
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Terrylene on monolayer WS2: coverage-dependent molecular re-orientation and interfacial electronic energy levels†
Qiang
Wang
a,
Sifan
You
b,
Björn
Kobin
c,
Patrick
Amsalem
ac,
Fengshuo
Zu
 ad,
Rongbin
Wang
a,
Andreas
Opitz
a,
Stefan
Hecht
c,
Lifeng
Chi
b and
Norbert
Koch
*ad
ad,
Rongbin
Wang
a,
Andreas
Opitz
a,
Stefan
Hecht
c,
Lifeng
Chi
b and
Norbert
Koch
*ad
aInstitut für Physik & Center for the Science of Materials Berlin (CSMB), Humboldt-Universität zu Berlin, Berlin 12489, Germany. E-mail: norbert.koch@physik.hu-berlin.de
bInstitute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon Based Functional Materials & Devices, Soochow University, Suzhou 215123, P. R. China
cInstitut für Chemie & Center for the Science of Materials Berlin (CSMB), Humboldt-Universität zu Berlin, Berlin 12489, Germany
dHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Berlin 12489, Germany
First published on 26th October 2024
Abstract
The electronic, optical, and functional properties of van der Waals heterostructures comprising organic and two-dimensional inorganic semiconductors depend on the structure of the molecular assembly at and near the interface. Despite the rising interest in such heterostructures, very little is known about the, potentially complex, interplay between the structure and resulting (opto)electronic properties. Herein, we demonstrate with photoemission spectroscopy and scanning tunneling microscopy experiments a coverage dependence of the molecular assembly of terrylene deposited onto monolayer WS2 (with sapphire serving as the substrate) and show how this impacts interfacial electronic properties. Up to monolayer coverage, terrylene molecules adapt a flat-lying orientation, which changes to an inclined orientation for higher coverages. This re-orientation is accompanied with a reduction in terrylene ionization energy by over 400 meV and an accordingly larger energy level offset of frontier energy levels of the two semiconductors and shift of the highest occupied molecular orbital energy level away from the WS2 valence band. This can, for instance, reduce the charge-separation efficiency of the heterostructure with molecular multilayer coverage compared to that with only monolayer coverage. Furthermore, the modification of monolayer WS2 excitonic features through molecular film deposition was evaluated using optical spectroscopy, yielding effective dielectric constants for a series of Rydberg excitons and exciton binding energies for bare and terrylene-covered monolayer WS2 supported by sapphire. Altogether, these findings allow a comprehensive and detailed understanding of the (opto)electronic properties of this prototypical van der Waals heterostructure.
Introduction
van der Waals (vdW) heterostructures comprising π-conjugated organic molecules and two-dimensional (2D) transition metal dichalcogenides (TMDCs) have recently gained increasing attention, notably in view of their applications in advanced electronics and optoelectronics.1,2 Layered materials, such as 2D-TMDCs, feature dangling-bond-free surfaces, which support the formation of clean interfaces with various other functional materials via weak vdW forces. The formation of vdW heterostructures from organic and 2D inorganic semiconductors may preserve the intrinsic properties of individual components but may also facilitate the emergence of distinct novel interfacial properties and functionalities,3 resulting from ground-state or excited-state charge transfer. In general, interface electronic properties are strongly influenced by the structural properties of adsorbed molecular layers. Upon adsorption, many molecular materials tend to form well-ordered 2D structures, whose details depend on the fine balance between intermolecular and molecule–substrate interactions.4 For instance, on metals or graphene as a substrate, it has been documented that organic molecules initially form wetting layers, which are characterized by either a flat-lying or edge-on configuration.5 With increasing coverage, molecules reorient into structures that often resemble their bulk form. It should be noted that the ionization energy (IE) and electron affinity (EA) of ordered molecular layers can vary by several 100 meV depending on their molecular orientation with respect to a surface or interface owing to the specific electrostatic potential landscape created by the molecular arrangement.6–8 Molecular orientation thus plays a crucial role in defining the optoelectronic functionality of heterostructures. Therefore, in order to develop an improved overall-picture of heterostructures formed between organic and 2D semiconductors, a detailed understanding of possible molecular orientation changes as a function of coverage, and how this impacts other properties, is highly desirable. To date, studies on organic molecule/TMDC-monolayer vdW heterostructures have predominantly focused on employing multilayer organic films,4,9–11 and only a few reports have attended to the structural and electronic properties of molecular monolayers and few-layers at the interface.12–142D materials introduce another level of complexity in semiconductor heterostructures compared to their three-dimensional counterparts, specifically pertaining to the energy levels. Even when an organic/TMDC interface formation does not involve covalent interactions or ground-state charge transfer, the energy level alignment at the interface may not only result from the difference in the frontier energy levels of the pristine individual components, because the TMDC's energy levels may be subject to renormalization due to the presence of additional dielectric screening by the organic overlayer.15 This would implicate deviations compared to the energy level alignment assumed from the Schottky–Mott limit, which is the usual approach employed in the literature. This effect of molecular adsorption on the electronic and optical/excitonic properties of the TMDCs has so far received little attention.16
π-conjugated molecules, in general, offer opportunities for engineering the properties of interfaces, such as facilitating energy or charge transfer, or minimizing energy barriers for charge injection. They are commonly employed to form ordered mono- or few-layers by self-assembly, as their extended π-conjugated system facilitates the formation of well-organized and stable molecular layers.2,17 For these molecules, the atomically flat and dangling-bond-free surfaces of TMDCs can serve as an ideal template for the formation of ordered self-assembled layers.18
Here, we investigate self-assembled mono- and few-layers of the π-conjugated molecule terrylene (C30H16) on monolayer (ML) WS2 supported by different substrates, namely sapphire, highly oriented pyrolytic graphite (HOPG), and Au(111). The choice of these substrates was motivated, on the one hand, by the desire to use an insulating substrate that does not couple strongly to the semiconductor, and, on the other hand, to allow contrasting the results with those of conductive substrates and to facilitate performing scanning tunneling microscopy (STM) experiments. Angle-resolved ultraviolet and X-ray photoelectron spectroscopy (ARUPS and XPS) measurements were carried out on the heterostructures to unravel the evolution of the electronic properties as a function of the molecular coverage. Complementarily, the orientation and packing of the molecules were unveiled by STM. We found that terrylene in the monolayer (1L) on ML-WS2 was flat-lying, while in the multilayer it exhibited tilted orientations in a dimerized herringbone-like structure resembling that of the single crystal. This change in orientation was accompanied by a change in the IE and overall interfacial energy level alignment by >400 meV, in both situations forming a staggered type-II semiconductor heterojunction that should enable interfacial charge transfer upon optical excitation. Additionally, the impact of molecular adsorption on the excitonic features of the ML-WS2 was investigated by optical absorption spectroscopy. While no change in the A and B exciton energies was noticed upon molecular adsorption, the higher Rydberg states of the A exciton allowed determining an increased effective dielectric constant for the terrylene-covered ML-WS2 compared to the bare ML-WS2 on sapphire, thereby decreasing the ML-WS2 quasiparticle band gap by ca. 60 meV. The detailed and comprehensive knowledge obtained in this study constitutes a benchmark for the terrylene/ML-WS2 vdW heterostructure and provides useful insights for future studies of this system as well as of similar heterostructures.
Results and discussion
Electronic properties
To explore the electronic properties of the terrylene/ML-WS2 interface, sapphire as an isolating substrate was chosen to avoid direct coupling between ML-WS2 and the conducting substrate and possible charge transfer between the substrate and the molecular overlayer.3,19,20 The ML-WS2/sapphire sample was annealed in situ after being introduced into ultrahigh vacuum to obtain a clean surface for the ARUPS measurements.21 As shown in Fig. S1 of the ESI,† ARUPS revealed the electronic band dispersions of ML-WS2/sapphire, with the valence band maximum located at the K point of the Brillouin zone (BZ), substantiating the ML nature of the employed WS2. The observation of band dispersion from an azimuthally disordered polycrystalline ML-WS2 (as used here) was enabled by the dominant photoelectron intensity coming from the two high-symmetry directions (along Γ–K and Γ–M), due to the dihedral group symmetry of the 2H-phase WS2.3,22 The annealed ML-WS2/sapphire, as shown in Fig. 1a, exhibited a work function (Φ) of 4.20 eV, as determined from the secondary electron cutoff (SECO) region, and the valence band maximum (VBM) was found to be at 1.71 eV binding energy (BE) below the Fermi level (EF) at the K point (Fig. 1b). The latter observation indicated the pronounced n-type character of the ML-WS2/sapphire. The “native” n-doping resulted in free charge carriers partially filling the conduction band.3,21,22 In the present case, a clear density of states at EF (at the K point) was observed, as shown in Fig. 1d. We remark that the role of sulfur vacancies to explain the n-type behavior of WS2 or MoS2 is still debated, because theoretically sulfur vacancies should not behave as a donor state,23 as confirmed by experiments using HOPG or graphene as a supporting substrate.14,24 However, when using sapphire as a substrate, experimental work using ARUPS and scanning tunneling spectroscopy has reported a partial filling of the MoS2 conduction band together with the formation of sulfur vacancies.21,25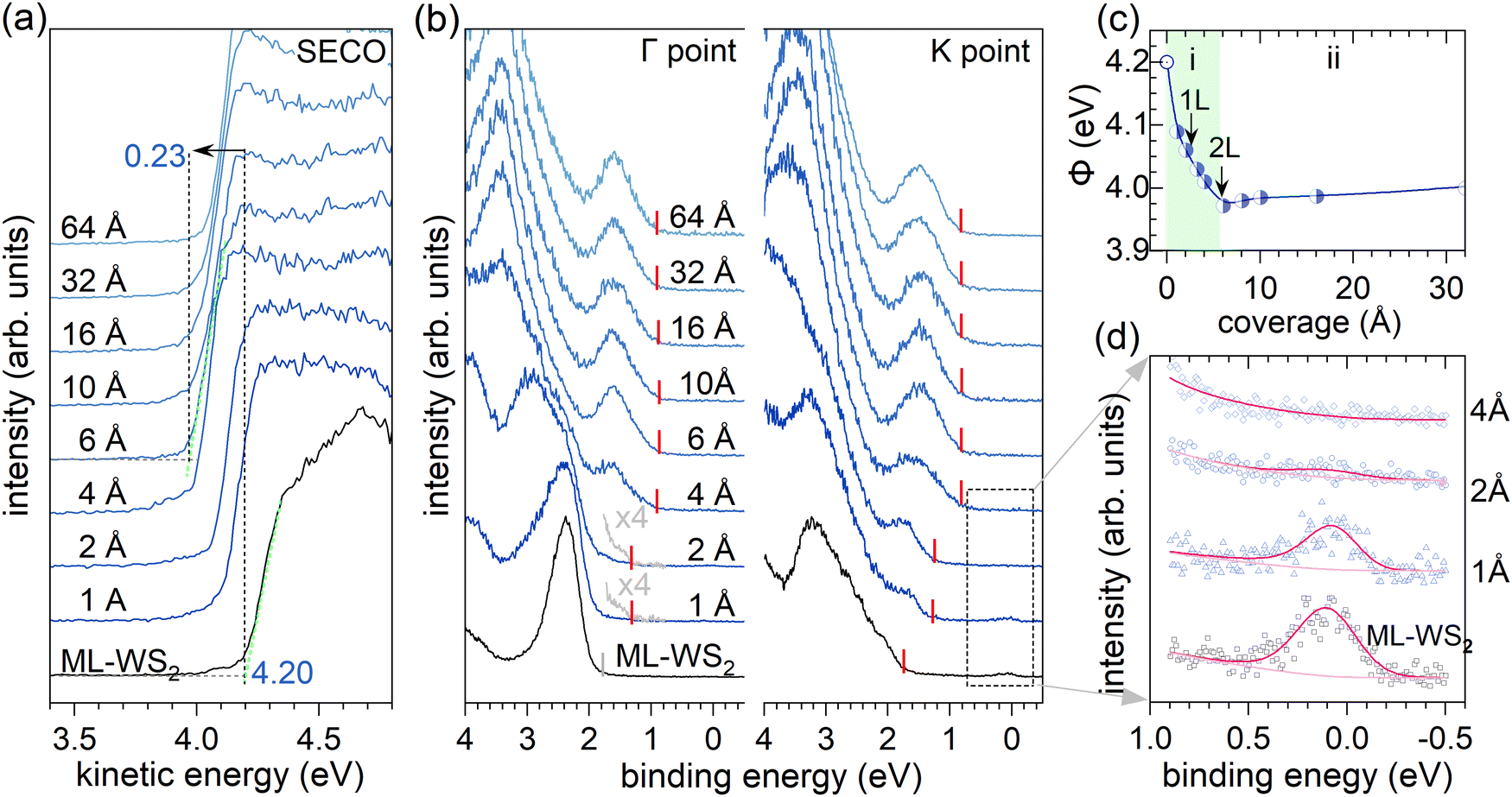 | ||
| Fig. 1 Nominal coverage-dependent ARUPS spectra of terrylene/ML-WS2 on sapphire showing (a) the secondary electron cutoff (SECO) and (b) valence electron region at the Γ and K points of the ML-WS2 Brillouin zone. The Fermi level was at zero binding energy. At the K point, the terrylene HOMO level was clearly visible already from 1 Å coverage. (c) Plot of the work function (Φ) as a function of terrylene coverage. The two regimes (i) and (ii) discussed in the text are indicated. (d) Zoomed-in valence region near the Fermi level. | ||
Next, terrylene was incrementally deposited onto the ML-WS2/sapphire and ARUPS measurements were performed at the Γ and K points after each step. For very low coverages of 1–2 Å, as shown in Fig. 1b, an additional state, corresponding to the highest occupied molecular orbital (HOMO) level, emerged at ca. 1.5 eV at the K point (ca. 40° emission angle) with its emission onset at 1.28 eV; this state was noticeable also at the Γ point (normal emission), but with a much lower intensity. Increasing the coverage beyond 2 Å led to a more substantial contribution of the molecules’ features to the valence region signal at both emission angles. Notably, the HOMO level of terrylene became more prominent and its onset shifted to a significantly lower BE, settling at 0.80 eV BE. The first molecular layer (1L) completion was indicated by the attenuation of the partially filled WS2 conduction band signal at EF, which was completely suppressed at a nominal 4 Å coverage.
For terrylene in the low coverage regime (<4 Å), the VB spectra displayed substantial differences in spectral shape and intensity at the Γ and K points, indicating a pronounced angular dependence of the photoelectron emission. Notably, the valence spectra of 2 Å terrylene/ML-WS2 at the Γ point resembled the momentum-integrated spectrum of bare ML-WS2. This likely originated from Umklapp processes, in which photoelectrons emitted from the ML-WS2 undergo elastic scattering by the molecular superlattice on top.9,26,27 This is supported by Fig. 2b, where a momentum-integrated ML-WS2 spectrum and terrylene HOMO spectrum (obtained from the fitting of terrylene on Au) were used to satisfactorily reconstruct the 2 Å terrylene/ML-WS2 spectra at Γ and K. At the K point, the spectral weight of the terrylene HOMO was about three times larger compared to that at the Γ point, as shown in Fig. 2c. Such a behavior has previously been observed for several π-conjugated molecules with a flat-lying orientation, and explained by the fact that the photoelectrons are emitted at angles associated with the Fourier transform of the real-space distribution of the probed molecular orbital.28,29 The maximum in emission from delocalized π-orbitals is then generally close to 40° for flat-lying molecules.30,31 This implies, therefore, the formation of a first (well-ordered) wetting layer of terrylene with a probably flat-lying orientation. The lack of significant angle-dependence in the HOMO intensity for higher coverage (>4 Å), as evidenced by the convergence of the HOMO intensity ratio SΓHOMO/SKHOMO to 1 as shown in Fig. 2c, indicated a change in the molecular orientation beyond the terrylene wetting layer (1L). We also note that the density of states from the presumably flat-lying molecules of the 1L with its peak maximum at ca. 1.8 eV remained discernable as a high BE shoulder of the main HOMO feature in the 4 Å coverage spectrum, while it was totally absent in the 6 Å spectrum (Fig. S2, ESI†). This suggests that the wetting itself may undergo a reorientation upon increasing coverage, instead of just being buried below differently oriented subsequent layers; this presumption is consistent with the findings from the STM measurements described further below.
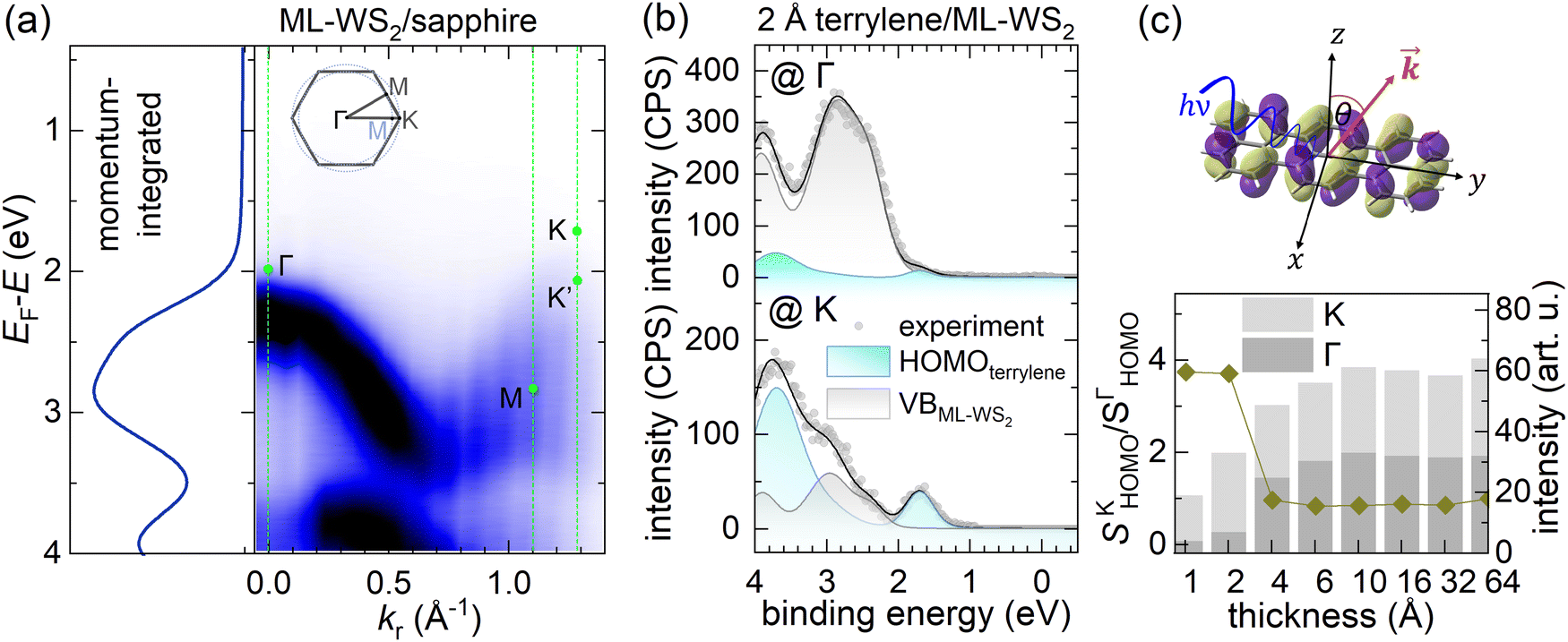 | ||
| Fig. 2 (a) ARUPS map of ML-WS2 and EDC curve of momentum-integrated spectra over the Γ–M–K direction. (b) Fitting of the VB spectra (Shirley background subtracted) of the 2 Å terrylene/ML-WS2 sample at the Γ and K points. The terrylene HOMO component was taken from the fitting of the 2.5 Å terrylene/Au(111) spectrum. (c) Relative intensity of the HOMO peak (1.1–2.1 eV) area. The ratio of the fitting area of emissions at Γ and K points is defined as SΓHOMO /SKHOMO. | ||
The SECO spectra of terrylene with increasing coverage are shown in Fig. 1a. The work function Φ change is depicted in Fig. 1c and can be divided into two regimes: (i) for coverages up to 4 Å, where a sharp Φ decrease from 4.20 eV to 3.97 eV (i.e., by 0.23 eV) occurs; (ii) with further increasing the coverage, Φ first slightly increases then settles at a constant value of about 4.02 eV. The initial Φ decrease in regime (i) apparently occurs upon completion of the terrylene 1L and is attributed to the Pauli pushback effect32,33 of the ML-WS2 with electron density spilling out of the layer, causing charge redistribution at the interface, which lowers the sample Φ. Beyond the 1L-regime of terrylene, surface de-wetting or structural defect formation34 in the organic films might lead to the observed increase in Φ in regime (ii), as shown in Fig. 1c. However, this slight Φ increase was more likely due to a decrease in the pushback effect as the terrylene molecules change from a flat-lying to inclined orientation when multilayer formation becomes possible (vide infra for STM support of this presumption). The HOMO onset of the 1L terrylene was determined to be at 1.28 eV BE, giving an ionization energy (IE) of 5.30 eV for flat-lying molecules. Upon increasing the coverage beyond 6 Å, the HOMO onset shifted to 0.8 eV BE, and with the small Φ change, an IE of 4.77 eV resulted for the multilayer terrylene coverage. As explained previously, such a trend in IE variation upon the molecular orientation change was expected.6–8
Next, we examined the core levels by XPS in order to gain further insights into the molecule–substrate interaction, the terrylene growth mode, and the anticipated changes in molecular orientation. Fig. 3a shows the relative XPS intensities of W 4f7/2 (ML-WS2) and C 1s (terrylene) against the nominal film coverage, revealing three distinct linear regions. The W 4f (C 1s) intensity decreased (increased) linearly up to 2.5 Å, from which point on the slope changed. Another change in slope was observed at 6 Å coverage. These characteristics suggest that the nominal thicknesses corresponding to 1L and a bilayer (2L) were 2.5 Å and 6 Å, respectively. Furthermore, the observed behavior is reminiscent of the characteristics of the Stranski–Krastanov growth mode, where the second layer starts growing only after the closure of the first layer, and weak islanding likely starts during the growth of the second layer.35,36 As shown in Fig. 3b, the XPS W 4f spectrum of bare ML-WS2 displayed the W 4f7/2 (33.89 eV) and W 4f5/2 (36.01 eV) doublet of W4+ due to the W–S bonds, together with faint contributions at 37.25 eV and 39.37 eV (3.8% of the spectral weight) corresponding to W6+ belonging to tungsten oxides. For the C 1s, two components were fitted, the main peak being attributed to the sp2 carbons of the molecule and the higher BE shoulder at 1.95 eV away from the main peak to a shake-up process, likely corresponding to the HOMO–LUMO transition. Upon increasing the terrylene coverage, the C 1s peak for 2L (284.78 eV) shifted toward a lower BE by 0.47 eV with respect to that of 1L (285.25 eV) (Fig. S3, ESI†). The observed BE shift supports the molecular reorientation suggested above, because the direction and magnitude of the C 1s peak are the same as the HOMO-onset change, underpinning the electrostatic origin of the shifts. The W 4f peaks exhibited a comparably small shift to lower BE by 90 meV upon terrylene deposition, in line with the improved screening of the final state by the adsorption of the molecular overlayer, in qualitative agreement with the change in dielectric screening found by optical spectroscopy in a related molecule/2D semiconductor system.3
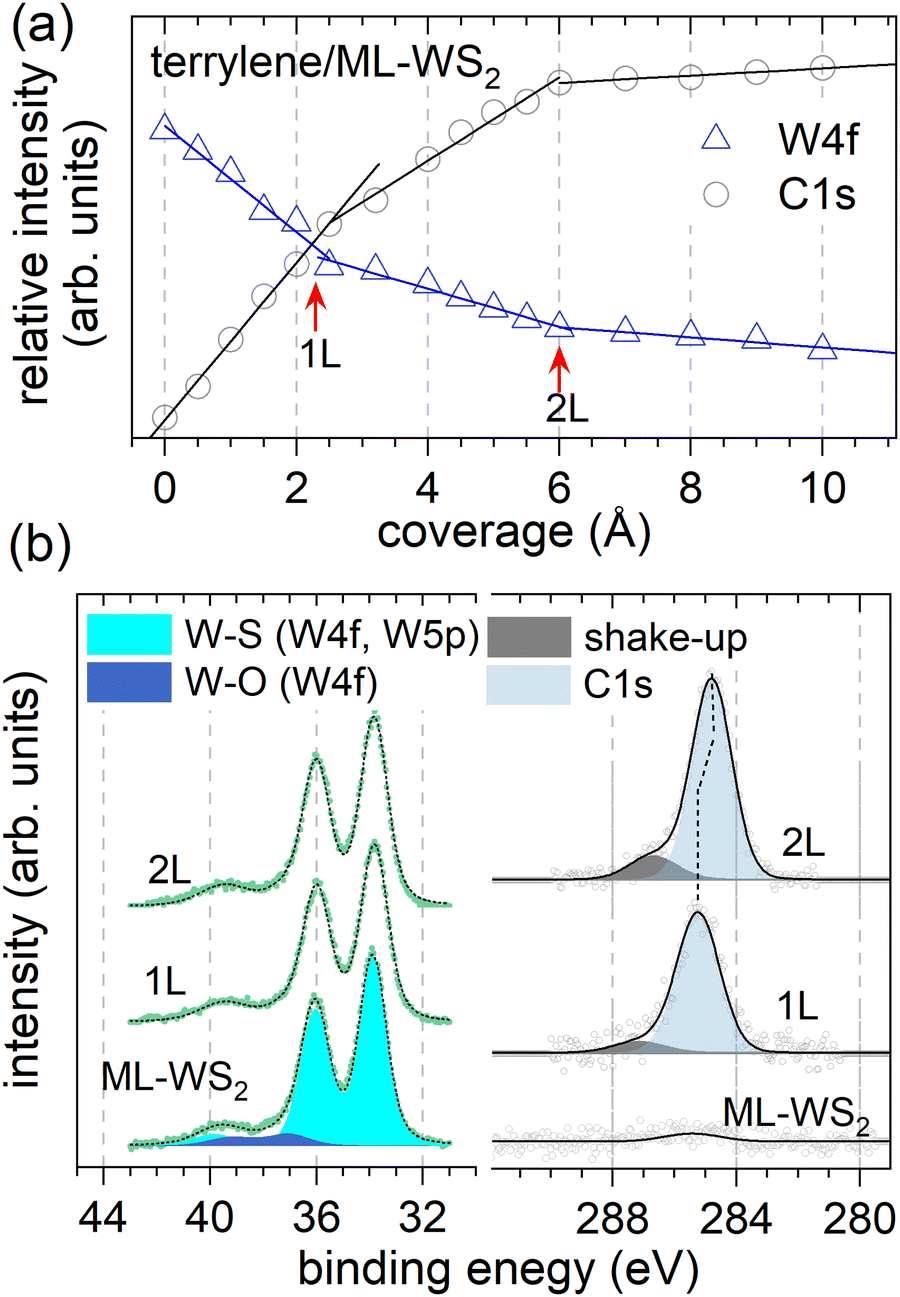 | ||
| Fig. 3 (a) Variation of the relative XPS intensities of W 4f and C 1s of terrylene/ML-WS2 as a function of coverage based on XPS data (Fig. S4, ESI†). For W 4f (C 1s), three linear decreasing (increasing) intensity regions could be fitted, with corresponding slope changes (red arrows). (b) W 4f and C 1s XPS spectra (Shirley background subtracted) of bare ML-WS2 and 1L and 2L of terrylene/ML-WS2. | ||
Structural properties
To substantiate the molecular orientation changes inferred from the photoemission results, STM measurements were performed on ML-WS2/HOPG (note: STM with sapphire as a support for the heterostructure was not feasible, probably due to the moderate conductivity of ML-WS2 and the high current density in the STM tip vicinity), which showed a similar terrylene IE change as observed in photoemission when using the sapphire substrate as the support (Fig. S5, ESI†).Prior to terrylene deposition, a well-defined and atomically clean ML-WS2 surface was observed, as shown in Fig. 4a. The unit cell parameters were measured to be a = (0.315 ± 0.006) nm, and θ = 59.5 ± 2°, which were consistent with the 2H phase of WS2.37 As illustrated in Fig. 4b and c, the terrylene molecules were arranged into distinct layered configurations, with the first one (1L) displaying an oblique lattice, and the second (2L) and third layers (3L) featuring hexagonal arrangements. The unit cell parameters of the 1L [a = (1.10 ± 0.04) nm, b = (1.84 ± 0.06) nm and θ = (60.0 ± 2.5)°] support the flat-lying orientation of closely packed terrylene molecules, thereby maximizing the substrate area coverage with the molecular area, a feature also observed on Au(111) (Fig. S6, ESI†). As shown in Fig. 4e, an inclined and seemingly dimerized molecular orientation, resembling the terrylene single crystal configuration (Fig. S6d, ESI†),38 was suggested by STM for higher coverage (2L and 3L), with unit cell parameters of a = (1.93 ± 0.04) nm, b = (1.84 ± 0.05) nm, and θ = (59.0 ± 3.0)°. Such a transition from a flat-lying to inclined orientation was further supported by the relative height and corrugation of each layer, as depicted in Fig. 4g. Notably, the apparent heights with respect to ML-WS2 were ca. (3.0 ± 0.1) Å for 1L, (9.4 ± 0.1) Å for 2L, and (19.0 ± 0.2) Å for 3L, accompanied by an increase in the in-plane corrugation [0.08 Å for 1L, and 0.23 Å for 3L]. We note that the apparent height of a terrylene layer lying flat on top of a first flat-lying monolayer was about 3.0 Å, as determined on Au(111) (see Fig. S6, ESI†). Notably, on ML-WS2/HOPG, the height of the 3L corresponded to twice the height of the 2L, indicating that 3L likely corresponded to two stacked 2Ls, while the height of the 2L was about three times that of the 1L. This therefore suggests that the flat-lying molecules of the 1L undergo reorientation upon multilayer formation, transitioning to form part of the 2L with inclined molecular orientation. The STM analysis thus unambiguously revealed the change in the molecular orientation and packing from 1L to 2L. Specifically, the results confirmed a flat-lying orientation for 1L, in contrast to the inclined configuration observed for 2L and beyond.
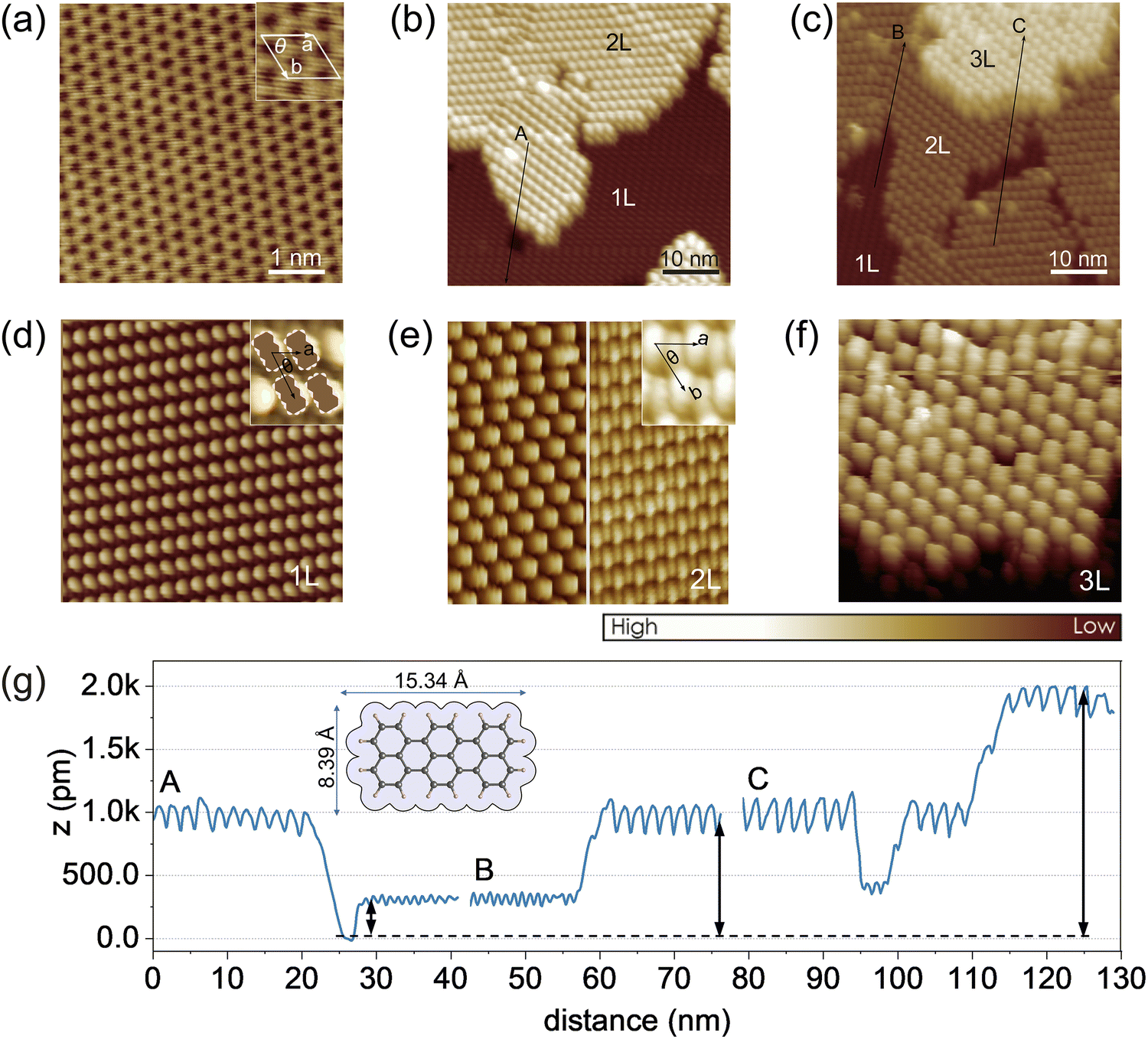 | ||
| Fig. 4 STM images of the orientation change in terrylene multilayer on ML-WS2/HOPG. (a) Clean surface of ML-WS2 before terrylene deposition. (b) Bare ML-WS2 surface (black area) next to molecular layers. (c) Multilayer terrylene on ML-WS2/HOPG, with a flat-lying orientation in 1L (first layer), and inclined dimerized structures in 2L (second layer) and 3L (third layer). High resolution zoomed-in images of 1L (d), 2L (e), and 3L (f) with the corresponding unit cell marked. Two different configurations are visualized as two distinct symmetries in the STM images. Each figure is 20 nm × 20 nm. (g) Line profile in (b) and (c), marked as A, B, and C. Lower relative height and small corrugation in 1L transform to a large step height and corrugation in 3L. Set point: (a) 500 mV, 20 pA, (b), (c), and (d)–(f) 3 V, 10 pA. | ||
To better visualize the molecular arrangement on ML-WS2, a schematic representation of the molecular configurations, as derived from the STM data, is depicted in Fig. 5. Upon superimposing the unit cells across the interface between 1L and 2L (Fig. 5a), it was evident that 2L follows the orientation of the lattice of 1L, albeit with a different periodicity. Specifically, the volume ratio between unit cells of 1L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2L was 4:7, where the dimers in 2L adopt a herringbone packing motif.39 The lattice vectors of the terrylene 1Ls were oriented along the [120] and [1
:2L was 4:7, where the dimers in 2L adopt a herringbone packing motif.39 The lattice vectors of the terrylene 1Ls were oriented along the [120] and [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] directions of the ML-WS2 lattice and formed what resembles a (
0] directions of the ML-WS2 lattice and formed what resembles a (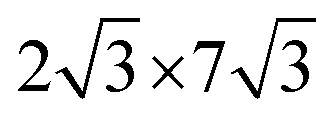 )R30° superlattice (Fig. 5b), otherwise described by the superstructure matrix
)R30° superlattice (Fig. 5b), otherwise described by the superstructure matrix 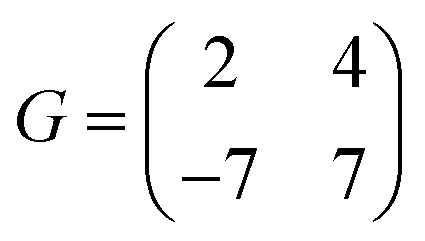 . For the 2L terrylene, the assembly continued along the [120] direction and changed into a dimerized structure, now resembling the single crystal structure (Fig. S6d, ESI†), which rearranged into a (
. For the 2L terrylene, the assembly continued along the [120] direction and changed into a dimerized structure, now resembling the single crystal structure (Fig. S6d, ESI†), which rearranged into a (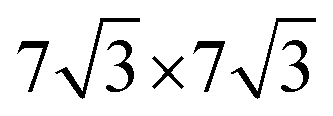 )R30° overlayer with
)R30° overlayer with 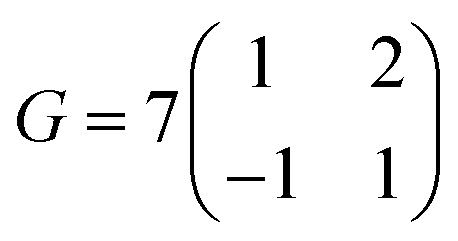 . Based on the structural model, the calculated molecular densities of 2L to 1L was 2.3, which corresponds to the thickness (molecular weight) ratio of 2.4 obtained from the PES data (Note 2, ESI†).
. Based on the structural model, the calculated molecular densities of 2L to 1L was 2.3, which corresponds to the thickness (molecular weight) ratio of 2.4 obtained from the PES data (Note 2, ESI†).
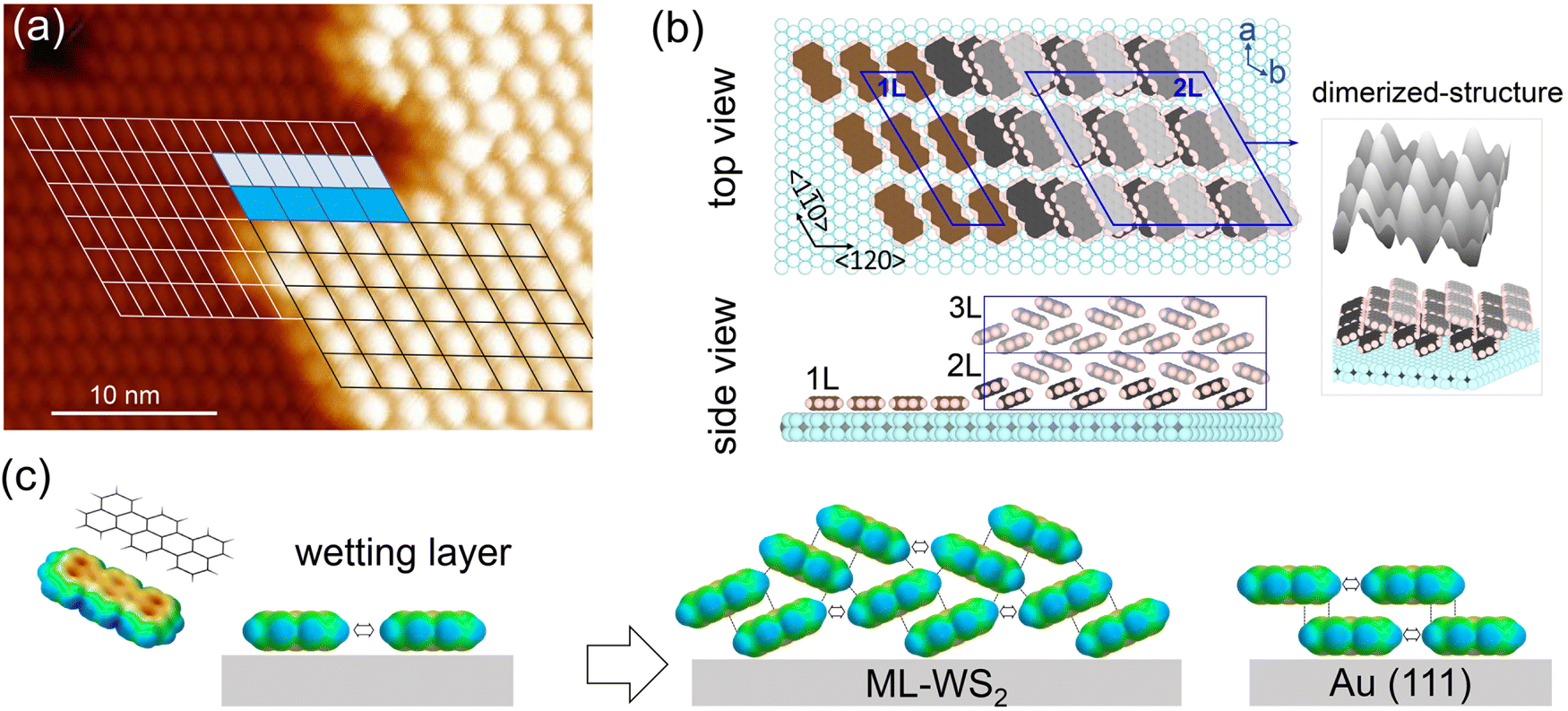 | ||
Fig. 5 (a) High-resolution STM images for 1L and 2L. The arrangement of self-assembled terrylene in 1L and 2L are marked as white and black lines, respectively. Set point: 3 V, 20 pA. (b) Top view and side view along the [100] direction of a reconstruction model based on STM results, with unit cells of (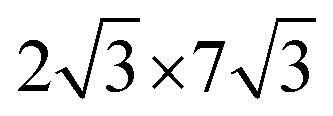 ) adlayer of 1L and ( ) adlayer of 1L and (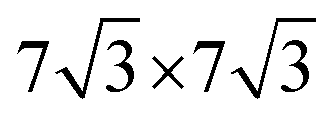 ) overlayer of 2L on ML-WS2. The unit cell dimensions of ML-WS2 are a = b = 0.316, θ = 120°. (c) Formation of 2L after the completion of a wetting layer on ML-WS2 and Au(111). Blue stands for positive charge and yellow stands for negative charge. ) overlayer of 2L on ML-WS2. The unit cell dimensions of ML-WS2 are a = b = 0.316, θ = 120°. (c) Formation of 2L after the completion of a wetting layer on ML-WS2 and Au(111). Blue stands for positive charge and yellow stands for negative charge. | ||
Optical properties
UV-vis spectroscopy was performed to evaluate the impact of the molecular adlayer atop the ML-WS2 TMDC on its excitonic properties. Detailed analysis of the spectra shown in Fig. 6 revealed that the characteristic A exciton peak maxima of ML-WS2 without and with terrylene differed by only ca. 2 meV.40 The spectral features of Rydberg states (n = 1s–5s) below the ionization threshold were enhanced by taking the secondary derivatives of the UV-vis spectra, as shown in Fig. 6. The Rydberg series can be approximated as s-type hydrogenic excitons,41,42 following a characteristic sequence of 1/(n − 1/2)2. The effective dielectric constant experienced by the excitons, kneff, with the overlayer for the nth exciton, can be calculated as:| kneff = [μe4/8h2ε02Enb,exp(n − 1/2)2]1/2 | (1) |
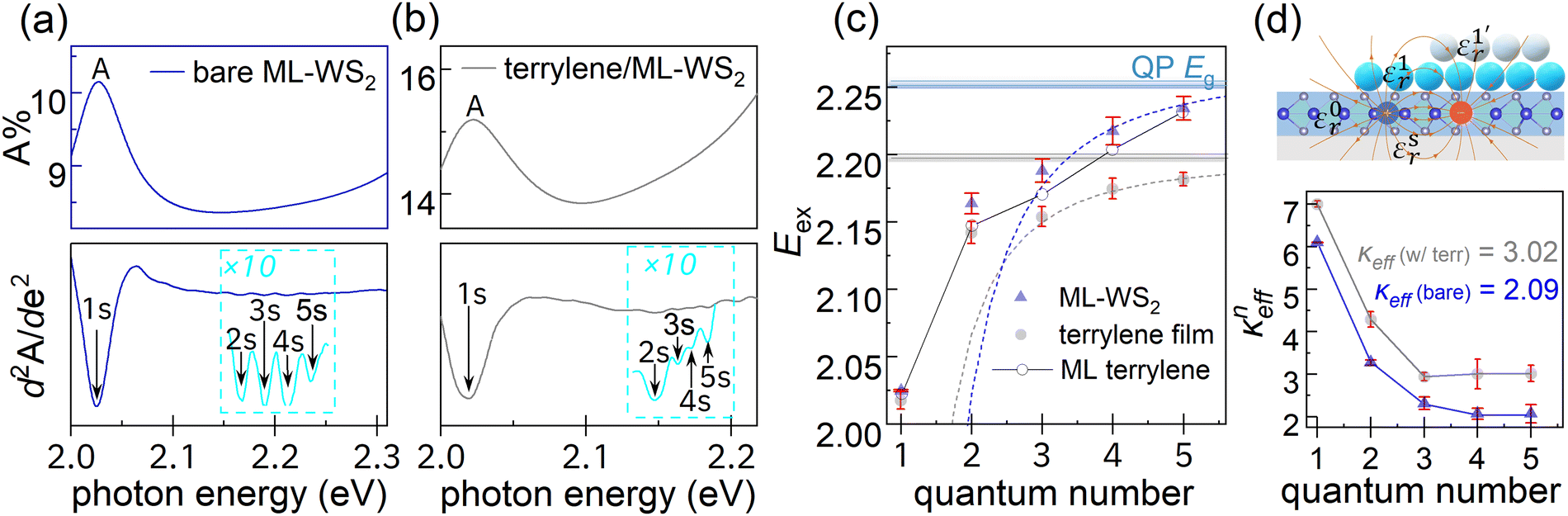 | ||
| Fig. 6 UV-vis absorption spectra of (a) ML-WS2/sapphire and (b) multilayer (64 Å) terrylene/ML-WS2/sapphire, with the second-order numerical derivative drawn below. The Rydberg states are indicated as 1s to 5s. (c) Experimentally obtained transition energies for the exciton states as a function of the quantum number n. The error bars resulted from the fitting procedure. The absorption spectrum of sub-ML (2 Å) terrylene is shown in hollow circles. The corresponding 2D hydrogenic models before or after terrylene deposition are depicted in dashed lines. (d) Calculated effective dielectric constant as a function of n using the hydrogenic model. | ||
A reduced exciton mass μ = 0.16 m0 for ML-WS2 was adopted.42,43 The exciton BE (Enb,exp) was experimentally determined by the difference between Eg and Eex. In Fig. 6c, the quasiparticle Eg values were obtained by fitting the Rydberg states (n = 3–5) with the hydrogenic model, resulting in values of (2.26 ± 0.01) eV and (2.20 ± 0.01) eV for the bare and terrylene-covered ML-WS2, respectively. Correspondingly, the ML-WS2 A-exciton BE (En=1b,exp) was found to decrease from 234 meV to 173 meV after terrylene deposition. As shown in Fig. 6d, the larger exciton size for the higher n resulted in enhanced dielectric screening from the surrounding material, causing the keff to decrease and eventually stabilize. Notably, the anisotropic screening of the tightly bound 1s exciton was mainly due to WS2, while the higher-order (3s–5s) e–h pairs with larger radii were primarily screened by the surrounding materials as Wannier-type excitons, where keff is equal to (εtopr + εbottomr)/2. Using εsapphirer = 3.18, the keff values for the A-exciton (n = 1) and higher n were approximately 6.10 and 2.09, respectively. With a terrylene film (64 nm, approx. 17-MLs), kn>3eff for higher n was fitted to 3.02.
Final heterostructure energy level diagram
We first remark that the ML-WS2/sapphire single-particle energy gap (Eg) could not be determined by ARUPS, because of the partial CB occupation after annealing in vacuum, i.e., the Eg of ML-WS2 without this strong native n-doping would be above 2 eV. The UV-vis analysis discussed above allowed estimating Eg at ca. 2.26 eV, by summing the optical gap and the exciton binding energy. This was likely because of air exposure of the UV-vis samples, which can significantly reduce the CB filling as oxygen inactivates a large part of the n-doping behavior in UHV. In Fig. 7, we summarize the energy level alignment diagram based on our ARUPS data including the occupied CB states, and use the Eg values obtained from the optical analysis to represent the unoccupied states. The optical bandgap of terrylene films was determined to be 1.95 eV on sapphire, as obtained from a Tauc plot (Fig. S7b, ESI†). Accounting for a representative value of the exciton binding energy of conjugated molecules of 0.55 eV,40,44,45 we obtained a terrylene electronic Eg of 2.5 eV. The resulting energy level alignment shows that a type-II heterojunction was formed both for the mono- and multilayer terrylene films, for which the HOMO lay at ca. 1.28 eV and 0.80 eV below EF, respectively, due to the transition from a flat-lying to an inclined molecular orientation when entering the multilayer regime, and including a reorientation of the first wetting layer, as depicted in Fig. 5b. In addition, upon increasing the terrylene thickness, dielectric screening intensified and reduced the Eg of ML-WS2 by about 0.1 eV, a value which we expect to be a reasonable guidance to estimate bandgap renormalization due to dielectric screening by organic semiconductor overlayers. This value was also in line with the 90 meV shift of the W 4f core levels observed upon the deposition of terrylene.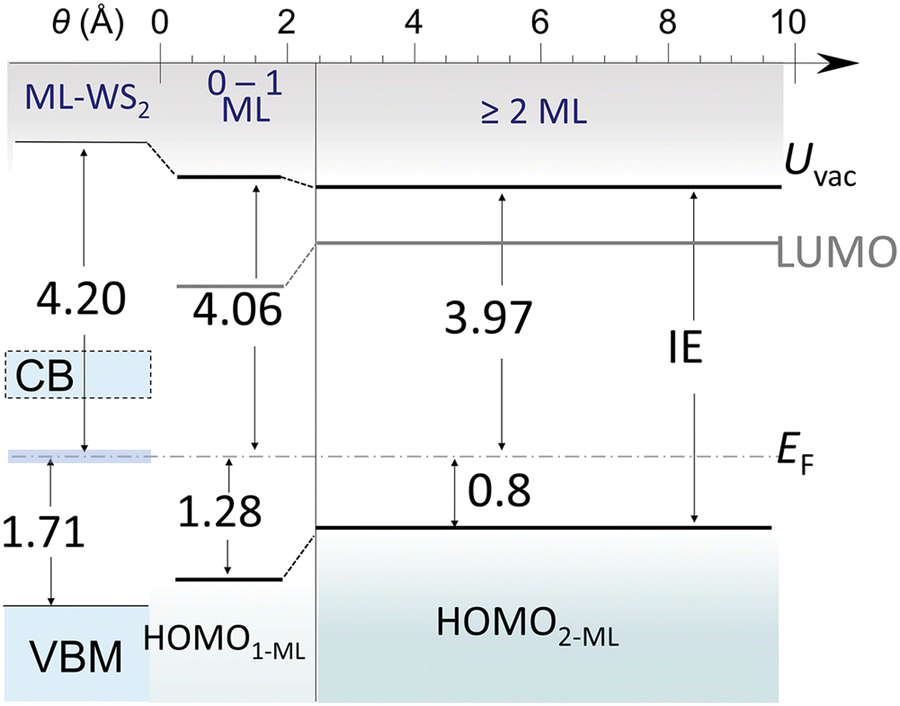 | ||
| Fig. 7 Schematic energy level alignment in terrylene/ML-WS2 heterostructures. The energy difference between energy levels and EF is shown by arrows and numbers (unit, eV). The partially filled CB in ML-WS2 is indicated by blue shading. Note that the “0–1 ML” region was no longer present as soon as the 2 ML coverage region has formed because the 1L flat-lying wetting layer of terrylene was consumed to form the 2L inclined layer (see Fig. 5 and associated text). | ||
Conclusion
In conclusion, we examined the molecular layer thickness-dependent structural and optoelectronic properties of the terrylene/ML-WS2 interface supported on sapphire, a prototypical insulator, and found a type-II energy level alignment at this interface. Thickness-dependent ARUPS and STM measurements allowed for correlating the observed change in molecular ionization energy with the molecular orientation: the reorientation from flat-lying adsorbed terrylene in the monolayer to the edge-on orientation in the multilayer film was accompanied by a reduction in terrylene IE by 0.48 eV. Furthermore, our results also suggest that the monolayer molecules change their orientation upon multilayer formation in order to form the bulk-like dimerized structure, which was possibly due to the weak molecule–substrate interaction. Additionally, we quantified the change in the TMDC quasiparticle bandgap and exciton binding energy resulting from the increased dielectric screening promoted by the molecular adlayer. The higher-order Rydberg states were found to be strongly affected due to the large e–h radius, while the 1s exciton remained largely unchanged due to its small spatial extend. These results demonstrate the importance of coupling the structural and optoelectronic characterization of these hybrid interfaces in order to extract the relevant energy level offset occurring upon the formation of device-relevant organic/TMDC hybrid heterostructures.Experimental section
Preparation of the ML-WS2 samples
The ML-WS2 films were synthesized by chemical vapor deposition using WO3, Na2WO4 and sulfur as precursors on a sapphire substrate. The as-grown ML-WS2 was transferred to a clean sapphire substrate and freshly cleaved HOPG substrate. The detailed synthesis and transfer for ML-WS2 is described in the ESI† (note). The after-transfer ML-WS2/sapphire samples were fixed to a sample holder and electrically connected to ground with screws fixing the film edges. The ML-WS2/sapphire or WS2/HOPG samples were loaded into an ultrahigh vacuum chamber (base pressure of 1 × 10−9 mbar) and annealed for 10 h at 350 °C to remove transfer-induced contamination.Terrylene synthesis
The synthesis of terrylene was carried out following a route published previously.46 Final purification was carried out by multiple subsequent steps of crystallization from 1,2,4-trichlorobenzene and sublimation to achieve a purity of 99.5%.Photoemission measurements
The samples were annealed at high temperature (500–600 °C) for 1 h in repeated cycles to create enough sulfur vacancies, prior to transferring to the analyzing chamber equipped with a hemispherical electron analyzer (SPECS Phoibos 100). The photoemission spectra were collected using a monochromatic He Iα source (HIS-13 lamp mounted on a VUV5046 UV monochromator) and Al Kα (1486.6 eV) as the excitation source for UPS and XPS analyses, respectively, ensuring no sample charging was observed. The overall energy resolution was determined by a polycrystalline gold sample, to be 0.07 eV and 0.8 eV for UPS and XPS, respectively. For the ARPES measurements, the spectra were collected by rotating the manipulator at an angular step of 2°.STM measurements
All the samples were prepared with a commercial low-temperature scanning tunneling microscopy system (Unisoku LT-STM 1500s) with a base pressure better than 1 × 10−10 torr. The WS2/HOPG sample was annealed to 823 K for 2 h before the deposition of terrylene molecules. Terrylene molecules were evaporated at 543 K for 5 min on ML-WS2/HOPG held at room temperature using a commercial Knudsen cell (Kentax, Germany). The STM tips were cleaned by e-beam heating under UHV. All the images were acquired at 77 K in the constant current mode. The STM images were processed using the WSxM software.47Optical measurements
Optical absorption spectra were obtained for the large area (>92% coverage) ML-WS2 films on sapphire using a Lambda 950 UV/Vis/NIR spectrophotometer (Perkin Elmer Inc.) under ambient conditions.Data availability
Data for this article are available at the Open-Access-Publikationsserver of Humboldt-Universität at https://doi.org/10.18452/28966.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, Projektnummer 182087777 – SFB951), National Natural Science Foundation of China (21790053, 51821002, 22072103, 22102109 and 22161132026), the Collaborative Innovation Center of Suzhou Nano Science and Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). Q. Wang acknowledges funding by the Chinese Scholarship Council. S. You acknowledges the support from Jiangsu Excellent Postdoctoral Program (2024ZB117).References
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- D. Jariwala, T. J. Marks and M. C. Hersam, Nat. Mater., 2017, 16, 170–181 CrossRef CAS PubMed.
- J. Ma, P. Amsalem, T. Schultz, D. Shin, X. Xu and N. Koch, Adv. Electron. Mater., 2021, 7, 2100425 CrossRef CAS.
- M. Dreher, D. Günder, S. Zörb and G. Witte, Chem. Mater., 2020, 32, 9034–9043 CrossRef CAS.
- B. Nickel, M. Fiebig, S. Schiefer, M. Göllner, M. Huth, C. Erlen and P. Lugli, Phys. Status Solidi A, 2008, 205, 526–533 CrossRef CAS.
- S. Duhm, G. Heimel, I. Salzmann, H. Glowatzki, R. L. Johnson, A. Vollmer, J. P. Rabe and N. Koch, Nat. Mater., 2008, 7, 326–332 CrossRef CAS PubMed.
- M. L. Tietze, W. Tress, S. Pfützner, C. Schünemann, L. Burtone, M. Riede, K. Leo, K. Vandewal, S. Olthof, P. Schulz and A. Kahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 085119 CrossRef.
- M. Schwarze, K. S. Schellhammer, K. Ortstein, J. Benduhn, C. Gaul, A. Hinderhofer, L. Perdigon Toro, R. Scholz, J. Kublitski, S. Roland, M. Lau, C. Poelking, D. Andrienko, G. Cuniberti, F. Schreiber, D. Neher, K. Vandewal, F. Ortmann and K. Leo, Nat. Commun., 2019, 10, 2466 CrossRef PubMed.
- S. Park, N. Mutz, S. A. Kovalenko, T. Schultz, D. Shin, A. Aljarb, L. J. Li, V. Tung, P. Amsalem, E. J. W. List-Kratochvil, J. Stähler, X. Xu, S. Blumstengel and N. Koch, Adv. Sci., 2021, 8, 2100215 CrossRef CAS PubMed.
- H. G. Shin, D. Kang, Y. Jeong, K. Kim, Y. Cho, J. Park, S. Hong, Y. Yi and S. Im, ACS Nano, 2020, 14, 15646–15653 CrossRef CAS PubMed.
- S. Park, T. Schultz, D. Shin, N. Mutz, A. Aljarb, H. S. Kang, C. H. Lee, L. J. Li, X. Xu, V. Tung, E. J. W. List-Kratochvil, S. Blumstengel, P. Amsalem and N. Koch, ACS Nano, 2021, 15, 14794–14803 CrossRef CAS PubMed.
- Z. Song, T. Schultz, Z. Ding, B. Lei, C. Han, P. Amsalem, T. Lin, D. Chi, S. L. Wong, Y. J. Zheng, M. Y. Li, L. J. Li, W. Chen, N. Koch, Y. L. Huang and A. T. S. Wee, ACS Nano, 2017, 11, 9128–9135 CrossRef CAS PubMed.
- S. Park, H. Wang, T. Schultz, D. Shin, R. Ovsyannikov, M. Zacharias, D. Maksimov, M. Meissner, Y. Hasegawa, T. Yamaguchi, S. Kera, A. Aljarb, M. Hakami, L. J. Li, V. Tung, P. Amsalem, M. Rossi and N. Koch, Adv. Mater., 2021, 33, 2008677 CrossRef CAS PubMed.
- G. Reecht, N. Krane, C. Lotze and K. J. Franke, ACS Nano, 2019, 13, 7031–7035 CrossRef CAS PubMed.
- W.-T. Hsu, J. Quan, C.-Y. Wang, L.-S. Lu, M. Campbell, W.-H. Chang, L.-J. Li, X. Li and C.-K. Shih, 2D Mater., 2019, 6, 025028 CrossRef CAS.
- P. A. Markeev, E. Najafidehaghani, G. F. Samu, K. Sarosi, S. B. Kalkan, Z. Gan, A. George, V. Reisner, K. Mogyorosi, V. Chikan, B. Nickel, A. Turchanin and M. P. de Jong, ACS Nano, 2022, 16, 16668–16676 CrossRef CAS PubMed.
- W. H. Lee, J. Park, S. H. Sim, S. Lim, K. S. Kim, B. H. Hong and K. Cho, J. Am. Chem. Soc., 2011, 133, 4447–4454 CrossRef CAS PubMed.
- C.-H. Lee, T. Schiros, E. J. G. Santos, B. Kim, K. G. Yager, S. J. Kang, S. Lee, J. Yu, K. Watanabe, T. Taniguchi, J. Hone, E. Kaxiras, C. Nuckolls and P. Kim, Adv. Mater., 2014, 26, 2812–2817 CrossRef CAS PubMed.
- S. Park, N. Mutz, T. Schultz, S. Blumstengel, A. Han, A. Aljarb, L.-J. Li, E. J. W. List-Kratochvil, P. Amsalem and N. Koch, 2D Mater., 2018, 5, 025003 CrossRef.
- Y. Liu, J. Guo, E. Zhu, L. Liao, S. J. Lee, M. Ding, I. Shakir, V. Gambin, Y. Huang and X. Duan, Nature, 2018, 557, 696–700 CrossRef CAS PubMed.
- F. Caruso, P. Amsalem, J. Ma, A. Aljarb, T. Schultz, M. Zacharias, V. Tung, N. Koch and C. Draxl, Phys. Rev. B, 2021, 103, 205152 CrossRef CAS.
- S. Park, T. Schultz, A. Han, A. Aljarb, X. Xu, P. Beyer, A. Opitz, R. Ovsyannikov, L. J. Li, M. Meissner, T. Yamaguchi, S. Kera, P. Amsalem and N. Koch, Commun. phys., 2019, 2, 68 CrossRef.
- J.-Y. Noh, H. Kim and Y.-S. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 205417 CrossRef.
- B. Schuler, D. Y. Qiu, S. Refaely-Abramson, C. Kastl, C. T. Chen, S. Barja, R. J. Koch, D. F. Ogletree, S. Aloni, A. M. Schwartzberg, J. B. Neaton, S. G. Louie and A. Weber-Bargioni, Phys. Rev. Lett., 2019, 123, 076801 CrossRef CAS PubMed.
- P. Vancso, G. Z. Magda, J. Peto, J. Y. Noh, Y. S. Kim, C. Hwang, L. P. Biro and L. Tapaszto, Sci. Rep., 2016, 6, 29726 CrossRef CAS PubMed.
- L. Giovanelli, P. Amsalem, T. Angot, L. Petaccia, S. Gorovikov, L. Porte, A. Goldoni and J. M. Themlin, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125431 CrossRef.
- M. Krivenkov, D. Marchenko, M. Sajedi, A. Fedorov, O. J. Clark, J. Sanchez-Barriga, E. D. L. Rienks, O. Rader and A. Varykhalov, Nanoscale, 2022, 14, 9124–9133 RSC.
- P. Puschnig, S. Berkebile, A. J. Fleming, G. Koller, K. Emtsev, T. Seyller, J. D. Riley, C. Ambrosch-Draxl, F. P. Netzer and M. G. Ramsey, Science, 2009, 326, 702–706 CrossRef CAS PubMed.
- X. Yang, M. Jugovac, G. Zamborlini, V. Feyer, G. Koller, P. Puschnig, S. Soubatch, M. G. Ramsey and F. S. Tautz, Nat. Commun., 2022, 13, 5148 CrossRef CAS PubMed.
- O. Endo, F. Matsui, S. Kera, W.-J. Chun, M. Nakamura, K. Amemiya and H. Ozaki, J. Phys. Chem. C, 2022, 126, 15971–15979 CrossRef CAS.
- Y. Nakayama, S. Kera and N. Ueno, J. Mater. Chem. C, 2020, 8, 9090–9132 RSC.
- P. S. Bagus, V. Staemmler and C. Woll, Phys. Rev. Lett., 2002, 89, 096104 CrossRef PubMed.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605–625 CrossRef CAS.
- T. Hosokai, H. Machida, A. Gerlach, S. Kera, F. Schreiber and N. Ueno, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195310 CrossRef.
- C. Argile and G. E. Rhead, Surf. Sci. Rep., 1989, 10, 277–356 CrossRef CAS.
- P. Schroeder, C. France, B. Parkinson and R. Schlaf, J. Appl. Phys., 2002, 91, 9095–9107 CrossRef CAS.
- H. M. Hill, A. F. Rigosi, K. T. Rim, G. W. Flynn and T. F. Heinz, Nano Lett., 2016, 16, 4831–4837 CrossRef CAS PubMed.
- C. L. Hall, I. Andrusenko, J. Potticary, S. Gao, X. Liu, W. Schmidt, N. Marom, E. Mugnaioli, M. Gemmi and S. R. Hall, Chem. Phys. Chem., 2021, 22, 1631–1637 CrossRef CAS PubMed.
- S. M. Ryno, C. Risko and J. L. Bredas, J. Am. Chem. Soc., 2014, 136, 6421–6427 CrossRef CAS PubMed.
- P. Navarro, F. C. Bocquet, I. Deperasińska, G. Pirug, F. S. Tautz and M. Orrit, J. Phys. Chem. C, 2014, 119, 277–283 CrossRef.
- T. Olsen, S. Latini, F. Rasmussen and K. S. Thygesen, Phys. Rev. Lett., 2016, 116, 056401 CrossRef PubMed.
- A. Chernikov, T. C. Berkelbach, H. M. Hill, A. Rigosi, Y. Li, O. B. Aslan, D. R. Reichman, M. S. Hybertsen and T. F. Heinz, Phys. Rev. Lett., 2014, 113, 076802 CrossRef CAS PubMed.
- T. C. Berkelbach, M. S. Hybertsen and D. R. Reichman, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 045318 CrossRef.
- Y. Tanaka, N. Fukui and H. Shinokubo, Nat. Commun., 2020, 11, 3873 CrossRef CAS PubMed.
- P. K. Nayak, Synth. Met., 2013, 174, 42–45 CrossRef CAS.
- Y. Avlasevich, C. Kohl and K. Müllen, J. Mater. Chem., 2006, 16, 1053–1057 RSC.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00688g |
| This journal is © The Royal Society of Chemistry 2024 |