
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of single-atom catalysts for efficient H2O2 production via a four-step strategy†
Shu-Long
Li
abc,
Xiaogui
Song
a,
Zuhui
Zhou
a,
Hongyuan
Zhou
a,
Liang
Qiao
 *b,
Yong
Zhao
*ad and
Li-Yong
Gan
*e
*b,
Yong
Zhao
*ad and
Li-Yong
Gan
*e
aInstitute for Advanced Study, Chengdu University, Chengdu 610106, China. E-mail: zhaoyong@cdu.edu.cn
bSchool of Physics, University of Electronic Science and Technology of China, Chengdu 611700, China. E-mail: liang.qiao@uestc.edu.cn
cWestern Superconducting Technologies Co, Ltd., Xi’an 710018, China
dCollege of Physics and Energy, Fujian Normal University, Fuzhou 350117, China
eCollege of Physics and Center of Quantum Materials and Devices, Chongqing University, Chongqing 401331, China. E-mail: ganly@cqu.edu.cn
First published on 18th September 2024
Abstract
Electrocatalysis presents an efficient and eco-friendly approach for the two-electron oxygen reduction reaction (2e− ORR) to produce hydrogen peroxide (H2O2). However, challenges persist in enhancing catalyst activity and refining design strategies. In this study, a general four-step strategy is introduced to develop efficient single-atom catalysts (SACs) for H2O2 production based on transition metals and nonmetals embedded into γ-graphyne monolayers (TM–NM–GY) through first-principles calculations. Our results indicate that the intrinsic activity for the 2e− ORR can be properly and handily evaluated using the robust intrinsic electronegativity descriptor. On this foundation, we propose two strategies of B doping and creating C vacancies (v) to further enhance catalytic activity. Remarkably, Ni–B–GY and Ag–v–GY exhibit exceptional selectivity, stability, and activity with overpotentials as low as 0.08 V and 0.15 V, respectively, approaching the ideal limit of H2O2 catalysts. Mechanistic investigations reveal that B doping facilitates electron transfer and strengthens the hybridization between Ni 3d and O 2p orbitals, leading to stronger adsorption strength of *OOH and thus enhancing the 2e− ORR catalytic performance. These findings not only present several promising SAC candidates for H2O2 production, but also pave the way for the rational design of highly efficient SACs for various catalytic reactions.
1. Introduction
Hydrogen peroxide (H2O2) is a versatile industrial chemical used in various applications such as medical treatment, bleaching, disinfection, chemical synthesis, wastewater treatment, and energy production.1–3 A conventional method for producing H2O2, known as the anthraquinone process, is complex and energy-intensive, and in particular, generates toxic by-products, and thus poses severe safety risks.4,5 In contrast, electrochemical synthesis offers a more sustainable and energy-efficient approach, drawing significant interest from the community. This process involves the two-electron (2e−) oxygen reduction reaction (ORR), where oxygen (O2) is directly converted to H2O2 (O2 + 2H+ + 2e− → H2O2).6,7 While noble metals like Pd or Au have been employed as catalysts for H2O2 production, their scarcity and high cost limit their widespread commercial application.8–10 Challenges include the selectivity for the 2e− ORR pathway to H2O2 competing with the four-electron (4e−) ORR pathway to H2O, as well as the poor stability of most catalysts at thermodynamic synthesis potentials.2 Therefore, it is crucial to develop efficient electrocatalysts with high stability, activity, selectivity, and atomic utilization rate for H2O2 production.Transition metal (TM) single-atom catalysts (SACs) are emerging as promising catalysts due to their nearly 100% utilization of active metals, high activity, durability, low coordination environment, and strong interaction with supports.11–14 These benefits have enabled the successful use of SACs in various reactions such as the ORR,15–21 oxygen evolution reaction (OER),22,23 nitrogen reduction reaction (NRR)24–29 and carbon dioxide reduction reaction (CO2RR).30,31 Currently, the application of TM-SAC in H2O2 production is still in its infancy, and its potential has not yet been fully demonstrated. Creating a complete picture that not only reveals trends and origin of activity, but also facilitates the development of efficient catalysts is critical for H2O2 production. However, major efforts have mainly centered on understanding the trends and origins of activity, rendering the design still far from satisfactory.
In recent years, numerous two-dimensional (2D) materials, including graphene,32 g-C3N4,33 C2N,34 MoS2,35 porphyrin,36 and phthalocyanine,37 have been reported as carriers for SACs. However, the selection of all-carbon materials as carriers for SACs presents significant challenges. The drift of graphene and MoS2, along with the weak interaction between transition metal (TM) and carbon (C), hinders the effective dispersion of TM atoms. To address this limitation, researchers must employ various complex and precise methods, such as constructing defects,29 applying strain,18 or forming TM–Nx complexes.20 Additionally, the high cost and limited solubility of porphyrin and phthalocyanine organic molecules restrict their application under specific solvents or reaction conditions.38 In contrast, exploring all-carbon materials with uniformly porous structures that can securely anchor active TM atoms in their pore positions may present a promising approach for SACs.
Graphyne (GY) is a newly discovered 2D porous material, similar to g-C3N4 and C2N, which has garnered significant attention following its successful synthesis.39–41 Composed of sp2 and sp hybridized C units (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– and –C
C– and –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–), GY exhibits advantageous properties, including enhanced conjugation, porosity and stability.42–44 These significant characteristics of GY endow TM-anchored GY (TM–GY) with strong structural stability and demonstrate excellent catalytic potential in the ORR and NRR.25,45 However, the exploration of TM–GY, as well as TM and non-metal (NM) co-doped GY (TM–NM–GY), as potential SACs for H2O2 production is still in its early stages.46 Several unresolved issues remain: (i) Is there a universal strategy to assess outstanding SACs for H2O2 production? (ii) Are there intrinsic descriptors that effectively characterize activity trends and origins? (iii) Are there some effective strategies for further optimizing activity?
C–), GY exhibits advantageous properties, including enhanced conjugation, porosity and stability.42–44 These significant characteristics of GY endow TM-anchored GY (TM–GY) with strong structural stability and demonstrate excellent catalytic potential in the ORR and NRR.25,45 However, the exploration of TM–GY, as well as TM and non-metal (NM) co-doped GY (TM–NM–GY), as potential SACs for H2O2 production is still in its early stages.46 Several unresolved issues remain: (i) Is there a universal strategy to assess outstanding SACs for H2O2 production? (ii) Are there intrinsic descriptors that effectively characterize activity trends and origins? (iii) Are there some effective strategies for further optimizing activity?
In this study, first-principles calculations were used to comprehensively assess the potential of 2e− ORR electrocatalysts among 30 types of TM–GY and 15 types of TM–NM–GY SACs via a general four-step strategy: (i) evaluating catalytic activity and designing intrinsic activity descriptors; (ii) enhancing activity through NM doping; (iii) assessing the selectivity; and (iv) examining the stability (Fig. 1(a)). Ag–, Cu–, Ni–, Pd–, and Pt–GY were identified as efficient SACs toward the 2e− ORR with outstanding catalytic activity. Two reliable intrinsic activity descriptors φ1 and φ2 were established for the screening and designing of 2e− ORR catalysts. Based on the descriptors, B doping and creating C vacancies were shown to be effective strategies for further enhancing catalytic activity, with Ni–B–GY and Ag–v–GY demonstrating outstanding activity. Particularly, Ni–B–GY exhibited superior activity, selectivity, and stability with a significantly lower overpotential of 0.08 V. The four-step screening strategy and descriptors introduced in this study are anticipated to offer insights and guidance for the design and prediction of other catalysts.
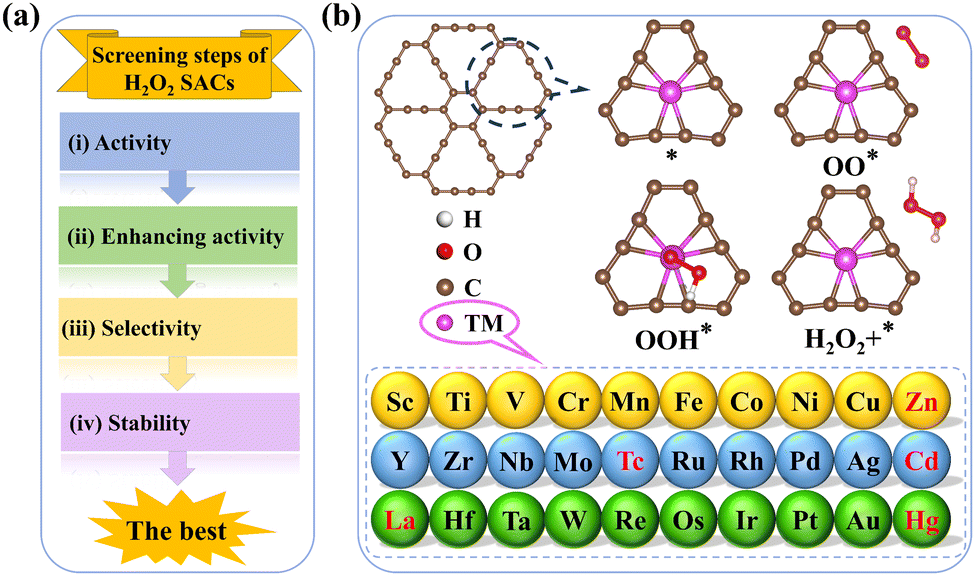 | ||
| Fig. 1 (a) Schematic diagram of screening H2O2 SACs. (b) 2e− ORR pathways on TM–GY. | ||
2. Computational details
All spin-polarized density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package.47 The electron-ion interactions were described using the projector augmented-wave-method.48 For accurate electron exchange–correlation, the Perdew–Burke–Ernzerhof functional within the generalized gradient approximation framework was employed.49 Additionally, the DFT+D2 method was applied to correct for van der Waals forces between the adsorbent carrier and the adsorbate.50 The lattice parameter of GY was 6.89 Å. A 2 × 2 × 1 GY supercell was employed to simulate electrocatalysis for H2O2 production, as the distance of 6.89 Å between the TM atoms is adequate for accurately evaluating the interactions between the TM and the adsorbates. A vacuum thickness of 15 Å was introduced to avoid preventing periodic interactions. The plane wave cut-off energy was set at 500 eV. The residual forces and energy convergence criteria were maintained below 0.02 eV Å−1 and 10−5 eV, respectively. Structural relaxation and electronic structure analyses were performed using 3 × 3 × 1 and 7 × 7 × 1 Γ-centered k-points to sample the Brillouin zone, respectively. The crystal orbital Hamilton population (COHP) method was used to explore the interaction between the active center and the intermediates.51,52 The thermodynamic stability of the catalyst was assessed through ab initio molecular dynamics (AIMD) simulations and the radial distribution function (RDF) analysis at 300 K.The binding energy (Ebind), adsorption energy (ΔE), Gibbs free energy (ΔG) of intermediates, limiting potential (UL) and the overpotential (η) were calculated using the following equations:
| Ebind = ETM–GY − EGY − ETM | (1) |
| ΔE = Eads-TM–GY − ETM–GY − Eads | (2) |
| ΔG = ΔE + ΔZPE − TΔS + ΔGU | (3) |
UL = max![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ΔG/e ΔG/e | (4) |
| η = UL − Ueq | (5) |
3. Results and discussion
The sp2 and sp3 hybridized C sites in GY lack the ability to adsorb O2 due to their limited positive charge or the absence of empty orbitals for accepting lone pair electrons from O2 (Fig. S1, ESI†). Conversely, the sp hybridized C site (–CC–) demonstrates strong O2 adsorption capacity, resulting in the dissociation of the OOH* intermediate into O* + OH* (* represents the catalyst), ultimately hindering the generation of H2O2. Thus, pure GY is ineffective for H2O2 production. Our focus then shifted towards developing TM–GY as a highly active H2O2 SACs. Previous studies have demonstrated TM atoms can be effectively incorporated into GY cavities due to the reactivity of sp-hybridized –CC– bonds.54 Therefore, TM was directly bound to a 2 × 2 × 1 GY supercell for 2e− ORR in this study.
To efficiently identify whether SACs are suitable for electrochemical H2O2 synthesis with high activity, stability, and selectivity, we established a general four-step strategy (Fig. 1(a)): (a) evaluating the catalytic activity and designing intrinsic activity descriptors of TM–GY SACs for H2O2 production; (b) enhancing the catalytic activity of TM–GY SACs through NM doping (TM–NM–GY) based on intrinsic activity descriptors; (c) assessing the selectivity of TM–GY and TM–NM–GY; (d) examining the stability of TM–GY and TM–NM–GY.
3.1 Screening efficient catalytic activity of TM–GY SACs for H2O2
Thirty types of TM–GY (TM = 3d–5d) were studied to screen efficient SACs for H2O2 production, as shown in Fig. 1(b). After excluding radioactive or toxic elements (Tc, Cd, and Hg), lanthanides (La), and metals that cannot bind to GY (Zn),54 only 25 types of TM–GY remained on the screening list.The adsorption of O2 molecules on TM–GY is a crucial prerequisite for the 2e− ORR to generate H2O2. Failure to adsorb O2 would hinder the subsequent hydrogenation reaction to produce H2O2. Therefore, both side-on and end-on adsorption configurations of O2 on all TM–GY catalysts were examined. Results indicate that O2 cannot be adsorbed on Au–GY due to its positive Gibbs free energy change (ΔGOO) of 0.24 eV, leading to the exclusion of Au–GY from further screening. Among the other TM–GY catalysts, O2 is adsorbed predominantly in the side-on configuration, except on Cu–, Ni– and Ag–GY, as shown in Fig. S2 (ESI†). Following successful O2 adsorption, the 2e− ORR progresses to a hydrogenation step, forming the OOH* intermediate without breaking the O–O bond. A linear relationship between ΔGOOH and ΔGOO, is observed with a coefficient of 0.81 (Fig. S3, ESI†), indicating that the ΔGOO and the catalytic activity of H2O2 can be represented by a single intermediate free energy descriptor of ΔGOOH.
The overpotential (η) is a critical parameter to assess the catalytic activity for H2O2 production. An effective electrocatalyst should have an ideal η close to 0 V (UL = Ueq = 0.7 V).40 Subsequently, the η on all TM–GY was determined using the standard hydrogen electrode method.55 As shown in Fig. 2(a), the activity trend picture illustrated a linear relationship resembling a volcano plot between ΔGOOH and η of TM–GY. A closer alignment of ΔGOOH and η values towards the peak of the volcano plot, indicates superior catalytic performance. Particularly, the significantly negative ΔGOOH values at the bottom of the left leg suggest strong interactions between OOH and TM–GY. Following the Sabatier principle,56 excessively strong intermediates may hinder the formation and release of the H2O2 product, leading to the potential determining step (PDS) occurring in the final step of (OOH* → H2O2). Conversely, weak ΔGOOH values correspond to weak O2 adsorption, which is unfavorable for subsequent hydrogenation steps, resulting in the PDS occurring in the initial step of (O2 → OOH*). Upon evaluation, Ir–, Rh–, Ag–, Co–, Ni–, Cu–, Pd–, and Pt–GY exhibit moderate ΔGOOH values close to 4.22 eV, indicating high catalytic activity. Additionally, we conducted a comparative study on the adsorption of OOH on GY, isolated Ni atom, and Ni–GY (Fig. S4, ESI†). The results indicate a synergistic interaction between GY and Ni atoms, which endows Ni–GY with an appropriate OOH adsorption strength, thereby facilitating the generation of H2O2. This may be the original cause of GY as a substrate for constructing SACs to produce H2O2.
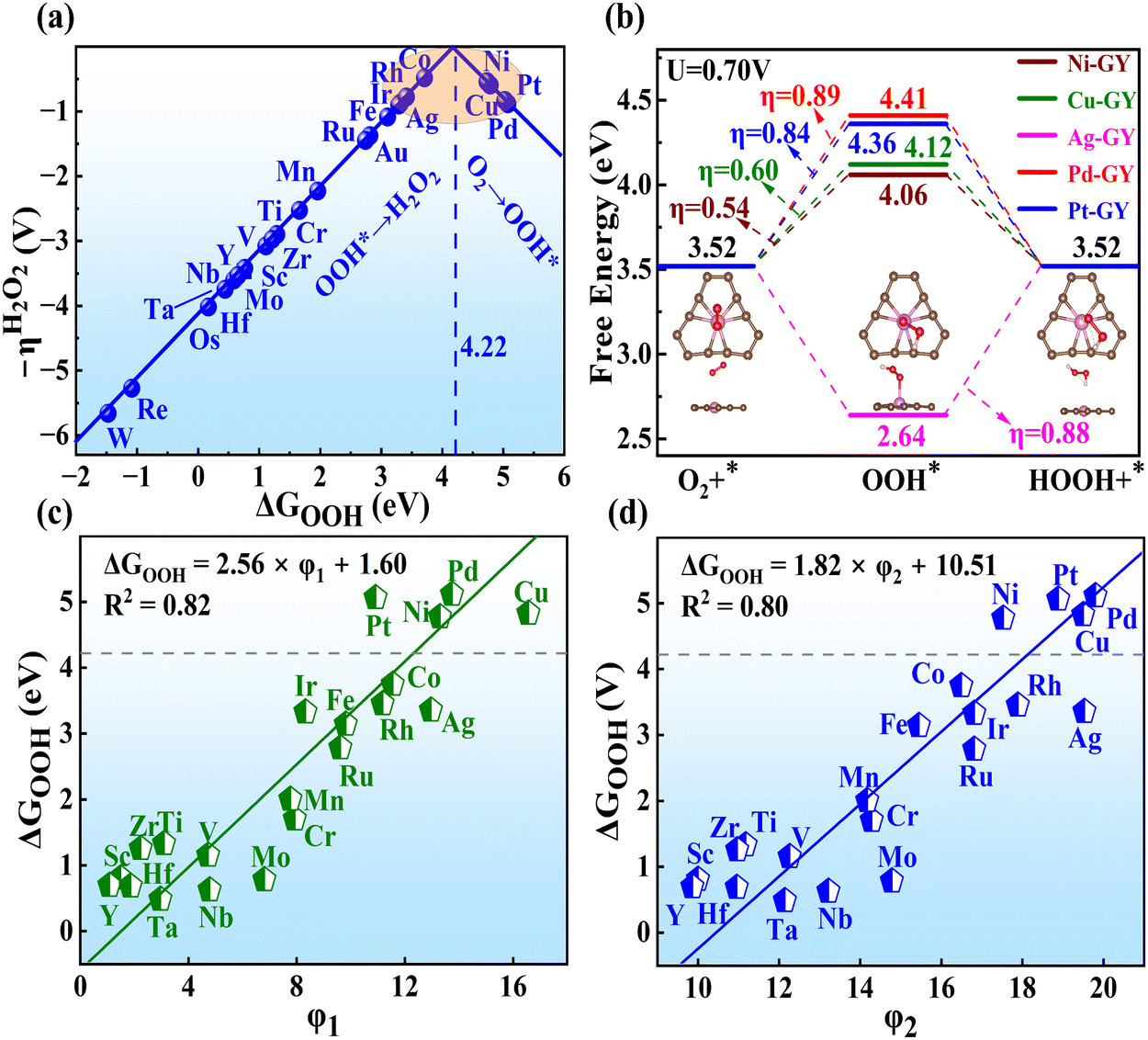 | ||
| Fig. 2 (a) The volcano plot of ΔGOOH and the overpotential. (b) Free energy diagram for 2e− ORR on TM–GY candidates (TM = Ni, Cu, Ag, Pd and Pt). (c) and (d) The scaling relationship between ΔGOOH and φ1, φ2 for TM–GY. | ||
It is noteworthy that the O–O bonds in the OOH* intermediates of Co–, Rh–, and Ir–GY underwent cleavage during the OOH* → H2O2 process, facilitating the formation of the O* intermediate and H2O, as shown in Fig. S5 (ESI†). This suggests that they are unsuitable as SACs for H2O2 production. Conversely, Ni–, Cu–, Pd–, Ag–, and Pt–GY had stable O–O bonds in the OOH* intermediates, enabling the successful synthesis of H2O2. Following screening, only five TM–GY (TM = Ag, Cu, Ni, Pd, and Pt) were selected as potential candidates for further investigation.
To achieve a deeper comprehension of the reaction mechanisms, free energy diagrams for the 2e− ORR process with UL of 0.7 V were plotted in Fig. 2(b). The initial steps of O2 → OOH* for Ni–, Cu–, Pd– and Pt–GY exhibit slight uphill trends, indicating a requirement for a small energy input at this stage. Conversely, Ag–GY shows a thermodynamically downhill initial step, suggesting a spontaneous exothermic process. For the final step of OOH* → H2O2, all are spontaneous except for Ag–GY. The overall reaction pathway for H2O2 production reveals overpotentials of 0.54, 0.60, 0.89, 0.84 and 0.88 V for Ni–, Cu–, Pd–, Pt– and Ag–GY, respectively.
After confirming the catalytic activity of these TM–GY SACs, it is crucial to establish a reliable and simple intrinsic activity descriptor to gain deeper insights into the underlying activity trends and to facilitate the design of improvements for the most promising candidates. Given that electronegativity reflects the ability of atoms to attract electrons in chemical bonds, and considering that the number of d-electrons and atomic radius may influence adsorption performance, we summarized these factors in Table S1 (ESI†) and plotted their relationships with activity in the Fig. S6a–c (ESI†). The results indicate that d-electrons and electronegativity are negatively correlated with activity, while atomic radius shows a positive correlation with activity. In this study, we introduce two intrinsic electronegativity modified descriptors, φ1 and φ2 inspired by Zeng et al. for 4e− ORR57 and Ren et al. for CO2 reduction,58 as well as one electronic spin moment modified descriptor φ3, inspired by Zhang et al. for NRR.59 The calculations are as follows:
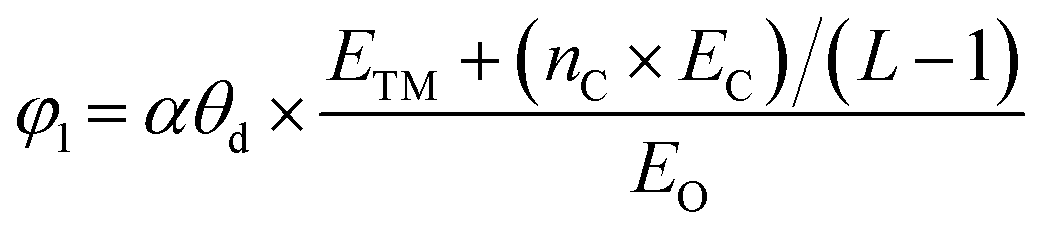 | (6) |
| φ2 = (ETM + nC × EC) + θd | (7) |
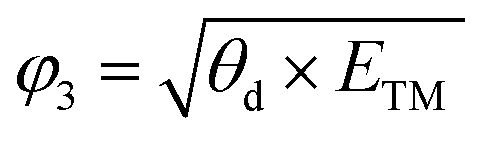 | (8) |
3.2 Further enhancement of activity through NM modification
After identifying five TM–GY (TM = Ag, Cu, Ni, Pd, and Pt) as promising SAC candidates for 2e− ORR to produce H2O2, it has been noted that there is significant space for improving their catalytic activity. By analyzing intrinsic activity descriptors, it is evident that the φ1 or φ2 values of these materials exceed the optimal activity point (intersection point φ1 or φ2 with 4.22), suggesting that reducing or adjusting the coordination number could be beneficial for enhancing the activity of these TM–GY. Previous studies have demonstrated that modification with NM such as B doping, N doping or creating C vacancies (v) can alter the coordination environment, thereby enhancing catalytic activity.60–62 Therefore, TM–NM–GY (NM = B, N, and v) were used to enhance the 2e− ORR activity, as shown in Fig. 3(a).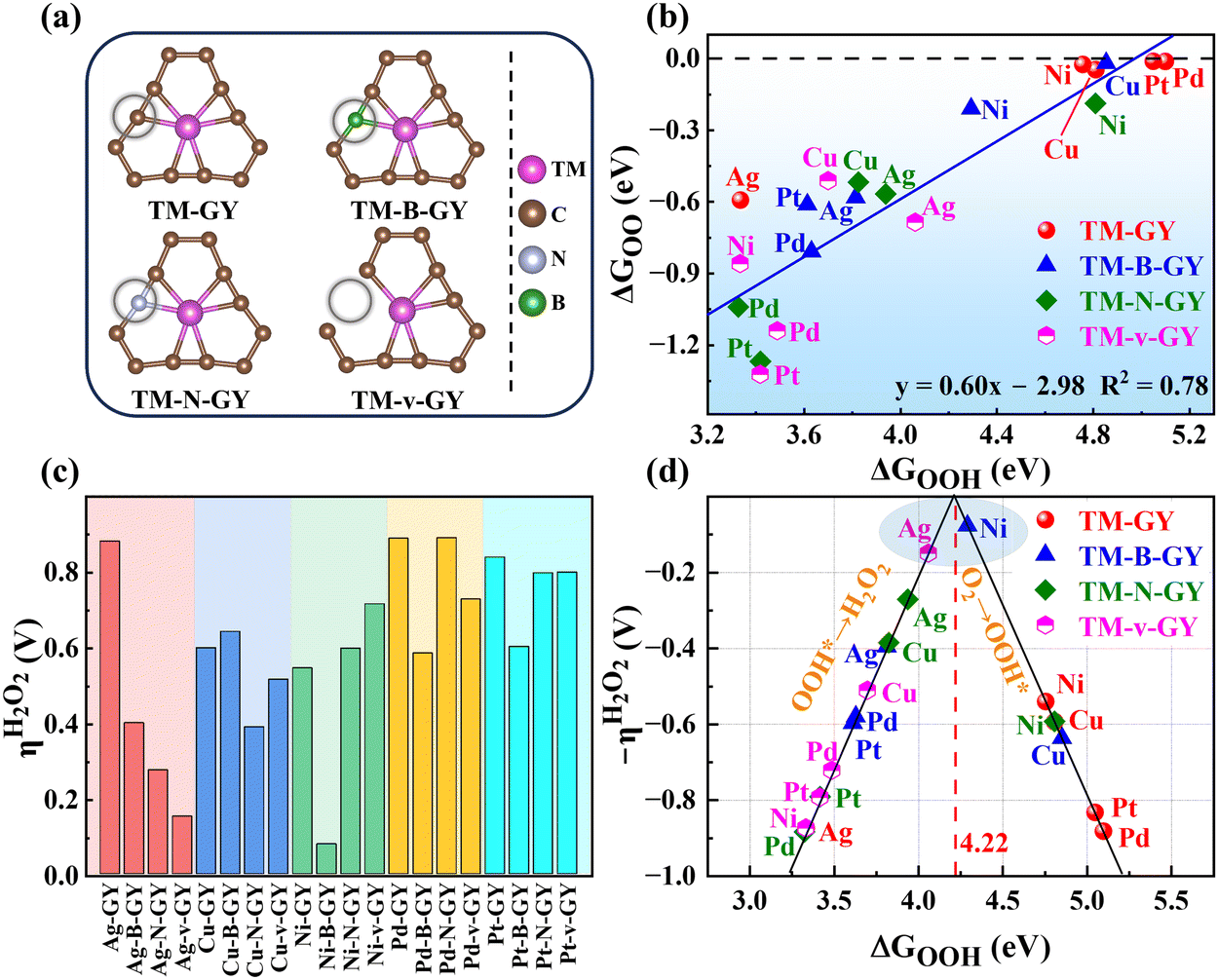 | ||
| Fig. 3 (a) The configuration of TM–NM–GY (NM = B, N doping or C vacancy). (b) The linear relationship between ΔGOOH and ΔGOO. (c) The ηH2O2 for TM–NM–GY. (d) Volcano plot of ΔGOOH as a function of overpotential ηH2O2. | ||
Our findings reveal that TM atoms serve as the active sites for capturing O2 with a side-on configuration on Pt–NM–GY and Pd–NM–GY, while they tend to prefer an end-on configuration on other materials (Fig. S7, ESI†). After undergoing the NM modification, TM–NM–GY (TM = Cu, Ni, Pd, and Pt) demonstrates a significantly improved capacity for O2 adsorption in comparison to TM–GY, as is evident from Fig. S8 (ESI†). Moreover, the C vacancy demonstrates a higher O2 adsorption capacity than B or N doping. Furthermore, a linear correlation was observed between ΔGOO and O–O bond length (Fig. S9, ESI†), indicating that stronger O2 adsorption strength corresponds to longer O–O bonds. The negative ΔGOO values and elongated O–O bonds indicate stable adsorption and activation of O2, preparing for the subsequent hydrogenation reaction to produce H2O2. Subsequently, the hydrogenation step forming the OOH* intermediate was investigated (Fig. S7, ESI†). A strong linear relationship was also identified between ΔGOOH and ΔGOO with a coefficient of 0.78 (Fig. 3(b)), indicating that the H2O2 catalytic activity can be represented by a single descriptor of ΔGOOH.
The catalytic activity of H2O2 on various TM–NM–GY catalysts was investigated through the analysis of free energy diagrams (Fig. S10, ESI†) and overpotentials (Fig. 3(c)). It was observed that NM modification significantly enhanced the catalytic activity of several TM–NM–GY catalysts. Particularly, Ag–v–GY and Ni–B–GY exhibited outstanding catalytic activity, with ultra-low overpotentials of 0.15 V and 0.08 V, approaching the theoretical limit. Additionally, a linear relationship resembling a volcano plot was observed between ΔGOOH and η of TM–NM–GY, as shown in Fig. 3(d). The positions of Ag–v–GY and Ni–B–GY are quite near the peak of the volcano plot, further validating their superior catalytic activity for H2O2. This improvement is attributed to the alteration of the local coordination structure through B doping and C vacancy, resulting in ΔGOOH values of Ni–B–GY (4.30 eV) and Ag–v–GY (4.07 eV), bringing them closer to the optimal catalyst with ΔGOOH value of 4.22 eV.
3.3 Selectivity of TM–NM–GY (H2O2vs. H2O)
In addition to catalytic activity, selectivity also plays a crucial role in determining catalytic performance. The ORR can proceed through either a 2e− pathway resulting in the production of H2O2 or a 4e− pathway leading to the formation of H2O, as shown in Fig. 4(a). The 4e− pathway competes and suppresses the production of H2O2. Therefore, it is essential to evaluate the selectivity towards H2O2 and H2O on TM–NM–GY based on their overpotentials (ηH2O2 and ηH2O) as previously reported in the literature.40 The Gibbs free energy diagrams for the 2e− and 4e− ORR pathways of all 20 types TM–NM–GY at zero potential and equilibrium potential are shown in Fig. S10 and S11 (ESI†). Furthermore, the corresponding ηH2O2 and ηH2O values are summarized in Fig. 4(b) and Fig. S12 (ESI†). It is observed that all TM–NM–GY exhibit lower ηH2O2 values compared to ηH2O except for Ag–GY, Pd–N–GY and Pd–B–GY, indicating their excellent selectivity towards H2O2 production. The introduction of B doping, N doping, and C vacancy in TM–GY results in a decrease in their overpotentials. Catalysts located above the dashed line in Fig. 4(b) demonstrate remarkable H2O2 selectivity, with darker colors indicating higher catalytic activity. Among these, Ni–B–GY demonstrates the lowest ηH2O2 (0.08 V) compared to ηH2O (0.61 V).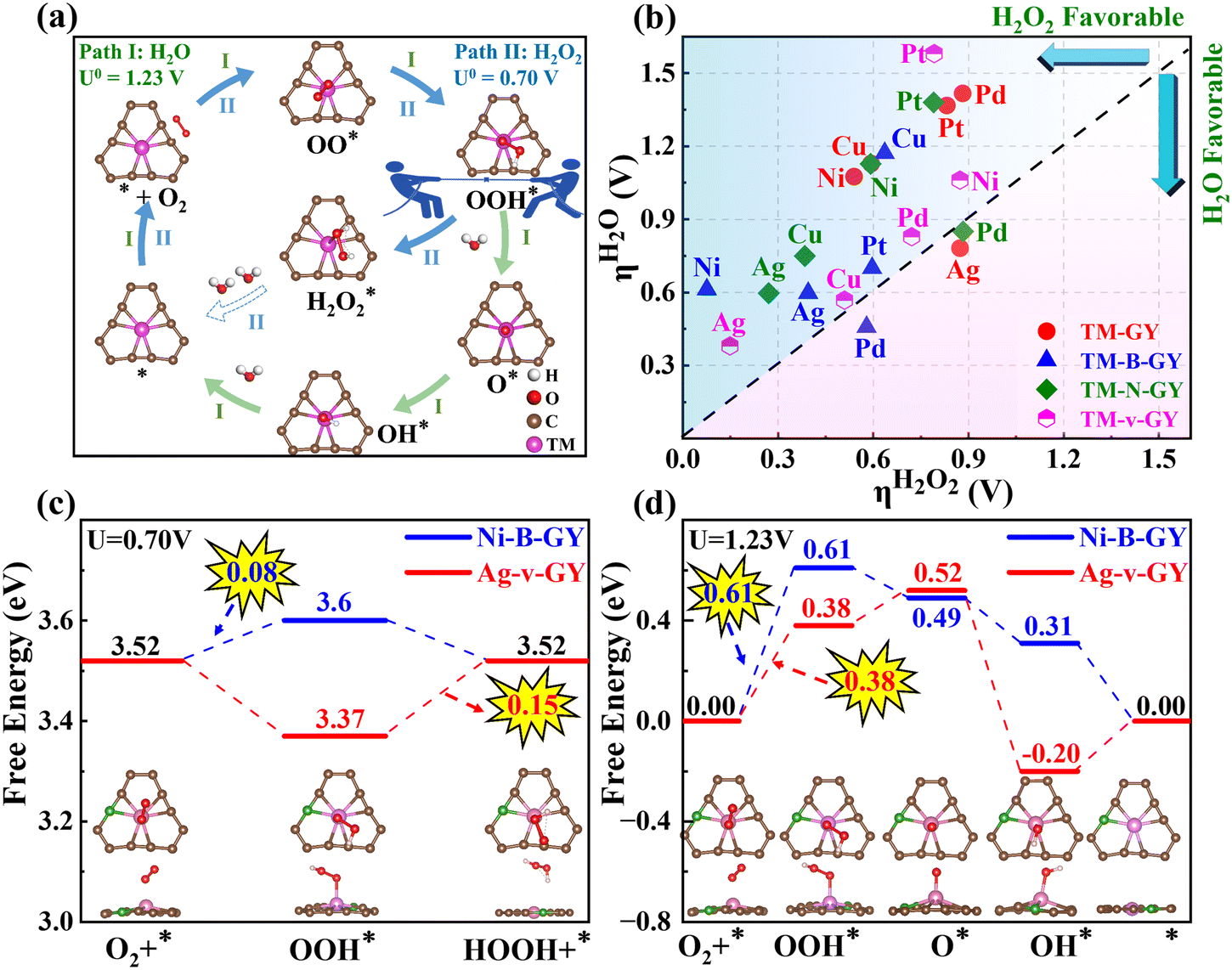 | ||
| Fig. 4 The selectivity of TM–NM–GY catalysts: (a) Schematic illustration of the 2e− and 4e− ORR pathways. (b) Overpotential for 2e− and 4e− ORR. (c) and (d) Free energy diagrams comparing 2e− and 4e− ORR for Ag–v–GY and Ni–B–GY, highlighting their superior catalytic activity. | ||
A further analysis of selectivity was performed on Ag–v–GY and Ni–B–GY, which exhibited the highest catalytic activity. The 2e− ORR at equilibrium potential (0.7 V) is shown in Fig. 4(c). On Ag–v–GY, the initial step (O2 → OOH*) is spontaneous, while the PDS occurs at the final step (OOH* → H2O2) with a ηH2O2 of 0.15 V. Conversely, on Ni–B–GY, the final step is spontaneous and the PDS occurs at the initial step with a ηH2O2 of 0.08 V. The 4e− ORR at the equilibrium potential (1.23 V) is shown in Fig. 4(d). For Ag–v–GY, only the third step (OOH* → O* + H2O) is spontaneous, while the other steps are endothermic. The PDS occurs at the initial step (O2 → OOH*) with an ηH2O value of 0.38 V. For Ni–B–GY, only the initial step (O2 → OOH*) is endothermic, while the remaining steps proceed spontaneously. Consequently, the PDS of Ni–B–GY (ηH2O is 0.61 V) is the same as that of Ag–v–GY. The ηH2O2 values on Ag–v–GY and Ni–B–GY (0.15 and 0.08 V) are less than half of ηH2O on them (0.38 and 0.61 V), indicating excellent selectivity.
3.4 Stability of TM–NM–GY
In addition to activity and selectivity, stability is another crucial factor for H2O2 electrocatalysts. The binding energy (Ebind) was used to preliminarily evaluate the structural stability of TM–NM–GY (TM = Ag, Cu, Ni, Pd, and Pt), as shown in Fig. 5(a). Sixteen types of TM–NM–GY (TM = Cu, Ni, Pd, and Pt) exhibited negative binding energies less than −2 eV, indicating relatively good stability. Furthermore, both B doping and C vacancy significantly enhance the stability of TM–GY, with C vacancy showing the most substantial stability improvement effect. The strongest Ebind value of −7.87 eV was observed in Pt–v–GY.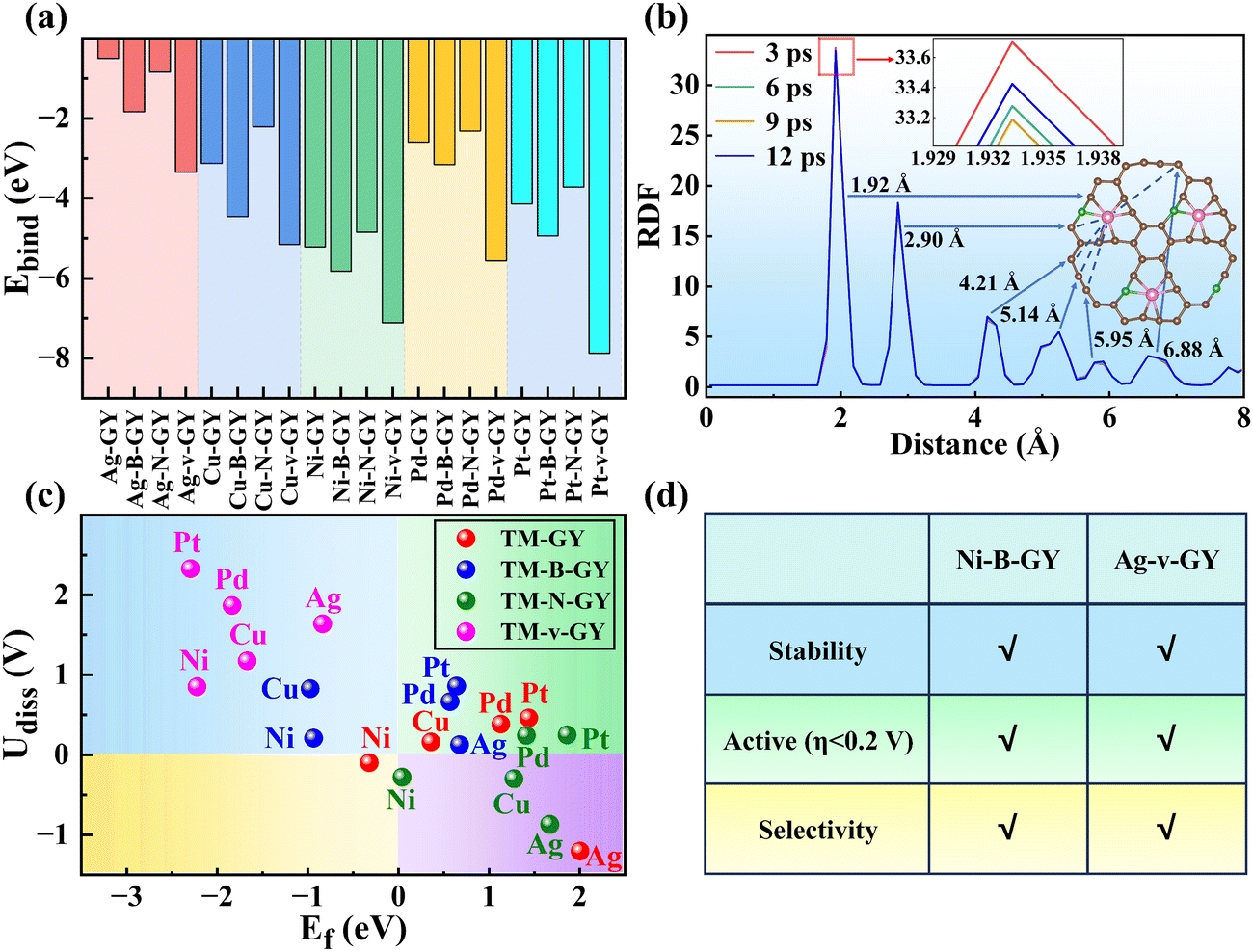 | ||
| Fig. 5 The stability of TM–NM–GY catalysts: (a) Binding energy (Ebind). (b) The RDF between the Ni and C for Ni–B–GY at 300 K and the configuration at 12 ps. (c) Dissolution potentials (Udiss) and formation energies (Ef). (d) The performances of Ni–B–GY and Ag–v–GY. | ||
The formation energy (Ef) is used to further evaluate thermodynamic stability:
| Ef = ETM–NM–GY − ENM–GY − ETM | (9) |
Furthermore, ab initio molecular dynamics simulations were conducted at 300 K for 12 ps to evaluate their thermodynamic stability (Fig. S13, ESI†). While Ag–GY and Ag–N–GY exhibited energy fluctuations suggesting limited stability, other TM–NM–GY catalysts showed minimal fluctuations and geometrical distortion, with TM atoms securely anchored within the GY cavity, indicating robust thermodynamic stability. Ni–B–GY was specifically studied due to its outstanding H2O2 catalytic activity (η = 0.08 V) and selectivity (ηH2O2 of 0.08 vs. ηH2O of 0.61 V). The radial distribution function (RDF) in Fig. 5(b) analyzed the Ni–C interactions, revealing multiple peaks corresponding to Ni–C spacing, with a higher peak indicating a stronger interaction.64,65 The consistent RDF patterns at different time points (3 ps, 6 ps, 9 ps, and 12 ps) suggest that the Ni–C bond remains stable throughout the kinetic process, further confirming its excellent thermodynamic stability.
After assessing the thermodynamic stability, the electrochemical stability was further examined through the dissolution potential Udiss40,63 as follows:
| Udiss = U0diss − (Ef)/ne | (10) |
By conducting a comprehensive evaluation of the stability for the TM–NM–GY using thermodynamics, molecular dynamics, and electrochemical analyses, we confirmed that Ni–B–, Cu–B–, Ni–v–, Cu–v–, Ag–v–, Pt–v– and Pd–v–GY exhibit good stability, as shown in Fig. S14 (ESI†). Subsequently, after a comprehensive evaluation of activity, selectivity, and stability, we identified Ni–B–GY, Cu–v–GY and Ag–v–GY with excellent comprehensive catalytic performance, as shown in Fig. S15 (ESI†). Notably, Ni–B–GY and Ag–v–GY demonstrated exceptional catalytic performance with remarkably low overpotentials of 0.08 and 0.15 V (Fig. 5(d)).
3.5 Origin of high activity for H2O2 production on Ni–B–GY
Having confirmed the activity, selectivity, and stability of TM–NM–GY, we moved on to explore the origin of its activity. To achieve a deeper comprehension, we chose the Ni–B–GY with the best catalytic activity for analysis. The pristine GY material is a semiconductor with a bandgap of 0.46 eV (see Fig. 6(a)). Upon the introduction of Ni atoms, the band gap decreases to 0.35 eV for Ni–GY. Further introduction of B atoms causes the valence band maximum to shift upwards and cross the Fermi level (EF), resulting in Ni–B–GY showing metallic properties. This combined introduction of TM and B significantly boosts the electrical conductivity, which is beneficial for electrocatalysis. At an equilibrium potential of 0.7 V, both Ni–B–GY and Ni–GY exhibit PDS in the first step, as shown in Fig. 6(b). The introduction of B atoms significantly enhances ΔGOOH, which is the key reason for the improved catalytic activity.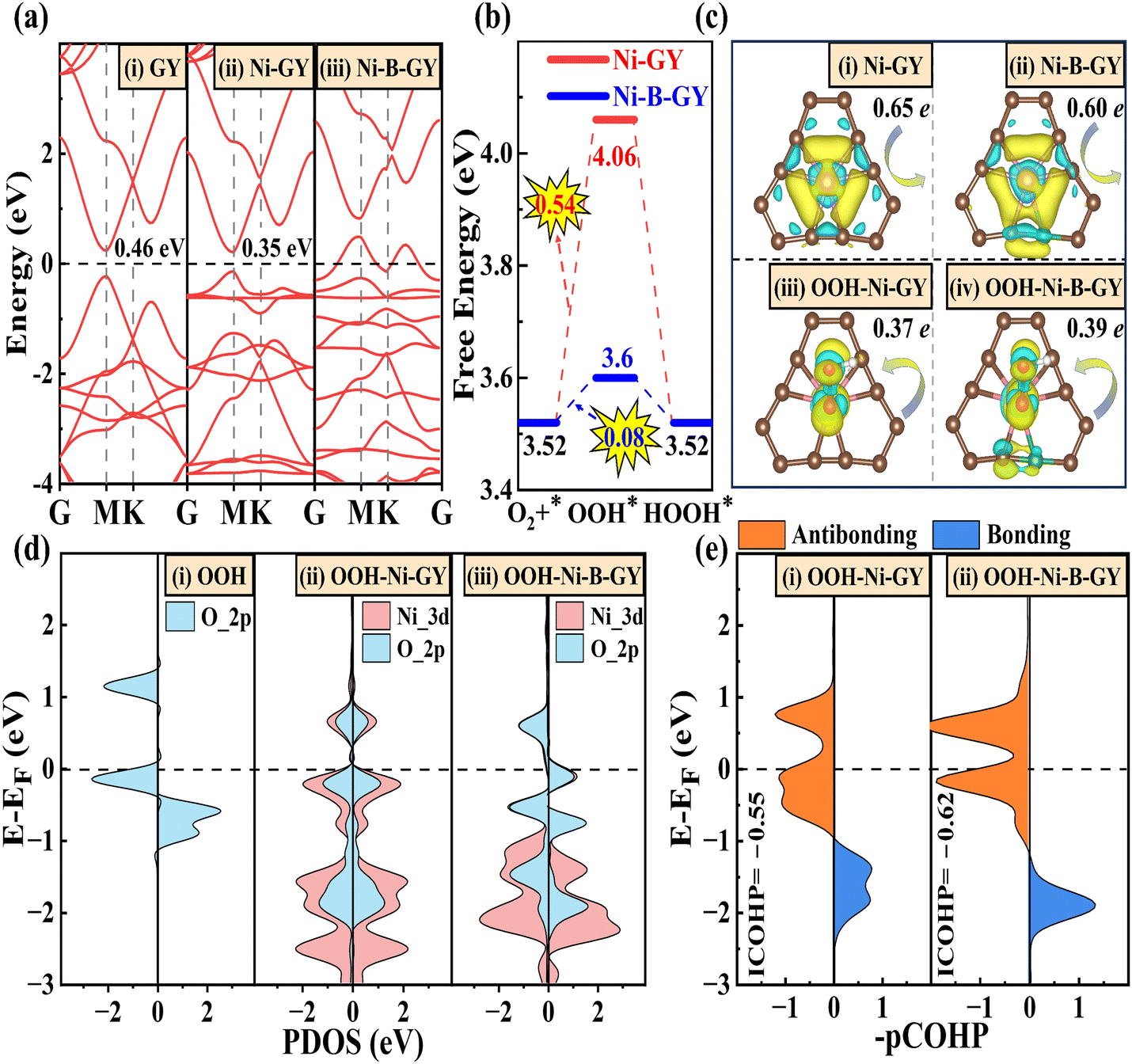 | ||
| Fig. 6 (a) The band structure of GY, Ni–GY and Ni–B–GY. (b) Free energy diagrams comparing 2e− ORR for Ni–GY and Ni–B–GY. (c) Charge density differences with and without OOH adsorbed on Ni–GY and Ni–B–GY. (d) The PDOS for OOH, OOH-Ni–GY and OOH-Ni–B–GY. (f) The COHP between the O (in OOH) and Ni for Ni–GY and Ni–B–GY. | ||
Then, we explored the reasons behind the enhanced OOH* adsorption due to B doping. Moreover, charge transfer plays a crucial role in electrocatalysis. The charge density difference in Fig. 6(c) suggests that –CC– structure offers a greater number of empty orbitals compared to the –CC– structure, which primarily facilitates charge transfer. During the reaction process, –CC– acts as an electron donor, while –CC– not only serves as an anchor site for active TM, but also shoulders the roles of electron donation and transfer (Fig. S16, ESI†). The introduction of B leads to a charge transfer from Ni to OOH of 0.39 e for Ni–B–GY, surpassing the 0.37 e for Ni–GY. This increased charge transfer enhances the adsorption strength of OOH, thereby facilitating H2O2 production. The higher charge transfer is attributed to the lower electronegativity of B (2.04) compared to C (2.55). These differences in atoms disrupt the original charge balance and coordination structure symmetry, leading to the transformation from Ni–6C to Ni–B–5C and the formation of a C–B bond by breaking the conjugated C–C or CC bonds. Consequently, the local coordination of B atoms effectively reduces the positive charge density of Ni-d-orbit-related electronic structures in Ni–B–GY. This results in a higher charge transfer from Ni to OOH, strengthening the adsorption strength of OOH and ultimately enhancing activity and selectivity for H2O2.
The projected density of states (PDOS) was used to elucidate the interaction between O 2p orbitals in OOH and Ni 3d orbitals, as shown in Fig. 6(d). The introduction of B atoms alters the symmetry of the coordination environment and charge density (Fig. S18, ESI†), leading to a stronger hybridization effect between Ni dx2−y2 and O pz orbitals near −1.8 eV below the Fermi level (EF), as shown in Fig. S19 (ESI†). This results in an increased charge transfer from the 2π* and 3σ molecular orbitals of OOH to the unoccupied d orbitals of Ni, as shown in Fig. 6(c) and the donation and back-donation model in Fig. S17 (ESI†). The enhanced donation effect creates a lower bonding state, facilitating the adsorption of OOH. Furthermore, the introduction of B will boost the number of back-donated electrons from the partially occupied Ni 3d orbitals to the OOH 2π* molecular orbitals. Consequently, this conversion transforms some initially empty orbitals into occupied ones, favoring the formation of partially occupied orbitals in an antibonding state.
To further validate the interaction between Ni and O (Ni–O), we employed the COHP method to assess the Ni–O bonding strength of with and without B doping. This assessment considers both bonding states (blue region) and antibonding states (red region), as shown in Fig. 6(e). Our findings indicate that following B doping, the bonding state shifts downward, occupying lower energy levels, while the occupancy of bonded states above the EF increases. The combined effects of bonding and antibonding states indicate that B doping enhances the OOH adsorption strength, thereby facilitating charge transfer during the catalytic reaction process and ultimately improving catalytic activity. Similar alterations in adsorption strength, charge transfer, and bonding state properties are also evident in OO* (see Fig. S20, ESI†). Ultimately, B doping not only enhances the d-2π* coupling facilitating the adsorption and activation of O2 in preparation for subsequent H2O2 reactions,4 but also contributes to the formation of lower bonding states for Ni–O, strengthening the adsorption of OO and OOH. The integrated COHP (ICOHP) was employed to quantitatively assess the Ni–O bonding interactions in OOH, with a lower value indicating stronger bond interactions.4,66 After B doping, the ICOHP for Ni–O decreases from −0.55 to −0.62, indicating an enhancement in the Ni–O interaction, leading to a stronger adsorption of OOH and ultimately enhancing the catalytic activity.
4. Conclusions
In summary, this study presented a full picture (activity trends, electronic origins, and design strategies) for effectively screening and designing potential 2e− ORR electrocatalysts for H2O2 production using TM–GY and TM–NM–GY SACs through a general four-step strategy. Initially, Ag–, Cu–, Ni–, Pd–, and Pt–GY were identified as promising SACs for H2O2 production out of 30 types of TM–GY. Subsequently, two reliable intrinsic activity descriptors φ1 and φ2 were established for screening and designing 2e− ORR catalysts. Based on these descriptors, B doping and creating C vacancies were found to be effective strategies for further enhancing catalytic activity. Particularly, Ag–v–GY and Ni–B–GY demonstrated remarkable activity, selectivity, and stability with extremely low overpotentials of 0.15 V and 0.08 V, respectively. The analysis of the activity mechanism for Ni–B–GY revealed that the remarkable catalytic performance can be attributed to the doping of B atoms, which alters the coordination environment, enhances conductivity, facilitates electron transfer, and strengthens the hybridization between TM 3d and O 2p orbitals, leading to improved adsorption strength of OOH and enhanced catalytic performance. These findings are expected to inspire further exploration and design of high-performance SACs.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (12074048 and 12347101), the China Postdoctoral Science Foundation (2023M732903 and 2023T160537), the Sichuan Science and Technology Program (2024NSFSC1382) and the Chengdu Municipal Science and Technology Program (2022-YF05-00886-SN). The numerical calculations in this paper have been carried out in the Hefei advanced computing center.References
- M. Hu, W. Wu and H. Jiang, ChemSusChem, 2019, 12, 2911–2935 CrossRef CAS PubMed.
- X. Shi, S. Siahrostami, G. L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- Y. Xia, X. Zhao, C. Xia, Z. Y. Wu, P. Zhu, J. Y. T. Kim, X. Bai, G. Gao, Y. Hu, J. Zhong, Y. Liu and H. Wang, Nat. Commun., 2021, 12, 4225 CrossRef CAS PubMed.
- W. Zhang, F.-L. Sun, Q.-J. Fang, Y.-F. Yu, J.-K. Pan, J.-G. Wang and G.-L. Zhuang, J. Phys. Chem. C, 2022, 126, 2349–2364 CrossRef CAS.
- C. Zhang, W. Shen, K. Guo, M. Xiong, J. Zhang and X. Lu, J. Am. Chem. Soc., 2023, 145, 11589–11598 CrossRef CAS PubMed.
- Q. Chang, P. Zhang, A. H. B. Mostaghimi, X. Zhao, S. R. Denny, J. H. Lee, H. Gao, Y. Zhang, H. L. Xin, S. Siahrostami, J. G. Chen and Z. Chen, Nat. Commun., 2020, 11, 2178 CrossRef CAS PubMed.
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, Chem., 2018, 4, 106–123 CAS.
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef PubMed.
- X. Sheng, N. Daems, B. Geboes, M. Kurttepeli, S. Bals, T. Breugelmans, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2015, 176–177, 212–224 CrossRef CAS.
- E. Pizzutilo, O. Kasian, C. H. Choi, S. Cherevko, G. J. Hutchings, K. J. J. Mayrhofer and S. J. Freakley, Chem. Phys. Lett., 2017, 683, 436–442 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- J. Liu, ACS Catal., 2016, 7, 34–59 CrossRef.
- Z. Xue, M. Yan, X. Wang, Z. Wang, Y. Zhang, Y. Li, W. Xu, Y. Tong, X. Han, C. Xiong, W. Wang, M. Chen, B. Ye, X. Hong, L. Song, H. Zhang, L. M. Yang and Y. Wu, Small, 2021, 17, 2101008 CrossRef CAS PubMed.
- Q. Yao, S.-Y. Lv, Z. Yu, Y.-C. Chang, C.-W. Pao, Z. Hu, L.-M. Yang, X. Huang, Q. Shao and J. Lu, ACS Catal., 2023, 13, 11023–11032 CrossRef CAS.
- S.-L. Li, Q. Li, Y. Chen, Y. Zhao and L.-Y. Gan, Appl. Surf. Sci., 2022, 605, 154828 CrossRef CAS.
- F. Li, H. Shu, C. Hu, Z. Shi, X. Liu, P. Liang and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 27405–27413 CrossRef CAS PubMed.
- Z. Yu, Y. Chen, J. Xia, Q. Yao, Z. Hu, W.-H. Huang, C.-W. Pao, W. Hu, X.-M. Meng, L.-M. Yang and X. Huang, Nano Lett., 2024, 24, 1205 CrossRef CAS PubMed.
- J. Yang, Z. Wang, C. X. Huang, Y. Zhang, Q. Zhang, C. Chen, J. Du, X. Zhou, Y. Zhang, H. Zhou, L. Wang, X. Zheng, L. Gu, L. M. Yang and Y. Wu, Angew. Chem., Int. Ed., 2021, 60, 22722–22728 CrossRef CAS PubMed.
- M. Zhu, C. Zhao, X. Liu, X. Wang, F. Zhou, J. Wang, Y. Hu, Y. Zhao, T. Yao, L.-M. Yang and Y. Wu, ACS Catal., 2021, 11, 3923–3929 CrossRef CAS.
- Z. Yang, X. Wang, M. Zhu, X. Leng, W. Chen, W. Wang, Q. Xu, L.-M. Yang and Y. Wu, Nano Res., 2021, 14, 4512–4519 CrossRef CAS.
- Z. Yu, S. Lv, Q. Yao, N. Fang, Y. Xu, Q. Shao, C. W. Pao, J. F. Lee, G. Li, L. M. Yang and X. Huang, Adv. Mater., 2022, 35, 2208101 CrossRef PubMed.
- T. Gao, S. Yu, Y. Chen, X. Li, X. Tang, S. Wu, B. He, H. Lan, S. Li, Q. Yue and D. Xiao, J. Colloid Interface Sci., 2023, 642, 120–128 CrossRef CAS PubMed.
- S. Zhou, N. Liu, Z. Wang and J. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 22578–22587 CrossRef CAS PubMed.
- T. Liu, X. Qu, Y. Zhang, X. Wang, Q. Dang, X. Li, B. Wang, S. Tang, Y. Luo and J. Jiang, Chem. Eng. J., 2023, 457, 141187 CrossRef CAS.
- S. Liu and J.-y Liu, J. Power Sources, 2022, 535, 231449 CrossRef CAS.
- M.-R. Zhao, B. Song and L.-M. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 26109–26122 CrossRef CAS PubMed.
- D. Deng and L.-M. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 22012–22024 CrossRef CAS PubMed.
- X. Wang and L.-M. Yang, Appl. Surf. Sci., 2022, 576, 151839 CrossRef CAS.
- X. Wang and L.-M. Yang, J. Mater. Chem. A, 2022, 10, 1481–1496 RSC.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S.-Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- S. Yadav, Z. Zhu and C. V. Singh, Int. J. Hydrogen Energy, 2014, 39, 4981–4995 CrossRef CAS.
- S. L. Li, H. Yin, X. Kan, L. Y. Gan, U. Schwingenschlogl and Y. Zhao, Phys. Chem. Chem. Phys., 2017, 19, 30069–30077 RSC.
- S. Ji, Z. Wang and J. Zhao, J. Mater. Chem. A, 2019, 7, 2392–2399 RSC.
- Z. He, Q. Liu, Y. Zhu, T. Tan, L. Cao, S. Zhao and Y. Chen, ACS Appl. Energy Mater., 2020, 3, 7039–7047 CrossRef CAS.
- X. Yao, Z.-W. Chen, Y.-R. Wang, X.-Y. Lang, Y.-F. Zhu, W. Gao and Q. Jiang, Appl. Surf. Sci., 2020, 529, 147183 CrossRef CAS.
- C. Wang, Y. Wang, Q. Guo, E. Dai and Z. Nie, ACS Omega, 2022, 7, 21994–22002 CrossRef CAS PubMed.
- W. Ji, T.-X. Wang, X. Ding, S. Lei and B.-H. Han, Coord. Chem. Rev., 2021, 439, 213875 CrossRef CAS.
- Q. Li, Y. Li, Y. Chen, L. Wu, C. Yang and X. Cui, Carbon, 2018, 136, 248–254 CrossRef CAS.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, ACS Catal., 2019, 9, 11042–11054 CrossRef CAS.
- X. Chen, X. Jiang and N. Yang, Small, 2022, 18, 2201135 CrossRef CAS PubMed.
- C. Wang and C. Guo, Int. J. Hydrogen Energy, 2021, 46, 33409–33419 CrossRef CAS.
- B. Li, S. Yu, M. Zhou, C. Chen, C. Lai, M. Zhang and H. Lin, J. Mater. Chem. A, 2022, 10, 3905–3932 RSC.
- W. Song, Z. Fu, P. Ma, X. Liu, Y. Guo, L. Fu and C. He, Appl. Surf. Sci., 2023, 617, 156550 CrossRef CAS.
- Y. Fu, D. Xu, Y. Wang, X. Li, Z. Chen, K. Li, Z. Li, L. Zheng and X. Zuo, ACS Sustainable Chem. Eng., 2020, 8, 8338–8347 CrossRef CAS.
- W. Zhang, Y. J. Gao, Q. J. Fang, J. K. Pan, X. C. Zhu, S. W. Deng, Z. H. Yao, G. L. Zhuang and J. G. Wang, J. Colloid Interface Sci., 2021, 599, 58–67 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- T. Bučko, J. Hafner, S. Lebègue and J. G. Ángyán, J. Phys. Chem. A, 2010, 114, 11814–11824 CrossRef PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- J.-S. Kim, J.-S. Kug and S.-J. Jeong, Nat. Commun., 2017, 8, 1674 CrossRef PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2024, 7, 207–218 CrossRef CAS.
- C. Ren, S. Lu, Y. Wu, Y. Ouyang, Y. Zhang, Q. Li, C. Ling and J. Wang, J. Am. Chem. Soc., 2022, 144, 12874–12883 CrossRef CAS PubMed.
- Y. Zhang, D. Wang, G. Wei, B. Li, Z. Mao, S.-M. Xu, S. Tang, J. Jiang, Z. Li, X. Wang and X. Xu, JACS Au, 2024, 4, 1509–1520 CrossRef CAS PubMed.
- H. W. Kim, H. Park, J. S. Roh, J. E. Shin, T. H. Lee, L. Zhang, Y. H. Cho, H. W. Yoon, V. J. Bukas, J. Guo, H. B. Park, T. H. Han and B. D. McCloskey, Chem. Mater., 2019, 31, 3967–3973 CrossRef CAS.
- D. Navadeepthy, A. Rebekah, C. Viswanathan and N. Ponpandian, Mater. Res. Bull., 2017, 95, 1–8 CrossRef CAS.
- E. Jung, H. Shin, B. H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K. S. Lee, S. P. Cho, J. S. Yoo, Y. E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed.
- K.-y Chen, J. Xia, S.-q Wang, R.-j Wu, W.-l Min, J.-y Wei, D.-s Hou and S. Mu, Cem. Concr. Res., 2024, 146, 105375 CrossRef CAS.
- Z. Yang, J. He, W. H. Lai, J. Peng, X. H. Liu, X. X. He, X. F. Guo, L. Li, Y. Qiao, J. M. Ma, M. Wu and S. L. Chou, Angew. Chem., Int. Ed., 2021, 133, 27292–27300 CrossRef.
- H. Niu, X. Wang, C. Shao, Y. Liu, Z. Zhang and Y. Guo, J. Mater. Chem. A, 2020, 8, 6555–6563 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00732h |
| This journal is © The Royal Society of Chemistry 2024 |