
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visualization of mechanochemical polymer-chain scission in double-network elastomers using a radical-transfer-type fluorescent molecular probe†
Takumi
Yamamoto
 a,
Akira
Takahashi
a and
Hideyuki
Otsuka
*ab
a,
Akira
Takahashi
a and
Hideyuki
Otsuka
*ab
aDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayma, Meguro-ku, Tokyo 152-8550, Japan. E-mail: otsuka@mac.titech.ac.jp
bLiving Systems Materialogy (LiSM) Research Group, International Research Frontiers Initiative (IRFI), Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
First published on 29th January 2024
Abstract
Double-network (DN) elastomers are renowned for combining stiffness and toughness. Their exceptional physical properties have garnered significant attention in recent years. However, the fracture phenomena in DN elastomers are much less understood than those in DN gels due to the limited scope of visualization methods. Here, we demonstrate the visualization of sacrificial bond cleavage in DN elastomers during elongation by adding a diarylacetonitrile (DAAN) derivative as a radical-transfer-type fluorescent molecular probe, which enables the visualization of polymer-chain scission without altering the mechanical properties. A tensile test of the DN elastomers that contain DAAN revealed that mechanoradicals are generated from the entire elongated region of the elastomers in the strain-hardening region. In contrast, DN gels generate mechanoradicals only at the necked region. This method is expected to accelerate the investigation of the fracture properties of various DN elastomers.
Introduction
The double-network (DN) method is an effective and widely used approach to enhance the toughness of polymeric materials, and has been successfully applied to numerous hydrogel and elastomer systems.1–6 A comprehensive examination of the fracture mechanisms in DN gels and elastomers is important not only from a fundamental chemical point of view, but also from an application perspective, as it has the potential to propel the development of advanced functional materials. However, analysing the fracture phenomena in polymeric materials still poses a significant challenge due to the short lifetime of the mechanoradicals generated by polymer-chain scission,7 which makes them difficult to detect even using specialized equipment.8,9 In the solution, mechanoradicals can be detected using spin-trapping agents, such as nitroso species,10–17 nitrones,18 and hydrazyl radicals.19–21 In DN hydrogels, the mechanoradicals generated by the cleavage of the sacrificial polymer chains can be visualized by using the Fenton reaction,22 a combination of mechanoradical-induced polymerization and an environment-responsive fluorescent probe,23 or the use of prefluorescent probes for detecting oxygen-relay radical-trapping reactions.24 The results of these pioneering studies have greatly advanced research into the fracture and toughening mechanisms of DN hydrogels. However, these mechanoradical-visualization methods theoretically require the presence of water and/or dissolved oxygen in water, and thus cannot be directly applied to DN elastomer systems. Therefore, color-changing mechanophores, which are known as mechanochromophores, have been used to visualize sacrificial bond cleavage in DN elastomers.4,25–28 Mechanochromophores are molecules wherein chemoselective rearrangements proceed accompanied by optical changes in response to mechanical stress.29–33 As such, they are ideal tools to obtain crucial information on sacrificial bonds by virtue of their self-reporting capability.34–37 However, introducing mechanophores into elastomers alters the properties of the original materials, as the activation energy of mechanophores is typically lower than those for the cleavage of representative C–C bonds, which means that the results may not accurately reflect the intrinsic properties of the DN elastomers.38,39 Moreover, covalent introduction of mechanophores often complicates the synthetic process.Against this background, we propose here a simple and versatile strategy to evaluate polymer-chain scission in DN elastomers using a radical-transfer-type fluorescent molecular probe that can detect mechanoradicals without affecting the physical properties of the elastomers. For this purpose, we chose a diarylacetonitrile (DAAN) derivative with a methoxy group at the para-position of the aromatic rings as the fluorescent molecular probe, because it can be easily synthesized in large quantities via a one-step reaction (Fig. 1a).40,41DAAN derivatives react with the highly reactive polymeric mechanoradicals generated by mechanical scission of polymer chains to produce the corresponding DAAN radicals,42,43 which are relatively stable under atmospheric conditions and fluoresce under UV irradiation. As such, DAAN is expected to be an ideal probe to obtain information on sacrificial bond cleavage in elastomers without altering their physical properties (Fig. 1b). In addition, the ability to post-synthetically endow DN elastomers with fracture-visualization properties should accelerate the evaluation of their physical properties.
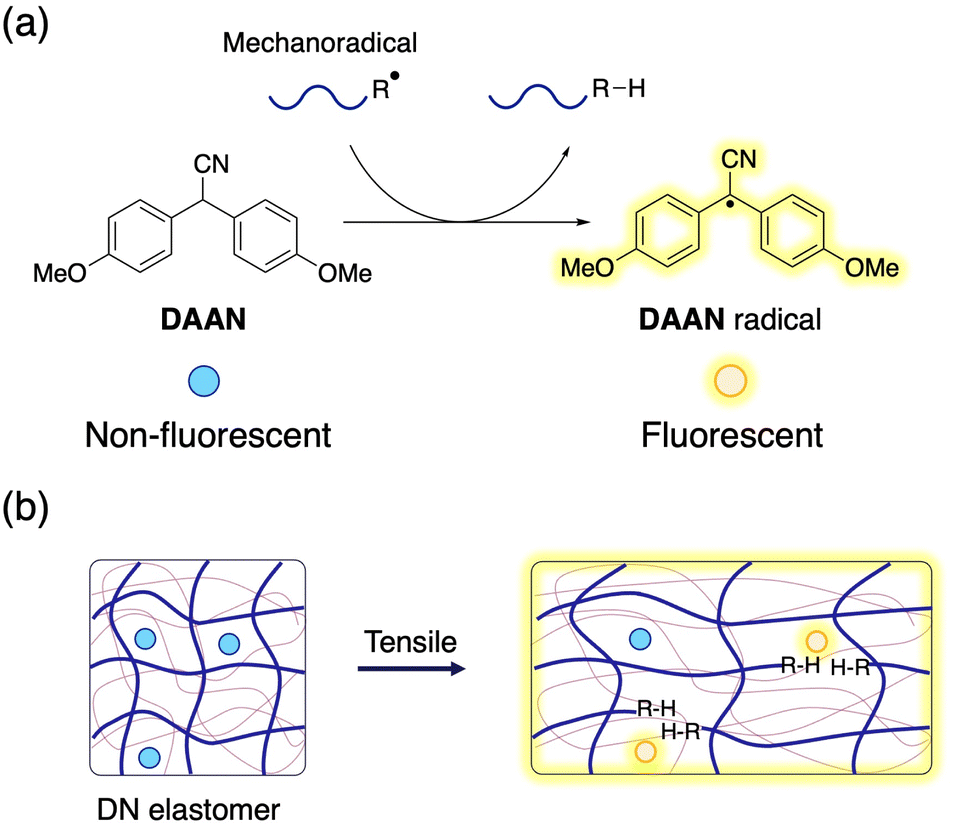 | ||
| Fig. 1 (a) Chemical reaction of DAAN with mechanoradicals. (b) Schematic illustrations of sacrificial bond scission and its visualization using DAAN in a DN elastomer. | ||
Experimental
Preparation of DN elastomers
The DN elastomers were synthesized according to a previously reported procedure (Fig. 2a).28 The first networks (FNs) were prepared using ethyl acrylate (EA) and triethylene glycol dimethacrylate (TGD) as cross-linkers via free-radical polymerization at room temperature. After washing and drying the resulting films to remove any unreacted components, the obtained FN films were then immersed in a mixture of methyl acrylate (MA), TGD, and the photoinitiator Irgacure 184. The films were swollen to their equilibrium state and then exposed to UV light while maintaining the film temperature <40 °C. Detailed synthetic procedures are provided in the ESI.† We also prepared several DN elastomers with different crosslinker loadings or FN polymer species using the same procedure (Table 1). Here, the weight fraction of the FNs in the DN elastomers varied according to the crosslink density of the FNs. For example, the swelling degree of the FNs in the monomer solution decreases with increasing crosslink density, which in turn led to an increase in the FN weight fraction in the resulting DN elastomers. The detailed properties of the obtained elastomers are summarized in Table S1.†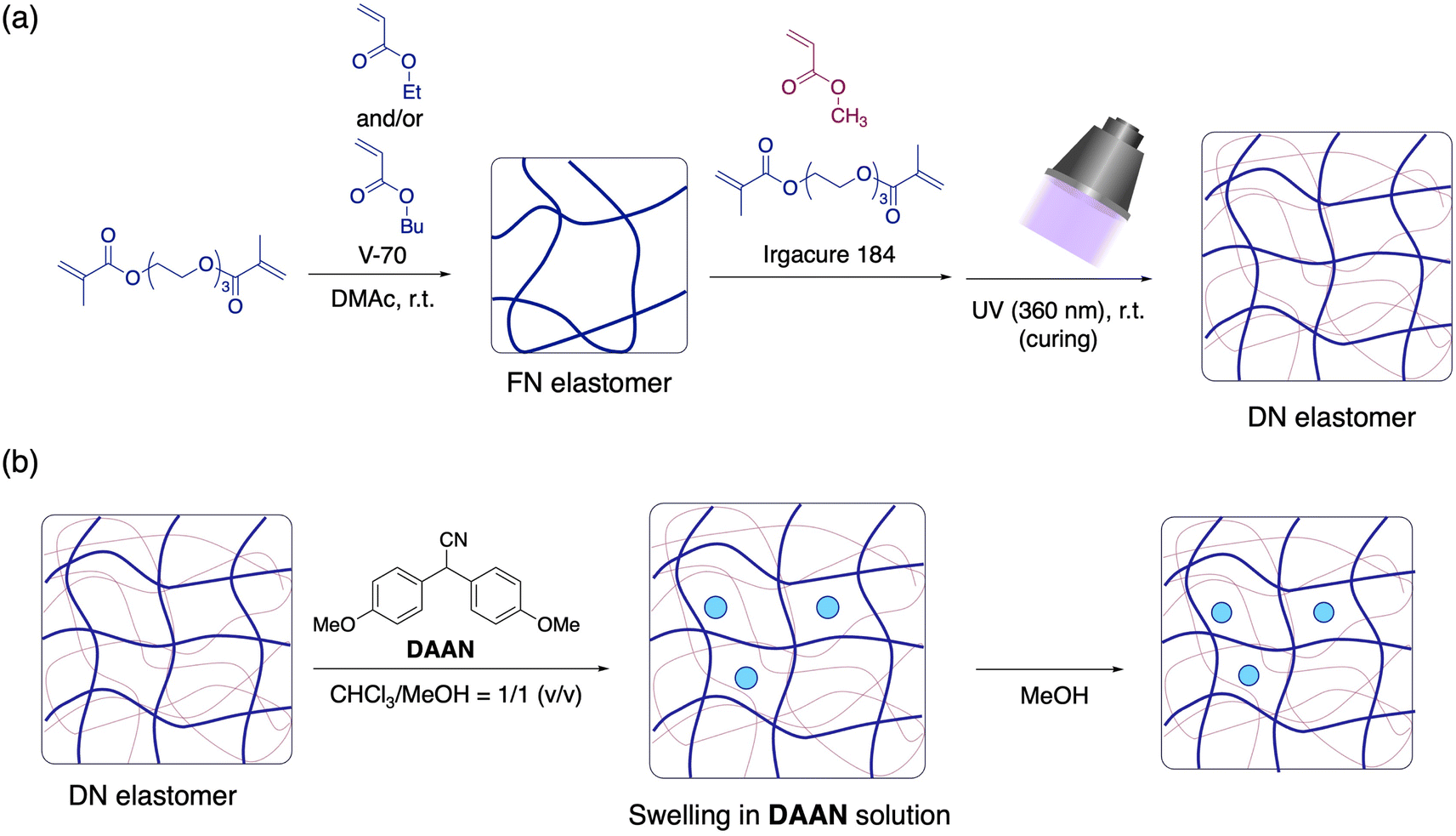 | ||
| Fig. 2 (a) Synthesis of DN elastomers. (b) Preparation of DAAN-containing elastomers (V-70: 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile); DMAc: N,N-dimethylacetamide). | ||
| Entry | Elastomer | FN | DN | ||
|---|---|---|---|---|---|
| Monomera | Crosslink density | Monomera | Crosslink density | ||
| a MA: methyl acrylate; EA: ethyl acrylate; BA: butyl acrylate; BAEA: mixture of BA and EA (1/1, mol/mol). | |||||
| 1 | EA-1/MA | EA | 1 mol% | MA | 1 mol% |
| 2 | EA-2/MA | EA | 2 mol% | MA | 1 mol% |
| 3 | EA-3/MA | EA | 3 mol% | MA | 1 mol% |
| 4 | BAEA-3/MA | BA & EA | 3 mol% | MA | 1 mol% |
| 5 | BA-3/MA | BA | 3 mol% | MA | 1 mol% |
Then, DAAN was incorporated into the synthesized DN elastomers by first swelling them in a saturated solution of DAAN in a mixture of chloroform/methanol (1/1, v/v; ∼2.46 × 10−2 mol L−1) for 3 h and subsequently shrinking them by immersion in methanol for 3 h (Fig. 2b). The shrunken elastomers were vacuum-dried at 40 °C for 24 h to completely remove the solvent. The thus-prepared DAAN-doped elastomers are denoted with the prefix “DAAN-”. The amount of DAAN incorporated was ∼1.05 ± 0.15 wt% for all elastomers, as determined by evaluating the weight difference before and after the incorporation process.
Tensile tests
Tensile tests were performed using a SHIMADZU EZ-L instrument equipped with a 100 N load cell at room temperature. For the tests dog-bone-shaped pieces were fabricated from the films in accordance with the JIS-7 standard (12 mm × 2 mm gauge section) with a thickness of 0.62–0.78 mm.Fluorescence spectroscopy
Fluorescence-spectroscopy measurements were carried out using a spectrofluorometer (JASCO FP-8550) between 500 nm and 750 nm.Results and discussion
Mechanical and mechanofluorescence properties
To investigate the mechanoradical-visualization ability of DAAN in the DN elastomers, DAAN-EA-3/MA films were uniaxially stretched at 10 mm min−1. As shown in Fig. 3a, the elastomer exhibited yellow emission under UV irradiation (λex = 365 nm). The fluorescence spectrum of DAAN-EA-3/MA after the tensile tests was in good agreement with the spectra derived from the DAAN radicals (Fig. 3b).40,42 These results suggest that mechanoradicals are generated by sacrificial FN cleavage in the DN elastomer during tensile testing, and that the DAAN radicals are generated by the radical-transfer reaction between these mechanoradicals and DAAN. | ||
| Fig. 3 (a) Digital images of DAAN-EA-3/MA during stretching (10 mm min−1) used for fluorescence-intensity analysis. (b) Fluorescence spectra of DAAN-EA/MA before and after tensile test. (c) Stress–strain curves of EA-3/MA and DAAN-EA-3/MA. | ||
Tensile tests of EA-3/MA and DAAN-EA-3/MA were conducted to evaluate the effect of the addition of DAAN, and the results confirmed that DAAN does not affect the stress–strain (S–S) curve (Fig. 3c). These results support the idea that the addition of DAAN derivatives enables mechanoradical detection in DN elastomers without altering their mechanical properties.
The mechanoradical generation of the prepared DAAN-containing DN elastomers was investigated using uniaxial tensile tests under UV irradiation (λex = 365 nm). All measurements were performed three times for each sample, which confirmed the good reproducibility of the S–S curves (Fig. S1†). All samples begin to show yellow emission at a certain strain, which then increases upon applying the tension (cf. e.g., Movie S1†). For the DAAN-EA/MA series with different FN crosslink densities, strain-hardening occurs at lower strain for the higher-crosslink-density samples (Fig. 4a). The onset points coincide well with the onset of fluorescence for DAAN-EA-2/MA and DAAN-EA-3/MA (Fig. 4b), suggesting that the sacrificial bond cleavage occurs after the strain-hardening begins. However, DAAN-EA-1/MA only begins to show emission in the latest stage, long after the onset of strain-hardening. This result suggests that the degree of sacrificial bond cleavage is insufficient to generate DAAN radicals at the strain-hardening point in DN elastomers with low crosslink density. In the case of DN gels, it is well established that large numbers of mechanoradicals are generated only in the neck region in the constant-stress regime above the yielding point.22,24 However, the DAAN-doped DN elastomers exhibited fluorescence over almost the entire film. This result indicates that the mechanoradicals are generated over a larger area in the DN elastomers than in the DN gels, and thus suggests a mechanoradical-generation mechanism is different from that of the DN gels. Specifically, the collapse of the first network at a specific area with a concomitant necking event is considered less likely in DN elastomers than in DN gels because of the higher weight fraction of the first network and the friction between the polymer chains during deformation due to the lack of solvent. Consequently, the first network in a DN elastomer is initially elongated without collapse until the strain-hardening point is reached, after which it begins to break over the entire elongated region without necking. In addition, we evaluated the shape recovery of DAAN-EA-3/MA after elongating to either 100% or 200% strain, the latter of which exhibited fluorescence due to the polymer chain scission while the former did not. As a result, both specimens fully recovered to their original shapes after the tension was removed irrespective of the chain scission (Fig. S2†). These results suggest that most of the polymer-chain scission in the DN elastomers originated from the first network while little cleavage occurred for the second network that constitutes the majority of the DN elastomer to maintain the overall shape.
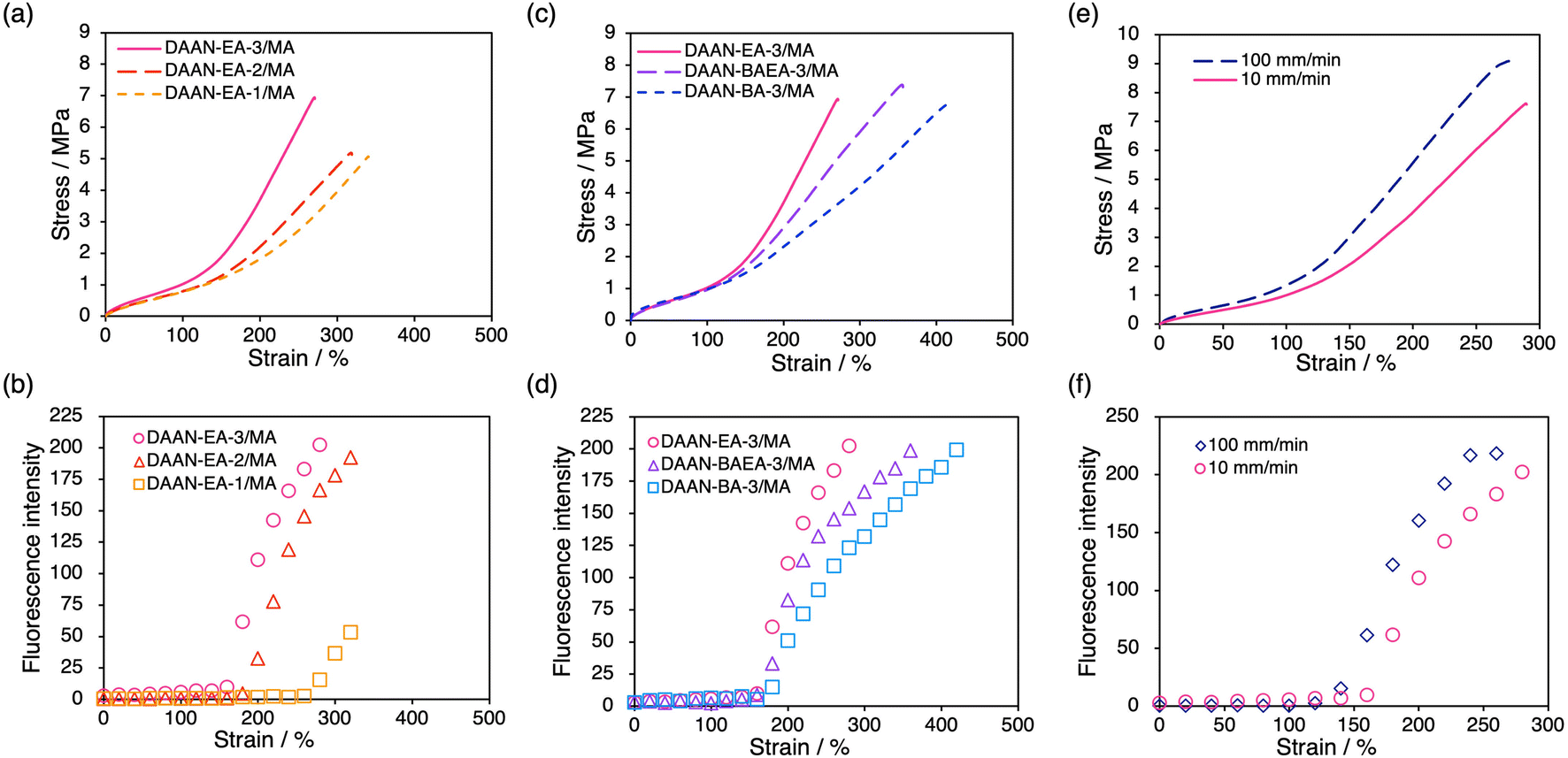 | ||
| Fig. 4 (a) Stress–strain curves of DAAN-EA-3/MA, DAAN-EA-2/MA, and DAAN-EA-1/MA (10 mm min−1). (b) Fluorescence intensity of DAAN-EA-3/MA, DAAN-EA-2/MA, and DAAN-EA-1/MA. (c) Stress–strain curves of DAAN-EA-3/MA, DAAN-BAEA-3/MA, and DAAN-BA-3/MA (10 mm min−1). (d) Fluorescence intensity of DAAN-EA-3/MA, DAAN-BAEA-3/MA, and DAAN-BA-3/MA. (e) Stress–strain curves of DAAN-EA-3/MA (100 mm min−1) and DAAN-BA-3/MA (10 mm min−1). (f) Fluorescence intensity of DAAN-EA-3/MA (100 mm min−1) and DAAN-BA-3/MA (10 mm min−1). | ||
Next, we evaluated the effect of the polymer species in the FNs by conducting the same tensile tests on DAAN-BAEA-3/MA and DAAN-BA-3/MA. As shown in Fig. 4c, their S–S curves were almost identical up to the strain-hardening region, despite the different polymer species in the FN. This is because the mechanical properties up to the strain-hardening region of the DN elastomers are derived from the second network, which accounts for >80% of their mass. Fluorescence was observed starting from the onset of strain-hardening, in the same manner as for DAAN-EA-3/MA (Fig. 3d), irrespective of the polymer species in the FN. Interestingly, the fracture stresses and fluorescence intensity at the fracture points are very similar for all the elastomers tested. Since approximately the same amount of DAAN is incorporated into each film, the fluorescence intensity should correlate with the amount of mechanoradicals generated. These similarities are likely due to their comparable cross-link densities, which can be expected to result in a similar number of mechanoradicals generated prior to fracture.
To investigate the effect of the strain rate on the fracture behavior of the sacrificial FN, DAAN-EA-3/MA was subjected to tensile tests at 100 mm min−1, and the results were compared to those obtained from 10 mm min−1. Strain-hardening occurred earlier in the 100 mm min−1 test than in the 10 mm min−1 test, and the strain at which the fluorescence emission started was accordingly smaller (Fig. 4e and f). The elastomer properties were further analyzed using Mooney–Rivlin (eqn (1)) plots (Fig. S4 and Table S2†).44–46
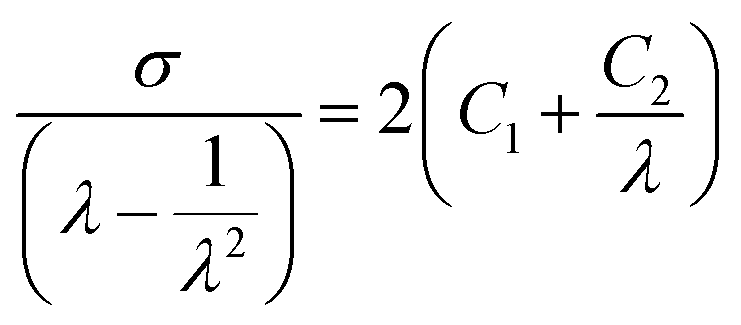 | (1) |
Fluorescence decay
We further examined the decay of the fluorescence intensity after stretching the DAAN-doped DN elastomers. The fluorescence intensity of the DAAN-EA-3/MA films at 556 nm after stretching was monitored over time. We observed that the fluorescence intensity diminished to half of its original value in ∼2 min, and that the fluorescence intensity became almost constant after 20 min (Fig. 5). This decay rate is significantly higher than that of DAAN-doped polystyrene.40 We attribute this difference to the higher mobility of the DAAN radicals in the elastomeric matrix with a low Tg, considering that DAAN radicals generally prefer to dimerize at room temperature.48 The fast decay hindered our efforts to detect the DAAN radicals using electron paramagnetic resonance. However, while the fluorescence intensity decay gradually converged, it did not completely reach zero. This can be interpreted in terms of a diminished probability of the coupling reaction to occur with decreasing concentration of DAAN radicals in the DN elastomers. Importantly, such time-dependent emission-quenching properties are advantageous for applications in the spatiotemporal characterization of the fracture of materials.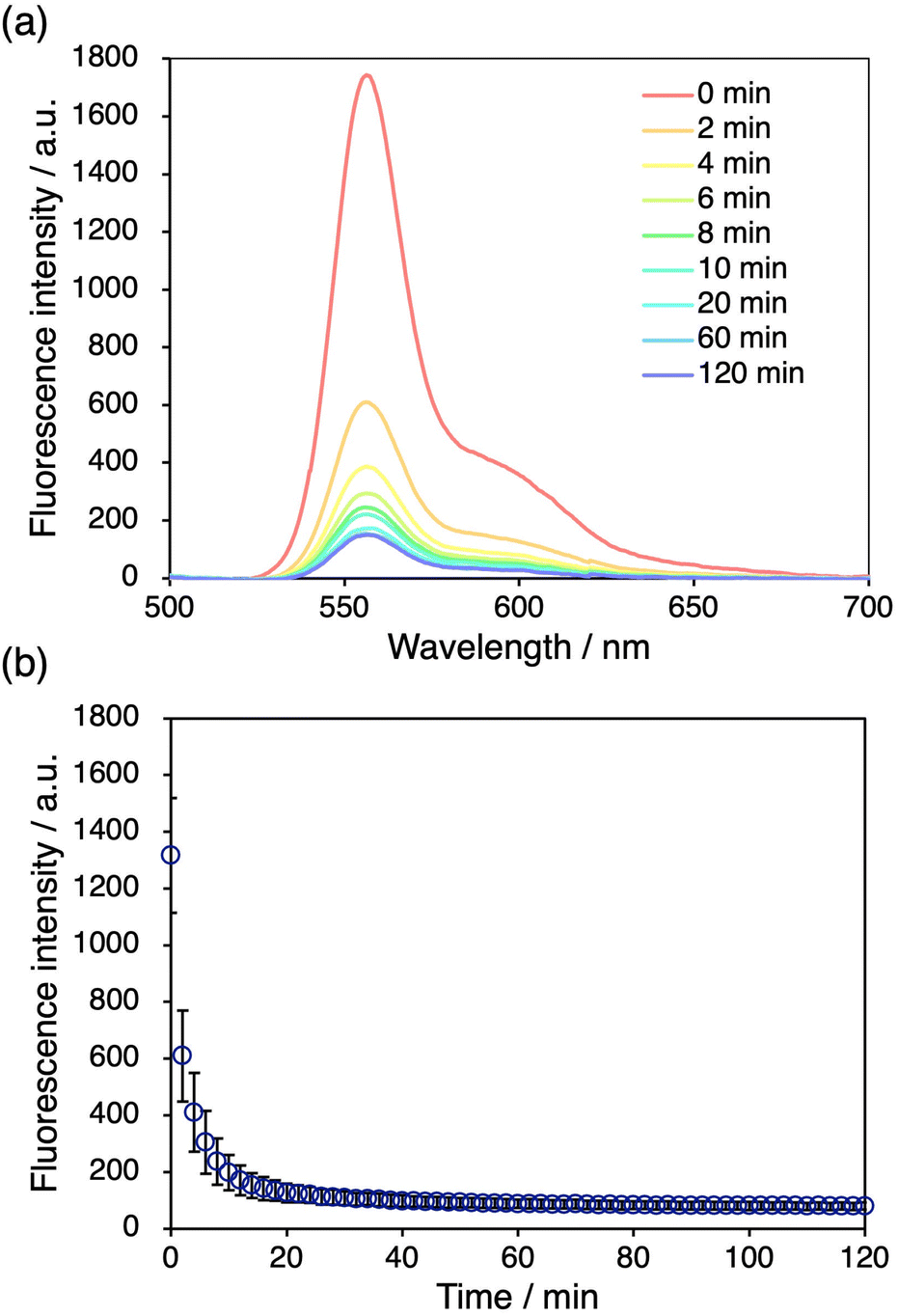 | ||
| Fig. 5 (a) Time-dependent fluorescence spectra of DAAN-EA/MA after tensile tests. (b) Time-dependent fluorescence intensity at 556 nm after tensile tests. | ||
Conclusions
In summary, we have demonstrated the visualization of sacrificial bond cleavage inside generic DN elastomers by post-synthetically adding a DAAN derivative and evaluated the fracture phenomena of the obtained elastomers. Tensile tests showed that sacrificial bond cleavage occurs after the onset of strain-hardening. Increasing the cross-link density of the first network causes the onset strain of mechanoradical generation to decrease and the amount of mechanoradicals generated in the elastomer to increase. Furthermore, very similar fluorescence intensity was observed for elastomers with different acrylate monomers in the first network, indicating that the amount of mechanoradicals generated at rupture is almost identical. Higher strain rates were found to make the network behave more elastically and induce molecular chain scission in the lower-strain region. The present method represents a simple and versatile technique for visualizing covalent bond cleavage in elastomers without altering their physical properties. We expect that these findings will accelerate the elucidation of the fracture mechanisms of elastomers.Author contributions
T. Y., A. T., and H. O. conceived the concept and designed the experiments. T. Y. performed the experiments and analyzed the data. T. Y., A. T., and H. O. wrote the manuscript.Conflicts of interest
There are no potential conflicts of interest to declare.Acknowledgements
This work was supported by KAKENHI grants 21H04689 (to H. O.) from the Japan Society for the Promotion of Science (JSPS), a JSPS Research fellowship for Young Scientists (22J21853 to T. Y.), the ANRI Fellowship (to T. Y.), and JST CREST via grant JPMJCR1991 (to H. O.).Notes and references
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC
.
- W. Zhang, S. Chen, W. Jiang, Q. Zhang, N. Liu, Z. Wang, Z. Li and D. Zhang, Eur. Polym. J., 2023, 185, 111807 CrossRef CAS
- J. Yang, Y. Li, L. Zhu, G. Qin and Q. Chen, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 1351–1362 CrossRef CAS
- J. Slootman, V. Waltz, C. J. Yeh, C. Baumann, R. Göstl, J. Comtet and C. Creton, Phys. Rev. X, 2020, 10, 041045 CAS
- J. Yang, K. Li, C. Tang, Z. Liu, J. Fan, G. Qin, W. Cui, L. Zhu and Q. Chen, Adv. Funct. Mater., 2022, 32, 2110244 CrossRef CAS
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed
- M. Sakaguchi and J. Sohma, J. Polym. Sci., Polym. Phys. Ed., 1975, 13, 1233–1245 CrossRef CAS
- M. Tabata and J. Sohma, Polym. Degrad. Stab., 1979, 1, 139–154 CrossRef CAS
- T. Hayashi, K. Kinashi, W. Sakai, N. Tsutsumi, A. Fujii, S. Inada and H. Yamamoto, Polymer, 2021, 217, 123416 CrossRef CAS
- M. Sono, K. Kinashi, W. Sakai and N. Tsutsumi, Macromolecules, 2018, 51, 1088–1099 CrossRef CAS
- S. Vivatpanachart, H. Nomura and Y. Miyahara, Polym. J., 1981, 13, 481–486 CrossRef CAS
- M. Sono, K. Kinashi, W. Sakai and N. Tsutsumi, Macromolecules, 2017, 50, 254–263 CrossRef CAS
- E. G. Janzen, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS
- E. G. Janzen and B. J. Blackburn, J. Am. Chem. Soc., 1968, 90, 5909–5910 CrossRef CAS
- N. J. Bunce, J. Chem. Educ., 1987, 64, 907 CrossRef CAS
- M. Tabata and J. Sohma, Eur. Polym. J., 1980, 16, 589–595 CrossRef CAS
- F. Wang, M. Burck and C. E. Diesendruck, ACS Macro Lett., 2017, 6, 42–45 CrossRef CAS PubMed
- S. L. Malhotra, J. Macromol. Sci., Part A: Pure Appl.Chem., 1986, 23, 729–748 CrossRef
- N. Willis-Fox, E. Rognin, C. Baumann, T. A. Aljohani, R. Göstl and R. Daly, Adv. Funct. Mater., 2020, 30, 2002372 CrossRef CAS
- Ö. Laçin, J. Kwiczak-Yiğitbaşı, M. Erkan, Ş. C. Cevher and B. Baytekin, Polym. Degrad. Stab., 2019, 168, 108945 CrossRef
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504–508 CrossRef CAS PubMed
- T. Matsuda, R. Kawakami, T. Nakajima and J. P. Gong, Macromolecules, 2020, 53, 8787–8795 CrossRef CAS
- Y. Zheng, J. Jiang, M. Jin, D. Miura, F. X. Lu, K. Kubota, T. Nakajima, S. Maeda, H. Ito and J. P. Gong, J. Am. Chem. Soc., 2023, 145, 7376–7389 CrossRef CAS PubMed
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed
- H. Zhang, D. Zeng, Y. Pan, Y. Chen, Y. Ruan, Y. Xu, R. Boulatov, C. Creton and W. Weng, Chem. Sci., 2019, 10, 8367–8373 RSC
- W. Qiu, P. A. Gurr and G. G. Qiao, Macromolecules, 2020, 53, 4090–4098 CrossRef CAS
- T. Watabe, D. Aoki and H. Otsuka, Macromolecules, 2022, 55, 5795–5802 CrossRef CAS
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, Nat. Rev. Mater., 2021, 6, 84–98 CrossRef CAS
- W. Qiu, J. M. P. Scofield, P. A. Gurr and G. G. Qiao, Macromol. Rapid Commun., 2022, 43, 2100866 CrossRef CAS PubMed
- S. Jung and H. J. Yoon, Synlett, 2022, 33, 863–874 CrossRef CAS
- M. Xuan, C. Schumacher, C. Bolm, R. Göstl and A. Herrmann, Adv. Sci., 2022, 9, 2105497 CrossRef CAS PubMed
- S. Shahi, H. Roghani-Mamaqani, R. Hoogenboom, S. Talebi and H. Mardani, Chem. Mater., 2022, 34, 468–498 CrossRef CAS
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed
- N. Willis-Fox, E. Rognin, T. A. Aljohani and R. Daly, Chem, 2018, 4, 2499–2537 CAS
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC
- M. Stratigaki and R. Göstl, ChemPlusChem, 2020, 85, 1095–1103 CrossRef CAS PubMed
- Z. Wang, X. Zheng, T. Ouchi, T. B. Kouznetsova, H. K. Beech, S. Av-ron, T. Matsuda, B. H. Bowser, S. Wang, J. A. Johnson, J. A. Kalow, B. D. Olsen, J. P. Gong, M. Rubinstein and S. L. Craig, Science, 2021, 374, 193–196 CrossRef CAS PubMed
- S. Wang, H. K. Beech, B. H. Bowser, T. B. Kouznetsova, B. D. Olsen, M. Rubinstein and S. L. Craig, J. Am. Chem. Soc., 2021, 143, 3714–3718 CrossRef CAS PubMed
- T. Yamamoto and H. Otsuka, Polym. Chem., 2023, 14, 2464–2468 RSC
- T. Yamamoto, D. Aoki, K. Mikami and H. Otsuka, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-ss0w5.
- T. Yamamoto, S. Kato, D. Aoki and H. Otsuka, Angew. Chem., Int. Ed., 2021, 60, 2680–2683 CrossRef CAS PubMed
- T. Yamamoto, D. Aoki and H. Otsuka, ACS Macro Lett., 2021, 10, 744–748 CrossRef CAS PubMed
- M. Mooney, J. Appl. Phys., 1940, 11, 582–592 CrossRef
-
R. S. Rivlin, Collected Papers of R. S. Rivlin, Springer-Verlag, New York, 1997, pp. 90–108 Search PubMed
- Y. Fukahori and W. Seki, Polymer, 1992, 33, 502–508 CrossRef CAS
- G. D. Spathis, J. Appl. Polym. Sci., 1991, 43, 613–620 CrossRef CAS
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mr00016h |
| This journal is © The Royal Society of Chemistry 2024 |