
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrimary photodegradation pathways of an exciplex-forming A–D molecular system†
Yeongcheol
Ki
 ac,
Jonghyun
Kim
b,
Yeri
Son
c,
Suhyun
Park
ac,
Won-jin
Chung
c,
Tae-Young
Kim
*b and
Hohjai
Lee
*ac
ac,
Jonghyun
Kim
b,
Yeri
Son
c,
Suhyun
Park
ac,
Won-jin
Chung
c,
Tae-Young
Kim
*b and
Hohjai
Lee
*ac
aInnovative Energy and Carbon Optimized Synthesis for Chemicals (Inn-ECOSysChem) Research Center (ERC), Gwangju Institute of Science and Technology (GIST), Gwangju 61005, Republic of Korea. E-mail: hohjai@gist.ac.kr
bSchool of Earth Sciences and Environmental Engineering, Gwangju Institute of Science and Technology (GIST), Gwangju 61005, Republic of Korea. E-mail: kimtaeyoung@gist.ac.kr
cDepartment of Chemistry, Gwangju Institute of Science and Technology (GIST), Gwangju 61005, Republic of Korea
First published on 25th September 2024
Abstract
The photodegradation process of pyrene–(CH2)12–O–(CH2)2-N,N-dimethylaniline (Py-DMA), serving as a model molecular system for exciplex-forming A–D systems, is meticulously examined in solution. The alkyl chain-linker ensures efficient electron transfer between Py and DMA, enabling exciplex formation at concentrations as low as ∼5 μM, free from the interferences dominant in solid-state devices (domain–electrode interface, domain morphological change, accumulation of defects, and so on). The photodegradation mechanism of Py-DMA is proposed for the first time based on chemical identification using steady-state spectroscopy and LC-UV-MS techniques. The mechanism predicts Py-MMA (N-monomethylaniline) and Py-MFA (N-methylformanilide) as primary products and is verified by crosschecking experimental data from FT-IR and 1H NMR, as well as quantum mechanical calculation data. The heavy involvement of molecular oxygen (O2) predicted in the mechanism is confirmed by a series of deoxygenated condition experiments. Although we focus on the two primary photodegradation products, secondary, tertiary, and subsequent photodegradation products are also reported, such as PyOH-MPCA (methylphenylcarbamic acid), Py-FA (formanilide), and even unspecified black carbon precipitates. With recent emerging evidence of a close correlation between the stabilities of optoelectronic devices and their active molecules, the molecular photodegradation pathways of Py-DMA will shed light on the molecular design for exciplex-based optoelectronic devices with longer lifespans.
Introduction
Exciplexes, excited complexes formed by a negatively charged electron acceptor (A) and a positive electron donor (D), arise through photo-induced electron transfer processes or direct electron/hole injections on electrode surfaces.1–3 These exciplex-forming A–D systems have garnered significant attention for their potential applications in sensors,4–6 imaging dyes,7–10 and optoelectronic devices such as OLEDs.11–15 Their unique optical properties, including sensitivity to microenvironments, electron spin-state variations, and a broad emission band with a large Stokes shift, make them promising candidates for various technological advancements. However, the transient nature of exciplexes renders them susceptible to unforeseen side reactions, posing challenges to their stability and performance. It is necessary to conduct a comprehensive investigation into these side reactions, termed photodegradation (PD) or electrodegradation (ED) depending on the exciplex generation method.Studies on ED processes have been performed under operational conditions with the aim of improving stability of the optoelectronic devices. Nonetheless, delving into the operational ED mechanisms presents formidable challenges and complexities. These are associated with morphological changes in active material layers on electrode surfaces, as well as the accumulation of impurities and defects resulting from degradation of the active molecules.16–24 Interestingly, there is increasing evidence that PD processes of active molecules in solution closely mirror device operational stability. For instance, correlations have been found between the operational lifespan of devices and the photochemical stability of their components.14,25–27 Insights derived from PD processes in solution are thus invaluable for understanding the intrinsic degradation mechanisms in solid state organic optoelectronic devices.
In this study, we investigated the PD process of a model exciplex-forming A–D system by employing pyrene (Py)–(CH2)12–O–(CH2)2–N,N-dimethylaniline (DMA) (Py-DMA, Fig. 1) in acetonitrile (ACN). The Py and DMA pair has undergone thorough examination in both unlinked and linked configurations, owing to the notable exciplex emission quantum yield.28–35 These investigations have focused on exciplex formation dynamics and the magnetic field's influence on exciplex emission. The chain-linked A–D pair systems offer several advantages compared to non-linked pair systems: the flexible chain facilitates efficient photoinduced electron transfer between a single donor molecule and a single acceptor molecule at low concentrations (∼micromolar) while maintaining minimal coupling in the ground state between them. This dilute condition minimizes the accumulation of damaged products near the active molecules, allowing for the disentanglement of the intrinsic photochemical degradation process from the complexities inherent in solid state devices. The chain-linked A–D pair also demonstrates the potential to exhibit an extend electron spin correlation time between A˙− and D˙+, making it a promising candidate for research focused on electron spin state manipulation.6,36,37
 | ||
| Fig. 1 Molecular structure of pyrene–(CH2)12–O–(CH2)2-N,N-dimethylaniline. Pyrene (Py) acts as an electron acceptor (A), and N,N-dimethylaniline (DMA) as an electron donor (D). | ||
Py-DMA exhibits two characteristic emission bands after photoexcitation of Py.7 (ESI,† Fig. S1) The first emission band stems directly from Py*, termed locally-excited emission (LE). The second emission band is exciplex emission (ExE) from the exciplex state denoted as (Aδ−⋯Dδ+)*, typically separated by 3–4 Å intermolecular distance.32,38 LE intensity peaks in the absence of DMA, while it diminishes in the presence of DMA, concurrent with the rise in ExE intensity. This indicates DMA quenching Py* through electron transfer, with the exciplex formed as the electron transfer product. Hence, LE intensity serves as an optical indicator for photoinduced electron transfer.
We quantified the extent of PD of Py-DMA by monitoring LE from Py* excited by a 355-nm laser. Especially, we focused on the primary photochemical reactions that occur early during irradiation. PD products were identified using LC-UV-MS, FT-IR, NMR methods and quantum mechanical calculation, allowing for the successful proposition of a detailed intrinsic PD mechanism of the Py-DMA for the first time. We confirmed the mechanism by crosschecking with the experimental data. Notably, our mechanism highlights the critical role of molecular oxygen (O2), a proposition confirmed through measurements under deoxygenated conditions.
Experimental
Synthesis of Py-DMA
Py-DMA was synthesized via a two-step reaction procedure outlined in a previous study.7 Initially, 1-bromopyrene underwent alkylation with 1,12-dibromododecane to yield 1-(12-bromododecyl)pyrene, which was subsequently etherified with 2-[4-(dimethylamino)phenyl]ethanol to produce Py-DMA.Sample preparation
The concentrations of Py-DMA solutions for absorption, emission, and LC–MS analyses were prepared at 5 μM. For other measurements, including FT-IR and NMR, the sample concentrations were adjusted according to the sensitivity of each analytic method to achieve a decent signal-to-noise ratio (S/N). HPLC-grade acetonitrile (ACN, Sigma-Aldrich) was selected as the working solvent due to its widespread use as an MS-compatible solvent, offering high solubility for Py-DMA and appropriate polarity for observing both exciplex and radical ion pairs. Given the unknown concentration and structure of the photodegradation products, we employed the same solvent for both the sample solutions and the LC–MS mobile phase. This approach was intended to maximize the reproducibility and reliability of the LC–MS analysis by ensuring high ionization efficiency and separation efficiency.Photodegradation setup
Samples were subjected to irradiation using a 355-nm continuous wave (CW) laser (Cobolt Zouk, Hübner Photonics) at a power density of 1.47 W cm−2. Irradiation time varied with sample concentration, analytical method sensitivity, and measurement purpose. For example, a 5 μM Py-DMA sample was irradiated for 15 min for optical measurements and LC-UV-MS, and up to 125 min to determine the optimal irradiation time. FT-IR involved 60 min on a 50 μM sample, and NMR involved 180 min on a 600 μM sample, due to their lower sensitivity compared to MS. Irradiation time was kept below 3 min μM−1 to minimize secondary, tertiary, and subsequent photodegradation processes.Samples were stirred with a magnetic stirrer (MaXtir MSH500, Daihan Science) to maintain the homogeneity of the solution during irradiation (ESI,† Fig. S2). The Py-DMA molecule is well-documented to exhibit a magnetic field effect (MFE) on its exciplex emission intensity,7 and the fringe field generated by the magnet stirrer's magnet, measured to be approximately 5 mT at the sample position, is anticipated to influence photodamage kinetics. However, our estimation indicates that the MFE on exciplex emission is only 1.4% or less at 5 mT under 355-nm excitation (ESI,† Fig. S3), suggesting that the effect is minimal.
Steady-state spectroscopy, NMR, and FT-IR for characterization
Absorption and PL spectra were recorded using a UV-Vis spectrometer (Ultraspec 2100 pro, Biochrom US) and a fluorometer (FLS980, Edinburgh Instrument), respectively. 1H NMR analyses in ACN-d3 (Cambridge Isotope) were conducted using a 400 MHz NMR spectrometer (JNM-ECS400, JEOL). FT-IR measurements were performed using an FT-IR microscope (Vertex 70v, Bruker) in ATR mode.LC-UV-MS for identification
LC-UV-MS analyses were conducted for chromatographic separation, quantitative analysis, and mass analysis of photodegradation products, utilizing an Agilent 1260 HPLC & 6546 Q-ToF system with a variable wavelength detector (VWD, 1290 Infinity II, Agilent) and diode array detector (DAD, 1260 DAD VL, Agilent) module. The mobile phase consisted of a mixture of 99% ACN and 1% H2O with 0.1% formic acid (v/v), with chromatographic separation performed using a C18 column (Waters Acquity UPLC BEH 1.7 μm, 2.1 mm × 100 mm) and an injection volume of 3 μL of 5-μM samples in ACN. Mass-to-charge ratios (m/z) of each product were determined from MS chromatograms, with additional structural information obtained from MS/MS chromatograms.Deoxygenation
For experiments conducted under deoxygenated conditions, samples were prepared using repeated freeze–pump–thaw (FPT) cycles until pressure stabilization at ∼7 mTorr (>5 cycles).Quantum mechanical calculation
DFT calculations were conducted using the Gaussian 16 program.39 HOMO energy level calculations for Py and donor of primary PD products were performed by using B3LYP and 6-31G(d) basis-set with self-consistent reaction field (SCRF) where ACN is the solvent.Results and discussion
Steady-state absorption and photoluminescence measurements
The steady-state absorption and photoluminescence (PL) spectra of Py-DMA were obtained, with their response to photodegradation (PD) depicted in Fig. 2. In the absorption spectrum, the dominant contributions are attributed to the S0 → S2 and S0 → S3 transitions of Py within the <400 nm range, except for around 300 nm, where DMA dominates absorption.7 The selection of a 355 nm irradiation wavelength for PD is based on Py‘s predominant photon absorption at this wavelength, aligning with widely available light sources. Fig. 2(a) illustrates a 8% overall decrease in absorption intensity after 15 min of irradiation, indicating Py-DMA degradation. Simultaneously, the observation of three isosbestic points (marked as black solid squares) suggests the formation of PD products.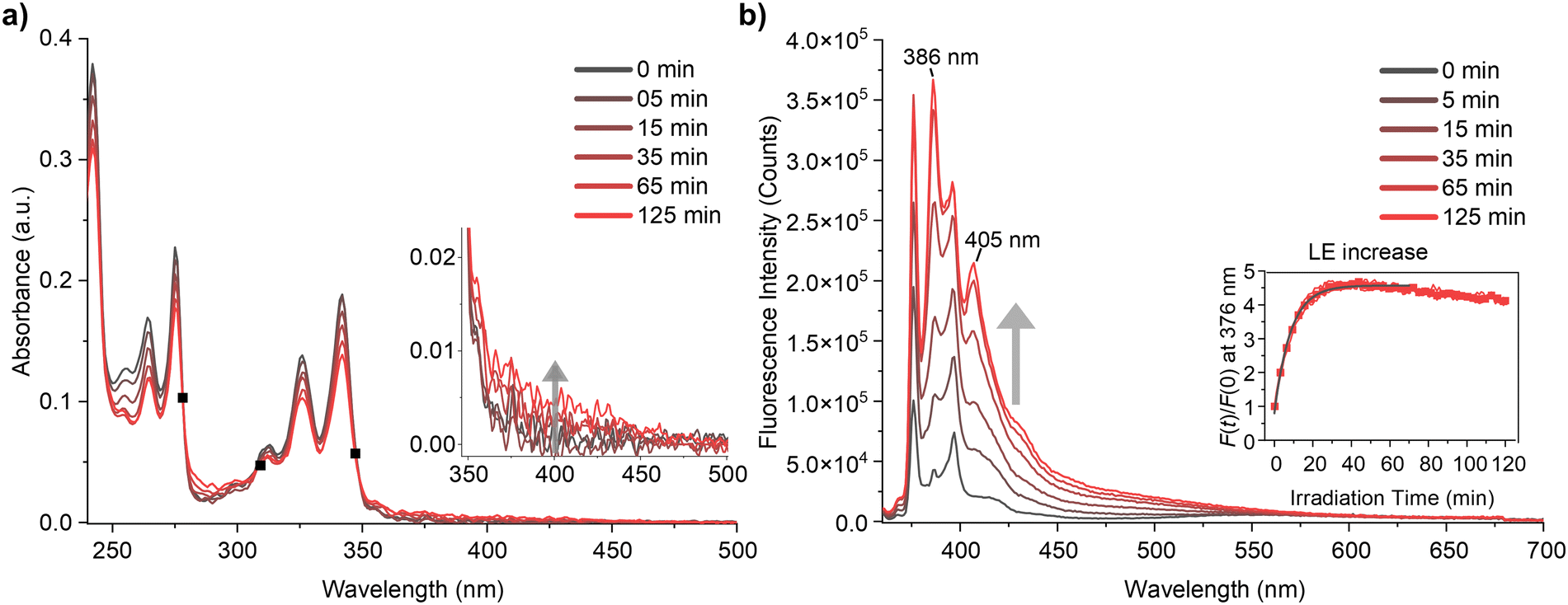 | ||
| Fig. 2 (a) Absorption spectra of Py-DMA over irradiation time. Isosbestic points are marked as black square (■). The inset plot reveals a broad and structureless absorption beyond 350 nm, indicating the formation of nonspecific black carbon. (b) Emission spectra of Py-DMA over irradiation time. The intensity of LE and peaks at 386 nm and 406 nm increased with irradiation time. The inset plot indicates the increase of LE through F(t)/F(0) at 376 nm over irradiation time. | ||
Moreover, weak, broad, structureless absorption beyond 350 nm (Fig. 2(a) inset) may arise from nonspecific black carbons possibly formed through multistep PD reactions under continuous irradiation.40,41 Additionally, precipitation of black particles was observed in concentrated Py-DMA solution (600 μM) after prolonged (180 min) irradiation (ESI,† Fig. S4).
The steady-state photoluminescence (PL) spectrum reveals a distinctive LE band within the 360–450 nm range and a broad ExE band spanning 450–700 nm (Fig. 2(b)). Notably, clearer effects of PD are observed in the PL spectra compared to absorption spectra (Fig. 2(a)). Primarily, the LE band undergoes a significant increase. To quantitatively assess this change, we measured the intensity of the peak at 376 nm at various irradiation times (t) relative to that without irradiation (F(t)/F(0)). The plot of F(t)/F(0) against irradiation time (t) reveals a steep increase before 30 min, plateauing thereafter with a slight decrease (Fig. 2(b) inset). Conversely, the intensity of the ExE band decreases, particularly evident in the emission spectrum normalized at 376 nm (ESI,† Fig. S5). However, the change in ExE band intensity is less pronounced compared to the LE band, attributed to photoluminescence from PD products occurring within a similar spectral region.
In Py-DMA, an excited state of pyrene (Py*) has the capability to emit LE and to form an exciplex upon quenching by DMA. The observed increase in LE and decrease in ExE as irradiation time increases suggest that PD hinders the quenching of Py* and the formation of an exciplex. This behavior was also observed in photoluminescence lifetime measurement: LE lifetime increases from 4 ns to 13.7 ns after irradiation. (ESI,† Fig. S6). Three potential PD pathways are conceivable: (1) degradation of DMA, (2) degradation of Py, and (3) cleavage of the chain-linker between them, leading to reduced collision frequency between Py* and DMA.
Consequently, the ratio F(t)/F(0) serves as a measure of the extent of PD in Py-DMA. For instance, the F(t)/F(0) inset plot in Fig. 2(b) indicates rapid PD within the first 30 min of irradiation under the experimental conditions outlined in this study. The subsequent decrease in F(t)/F(0) after 30 min can be attributed to degradation of the Py moiety with prolonged irradiation.
Another noteworthy feature of the PL spectra is the appearance of the new peaks at 386 nm and 405 nm that were not present before PD. We assign the origin of these peaks to hydroxypyrene (PyOH) as a PD product of Py-DMA. This assignment is supported by the fact that the peak positions exactly match those of a pure PyOH solution, as shown in ESI,† Fig. S7(a). Moreover, its absorption spectrum is slightly red-shifted compared to Py-DMA and has significantly larger absorbance at the excitation wavelength (355 nm) (ESI,† Fig. S7(b)). Therefore, the emission peaks of PD product PyOH grow rapidly even though the amount generated by the irradiation might be small. The formation of the PyOH is also detected by mass analysis, as described below. Sigman et al. reported that PyOH can be formed from Py via pyrene–oxygen contact charge transfer.42
Identification of photodegradation products with LC-UV-MS and FT-IR
In order to explore the abovementioned PD further, PD products of Py-DMA were identified using UV and MS. Notably, the Py moiety remained structurally stable through PD, allowing us to target a specific wavelength (342 nm) for UV analysis. This wavelength was selected due to its maximal absorption within the absorption spectrum range contributed by Py. For the LC-UV-MS analysis, a 15-min irradiation was selected for a 5 μM Py-DMA solution based on the F(t)/F(0) profiles in Fig. 2(b), which corresponds to the time when F(t)/F(0) reaches 80% of the maximum, to minimize the potential PD products from secondary and further photodegradation.The UV chromatograms revealed seven peaks that could be attributed to PD products with Py moiety (Fig. 3(b)). Most PD products were ionized effectively, enabling the detection of their corresponding MS peaks with unique m/z values. Interestingly, peak 1 in the UV chromatogram appeared singular, but MS analysis revealed the co-elution of two compounds with distinct masses, designated as peaks 1-1 and 1-2. Using the accurate m/z values obtained, the elemental composition of each product was determined with a mass accuracy below 1 ppm. Finally, the structural formulas of the PD products were proposed based on the structure and elemental composition of Py-DMA (Table 1 and Fig. 4).
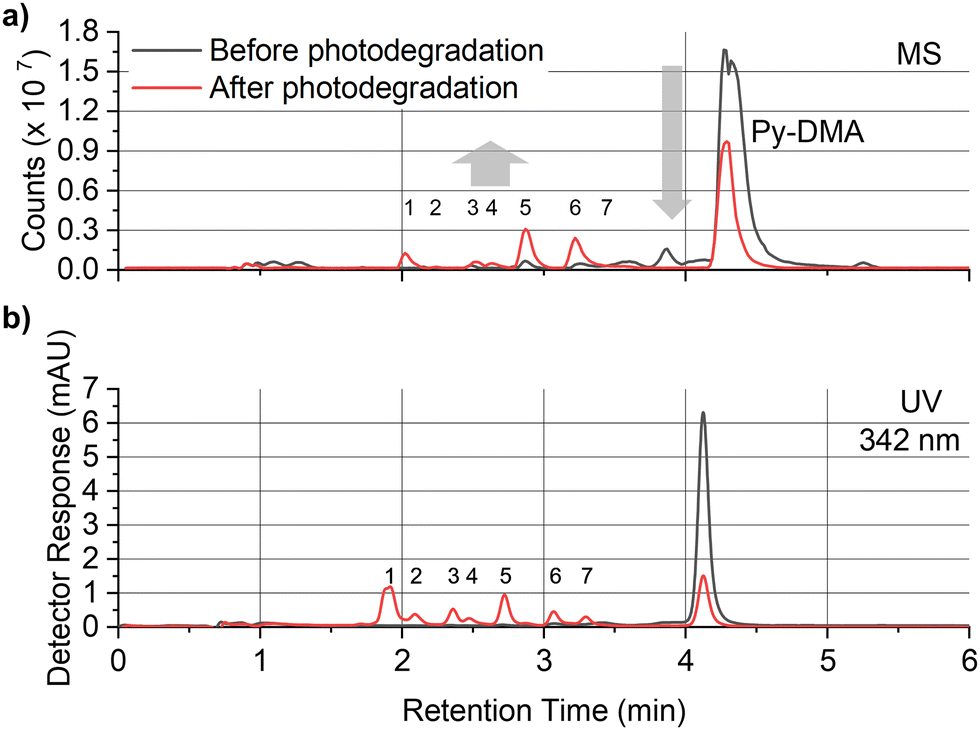 | ||
| Fig. 3 (a) LC–MS base peak chromatogram and (b) UV chromatogram of Py-DMA before/after irradiation for 15 min. A decrease in the peak for Py-DMA and an increase in the peaks for PD products are observed. The MS and UV chromatograms show a one-to-one matching for most peaks, except for peak 7, which was detected in the UV chromatogram, but was not detected in the MS chromatogram due to the absence of a protonation site in the ESI+ mode. | ||
| Peak number | Name | Retention time [min] | Theoretical m/z, [M + H]+ | Measured m/z, [M + H]+ | Mass error [ppm] |
|---|---|---|---|---|---|
| a Not detected in the MS. | |||||
| 1-1 | PyOH-MPU | 1.89 | 579.3582 | 579.3585 | 0.5 |
| 1-2 | PyOH-MPCA | 1.89 | 580.3422 | 580.3426 | 0.7 |
| 2 | Py-MPCA | 2.07 | 564.3473 | 564.3475 | 0.4 |
| 3 | Py-FA | 2.34 | 534.3367 | 534.3372 | 0.9 |
| 4 | Py-A | 2.44 | 506.3418 | 506.3421 | 0.6 |
| 5 | Py-MFA | 2.69 | 548.3524 | 548.3528 | 0.7 |
| 6 | Py-MMA | 3.04 | 520.3574 | 520.3578 | 0.8 |
| 7 | N.D.a | 3.27 | N.D. | N.D. | N.D. |
| Parent | Py-DMA | 4.09 | 534.3731 | 534.3739 | 1.5 |
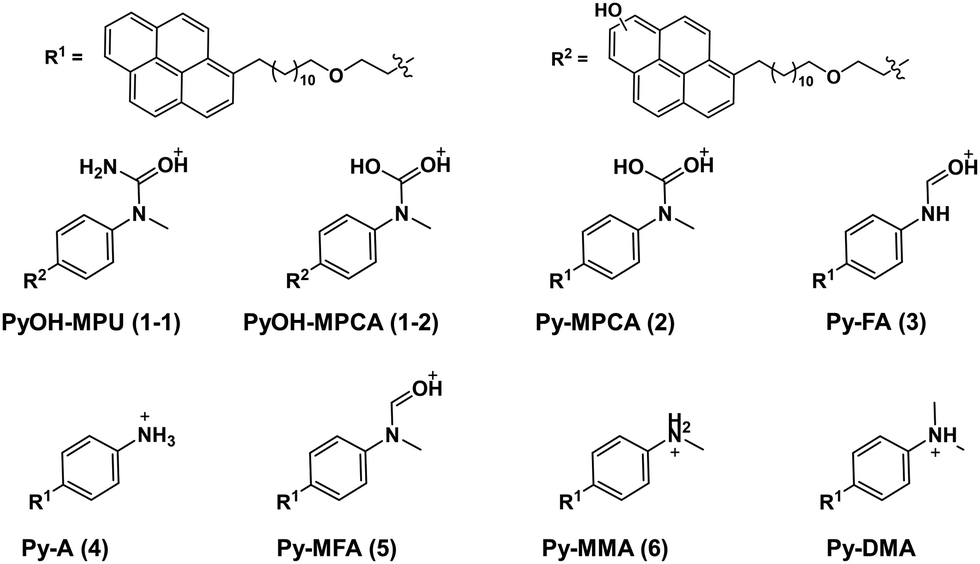 | ||
| Fig. 4 Molecular structures of proposed PD products are shown in the [M + H]+ ion form. Electron acceptor (pyrene) moiety and alkyl chain-linker (–(CH2)12–O–(CH2)2–) are omitted and indicated as R1 and R2. DMA: N,N-dimethylaniline, MMA: N-monomethylaniline, MFA: N-methylformanilide, A: aniline, FA: formanilide, MPCA: methylphenylcarbamic acid, MPU: methylphenyl urea, Py: pyrene, PyOH: hydroxypyrene. | ||
The LC-UV-MS analysis suggested various transformation pathways for the Py-DMA PD products. The PD products were primarily attributed to demethylation, formylation, and oxidation reactions occurring on the DMA moiety. The absence of peak 7 in the MS spectrum implies the loss of an ionizable amine or aniline group, potentially due to deamination reactions or cleavage of the chain-linker during PD. Shared functional groups among the proposed PD product structures contribute to the presence of common adduct ions in the MS spectrum (ESI,† Fig. S8). For instance, peaks 1-2 and 2, containing a carbamic acid moiety, exhibit a neutral loss of H2O, likely due to dehydration following the formation of two hydroxy groups upon protonation of the carbamic acid. Similarly, peaks 1-1, 1-2, 2, 3, and 5, all containing carbonyl groups, share a common ACN adduct ion form, [M + ACN + NH4]+. This observation aligns with previous findings where polarization in ACN facilitates the formation of carbonyl and nitrile clusters via dipole–dipole interactions,43 explaining the presence of carbonyl groups in these peaks. Furthermore, the presence of ammonium ions likely originates from the deamination of Py-DMA during the PD process. Following a 15-min irradiation period, a pronounced decline in Py-DMA (m/z = 534.3739, parent in Fig. 4 and Table 1) was observed, alongside the emergence of several new peaks at earlier retention times.
Additionally, one additional peak (RT 3.27 min, 7) was exclusively detected in the UV chromatogram. Given that the amine in DMA undergoes ionization during the ionization process, the lack of detection in MS suggests deamination or cleavage of the chain-linker during PD. Two primary products, Py-monomethylaniline (Py-MMA, m/z = 520.3578, 6) and Py-methylformanilide (Py-MFA, m/z = 548.3528, 5), were identified. Py-MMA is likely formed through the demethylation of Py-DMA, while Py-MFA arises from the oxidative formylation of Py-DMA. Subsequent analysis confirmed the remaining products as secondary products originating from primary degradation. Evidences of multiple instances of demethylations and formylations were also observed, along with additional oxidations and aminations. Py-aniline (Py-A, m/z = 506.3421, 4) arises from two demethylations of Py-DMA, whereas Py-formanilide (Py-FA, m/z = 534.3372, 3) results from both demethylation and formylation of Py-DMA. Py-methylphenylcarbamic acid (Py-MPCA, m/z = 564.3475, 2) is formed through further oxidation of the formyl group of Py-MFA. Hydroxypyrene-methylphenyl urea (PyOH-MPU, m/z = 579.3585, 1-1) and hydroxypyrene-methylphenylcarbamic acid (PyOH-MPCA, m/z = 580.3426, 1-2) are attributed to amination at the formyl group or oxidation of Py-MFA, respectively. Identification of PyOH-MPU and PyOH-MPCA strongly supports our assignment of the two emerging peaks at 386 and 405 nm in PL spectrum to PyOH spectrum (Fig. 2(b) and ESI,† Fig. S7(a)). However, we were unable to identify PyOH-DMA, which is attributed to the higher reactivity of DMA compared to Py. This suggests that the PD products, PyOH-MPCA and PyOH-MPU, are most likely formed via Py-MPCA and Py-MPU pathways, rather than through the PyOH-DMA pathway. No indication of cleavage of the linker was found in the MS chromatogram.
MS/MS analysis (Fig. 5(a)–(h)) further corroborated the structures of PD products outlined in Fig. 4, employing sample preparation and MS conditions identical to those used in the LC-UV-MS experiments. The dominant fragment ions for both Py-DMA and its PD products originate from the cleavage of the ether group at the chain-linker, enabling separate analysis of the donor and acceptor. Notably, Py-DMA, Py-MMA, and Py-A all exhibited a single strong fragment ion associated with aniline derivatives. For Py-MPCA and PyOH-MPCA, which contain carbamic acid, characteristic fragment ions arose from formaldehyde release or decarboxylation following ether linkage cleavage. Specifically, the fragment with an m/z of 245.0965 detected at peak 1 confirms the presence of PyOH. Structures containing a PyOH moiety displayed a more diverse range of fragment ion patterns, including ions with charged PyOH moieties. The observed fragmentation patterns in MS/MS support the initial product structures proposed in the MS analysis, thereby strengthening the validity of our suggested PD products.
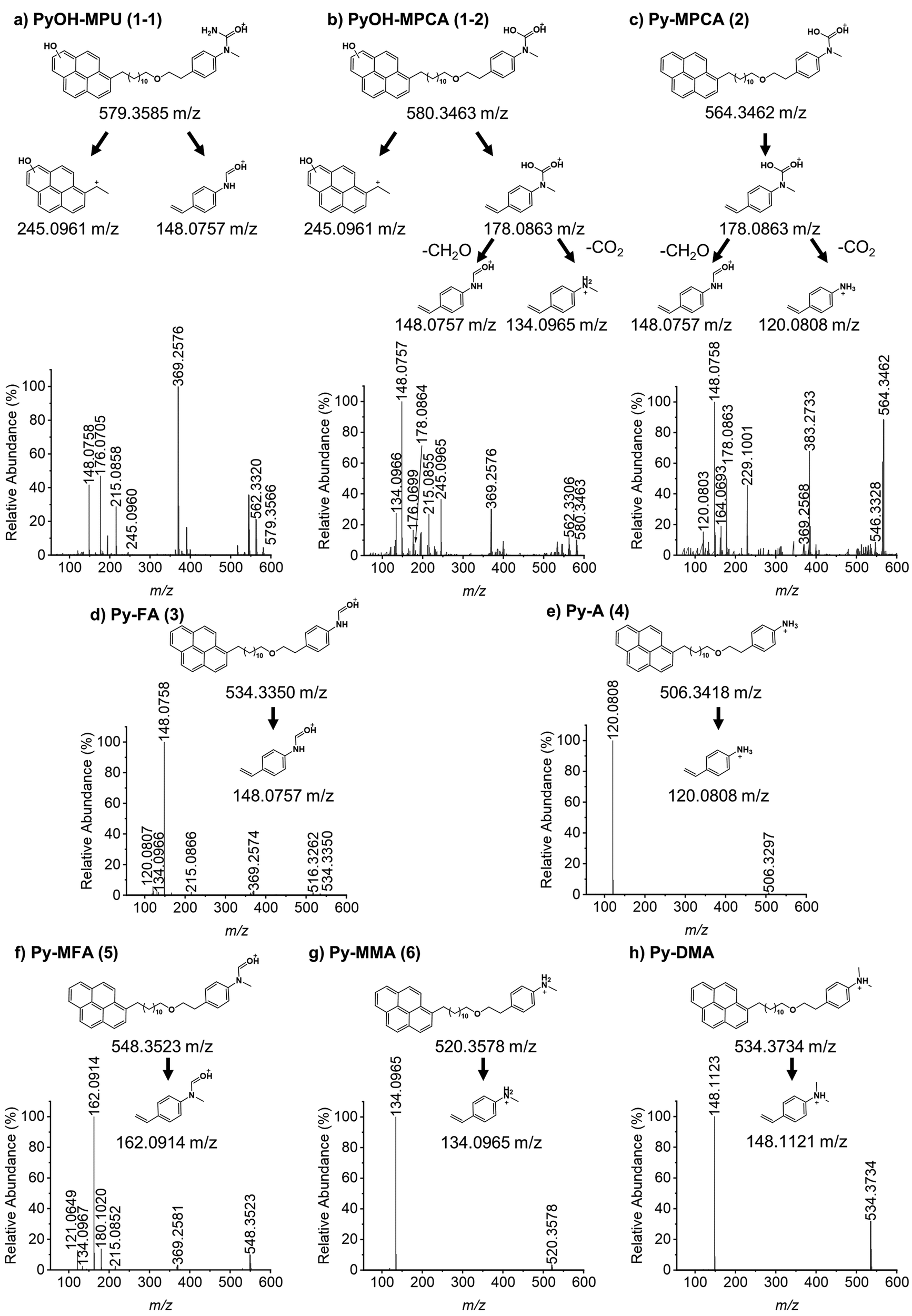 | ||
| Fig. 5 MS/MS spectra of Py-DMA and PD products after 15-min irradiation. DMA: N,N-dimethylaniline, MMA: N-monomethylaniline, MFA: N-methylformanilide, A: aniline, FA: formanilide, MPCA: methylphenylcarbamic acid, MPU: methylphenyl urea, Py: pyrene, PyOH: hydroxypyrene. | ||
FT-IR measurements were conducted to validate the presence of new functional groups in the proposed PD products as identified by MS. The FT-IR spectrum of the degraded Py-DMA solution revealed two distinct peaks not present in the Py-DMA sample before PD (ESI,† Fig. S9(a)): a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode at 1680 cm−1 from MFA and a CO stretching mode at 1720 cm−1 from formaldehyde, which is a byproduct for the demethylation process. These peaks support formylation and demethylation in DMA, respectively. Furthermore, the overall spectrum of photodegraded Py-DMA exhibited reduced intensity and broadening compared to Py-DMA before PD, indicating the generation of various PD products. Considering the nature of PD, it can be easily anticipated that the degradation will continuously proceed with extended irradiation until the starting materials are decomposed and aggregated into nonspecific black carbon particles, as previously discussed. Therefore, we will focus the discussion on the primary products, Py-MMA and Py-MFA, rather than other secondary, tertiary, and subsequent products.
O stretching mode at 1680 cm−1 from MFA and a CO stretching mode at 1720 cm−1 from formaldehyde, which is a byproduct for the demethylation process. These peaks support formylation and demethylation in DMA, respectively. Furthermore, the overall spectrum of photodegraded Py-DMA exhibited reduced intensity and broadening compared to Py-DMA before PD, indicating the generation of various PD products. Considering the nature of PD, it can be easily anticipated that the degradation will continuously proceed with extended irradiation until the starting materials are decomposed and aggregated into nonspecific black carbon particles, as previously discussed. Therefore, we will focus the discussion on the primary products, Py-MMA and Py-MFA, rather than other secondary, tertiary, and subsequent products.
Exciplex formation test for the primary photodegradation products
The subsequent investigation aims to ascertain whether the primary PD products, Py-MMA and Py-MFA, manifest the observed photochemical behaviors witnessed in the PL spectra presented in Fig. 2(b). Specifically, we seek to determine if there is impairment in either the quenching of Py* or the formation of emissive exciplexes due to the PD. To address this inquiry, we devised a non-linked Py + donor mixture under conventional exciplex-forming conditions, employing an excessive donor concentration ([Py]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : [donor] = 1:60). We used cyclohexane as the solvent, where the formation of exciplex is more favorable than in ACN, facilitating the observation of exciplex formation. The PL spectra of Py + DMA, Py + MMA, and Py + MFA are compared with that of Py-only solution in ESI,† Fig. S10(a). While DMA serves as an efficient quencher of Py* and an exciplex-forming partner of Py, MMA demonstrates more efficient quenching but is a poor exciplex-forming partner, and MFA exhibits neither efficient quenching nor exciplex-forming capabilities. Given that both Py + MMA and Py + MFA fail to form exciplexes, the PD of Py-DMA to these entities would lead to a reduction in ExE in the 500–700 nm range, aligning with the trend observed in Fig. 2(b). The anticipated contributions to LE from the two primary PD products are expected to be opposite. Specifically, the formation of Py-MMA is anticipated to decrease the LE due to MMA's effective quenching properties, whereas the formation of Py-MFA is expected to increase the LE emissions due to MFA's poor quenching capabilities. However, it is noteworthy that Py-MFA formation is nearly double that of Py-MMA at a 15-min irradiation duration, as per the quantitative analysis discussed later (Fig. 7). Considering the faster formation rate of Py-MFA, the overall observed increase in LE emissions in Fig. 2(b) can be reasonably elucidated by the findings of these Py + donor mixture experiments. However, it is important to acknowledge that the photophysics of a chain-linked Py-donor system and a non-linked Py + donor mixture is not directly interchangeable due to variations in their properties, such as relative distance, orientation, and collision frequency.
: [donor] = 1:60). We used cyclohexane as the solvent, where the formation of exciplex is more favorable than in ACN, facilitating the observation of exciplex formation. The PL spectra of Py + DMA, Py + MMA, and Py + MFA are compared with that of Py-only solution in ESI,† Fig. S10(a). While DMA serves as an efficient quencher of Py* and an exciplex-forming partner of Py, MMA demonstrates more efficient quenching but is a poor exciplex-forming partner, and MFA exhibits neither efficient quenching nor exciplex-forming capabilities. Given that both Py + MMA and Py + MFA fail to form exciplexes, the PD of Py-DMA to these entities would lead to a reduction in ExE in the 500–700 nm range, aligning with the trend observed in Fig. 2(b). The anticipated contributions to LE from the two primary PD products are expected to be opposite. Specifically, the formation of Py-MMA is anticipated to decrease the LE due to MMA's effective quenching properties, whereas the formation of Py-MFA is expected to increase the LE emissions due to MFA's poor quenching capabilities. However, it is noteworthy that Py-MFA formation is nearly double that of Py-MMA at a 15-min irradiation duration, as per the quantitative analysis discussed later (Fig. 7). Considering the faster formation rate of Py-MFA, the overall observed increase in LE emissions in Fig. 2(b) can be reasonably elucidated by the findings of these Py + donor mixture experiments. However, it is important to acknowledge that the photophysics of a chain-linked Py-donor system and a non-linked Py + donor mixture is not directly interchangeable due to variations in their properties, such as relative distance, orientation, and collision frequency.
Additionally, our quantum mechanical calculations corroborate the aforementioned trends in the quenching capabilities of the donors. The highest occupied molecular orbital (HOMO) energy levels of DMA and MMA are higher than that of Py by 0.313 and 0.152 eV, respectively, while the HOMO energy of MFA is lower than Py by 0.678 eV (ESI,† Fig. S10(b)). This implies that electron transfer from DMA to Py* is thermodynamically most favorable, whereas electron transfer from MFA to Py* is unfavorable, aligning with the observations in the Py + donor mixture experiments. The optimized molecular structure and electron density surface (mapped with electrostatic potential) of Py and donors are presented in ESI,† Table S1 and Fig. S11.
Suggested photodegradation mechanism
Based on the spectroscopic and mass spectrometric data provided, alongside prior investigations concerning the demethylation and oxidative formylation of N-methylamine,44–47 we propose a PD mechanism for the primary PD products Py-MMA and Py-MFA from Py-DMA (Fig. 6). Initially, Py-DMA (R1 = R2 = CH3) is excited by 355-nm light, resulting in Py*-DMA, which subsequently generate Py˙−–DMA˙+ through photo-induced electron transfer. Energy transfer from the triplet excited state pyrene (3Py*), acting as a photosensitizer, to ground state triplet oxygen (3O2) leads to the generation of excited-state singlet oxygen (1O2). Both triplet and singlet oxygen molecules serve as primary oxidizing agents throughout the PD process. The molecular oxygen oxidizes either a donor (D) or acceptor anion (A˙−) to a donor cation (D˙+) or neutral acceptor (A), respectively, subsequently forming the oxygen radical anion (O2˙−). Proton transfer between DMA˙+ (a) and O2˙− produces hydroperoxyl radical (HOO˙) and α-carbon radical in DMA (b). Compound b undergoes three degradation pathways. The first pathway involves demethylation, in which b is oxidized to form an iminium cation (c) which, upon hydration, yields compound d. After proton transfer, it undergoes fragmentation to form Py-MMA, producing formaldehyde as a by-product. This pathway was verified by comparing the 1H NMR spectra of the Py-DMA solution after PD and a diluted pure formaldehyde solution, both of which exhibited a singlet peak at 9.57 ppm (ESI,† Fig. S9(b)).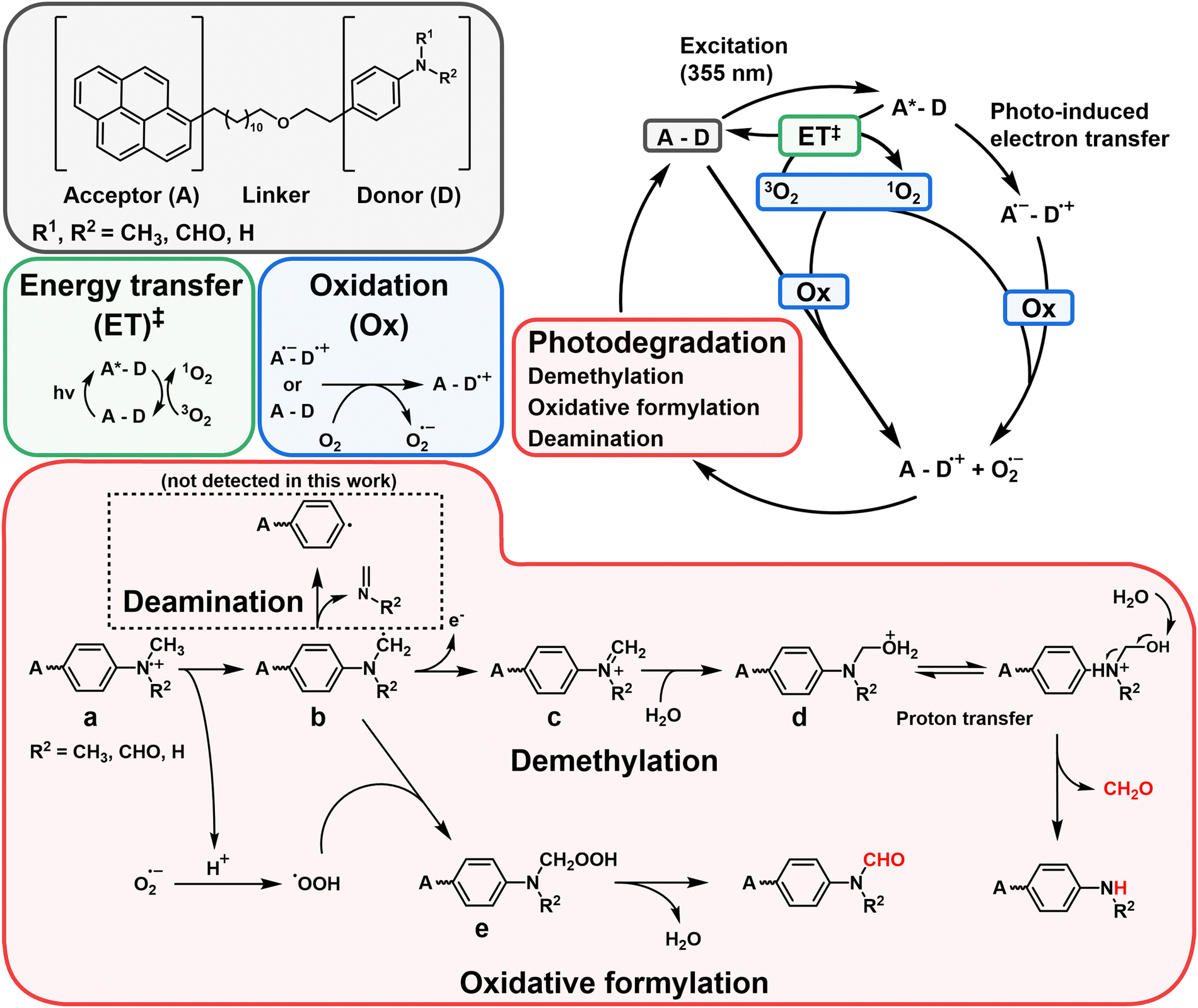 | ||
Fig. 6 Proposed photodegradation mechanism of Py-DMA. In the red box labeled “photodegradation”, the alkyl chain-linker is represented by a wavy line. ‡The intersystem crossing processes from 1A*–D to 3A*–D, followed by energy transfer (ET) with 3O2 to generate 1O2, include: (1)  , (2) , (2)  and (3) and (3)  . The direct formation pathway (1) is anticipated to be the major route due to the relatively high yield of triplet pyrene (38%, ref. 48) and the single-step formation of 3Py*–D from 1Py*–D. Pathways (2) and (3), which involve multiple steps, are omitted from the figure for simplicity. The molecules and functional groups that have been confirmed by spectroscopic methods (FT-IR and 1H NMR) are noted in red. . The direct formation pathway (1) is anticipated to be the major route due to the relatively high yield of triplet pyrene (38%, ref. 48) and the single-step formation of 3Py*–D from 1Py*–D. Pathways (2) and (3), which involve multiple steps, are omitted from the figure for simplicity. The molecules and functional groups that have been confirmed by spectroscopic methods (FT-IR and 1H NMR) are noted in red. | ||
The second pathway is the oxidative formylation of DMA. Hydroperoxyl radical (HOO˙) and carbon-centered radicals (b) in DMA react to yield methylene hydroperoxide (e), which can be dehydrated to yield formamide Py-MFA. The third possible pathway involves the deamination after rearrangement of b as reported in ref. 49 (black dashed box in Fig. 6).49 We obtained the evidence of this process in the absence of peak 7 in the MS spectrum (Fig. 3(a)) due to the loss of an ionizable amine. Primary products, Py-MMA (R1 = H, R2 = CH3) and Py-MFA (R1 = CHO, R2 = CH3), may undergo further PD following another excitation of Py. Considering that the LC–MS spectrum of photodegraded Py-DMA shows peaks corresponding to Py-A (R1 = R2 = H), Py-FA (R1 = H, R2 = CHO), and the other PD products, secondary and further reactions are also possible with a prolonged irradiation through subsequent degradation. However, tracking the latter part of the reaction lies beyond the scope of this work.
Quantitative analysis and photodegradation mechanism verification
In pursuit of delineating the predominant pathways between demethylation and oxidative formylation (Fig. 6), a quantitative analysis of the PD products was undertaken. Given the inherent limitation in in quantification using peak area in the MS chromatogram, we opted to leverage the absorbance of Py moiety measured by a diode array detector (DAD) module. We assumed uniform molar absorptivity at 342 nm for the primary PD products, Py-MMA and Py-MFA. Encouragingly, the absorption spectra obtained using the DAD module revealed consistent absorption spectrum shapes within 250–350 nm range across most MS-detected PD products, regardless of their counterparts’ identities (ESI,† Fig. S12(a)). Subsequently, an absorbance–concentration plot was constructed by measuring Py-DMA solutions’ absorbance across concentrations ranging from 0.01 μM to 10 μM in ACN (ESI,† Fig. S12(b)).The plot unveiled a linear correlation between peak area in the UV chromatogram and concentration with an R2 value of 0.9999. Using this linear fit as a standard curve facilitated the quantification of both PD products and residual Py-DMA. The concentrations of the primary products, Py-MMA and Py-MFA, over irradiation time are plotted in Fig. 7(a). Upon the onset of irradiation, Py-MMA and Py-MFA form at nearly identical rates. However, while the concentration of Py-MMA begins plateauing after 10 min, Py-MFA continues to steadily increase until the end of the measurement period at 30 min, at which point the concentration of Py-MFA surpasses that of Py-MMA by more than double. This plot indicates that the oxidative formylation process is more favorable than demethylation.
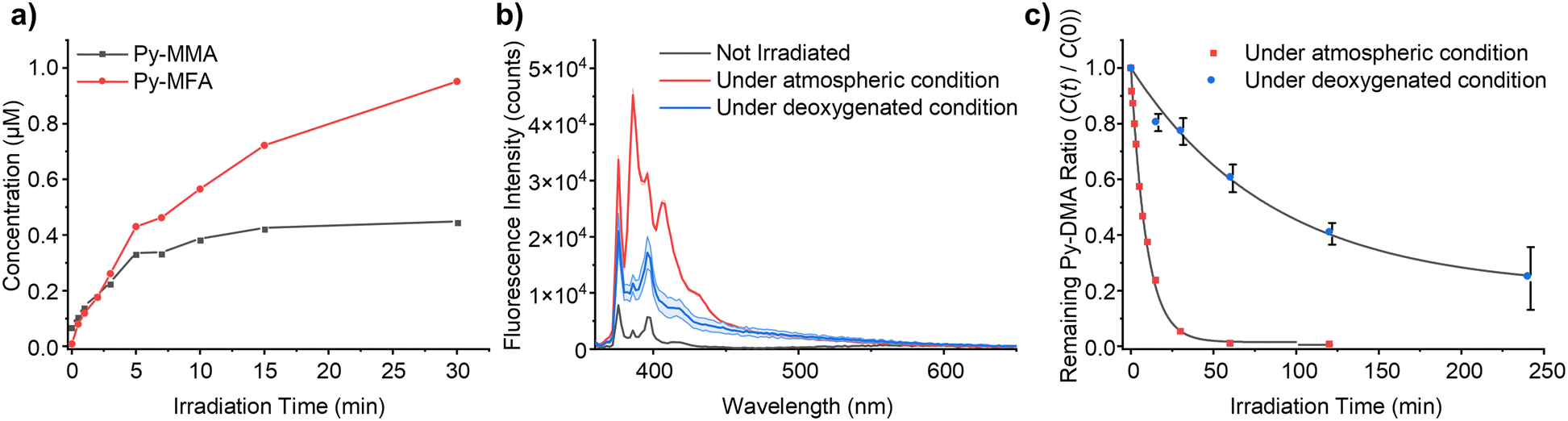 | ||
| Fig. 7 Quantitative analysis for determining the dominant photodegradation pathway and the importance of molecular oxygen on the PD process. (a) The concentration of primary products (Py-MMA and Py-MFA) over irradiation time. (b) The emission spectra of Py-DMA after PD under deoxygenated and atmospheric condition. Area represents standard deviation (n = 4). (c) The remaining Py-DMA ratio (C(t)/C(0)) over irradiation time under deoxygenated and atmospheric condition. | ||
The proposed mechanism depicted in Fig. 6 suggests that molecular oxygen (O2) plays a pivotal role in the PD process. Consequently, we conducted PD of Py-DMA under deoxygenated conditions and compared it with samples exposed to atmospheric conditions to validate the mechanism. The PL spectra of the deoxygenated and atmospheric samples after 120 min of irradiation are presented in Fig. 7(b). The PL spectrum of the atmospheric sample exhibits a similar trend to that shown in Fig. 2(b) (red lines), whereas the deoxygenated sample displays two notable differences. Firstly, the LE increased less compared to the atmospheric sample despite the same irradiation duration. This observation suggests that more Py-DMA remains undegraded, preserving the formation of an exciplex, and indicates that deoxygenation protects Py-DMA from degradation. This was further quantitatively confirmed by monitoring the concentration of Py-DMA obtained by the standard curve above as a function of irradiation time, as shown in Fig. 7(c), where C(0) represents the initial concentration of Py-DMA and C(t) denotes the concentration at a given time (t). In the atmospheric sample, approximately 80% of the Py-DMA was degraded within 15 min of irradiation, and Py-DMA was undetectable after 60 min. In contrast, in the deoxygenated sample, only about 13% was degraded within 15 min, and approximately 25% of Py-DMA remained intact even after 240 min. The second notable point is the disappearance of the steeply growing two PL peaks at 386 and 405 nm, which are assigned to PyOH. This observation supports the notion that the two peaks are linked to molecular oxygen, thereby re-verifying the assignment.
Furthermore, we observed disparities between the chromatograms of the atmospheric PD sample and the deoxygenated PD sample, leading us to draw similar conclusions as those derived from the PL analysis. Further details are provided in the ESI† (Fig. S13).
Conclusions
In this study, we prepared Py–(CH2)12–O–(CH2)2–DMA (Py-DMA) solution as a model exciplex-forming A–D and investigated the PD mechanism in molecular detail. The PD products were identified using absorption and photoluminescence spectra, LC-UV-MS, FT-IR, 1H NMR, as well as quantum mechanical calculations. Especially, the locally-excited emission (LE) band and exciplex emission (ExE) band of Py-DMA were extensively used to monitor electron transfer process from DMA to Py* and quantify the extent of PD. Based on the data, we successfully proposed a mechanism for the PD of Py-DMA. Py-MMA and Py-MFA appeared to be primary products via demethylation and oxidative formylation of the tertiary amine of DMA, respectively. We verified the mechanism by crosschecking with the experimental data. For example, formaldehyde, predicted as a byproduct of the demethylation reaction, was confirmed by the 1H NMR spectroscopy. Molecular oxygen (O2) was expected to play an important role according to the proposed mechanism, and we verified it through deoxygenated condition experiments. A quantitative analysis using the UV-chromatogram and absorbance–concentration calibration curve showed that Py-MFA formed twice as much as Py-MMA in 30-min 355-nm irradiation. Although we focused on the two primary products, we do not rule out other secondary, tertiary, and subsequent products that are likely predominant with prolonged irradiation, such as degradation of the Py moiety and unspecific black carbon precipitation. Since there is increasing evidence that the photostability of the OLED molecules is closely correlated with the operational device stability, the mechanism for the PD reaction in this work will provide useful information for designing organic optoelectronic device materials with a long lifespan. Our research indicates that the amine group of DMA in the Py-DMA exciplex system is the primary site of photodegradation, accelerated by oxygen molecules. We propose substituting the methyl groups on DMA with bulkier groups to prevent oxygen access, although this may reduce exciplex formation yield. Stringent deoxygenation and adding antioxidants like butylated hydroxytoluene (BHT) can also help by scavenging free radicals. Another potential strategy is to use a host molecule, such as cyclodextrin, to encapsulate Py-DMA and protect against photodegradation.50The solution-state PD study on the model exciplex-forming A–D system showed several benefits over the ED degradation study on a solid-state device. Firstly, the low concentration allows for the direct information of the degradation mechanism of the active molecules, without interference from electrode interfaces, domain–domain interface, accumulated defects, morphological changes, and so forth. Secondly, the solution sample enables the use of various conventional analytical tools, such as steady-state spectroscopy and LC–MS, whose capability for chemical identification is critical in PD studies. A polar organic solvent, acetonitrile (ACN, ε = 37.5), was exclusively used in this study. However, it is anticipated that a non-polar solvent would provide an environment more similar to actual devices. A non-polar solvent may result in different photodegradation product yields and kinetics, and a related further study will provide valuable information in parallel with the current work. For a more comprehensive understanding of the intrinsic degradation of exciplex-based organic optoelectronic materials, it would be desirable to integrate insights from additional studies on operational degradation mechanisms, kinetic measurements, computational investigations, and molecular mechanism studies, alongside the findings presented in this work.
Author contributions
Y. K. – conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing; J. K. – data curation, formal analysis, investigation, methodology, visualization, writing – original draft; Y. S. – methodology, writing – original draft; S. P. – conceptualization, methodology, validation; W.-J. C. – resources, supervision, validation, writing – original draft; T.-Y. K. – conceptualization, resources, supervision, validation, writing – original draft; H. L. – conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, writing – original draft, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Research Foundation of Korea (NRF-RS-2023-00279276 and NRF-2021R1A5A1028138 to Y. K. and H. L.) and Samsung Science and Technology Foundation under project number SSTF-BA2002-10. Y. K. and H. L. are grateful for financial support from the GIST Research Institute (GRI) grant funded by the GIST in 2024.References
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, 1991 Search PubMed.
- G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer, VCH Publishers, 1993 Search PubMed.
- C.-K. Moon, Molecular orientation and emission characteristics of Ir complexes and exciplex in organic thin films, Springer, Singapore, 2019 Search PubMed.
- G. Xie, Y. Sueishi and S. Yamamoto, J. Fluoresc., 2005, 15, 475–483 CrossRef CAS PubMed.
- E. V. Bichenkova, H. E. Savage, A. R. Sardarian and K. T. Douglas, Biochem. Biophys. Res. Commun., 2005, 332, 956–964 CrossRef CAS PubMed.
- D. Kim, Y. J. Lee, D. H. Ahn, J. W. Song, J. Seo and H. Lee, J. Phys. Chem. Lett., 2020, 11, 4668–4677 CrossRef CAS PubMed.
- D. Kim, M. Jung, H. Kim, W. J. Chung and H. Lee, Photochem. Photobiol. Sci., 2019, 18, 2688–2695 CrossRef CAS PubMed.
- H. Lee, N. Yang and A. E. Cohen, Nano Lett., 2011, 11, 5367–5372 CrossRef CAS PubMed.
- N. Yang and A. E. Cohen, Opt. Express, 2010, 18, 25461–25467 CrossRef CAS PubMed.
- H. Lee, D. Brinks and A. E. Cohen, Opt. Express, 2015, 23, 28022–28030 CrossRef CAS PubMed.
- M. Sarma and K. T. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 19279–19304 CrossRef CAS PubMed.
- M. Tanaka, R. Nagata, H. Nakanotani and C. Adachi, Commun. Mater., 2020, 1, 18 CrossRef.
- M. Colella, A. Danos and A. P. Monkman, J. Phys. Chem. Lett., 2019, 10, 793–798 CrossRef CAS PubMed.
- S. Hwang, Y. K. Moon, H. J. Jang, S. Kim, H. Jeong, J. Y. Lee and Y. You, Commun. Chem., 2020, 3, 53 CrossRef CAS PubMed.
- H. B. Kim, D. Kim and J. J. Kim, Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence, John Wiley & Sons, Ltd, 2018, pp. 331–376 Search PubMed.
- S. Sudheendran Swayamprabha, D. K. Dubey, Shahnawaz, R. A. K. Yadav, M. R. Nagar, A. Sharma, F. C. Tung and J. H. Jou, Adv. Sci., 2021, 8, 2002254 CrossRef CAS PubMed.
- B. Madushani, M. Mamada, K. Goushi, T. B. Nguyen, H. Nakanotani, H. Kaji and C. Adachi, Sci. Rep., 2023, 13, 7644 CrossRef CAS PubMed.
- S. Schmidbauer, A. Hohenleutner and B. König, Adv. Mater., 2013, 25, 2114–2129 CrossRef CAS PubMed.
- A. S. D. Sandanayaka, T. Matsushima and C. Adachi, J. Phys. Chem. C, 2015, 119, 23845–23851 CrossRef CAS.
- D. Y. Kondakov, W. C. Lenhart and W. F. Nichols, J. Appl. Phys., 2007, 101, 024512 CrossRef.
- W. R. Mateker and M. D. McGehee, Adv. Mater., 2017, 29, 1603940 CrossRef PubMed.
- N. Grossiord, J. M. Kroon, R. Andriessen and P. W. M. Blom, Org. Electron., 2012, 13, 432–456 CrossRef CAS.
- H. Aziz and Z. D. Popovic, Chem. Mater., 2004, 16, 4522–4532 CrossRef CAS.
- S. Scholz, D. Kondakov, B. Lüssem and K. Leo, Chem. Rev., 2015, 115, 8449–8503 CrossRef CAS PubMed.
- A. Hohenleutner, S. Schmidbauer, R. Vasold, D. Joosten, P. Stoessel, H. Buchholz and B. König, Adv. Funct. Mater., 2012, 22, 3406–3413 CrossRef CAS.
- S. Schmidbauer, A. Hohenleutner and B. König, Beilstein J. Org. Chem., 2013, 9, 2088–2096 CrossRef PubMed.
- Y. K. Moon, H. J. Jang, S. Hwang, S. Kang, S. Kim, J. Oh, S. Lee, D. Kim, J. Y. Lee and Y. You, Adv. Mater., 2021, 33, 2003832 CrossRef CAS PubMed.
- S. Richert, A. Rosspeintner, S. Landgraf, G. Grampp, E. Vauthey and D. R. Kattnig, J. Am. Chem. Soc., 2013, 135, 15144–15152 CrossRef CAS PubMed.
- D. Das, D. N. Nath, P. P. Parui and M. Chowdhury, Chem. Phys. Lett., 2006, 424, 300–306 CrossRef CAS.
- P. Roy, A. K. Jana, M. B. Ghanavi and D. N. Nath, Chem. Phys. Lett., 2012, 554, 82–85 CrossRef CAS.
- H. J. Werner, H. Staerk and A. Weiler, J. Chem. Phys., 1978, 68, 2419–2426 CrossRef CAS.
- G. N. Taylor, E. A. Chandross and A. H. Schiebel, J. Am. Chem. Soc., 1974, 96, 2693–2697 CrossRef CAS.
- M. J. Bertocchi, X. F. Zhang, A. Bajpai, J. N. Moorthy and R. G. Weiss, J. Photochem. Photobiol., A, 2018, 355, 467–478 CrossRef CAS.
- N. Mataga, T. Okada and K. Ezumi, Mol. Phys., 1966, 10, 203–204 CrossRef CAS.
- K. König, P. T. So, W. W. Mantulin, B. J. Tromberg and E. Gratton, J. Microsc., 1996, 183, 197–204 CrossRef.
- H. Cao, Y. Fujiwara, T. Haino, Y. Fukazawa, C.-H. Tung and Y. Tanimoto, Bull. Chem. Soc. Jpn., 1996, 69, 2801–2813 CrossRef CAS.
- Y. Tanimoto, K. Hasegawa, N. Okada, M. Itoh, K. Iwai, K. Sugioka, F. Takemura, R. Nakagaki and S. Nagakura, J. Phys. Chem., 1989, 93, 3586–3594 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallin, 2016 Search PubMed.
- H. Bové, C. Steuwe, E. Fron, E. Slenders, J. D’Haen, Y. Fujita, H. Uji-I, M. Vandeven, M. Roeffaers and M. Ameloot, Nano Lett., 2016, 16, 3173–3178 CrossRef PubMed.
- G. E. Lecroy, F. Messina, A. Sciortino, C. E. Bunker, P. Wang, K. A. S. Fernando and Y. P. Sun, J. Phys. Chem. C, 2017, 121, 28180–28186 CrossRef CAS.
- M. E. Sigman, P. F. Schuler, M. M. Ghosh and R. T. Dabestani, Environ. Sci. Technol., 1998, 32, 3980–3985 CrossRef CAS.
- K. Colizza, K. E. Mahoney, A. V. Yevdokimov, J. L. Smith and J. C. Oxley, J. Am. Soc. Mass Spectrom., 2016, 27, 1796–1804 CrossRef CAS PubMed.
- S. Yang, P. Li, Z. Wang and L. Wang, Org. Lett., 2017, 19, 3386–3389 CrossRef CAS PubMed.
- M. Pérez-Gilabert, A. Sánchez-Ferrer and F. García-Carmona, Free Radicals Biol. Med., 1997, 23, 548–555 CrossRef PubMed.
- E. Baciocchi, M. F. Gerini, O. Lanzalunga, A. Lapi, S. Mancinelli and P. Mencarelli, Chem. Commun., 2000, 393–394 RSC.
- E. Baciocchi, M. Bietti, M. F. Gerini and O. Lanzalunga, J. Org. Chem., 2005, 70, 5144–5149 CrossRef CAS PubMed.
- T. Medinger and F. Wilkinson, Trans. Faraday Soc., 1966, 62, 1785–1792 RSC.
- V. I. Stenberg, V. R. Srinivas, R. J. Baltisberger and N. F. Woolsey, J. Org. Chem., 1983, 48, 1107–1110 CrossRef CAS.
- J. Vishwakarma, D. Takkella, S. Sharma and K. Gavvala, J. Photochem. Photobiol., A, 2024, 449, 115411 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00532e |
| This journal is © The Royal Society of Chemistry 2024 |