F. Safciuc, I. Manduteanu, G. Fundueanu, M. Simionescu and M. Calin, Int. J. Biol. Macromol., 2021, 185, 604–619, DOI:10.1016/j.ijbiomac.2021.06.174.
- O. S. Yevlashevskaya, B. A. Scheven, A. D. Walmsley and R. M. Shelton, Sci. Rep., 2023, 13, 14472, DOI:10.1038/s41598-023-40835-w.
- G. Voicu, C. A. Mocanu, F. Safciuc, M. Anghelache, M. Deleanu, S. Cecoltan, M. Pinteala, C. M. Uritu, I. Droc, M. Simionescu, I. Manduteanu and M. Calin, Mater. Today. Bio, 2023, 20, 100620, DOI:10.1016/j.mtbio.2023.100620.
- E. Podstawka, A. Kudelski, P. Kafarski and L. M. Proniewicz, Surf. Sci., 2007, 601, 4586–4597, DOI:10.1016/j.susc.2007.07.013.
- E. F. Medvedev and A. Sh Komarevskaya, Glass Ceram., 2007, 64(1–2), 42–46, DOI:10.1007/s10717-007-0010-y.
- K. J. Payne and A. Veis, Biopolymers, 1988, 27, 1749–1760, DOI:10.1002/bip.360271105.
- C. Y. Liu, D. Goto, C. Hongo, T. Matsumoto and T. Nishino, ACS Appl. Bio. Mater., 2018, 1, 1362–1368, DOI:10.1021/acsabm.8b00302.
- J. R. Woodard, A. J. Hilldore, S. K. Lan, C. J. Park, A. W. Morgan, J. A. Eurell, S. G. Clark, M. B. Wheeler, R. D. Jamison and A. J. Wagoner Johnson, Biomaterials, 2007, 28, 45–54, DOI:10.1016/j.biomaterials.2006.08.021.
- I. Levental, P. C. Georges and P. A. Janmey, Soft Matter, 2007, 3, 299–306, 10.1039/b610522j.
- M. Wang, N. H. Ladizesky, K. E. Tanner, I. M. Ward and W. Bonfield, J. Mater. Sci., 2000, 35, 1023–1030, DOI:10.1023/A:1004731315328.
- R. Teimouri, K. Abnous, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Mater. Res. Technol., 2023, 24, 7938–7973, DOI:10.1016/j.jmrt.2023.05.076.
- I. Spiridon, N. Anghel, M. V. Dinu, S. Vlad, A. Bele, B. I. Ciubotaru, L. Verestiuc and D. Pamfil, Polymers, 2020, 12, 1191, DOI:10.3390/polym12051191.
- I. Spiridon, N. C. Anghel, R. N. Darie-Nita, A. Iwanczuk, R. G. Ursu and I. A. Spiridon, Int. J. Biol. Macromol., 2020, 156, 1435–1444, DOI:10.1016/j.ijbiomac.2019.11.185.
- E. M. Czekanska, M. J. Stoddart, J. R. Ralphs, R. G. Richards and J. S. Hayes, J. Biomed. Mater. Res., Part A, 2014, 102, 2636–2643, DOI:10.1002/jbm.a.34937.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00456f |
| This journal is © The Royal Society of Chemistry 2024 |