
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing uranium tetrafluoride microrods†
Harry
Jang
and
Frederic
Poineau
 *
*
Department of Chemistry & Biochemistry, University of Nevada, Las Vegas, Las Vegas, Nevada 89154, USA. E-mail: poineauf@unlv.nevada.edu
First published on 24th September 2024
Abstract
Anhydrous and hydrated UF4 microrods (5–25 μm) were prepared from the reactions of UO2 microrods (5–15 μm) with HF(g), produced from the decomposition of silver bifluoride (AgHF2, SBF). In order to optimize the preparation of UF4 mr, several experimental parameters including atmosphere (air or N2), temperature (150 or 250 °C) and amount of SBF were evaluated. In all reactions, rodlike morphologies were retained. At 250 °C, the reaction products always consist of an anhydrous UF4/hydrated UF4 mixture, while at 150 °C only hydrated UF4 was detected. Anhydrous UF4 microrods were obtained by dehydration of the anhydrous UF4/hydated UF4 mixture using TGA-DSC. Changing the atmosphere from air to N2 or reducing the amount of SBF by half did not affect the nature of the reaction products.
Introduction
At the nano- and microscale, materials can exhibit properties (e.g., optical, catalytic, electronic, mechanical, thermal, magnetic)1–3 that are not observed at the macroscale. The common applications of micro- and nano- materials (e.g. gas sensors, electrochromic devices, solar cells, batteries) are shared between many metal oxides (e.g. Ti,4 Co,1,5,6 Ni,2,7 Zn,8–10 Mo,11–15 and In16,17). The rod-morphology is one of the most studied and examples of microrods and nanorods respectively include In1−xGaxP,18 MnOOH,19 Bi2S3,20 and Ga2O3.21One element whose material chemistry of micro- and nanorods has been poorly studied is uranium. Research on uranium has primarily focused on the study of spherical particles of binary oxides, nitrides, carbides, and fluorides.22–28 Uranium microstructures can find applications as targets for medical isotope production,29–31 fuels for nuclear reactors,32–34 standards for nuclear forensics,35–37 and energy sources for space exploration.38 Morphological studies of uranium materials are also relevant to the field of nuclear forensics.39,40 One critical material for the nuclear industry is UF4, which is produced from the reaction of UO2 with HF gas at elevated temperatures (eqn (1)).
| UO2(s) + 4HF(g) → UF4(s) + 2H2O(g) | (1) |
Besides serving as an intermediate in UF6 production,41,42 UF4 has also found applications as targets for heavy ion production43 and the primary precursor material for U metal production.44 It is also proposed as a fuel for molten salt reactors.45 Anhydrous UF4, a green solid with low solubility in water,46 exhibits several hydrated forms (i.e., UF4·xH2O, x = 0.5, 0.7, 0.75, 1.2, 1.3, 1.5, 2.0, 2.5)47–49 which are formed upon reactions of the material with water (eqn (2)).
| UF4(s) + xH2O(g) → UF4·xH2O(s) | (2) |
As water is ubiquitous in the uranium industry, it is important to address environmental and industrial concerns pertaining to the hydrolytic behaviors of UF4.48 Though the physico-chemical properties of UF4 and its hydrates are well characterized at the macroscale,47–64 there is a lack of knowledge concerning their preparations and characterizations at the microscale. As the applications of UF4 expand, it is essential that information and accessibility to UF4 materials at the microscale become more readily available.
So far, the only UF4 morphology that can be prepared and controlled at the microscale is the spherical one. UF4 microspheres (ms) have already been produced and are commercially available,27,49,54,65 and although details on their production remain proprietary, high temperature hydrofluorination of UO2 was mentioned.66 Other UF4 morphologies such as microrods (mr) or microplates (mp) have not yet been prepared in a controlled manner.
Previously, we have reported on the preparation of UO2F2 microspheres, microrods and microplates using chemical transformation.28,67 In these works, uranium oxide micromaterials (UO3, U3O8) were fluorinated in an autoclave with HF(g) produced from the thermal decomposition of silver bifluoride (AgHF2, SBF). Scanning electron microscope (SEM) results concerning the morphology and particle size distribution of UO2F2 mp have shown high variance, whereas SEM results relating to UO2F2 mr have been consistent. As a continuation, investigating U(IV) fluoride micromaterials was a natural progression and microrods were selected as the primary microstructure.
Here, we report on the preparation of anhydrous and hydrated UF4 microrods. The materials were prepared by chemical transformation from the reaction of UO2 mr with SBF in autoclaves and were characterized by scanning electron microscopy and powder X-ray diffraction (PXRD).
Experimental
Caution! Uranium-238 is an α emitter (Emax = 4.26 MeV). All manipulations were performed in a designed radiochemistry laboratory equipped with HEPA filter hoods and by following approved radioisotope handling and monitoring procedures.Materials and methods
Silver bifluoride (≥99%, Alfa Aesar), glycerol (≥99.5%, Sigma-Aldrich), and urea (≥98%, Sigma-Aldrich) were used as received. UO2(NO3)2·6H2O (UNH) was prepared from the treatment of uranium metal dissolved in hot nitric acid followed by recrystallization. Fluorination and hydrothermal reactions were conducted within a Parr model 4749 autoclave placed in a Thermo Scientific Thermolyne Benchtop muffle furnace (model FB1315M). Fluorinations were conducted at 150–250 °C for 6–24 hours in the setup reported previously.67 For dry reactions, N2 was regarded as inert as it does not react with reactants at these temperatures. HF(g), the gaseous decomposition product of SBF, provides the source of fluorine for the reactions. In these reactions, SBF in excess molar quantity was placed on the Teflon liner of the vessel, while the uranium oxide material was placed in a 15 mL Teflon vial above the SBF.67Powder X-ray diffraction measurements were performed at room temperature on a Bruker D8 Advanced diffractometer equipped with Cu Kα X-rays (λ = 1.5406 Å) and a solid-state Si detector. Imaging was performed on the JEOL Tescan CLARA field emission scanning electron microscope, and samples were mounted on carbon tape without coating. TGA-DSC measurements were conducted with a TA instruments SDT 650 Discovery series TGA-DSC from 50–600 °C with a heating rate of 10 °C min−1. The measurements were performed in alumina crucibles under flowing argon gas with a sample and balance flow rate of approximately 100 mL min−1. Particle sizes were measured using ImageJ, and particle size distribution figures were generated using SciDAVis.
Sample preparation
| Reaction # | Mass of UO2 (mg) | Mass of SBF (mg) | T (°C) | Time (h) | Atmosphere | Yield (mg, %) | Reaction product |
|---|---|---|---|---|---|---|---|
| 1 | 50.8 | 303.9 | 250 | 24 | Air | 57.9 (98.0%) | UF4/UF4·2H2O |
| 2 | 30.1 | 400.0 | 250 | 24 | N2 | 29.5 (82.3%) | UF4/UF4·xH2O |
| 3 | 31.5 | 301.6 | 150 | 24 | Air | 35.0 (86.0%) | UF4·1.5H2O |
| 4 | 33.8 | 155.8 | 250 | 24 | Air | 35.2 (89.6%) | UF4/UF4·2H2O |
 | ||
| Fig. 1 SEM images of UO2 mr at (a) 4790× and (b) 6640×. | ||
Results and discussion
Here, the reactions were set up identically to previous UO2F2 microrod works except that the starting material (i.e., U3O8 mr) was replaced with UO2 mr.Various experimental parameters have been evaluated (atmosphere, temperature, amount of SBF). A total of four reactions were investigated. In each reaction, UO2 was weighed and placed in a Teflon vial and then placed in the Teflon liner of the autoclave containing SBF. The autoclave was sealed either in air (reaction 1, 3, and 4) or under N2 atmosphere (reaction 2) and thermally treated for 24 hours at 150 or 250 °C. Following the reaction, the autoclave was cooled for 2 hours to room temperature and opened, and the resulting product was weighed and characterized by PXRD and SEM. The conditions, reaction products, and yields for the four reactions are presented in Table 1, and the effects of atmosphere, temperature, and amount of SBF on the nature of the reaction products are discussed in the following sections.
Baseline reaction
Reaction 1 follows the same procedure as the one applied for the preparation of UO2F2 (24 hours at 250 °C using ∼300 mg SBF [U:HF = ∼3–5] prepared in air). Here the reaction of UO2 and HF(g) led to a mixture of anhydrous and hydrated UF4 (Fig. S2a, ESI†). The presence of hydrated UF4 in the reaction product is probably due to the reaction of UF4 and water (eqn (2)) that was formed as a byproduct (eqn (1)).Morphologically, the rodlike particles remained intact and experienced some visible surface roughing (Fig. 2a–c). Particle size distribution (PSD) analysis (Fig. 3) shows the average length of these particles have increased by ∼7 μm.
 | ||
| Fig. 2 SEM images of the reaction products of reaction 1 at (a) 2000×, (b) 4450×, (c) 6650×, and (d) after TGA-DSC at 2650×. | ||
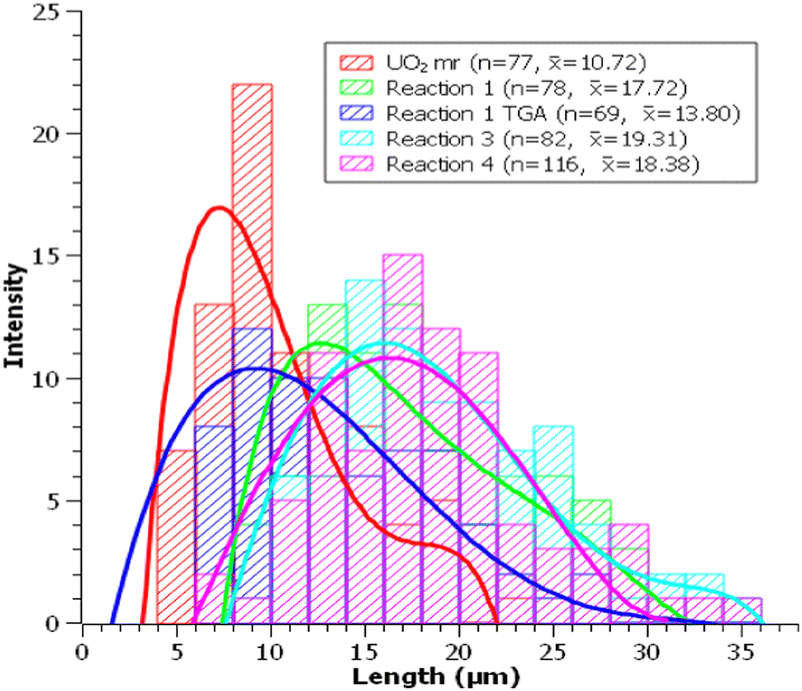 | ||
| Fig. 3 Particle size distribution of UO2 mr and particles from reaction 1, 3, and 4. | ||
Treatment of the reaction product (16.94 mg) by TGA-DSC up to 600 °C (ramp rate of 10 °C min−1) under argon converted the UF4/UF4·2H2O mixture to anhydrous UF4. The TGA-DSC curves (Fig. S3, ESI†) show a steady mass decrease followed by a plateau at ∼380 °C, indicating the point of complete dehydration to anhydrous UF4. Following TGA-DSC, the sample was characterized by SEM (Fig. 2d) and PXRD (Fig. S2b, ESI†). SEM analysis shows no morphological changes to the particles, and PSD analysis showed a decrease in average particle length by ∼4 μm. The dehydration of UF4/UF4 hydrate proved efficient for the preparation of anhydrous UF4 mr and could be transposed to other morphologies of UF4 hydrates.
Effect of atmosphere
The effect of atmosphere on the reaction product was investigated in reaction 2. Treatment of UO2 with HF(g) under N2 atmosphere at 250 °C for 24 hours yielded a mixture of anhydrous UF4 and hydrated UF4 (PXRD in Fig. S2c, ESI†). SEM shows the rods to exhibit rough surfaces that were not observed in the UO2 mr precursor (Fig. 4), and PXRD shows less hydrated UF4 than from the one from reaction 1 (Fig. S2c, ESI†). | ||
| Fig. 4 SEM images of the reaction products of reaction 2 at (a) 1260× and (b) 7360×. | ||
Effect of temperature
Reaction 3 was performed at 150 °C, while the other parameters (atmosphere, reaction time, and amount of SBF) were identical to the baseline reaction. Following the reaction, PXRD analysis (Fig. S2d, ESI†) shows the presence of UF4·1.5H2O as a single phase.SEM analysis (Fig. 5a and b) shows the UF4·1.5H2O mr to exhibit smoother surfaces than the reaction products from reactions 1 and 2. The particle size distributions (Fig. 3) showed no size disparities between UF4·1.5H2O mr and that of UF4/UF4·2H2O mr (reaction 1).
 | ||
| Fig. 5 SEM images of the reaction products of reaction 3 at (a) 1950× and (b) 3510×, and reaction 4 at (c) 3080×. | ||
The absence of anhydrous UF4 at 150 °C indicated that the hydrate was initially formed near this temperature and that the increase in temperature to 250 °C would initiate dehydration leading to the partially hydrated mixture. We hypothesized that the reaction at 400 °C should lead exclusively to anhydrous UF4.
Effect of SBF
In reaction 4, about half the amount of AgHF2 was used (155.8 mg, 1.061 mmol). Calculations indicated that at 250 °C, decreasing the amount of SBF by half would decrease the partial pressure of HF(g) in the autoclave from ∼4 atm to ∼2 atm (∼5 atm to ∼3 atm total pressure). PXRD analysis showed the presence of a UF4/UF4·2H2O mixture (Fig. S2e, ESI†) while SEM (Fig. 5c) indicated the presence of microrods. PSD observations (Fig. 4) were consistent with the results of reactions 1 and 3. Overall, decreasing the amount of SBF does not fundamentally change the nature of the reaction products as anhydrous UF4 and hydrated UF4 were obtained.Conclusions
For the first time, uranium tetrafluoride microrods were prepared by chemical transformation. The reactions of UO2 microrods (5–15 μm) with HF(g), produced from the decomposition of SBF, were investigated in autoclaves. Several experimental parameters including atmosphere, temperature, and amount of SBF were tested. In all reactions, rodlike morphologies were retained. At 250 °C, the reaction products always consist of an anhydrous UF4/hydrated UF4 mixture, while at 150 °C only the presence of hydrated UF4 was detected. Anhydrous UF4 microrods were obtained by dehydration of the anhydrous UF4/hydrated UF4 mixture via thermal treatment using TGA-DSC. Changing the atmosphere from air to N2 or reducing the amount of SBF by half did not fundamentally affect the nature of the reaction products. Using experimental set-up to 250 °C for the highest operational temperature indicated that the preparation of single phase hydrated UF4 or anhydrous UF4 is respectively a one-step (hydrofluorination) and two-step process (hydrofluorination and dehydration). The preparation of anhydrous UF4 in a single step process would require hydrofluorination in an autoclave at T > 250 °C.Currently, the preparation of UF4 nanospheres and microplates using the method presented here is under progress and results will be reported in due course. Finally, successful fluorinations of uranium oxide micromaterials will lay the groundwork for the development of other f-element fluoride micromaterials.
Author contributions
The manuscript was written through contributions of all authors. H. J.: investigation, methodology, validation, visualization, and writing – original draft. F. P.: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work performed under the auspices of the Consortium on Nuclear Security Technologies (CONNECT) supported by the Department of Energy/National Nuclear Security Administration under Award Number(s) DE-NA0003948. The authors would like to thank Mrs Wendee Johns for administrative support and Mr Quinn Summerfield for laboratory support.References
- S. Iravani and R. S. Varma, Green Chem., 2020, 22, 2643–2661 RSC.
- M. Imran Din and A. Rani, Int. J. Anal. Chem., 2016, 2016, 1–14 CrossRef PubMed.
- M. A. Albrecht, C. W. Evans and C. L. Raston, Green Chem., 2006, 8, 417–432 RSC.
- V. K. H. Bui, V. Van Tran, J. Y. Moon, D. Park and Y. C. Lee, Nanomaterials, 2020, 10, 1–31 CrossRef PubMed.
- D. Alburquenque, E. Vargas, J. C. Denardin, J. Escrig, J. F. Marco, J. Ortiz and J. L. Gautier, Mater. Charact., 2014, 93, 191–197 CrossRef CAS.
- N. Wu, J. Shen, L. Sun, M. Yuan, Y. Shao, J. Ma, G. Liu, D. Guo, X. Liu and Y. B. He, Electrochim. Acta, 2019, 310, 70–77 CrossRef CAS.
- S. S. Narender, V. V. S. Varma, C. S. Srikar, J. Ruchitha, P. A. Varma and B. V. S. Praveen, Chem. Eng. Technol., 2022, 45, 397–409 CrossRef CAS.
- J. H. Kim, D. Andeen and F. F. Lange, Adv. Mater., 2006, 18, 2453–2457 CrossRef CAS.
- T. Gao and T. H. Wang, Appl. Phys. A: Mater. Sci. Process., 2005, 80, 1451–1454 CrossRef CAS.
- Q. Deng, X. Duan, D. H. L. Ng, H. Tang, Y. Yang, M. Kong, Z. Wu, W. Cai and G. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6030–6037 CrossRef CAS PubMed.
- X. Hu, W. Zhang, X. Liu, Y. Mei and Y. Huang, Chem. Soc. Rev., 2015, 44, 2376–2404 RSC.
- J. Sun, Y. Dong, X. Wang, J. Cao, M. Gong and C. Li, J. New Mater. Electrochem. Syst., 2021, 24, 73–77 CrossRef CAS.
- J. Zhou, N. S. Xu, S. Z. Deng, J. Chen, J. C. She and Z. L. Wang, Adv. Mater., 2003, 15, 1835–1840 CrossRef CAS.
- T. S. Sian and G. B. Reddy, J. Appl. Phys., 2005, 98, 6104 CrossRef.
- C. D. A. Lima, J. V. B. Moura, G. S. Pinheiro, J. F. D. F. Araujo, S. B. S. Gusmão, B. C. Viana, P. T. C. Freire and C. Luz-Lima, Ceram. Int., 2021, 47, 27778–27788 CrossRef CAS.
- S. Luo, W. Zhou, W. Wang, Z. Zhang, L. Liu, X. Dou, J. Wang, X. Zhao, D. Liu, Y. Gao, L. Song, Y. Xiang, J. Zhou and S. Xie, Appl. Phys. Lett., 2005, 87, 3109 Search PubMed.
- J. Bartolomé, A. Cremades and J. Piqueras, J. Mater. Chem. C, 2013, 1, 6790–6799 RSC.
- M. K. K. Nakaema, M. P. F. Godoy, M. J. S. P. Brasil, F. Iikawa, D. Silva, M. Sacilotti, J. Decobert and G. Patriarche, J. Appl. Phys., 2005, 98, 3506 CrossRef.
- Y. Zhang, Y. Liu, F. Guo, Y. Hu, X. Liu and Y. Qian, Solid State Commun., 2005, 134, 523–527 CrossRef CAS.
- G. Xie, Z.-P. Qiao, M.-H. Zeng, X.-M. Chen and S.-L. Gao, Cryst. Growth Des., 2004, 4, 513–516 CrossRef CAS.
- J. Zhang, Z. Liu, C. Lin and J. Lin, J. Cryst. Growth, 2005, 280, 99–106 CrossRef CAS.
- J. H. Yang, Y. W. Rhee, D. Kim, J. H. Kim, K. W. Kang and K. S. Kim, Transactions of the Korean Nuclear Society Autumn Meeting, Gyeongju, Korea, 2006, pp. 1–2 Search PubMed.
- C. M. Silva, R. D. Hunt, L. L. Snead and K. A. Terrani, Inorg. Chem., 2015, 54, 293–298 CrossRef CAS PubMed.
- G. Ledergerber, Z. Kopajtic, F. Ingold and R. W. Stratton, J. Nucl. Mater., 1992, 188, 28–35 CrossRef CAS.
- T. B. Lindemer, S. L. Voit, C. M. Silva, T. M. Besmann and R. D. Hunt, J. Nucl. Mater., 2014, 448, 404–411 CrossRef CAS.
- W. Tian, H. Guo, D. Chen, M. A. Pouchon, A. Horwege, X. Yin, Q. Huang, J. Wang, S. Cao, D. Chen, J. Bai, C. Tan, F. Fan, X. Wu, T. Shen and Z. Qin, Ceram. Int., 2018, 44, 17945–17952 CrossRef CAS.
- B. J. Foley, J. H. Christian, C. A. Klug, E. Villa-Aleman, M. S. Wellons, M. DeVore, N. Groden and J. Darvin, Dalton Trans., 2022, 51, 6061–6067 RSC.
- H. Jang, J. Louis-Jean and F. Poineau, ACS Omega, 2023, 8, 21996–22002 CrossRef CAS PubMed.
- R. Henry and H. Saclay, The use of recoil for the separation of uranium fission products; Utilisation du recul pour la separation des produits de fission de l’uranium, France, 1959 Search PubMed.
- U. Passy and N. H. Steiger, Nucl. Sci. Eng., 1963, 15, 366–374 CrossRef CAS.
- J. M. Dorhout, M. P. Wilkerson and K. R. Czerwinski, J. Radioanal. Nucl. Chem., 2019, 320, 415–424 CrossRef CAS.
- L. R. Khanal, J. A. Sundararajan and Y. Qiang, Energy Technol., 2020, 8, 1–23 Search PubMed.
- E. A. Filippov, A. I. Karelin, O. P. Lobas, A. S. Papkov, A. N. Zhiganov, L. A. Mishina and V. I. Shamin, J. Radioanal. Nucl. Chem., 1990, 143, 53–60 CrossRef CAS.
- A. C. Robisson, S. Lemonnier and S. Granjean, Atalante, 2004, 1–4 Search PubMed.
- R. Middendorp, M. Dürr and D. Bosbach, Procedia Chem., 2016, 21, 285–292 CrossRef.
- S. Richter, J. Truyens, C. Venchiarutti, Y. Aregbe, R. Middendorp, S. Neumeier, P. Kegler, M. Klinkenberg, M. Zoriy, G. Stadelmann, Z. Macsik, A. Koepf, M. Sturm, S. Konegger-Kappel, A. Venzin, L. Sangely and T. Tanpraphan, J. Radioanal. Nucl. Chem., 2022, 1–5 CAS.
- P. Kegler, F. Pointurier, J. Rothe, K. Dardenne, T. Vitova, A. Beck, S. Hammerich, S. Potts, A. L. Faure, M. Klinkenberg, F. Kreft, I. Niemeyer, D. Bosbach and S. Neumeier, MRS Adv., 2021, 6, 125–130 CrossRef CAS.
- W. J. Carmack, W. C. Richardson, D. L. Husser and T. C. Mohr, AIP Conf. Proc., 2004, 699, 420–425 CrossRef CAS.
- I. J. Schwerdt, C. G. Hawkins, B. Taylor, A. Brenkmann, S. Martinson and L. W. McDonald, Radiochim. Acta, 2019, 107, 193–205 CrossRef CAS.
- L. W. McDonald, K. Sentz, A. Hagen, B. W. Chung, C. A. Nizinski, I. J. Schwerdt, A. Hanson, S. Donald, R. Clark, G. Sjoden, R. Porter, M. T. Athon, T. Tasdizen, V. Noel, S. M. Webb, A. Van Veelen, S. M. Hickam and C. Ly, J. Nucl. Mater., 2024, 588, 154779 CrossRef CAS.
- B. Morel and B. Duperret, J. Fluor. Chem., 2009, 130, 7–10 CrossRef CAS.
- K. J. Pastoor, R. S. Kemp, M. P. Jensen and J. C. Shafer, Inorg. Chem., 2021, 60, 8347–8367 CrossRef CAS PubMed.
- G. Sibbens, A. Moens and R. Eykens, J. Radioanal. Nucl. Chem., 2015, 305, 723–726 Search PubMed.
- H. Jang, J. Louis-Jean, B. Childs, K. Holliday, D. Reilly, M. Athon, K. Czerwinski, D. Hatchett and F. Poineau, R. Soc. Open Sci., 2022, 9, 211870 Search PubMed.
- J. McFarlane, P. Taylor, D. Holcomb and W. P. Poore, Review of Hazards Associated with Molten Salt Reactor Fuel Processing Operations (No. ORNL/TM-2019/1195), Oak Ridge National Laboratory, Oak Ridge, TN, 2019 Search PubMed.
- J. J. Katz and E. Rabinowitch, The chemistry of uranium, 1951 Search PubMed.
- J. K. Dawson, R. W. M. D’Eye and A. E. Truswell, J. Chem. Soc., 1954, 3922–3929 RSC.
- A. Miskowiec, K. J. Pastoor, J. H. Christian, J. L. Niedziela, B. J. Foley, S. Isbill, A. E. Shields, L. L. Daemen, E. Novak, E. Nykwest, T. Spano, M. S. Wellons, M. Jensen and J. Shafer, J. Phys. Chem. C, 2021, 125, 25007–25021 CrossRef CAS.
- J. H. Christian, C. A. Klug, M. Devore, E. Villa-Aleman, B. J. Foley, N. Groden, A. T. Baldwin and M. S. Wellons, Dalton Trans., 2021, 50, 2462–2471 RSC.
- J. G. Tobin, A. M. Duffin, S.-W. Yu, R. Qiao, W. L. Yang, C. H. Booth and D. K. Shuh, J. Vac. Sci. Technol., A, 2017, 35, E108 CrossRef.
- C. A. Klug and J. B. Miller, Solid State Nucl. Magn. Reson., 2018, 92, 14–18 CrossRef CAS PubMed.
- S. P. Gabuda, L. G. Falaleeva and Y. V. Gagarinskii, Phys. Status Solidi, 1969, 33, 435–438 CrossRef CAS.
- A. Miskowiec, A. E. Shields, J. L. Niedziela, Y. Cheng, P. Taylor, G. DelCul, R. Hunt, B. Spencer, J. Langford and D. Abernathy, Phys. B, 2019, 570, 194–205 CrossRef CAS.
- E. Villa-Aleman and M. S. Wellons, J. Raman Spectrosc., 2016, 47, 865–870 CrossRef CAS.
- A. Y. Teterin, Y. A. Teterin, K. I. Maslakov, A. D. Panov, M. V. Ryzhkov and L. Vukcevic, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 5101 CrossRef.
- D. T. Hodul, Spectrosc. Lett., 1983, 16, 181–191 CrossRef CAS.
- C. Görller-Walrand, M. P. Gos and W. D’Olieslager, Radiochim. Acta, 1993, 62, 55–60 CrossRef.
- W. H. Zachariasen, Acta Crystallogr., 1949, 2, 388–390 CrossRef CAS.
- A. C. Larson, R. B. Roof Jr and D. T. Cromer, Acta Crystallogr., 1964, 17, 555–558 CrossRef CAS.
- T. K. Keenan and L. B. Asprey, Inorg. Chem., 1969, 8, 235–238 CrossRef CAS.
- J. Shankar, P. G. Khubchandani and V. M. Padmanabhan, Anal. Chem., 1957, 210, 1374 CrossRef.
- B. Scheibe, J. Bruns, G. Heymann, M. Sachs, A. J. Karttunen, C. Pietzonka, S. I. Ivlev, H. Huppertz and F. Kraus, Chem. – Eur. J., 2019, 25, 7366–7374 CrossRef CAS PubMed.
- S. Kern, J. Hayward, S. Roberts, J. W. Richardson, F. J. Rotella, L. Soderholm, B. Cort, M. Tinkle, M. West, D. Hoisington and G. H. Lander, J. Chem. Phys., 1994, 101, 9333–9337 CrossRef CAS.
- A. Miskowiec, Phys. Chem. Chem. Phys., 2018, 20, 10384–10395 RSC.
- J. Plaue, Signatures of Chemical Process History in Uranium Oxides, University of Nevada, Las Vegas, 2013 Search PubMed.
- K. J. Pastoor, A. J. Miskowiec, J. L. Niedziela, J. H. Christian, B. J. Foley, S. B. Isbill, A. E. Shields, A. M. Manjón-Sanz, E. C. Nykwest, T. L. Spano, M. S. Wellons, J. C. Shafer and M. P. Jensen, J. Phys. Chem. C, 2022, 126, 13256–13267 CrossRef CAS.
- H. Jang and F. Poineau, ACS Omega, 2024, 9, 26380–26387 CrossRef CAS PubMed.
- L. Wang, R. Zhao, C. Z. Wang, L. Y. Yuan, Z. J. Gu, C. L. Xiao, S. A. Wang, X. W. Wang, Y. L. Zhao, Z. F. Chai and W. Q. Shi, Chem. – Eur. J., 2014, 20, 12655–12662 CrossRef CAS PubMed.
- J. Louis-Jean, H. Jang, A. J. Swift and F. Poineau, ACS Omega, 2021, 6, 26672–26679 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00796d |
| This journal is © The Royal Society of Chemistry 2024 |