
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen-bonded cocrystals via resonant acoustic mixing†
Alireza
Nari
 ,
Jeffrey S.
Ovens
and
David L.
Bryce
*
,
Jeffrey S.
Ovens
and
David L.
Bryce
*
Department of Chemistry and Biomolecular Sciences, Centre for Catalysis Research and Innovation, and Nexus for Quantum Technologies, University of Ottawa, Ottawa, Ontario, Canada K1N6N5. E-mail: dbryce@uottawa.ca; Fax: +1-613-562-5170; Tel: +1-613-562-5800
First published on 31st January 2024
Abstract
Resonant acoustic mixing is a relatively gentle mechanochemical technology that employs pressure waves to induce chemical and morphological transformations. We report here on the production of eleven halogen-bonded (XB) cocrystalline architectures via neat and liquid-assisted resonant acoustic mixing (RAM). Two strong iodinated XB donors, namely 1,4-diiodotetrafluorobenzene (p-DITFB, 1) and 1,3,5-trifluoro-2,4,6-triiodobenzene (sym-TFTIB, 2) each react with five XB donors, namely 2,3,5,6-tetramethylpyrazine (TMP, a), 4-dimethylaminopyridine (DMAP, b), 1,10-phenanthroline (o-Ph, c), 1,10-phenanthroline-5,6-dione (PheDON, d), and 4,5-diazafluoren-9-one (DIZFON, e) to form ten cocrystals. For these systems, it is shown that RAM is capable of producing the same products as are obtained via ball milling. Two novel cocrystals are obtained (of 2d featuring bifurcated XBs, and 2e featuring monofurcated XBs) and their single-crystal X-ray structures are reported. However, an eleventh stoichiomorphic cocrystal of p-DITFB and TMP is obtained exclusively via RAM, suggesting that the combination of RAM and milling approaches may afford a broader exploration of the polymorphic and stoichiomorphic landscape than the use of a single technique in isolation. All products are characterized via powder X-ray diffraction, and 13C cross-polarization magic angle spinning (CP/MAS) and 19F MAS NMR spectroscopy, providing further evidence for the phase purity of samples obtained from RAM experiments and for the degree of polymorphic control available when small volumes of liquid are employed in mechanochemical reactions. This work demonstrates the potential of RAM for the production of known and novel halogen-bonded cocrystalline assemblies, including polymorphic and stoichiomorphic structures.
Introduction
Halogen bonding (XB), a distinctive noncovalent interaction, has surged in interest due to its directional and tunable characteristics,1,2 enabling the deliberate design of frameworks3,4 and control of biomolecular structure.5,6 This interaction involves the electrophilic region of a covalently bonded halogen (termed the σ-hole)7 on the halogen bond donor engaging with a Lewis base as the halogen bond acceptor.8 The presence of this interaction is often assessed using the normalized contact distance ratio, RXB,9 which gauges the halogen bond distance (dX⋯Y) relative to the sum of the van der Waals radii of the atoms involved (∑dvdw):10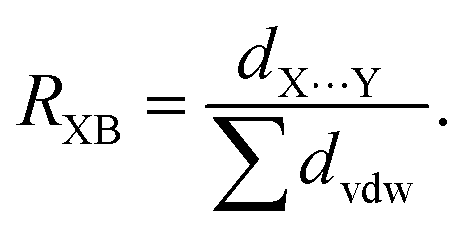
Over the past two decades, halogen bonding has emerged as a versatile tool, finding applications across diverse domains such as crystal engineering, medicinal chemistry,11,12 anion recognition,13,14 supramolecular systems,15 and catalysis.16,17 Its adaptable nature and the potential for precise modulation make halogen bonding an intriguing avenue for innovative molecular design and tailored interactions.
Mechanochemistry, a dynamic and evolving field, has emerged as an innovative approach to chemical synthesis and materials discovery. This methodology harnesses mechanical force, such as grinding or milling, as a key driver in chemical reactions, replacing or complementing traditional solution-based processes.18,19 The concept of mechanochemistry dates back to the 19th century when it was first noted that certain reactions occurred more readily under mechanical action.20–22 However, it wasn't until recent decades that the full potential of mechanochemistry began to be realized. Today, this field offers a versatile platform for green and sustainable chemistry,23,24 with applications ranging from pharmaceuticals25–27 to catalysis,28–33 and materials science.34,35
In addition to the absence of solvents offering advantages, solid-state reactions frequently yield highly pure products and can be conducted using uncomplicated equipment, such as basic manual grinding36 or mechanical milling.37–39 Occasionally, a small quantity of liquid is introduced into mechanochemical reactions to expedite the transformation or facilitate the reaction, which, in certain instances, may induce polymorphic variations.40 These reactions can be categorized as “minimal solvent” rather than purely “solvent-free”,18 and they are often referred to as kneading, solvent drop grinding, or, in a more comprehensive and contemporary context, liquid-assisted grinding (LAG).41,42
Mechanochemical reactions are often characterized by their unique ability to access reaction pathways and product formations that are often elusive through traditional methods.43 The fundamental principles behind mechanochemistry involve the application of mechanical energy to overcome activation barriers and promote bond cleavage, facilitating the synthesis of complex molecules and materials. As a result, mechanochemistry has garnered increasing interest within the scientific community for its potential to revolutionize the way we approach chemical synthesis and discover new materials. Recent advances in the field have demonstrated its efficacy in preparing cocrystals,44–49 polymorphs,50 and metal–organic frameworks,51,52 highlighting its versatility and potential in various applications.
Here, we provide a proof of principle study of direct mechanochemical halogen bond-induced cocrystallization by resonant acoustic mixing (RAM), focusing on the reduction of reaction time and the engineering of cocrystals featuring I⋯N and I⋯O interactions (Fig. 1). We show that acoustic mechanochemistry with RAM, leveraging acoustic wave pressure as the driving force, can efficiently provide the energy required for promoting halogen bond-induced cocrystallization. Cocrystal synthesis via RAM has been reported previously,53–58 but to our knowledge the production of halogen-bonded systems has not been systematically explored.
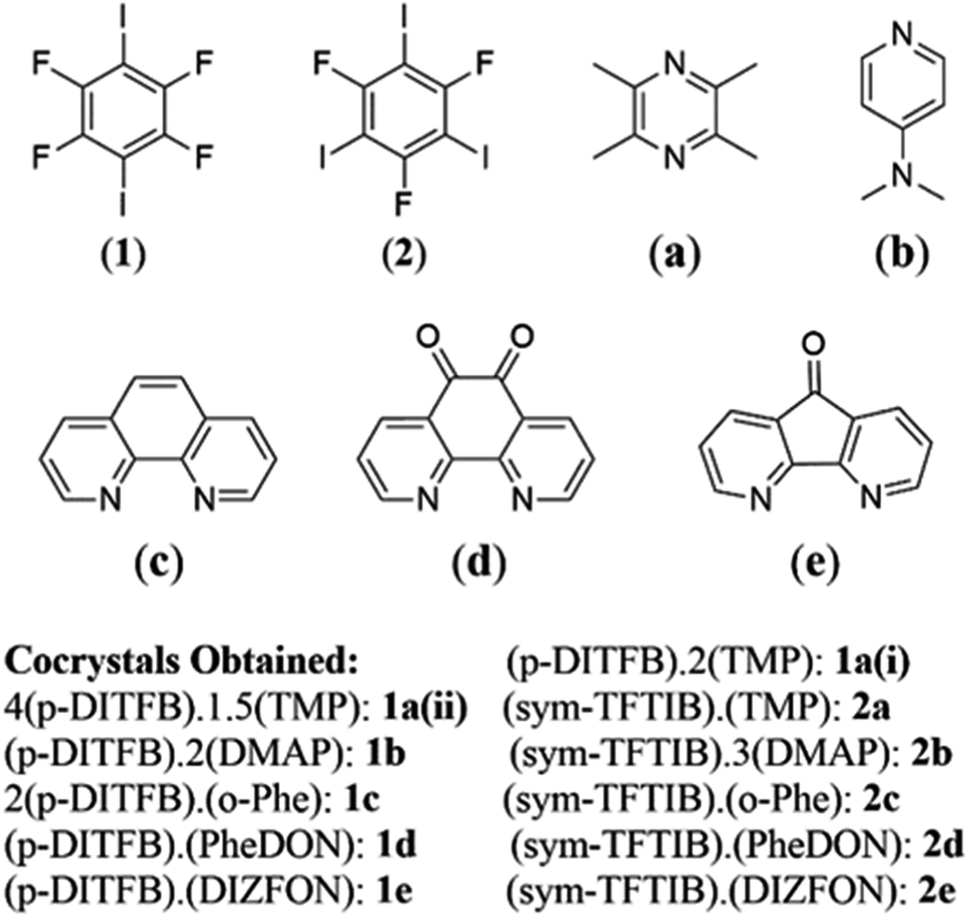 | ||
| Fig. 1 Molecular structure of halogen bond donors and acceptors studied herein with the cocrystals obtained. | ||
Furthermore, our ongoing work focuses on fine-tuning the coordination of XB interactions using RAM methodology. Notably, in a recent study by Michalczyk et al.,59 it was reported that only 2% of XB cocrystals exhibit bifurcated interactions, underscoring the rarity of such configurations obtained via traditional methods. The efficiency of RAM in forging XB interactions may hold the potential to unlock bifurcated and multi-coordinated XB interactions, thus broadening the possibilities for tailoring cocrystals in ways that were previously inaccessible through traditional mechanochemical approaches.
Experimental section
(i) General
1,4-Diiodotetrafluorobenzene (p-DITFB, 1), 2,3,5,6-tetramethylpyrazine (TMP, a), and 4-dimethylaminopyridine (DMAP, b), were purchased from Sigma-Aldrich. 1,10-Phenanthroline (o-Ph, c) and 1,10-phenanthroline-5,6-dione (PheDON, d) were purchased from Fisher Scientific. 4,5-Diazafluoren-9-one (DIZFON, e) was purchased from TCI. 1,3,5-Trifluoro-2,4,6-triiodobenzene (sym-TFTIB, 2) was purchased from SynQuest Laboratories. These compounds were utilized in their as-received state without further purification. Reagent grade solvents and doubly distilled water were used.(ii) Mechanochemistry and sample preparation
Mechanochemical transformations were performed using ball milling (Retsch MM 400) and resonant acoustic mixing (Resodyn PharmaRAM 1) devices.| Compound | Molar ratio (donor : acceptor) | Masses (donor : acceptor) (mg) | Ball milling | RAM | Refcode | ||||
|---|---|---|---|---|---|---|---|---|---|
| Time (min) | Liquid | η (μL mg−1) | Time (min) | Liquid | η (μL mg−1) | ||||
| a Not achievable. | |||||||||
| 1a(i) | (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
(241:82) |
45 | CHCl3 | 0.10 | 50 | CHCl3 | 0.20 | JAQMAQ60 |
| 1a(ii) | (2:1) |
(402:68) |
NA | NA | NA | 45 | None | — | KIHRAX61 |
| 1b | (1:2) |
(321:196) |
45 | None | — | 45 | None | — | RUYHID62 |
| 1c | (1:1) |
(241:108) |
45 | CHCl3 | 0.05 | 50 | CHCl3 | 0.10 | JAQMIY60 |
| 1d | (1:1) |
(205:105) |
30 | CHCl3 | 0.10 | 50 | THF | 0.5 | EXIFEX63 |
| 1e | (1:1) |
(205:91) |
30 | CHCl3 | 0.10 | 50 | THF | 0.5 | EXIFAT63 |
| 2a | (1:1) |
(357:95) |
30 | THF | 0.05 | 50 | THF | 0.1 | SAJCUE9 |
| 2b | (1:3) |
(255:183) |
45 | None | — | 50 | None | — | RUYJAX64 |
| 2c | (1:1) |
(255:90) |
45 | CHCl3 | 0.05 | 50 | THF | 0.10 | SAJDEP9 |
| 2d | (1:1) |
(255:105) |
45 | CHCl3 | 0.05 | 50 | CHCl3 | 0.7 | This work |
| 2e | (1:1) |
(255:94) |
45 | CHCl3 | 0.10 | 50 | CHCl3 | 0.7 | This work |
(iii) Powder X-ray diffraction
Powder X-ray diffraction (PXRD) data were collected using a Bruker D8 Endeavor instrument that features a 1 kW Cu Kα radiation source from a sealed tube and a LynxEye XE-T high-speed detector operating at a wavelength of λ = 1.54056 Å. These measurements were conducted under standard room temperature conditions. The samples were securely positioned within a well plate and underwent a rotational motion at 15 revolutions per minute throughout the data collection process.(iv) Single-crystal X-ray diffraction
Single cocrystals of compounds 2d and 2e were affixed onto a thin glass fiber using parabar oil. SCXRD analysis was conducted employing a Bruker AXS SMART single crystal diffractometer equipped with a sealed Mo tube source emitting radiation at a wavelength of 0.71073 Å. This analysis was carried out at a temperature of 213 K.The diffraction dataset revealed systematic absences, and the unit cell parameters were in accordance with the monoclinic P21/c crystal system for both compounds 2d and 2e. See ESI† for further details.
(v) Solid-state NMR spectroscopy
1H → 13C cross-polarization magic-angle spinning (CPMAS) SSNMR experiments were conducted using a Bruker Avance III spectrometer operating at 9.4 T. A Bruker 4 mm HXY MAS probe was employed, and the spinning frequency was typically set to 11 kHz (see ESI† for further details). The CPMAS technique involved a 6 to 8 ms contact time, a proton π/2 pulse duration of 3.60 μs, and 1H decoupling at a nutation frequency of 54.3 kHz. To determine chemical shifts, referencing was done relative to glycine at 176.4 ppm (13C = O) with respect to TMS.19F MAS SSNMR experiments were conducted at 11.7 T, utilizing a Bruker Avance III spectrometer. For these experiments, a Bruker 2.5 mm HX MAS probe was employed, with a spinning frequency set at 25 kHz. The data acquisition employed a Bloch decay sequence with a π/2 pulse duration of 1.75 μs and a recycle delay of 60–90 seconds. Chemical shifts are reported with respect to polytetrafluoroethylene at −122.2 ppm.
Results and discussion
(i) Mechanochemistry and PXRD
The preparation of XB cocrystals can typically be achieved through both solution crystal growth and solid-state mechanochemistry. Nevertheless, mechanochemistry offers a rapid and efficient means of cocrystal synthesis while minimizing solvent usage and by-product formation. However, for obtaining molecular structural information and geometry, the use of single crystals is indispensable, which can be achieved through solvent-based crystallization. Herein, the employed cocrystallization techniques encompass neat solid-state and liquid-assisted ball milling and RAM. Additionally, solution growth methods were employed to produce single cocrystals of two novel compounds, i.e., sym-TFTIB with PheDON and DIZFON (cocrystals 2d and 2e).We describe here the mechanochemical preparation, via ball milling and RAM, of cocrystals 1a, 2a, 1b, 2b, 1c, 2c, 1d, 2d, 1e, and 2e (Fig. 1). Except for two novel cocrystals (2d and 2e), the rest of these compounds had previously been cocrystallized through solution growth and characterized via SCXRD. This work therefore focusses on minimizing solvent usage and on exploring the potential of environmentally friendly and low energy consumption RAM mechanochemical methods for producing halogen-bonded compounds.
In all cases, the PXRD patterns depicted in Fig. 2 and in the ESI† reveal a notable similarity between the products obtained through ball milling and RAM methods when the same stoichiometry of reactants and identical synthesis conditions (e.g., reaction time and liquid additive) were applied. Pure and stable forms of these cocrystals were obtained using RAM, typically within less than an hour (in most cases, around 40 min), under both neat and LAG conditions.
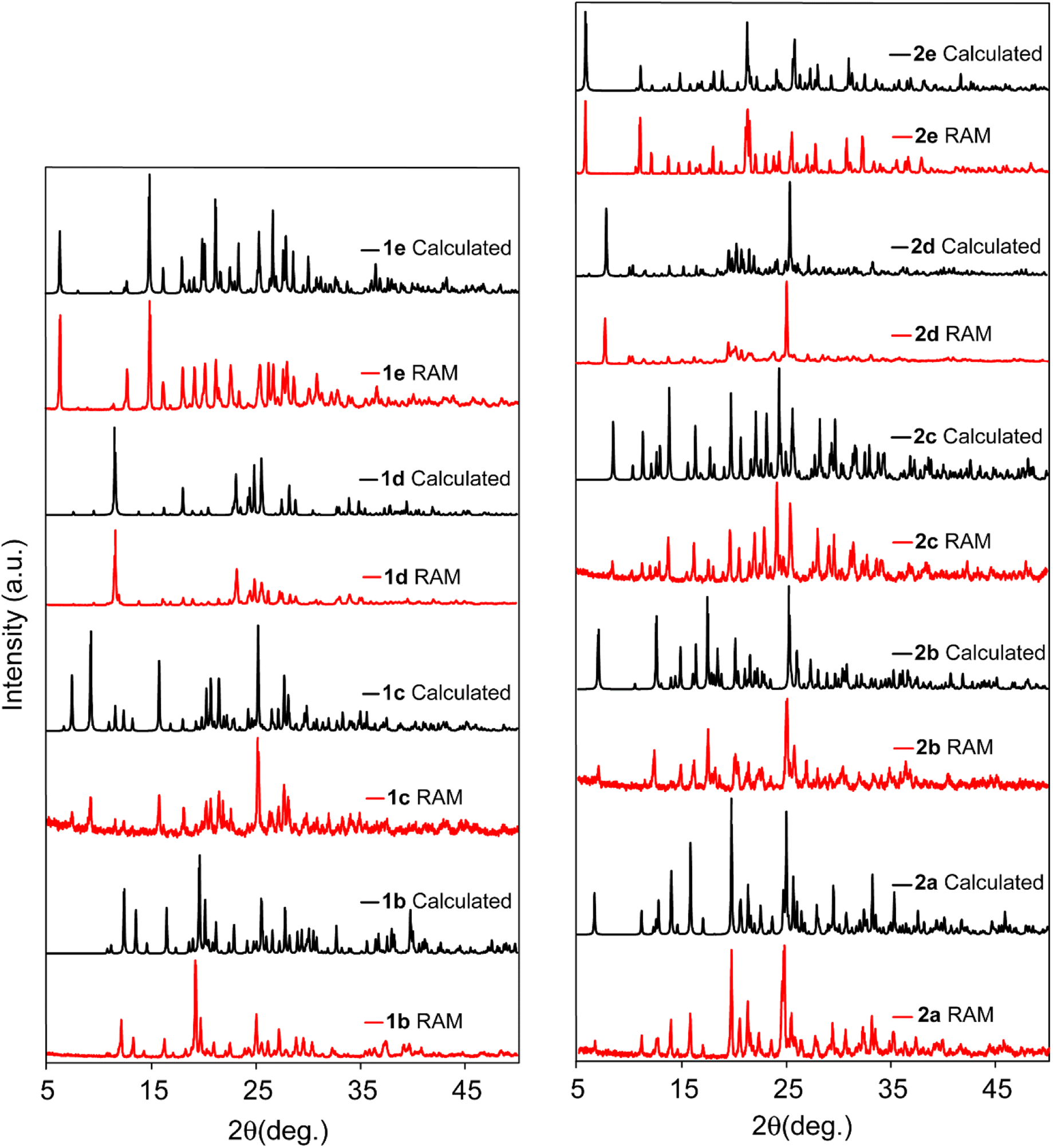 | ||
| Fig. 2 Comparison between experimental PXRD patterns of cocrystals prepared via RAM (red) and the simulated X-ray powder patterns (black). | ||
In the investigation of the application of RAM in the mechanochemical synthesis of halogen bonded cocrystals, eleven cocrystals were prepared, as outlined in Table 1. Specifically, cocrystal 1b was prepared via RAM by introducing a 1:2 molar ratio of the donor to the acceptor into a 1-dram glass vial, and subjecting it to pressure waves of 80 g for a duration of 45 min. The resulting PXRD pattern, as illustrated in Fig. 2, confirms the successful formation of the RUYHID cocrystal (Table 1) using the pristine solvent-free resonance acoustic mixing approach. To validate the reproducibility and versatility of RAM in producing halogen-bonded cocrystals and to make a comparative assessment with conventional mechanochemistry methods, we employed ball milling under identical synthesis conditions for the preparation of cocrystal 1b. Remarkably, the PXRD data indicate a striking similarity between the cocrystals obtained through these two distinct methods, confirming the formation of a pure and stable phase of cocrystal 1b.
Subsequently, cocrystal 2b was synthesized using RAM, employing a 1:3 molar ratio of donor 2 to acceptor b, with an 80 g acceleration for a duration of 50 min. Additionally, a parallel synthesis was carried out using ball milling with a frequency of 30 kHz and a duration of 45 min. The PXRD patterns, depicted in Fig. 2 (and ESI†), provide compelling evidence for the formation of a pure and stable RUYJAX phase in both cases.
It should be noted that our optimization process for RAM synthesis commenced with neat powders to avoid the use of solvents. Additionally, we explored various preparation times, including 15, 30, and 45 min. The results presented herein encompass the optimized synthesis conditions for each cocrystal via both RAM and ball milling techniques. It was observed that the exclusion of liquid additives yielded pure cocrystals only in the case of 1a(ii), 1b, and 2b. This phenomenon can be attributed to the superior properties of acceptors TMP(a) and DMP(b), characterized by their enhanced nucleophilicity and reduced steric hindrance, facilitating efficient halogen bond formation. This distinction is expected to result in shorter bond lengths and stronger XB interactions within these compounds. This is evident in the geometric parameters obtained from SCXRD data where the RXB values of 0.85, 0.76 and 0.78 were determined for the I⋯N interactions in 1a(ii), 1b, and 2b cocrystals, respectively.
To address this issue, we adopted a strategy involving the introduction of liquid additives33 within the range of 0.05 to 0.7 μL per milligram of reactants (η = 0.05 to 0.7) in the reaction jar. In this context, the RAM of 1 and c reactants in a 1:2 molar ratio, supplemented with chloroform at an η = 0.1 μL mg−1 ratio, yielded the pure and stable cocrystal 1c (refcode: JAQMIY). Similarly, the syntheses of cocrystals 1d and 1e, as confirmed by their PXRD patterns displayed in Fig. 2, were achieved by introducing equimolar quantities of starting materials, along with an η = 0.5 μL mg−1 of THF. Applying an 80 g acceleration for 50 min via RAM yielded the EXIFEX and EXIFAT phases for 1e and 1d, respectively. Comparison with the simulated PXRD pattern alongside those of the RAM and ball mill products unequivocally establishes the production of a pure phase of cocrystal 1e through both mechanochemical methods.
To further investigate the influence of the type and quantity of liquid additives on RAM-synthesized XB cocrystals, a screening experiment was conducted on cocrystal 2c. Samples were prepared under neat conditions, with 0.1 μL mg−1 CHCl3, and with 0.1 μL mg−1 THF in the RAM process. Fig. 3 showcases the PXRD results, revealing incomplete conversion in the neat and CHCl3 experiments while highlighting the formation of a pure SAJDEP phase when employing THF as the liquid additive in RAM cocrystallization for cocrystal 2c. This comprehensive exploration of mechanochemical synthesis conditions and the influence of liquid additives underscores the versatility and effectiveness of RAM in the preparation of diverse halogen-bonded cocrystals, offering valuable insights into the optimization of this synthesis approach. Friščić and co-workers have discussed the optimization of the η value in liquid-assisted RAM applied to catalysis.33
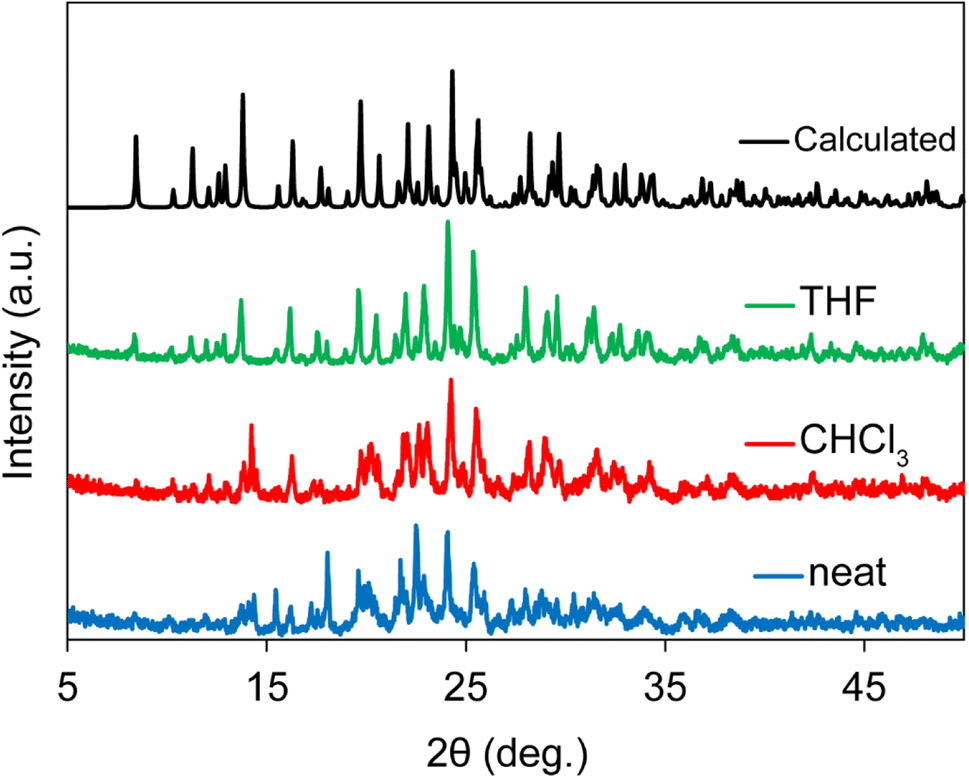 | ||
| Fig. 3 Powder X-ray diffractograms for samples of 2c produced using different RAM conditions: neat RAM (blue), 0.1 μL mg−1 CHCl3 RAM (red), and 0.1 μL mg−1 THF RAM (green). The calculated PXRD pattern based on the SCXRD structure is shown in black. | ||
In the preparation of cocrystals 2d and 2e, equimolar quantities of the starting materials were introduced into the reaction vessel, accompanied by the addition of 0.7 μL mg−1 of CHCl3. The resulting PXRD patterns, as illustrated in Fig. 2, unequivocally demonstrate the successful formation of these novel cocrystals after a reaction duration of 50 min at 80 g acceleration. This is in excellent agreement with the simulated pattern derived using Mercury 4.2.0 software65 based on newly acquired SCXRD data (vide infra). It is noteworthy that RAM proved to be an efficient and viable mechanochemical approach for the synthesis of these two novel XB cocrystals, eliminating the necessity for milling media and extensive mechanical grinding or abrasion processes.
During the RAM screening for cocrystal formation, with the compound 1a, we observed intriguing results. Neat RAM for 45 min with a 1:2 ratio (donor:acceptor) led to the formation of the stable phase of the 1a(ii) stoichiomorph (refcode: KIHRAX)61 which was not achievable with other mechanochemical methods like ball milling. Addition of 85 μL of chloroform (η = 0.2 μL mg−1) and mixing time of 50 min, however, transform the reactants (1:2 ratio (donor:acceptor)) instead to the 1a(i) stoichiomorph (refcode = JAQMAQ),60 the same product as obtained when ball milling for 45 min with same liquid additive. These results are presented in Fig. 4, showcasing PXRD patterns acquired for both neat and liquid-added RAM products. This finding affirms that varying RAM conditions can yield distinct stoichiomorphs of compound 1a and highlights the remarkable potential of RAM as a tool for controlling and influencing stoichiomorphism and polymorphism. Notably, our attempts to prepare the 1a(ii) stoichiomorph using ball milling were unsuccessful. It is important to mention that previous applications of mechanochemical ball milling, involving neat grinding and LAG, have resulted in the formation of different polymorphs, as reported elsewhere.66 To the best of our knowledge, the present work represents the first instance where the formation of different stable cocrystalline phases can be controlled using RAM; related work on controlling polymorphism with RAM has recently been reported.33
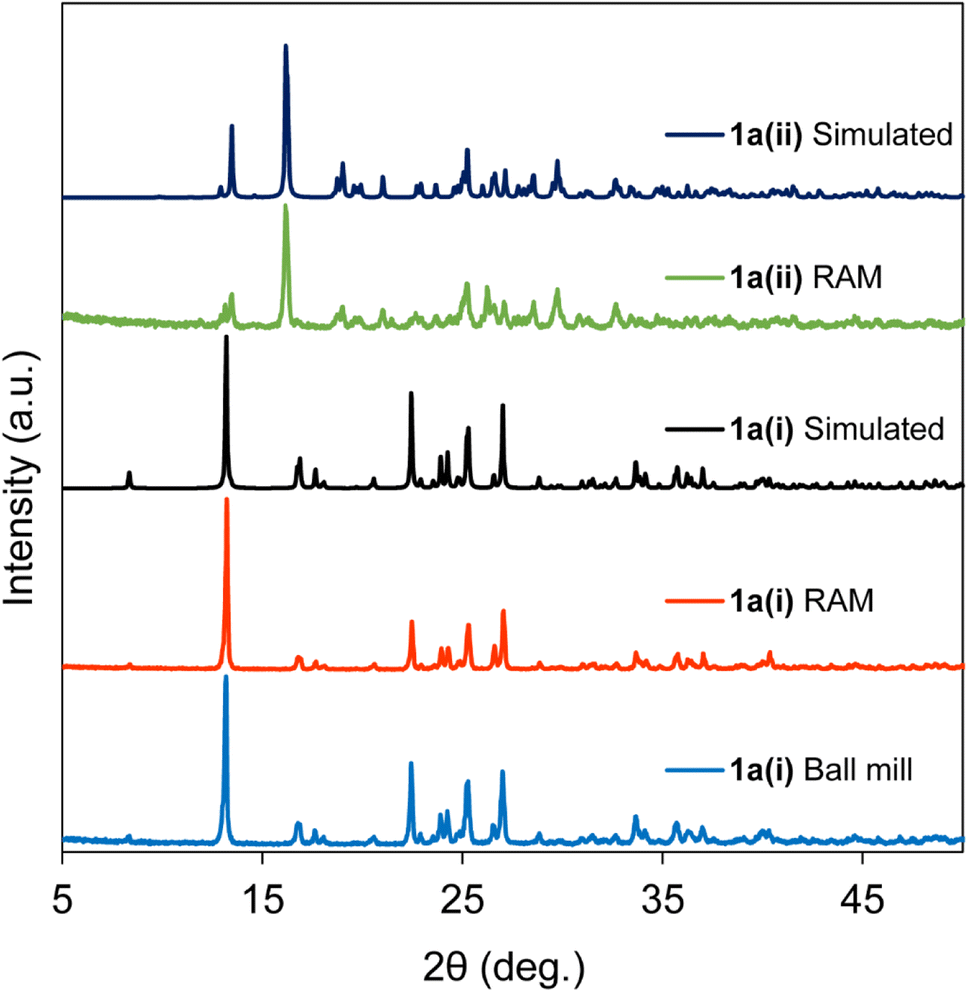 | ||
| Fig. 4 X-ray diffractograms of 1a(i) and 1a(ii) cocrystalline powders prepared under ball milling and RAM conditions. See Table 1 and the main text for details. Simulated diffractograms are generated from the known single-crystal X-ray structures with refcodes JAQMAQ (1a(i)) and KIHRAX (1a(ii)). | ||
While the exact mechanism underlying this phenomenon remains incompletely understood, two distinct factors appear to influence the formation of different products via RAM. Firstly, in the liquid-assisted approach, the presence of a liquid component can enhance the reaction rate by introducing degrees of orientational and conformational freedom to the molecules at various interfaces.67 Additionally, adding liquid may lead to the formation of a mobile surface layer at the molecule interface, which can enhance the reactivity of the reactants68 and affect the resulting polymorphs. The second factor could be particularly apposite for RAM, which operates as a milder mechanochemical methodology where the reaction occurs through the direct impact of the reactant particles.
(ii) Single crystal X-ray diffraction
The crystal structures of 1a(i) (CCDC 259702),601a(ii) (CCDC 1854044),612a (CCDC 1505893),91b (CCDC 773127),642b (CCDC 773130),641c (CCDC 259705),602c (CCDC 1505895),91d (CCDC 824297),63 and 1e (CCDC 824296)63 have been previously documented. In this study, we determined the single crystal X-ray diffraction (SCXRD) structures for compounds 2d and 2e to enhance our comprehension of halogen bonding interactions within these supramolecular assemblies.SCXRD analysis revealed a 1:1 stoichiometry of sym-TFTIB to PheDON in 2d. This compound crystallizes in a monoclinic crystal system, belonging to the P21/c space group. The volume of the unit cell is determined to be 15288(7) Å3, and the crystal contains 32 molecules in the unit cell. The unit cell parameters and the other relevant information are presented in Table 2. As can be seen in Fig. 5(C), in 2d, both nitrogen atoms of the bidentate PheDON interact with one of the sym-TFTIB iodine atoms, resulting in the observation of a slightly asymmetrical bifurcated halogen bond (XB) with I–N bond lengths of 2.997 and 3.297 Å (for example; there are 7 other pairs with similar distances as shown in Table 3). A straightforward layered packing arrangement is formed, where I–C and π—π interactions lead to a parallel stacking arrangement between sym-TFTIB and PheDON molecules as shown in Fig. 5(A), with intermolecular distances of 3.557 Å and 3.393 Å, respectively. Furthermore, the crystal structure reveals the presence of C–H⋯I and F⋯F interactions, which play a crucial role in extending the one-dimensional (1D) layer structures throughout the crystal network.
| Compound | 2d | 2e |
|---|---|---|
| Chemical formula | C18H6F3I3N2O2 | C17H6F3I3N2O |
| CCDC number | 2305189 | 2305188 |
| FW (g mol−1) | 719.95 | 691.94 |
| Crystal colour | Orange | Yellow |
| Crystal system | Monoclinic | Monoclinic |
| Crystal space group | P21/c | P21/c |
| Temperature (K) | 100 K | 200 K |
| a, b, c (Å) | 29.938(8), 18.399(5), 28.189(7) | 8.3178(7), 30.472(2), 7.4150(5) |
| α, β, γ (deg) | 90, 100.074(3), 90 | 90, 94.297(4), 90 |
| V (A3) | 15288(7) |
1874.1(2) |
| Z | 32 | 4 |
| R 1 (final) | 0.1051 | 0.0504 |
| wR2 (final) | 0.2484 | 0.0999 |
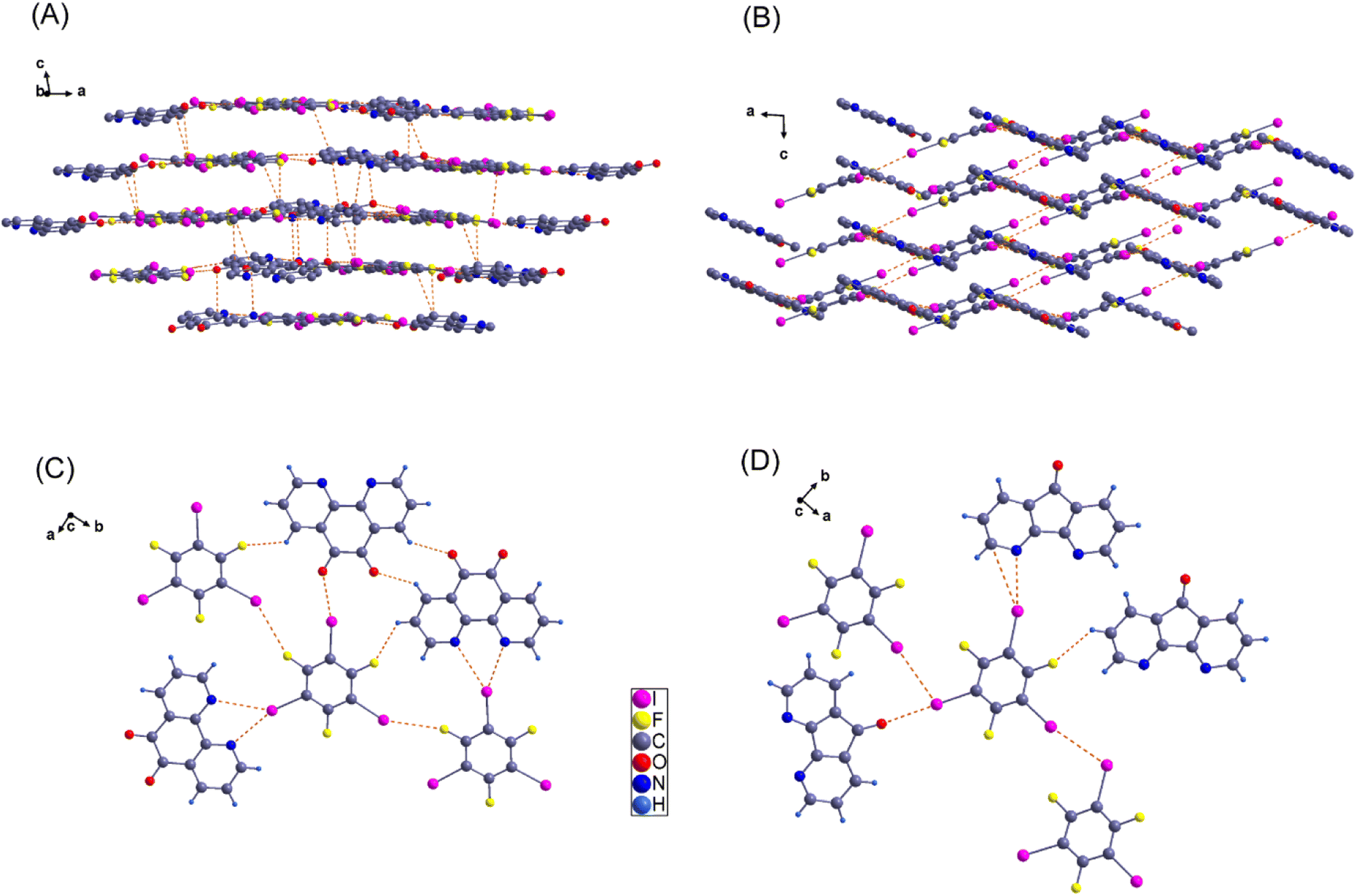 | ||
| Fig. 5 Two novel crystal structures of compound 2d (left, parts (A) and (C)) and 2e (right, parts (B) and (D)) showing halogen bonds (dashed lines) in compounds 2d and 2e. In cocrystal 2d, the iodine interacts with two nitrogen atoms making a bifurcated halogen bond and one oxygen with a monocoordinated interaction. In compound 2e, the iodine of the donor interacts with two crystallographically distinct acceptor moieties. | ||
| Cocrystal | d I⋯N (Å) | θ C–N⋯I | R XB (I) | d I⋯O (Å) | θ C–N⋯O | R XB (O) |
|---|---|---|---|---|---|---|
| a For this compound, we avoid detailing the entire list of I⋯O XB interactions and instead provide one example in this table. | ||||||
| 1a(i) (p-DITFB)·(TMP) | 3.067 | 177.15 | 0.869 | — | — | — |
| 1a(ii) (p-DITFB)·(TMP) | 3.001 | 177.51 | 0.850 | — | — | — |
| 2a (sym-TFTIB)·(TMP) | 2.991 | 178.90 | 0.847 | — | — | — |
| 2.993 | 179.81 | 0.849 | ||||
| 1b (p-DITFB)·(DMAP) | 2.677 | 179.25 | 0.756 | — | — | — |
| 2b (sym-TFTIB)·(DMAP) | 2.766 | 176.15 | 0.784 | — | — | — |
| 2.886 | 168.58 | 0.818 | ||||
| 1c (p-DITFB)·(o-Phe) | 3.01 | 159.03 | 0.853 | — | — | — |
| 3.274 | 149.76 | 0.927 | ||||
| 2c (sym-TFTIB)·(o-Phe) | 3.02 | 175.68 | 0.856 | — | — | — |
| 3.148 | 164.18 | 0.892 | ||||
| 1d (p-DITFB)·(PheDON) | 3.149 | 154.93 | 0.892 | 3.214 | 155.14 | 0.918 |
| 2d (sym-TFTIB)·(PheDON) | 3.297, 3.011, 3.215, 3.047, 3.225, 3.097, 2.997, 3.326, 3.316, 3.026, 3.406, 3.029, 3.039, 3.259, 3.094, 3.230 | 144.8, 165.1, 166.7, 143.3, 165.6, 143.9, 159.0, 149.5, 160.8, 149.0, 162.4, 146.5, 158.7, 149.9, 144.6, 164.1 | 0.934, 0.853, 0.911, 0.863, 0.914, 0.877, 0.849, 0.942, 0.939, 0.857, 0.965, 0.858, 0.861, 0.923, 0.876, 0.91 | 2.884a | 177.52 | 0.824 |
| 1e (p-DITFB)·(DIZFON) | 2.924 | 165.10 | 0.828 | 3.036 | 163.02 | 0.867 |
| 2e (p-DITFB)·(PheDON) | 2.968 | 158.66 | 0.841 | 2.931 | 177.52 | 0.824 |
sym-TFTIB and DIZFON were cocrystallized to form the 1:1 cocrystal 2e in the monoclinic space group P21/c. The supramolecular structure of 2e is characterized by a monocoordinated XB of one sym-TFTIB with two acceptor moieties featuring one I–N and one I–O XB interaction (Fig. 5(D)). As is depicted in Fig. 5(B), the 2e cocrystal structure propagates in zigzag chains where each 1D chain is packed on the next via I–I intermolecular interactions between donor moieties, generating a 2D layered structure.
It is not surprising that the structure of 2e lacks a bifurcated XB, consistent with conclusions reached in the study by Ji et al. on similar acceptor moieties.63 This finding can be attributed to the relatively greater distance between the two electron-rich atoms (N) in DIZFON molecules, exceeding the threshold for the formation of a three-centered XB involving I and two N atoms. The distance between I and N in the XB interaction is measured at 2.968 Å, which is shorter than the corresponding bifurcated I–N XB in the 2d cocrystal. This observation aligns with the concept that a monocoordinated XB is expected to be stronger than a bifurcated XB.63
(iii) Solid-state NMR spectroscopy
To further characterize the halogen-bonded cocrystals produced via RAM, we conducted 13C CP/MAS SSNMR spectroscopy on all of the samples. The use of 1H→13C cross-polarization has been demonstrated as a reliable and effective indirect method for characterizing the formation of XB cocrystals.9 This technique allows us to gain insights into changes in the crystallographic and electronic environment surrounding the carbon atom covalently bonded to the nitrogen atom (C–N) in the acceptor moieties. The principle underlying this technique lies in the polarization transfer from protons to the carbon atom covalently bonded to the nitrogen atom. This 13C nucleus is indirectly engaged in each XB (i.e., one bond removed), and is subjected to small changes in electronic and crystallographic environment upon cocrystallization. Consequently, this involvement results in changes in the chemical shifts, exemplified in Fig. 6 for the two stoichiomorphs of 1a (1a(i) and 1a(ii)). The distinct 13C chemical shifts observed for the two samples reflect differences in the local nuclear environment.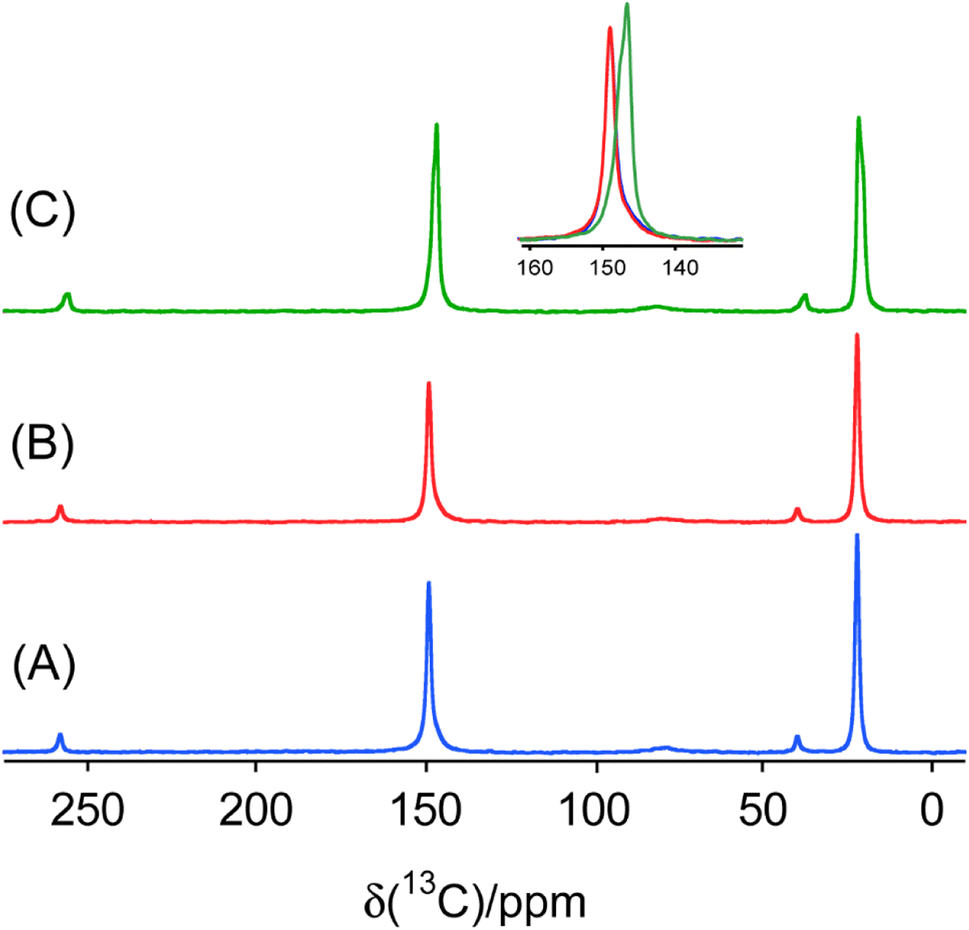 | ||
| Fig. 6 1H → 13C CP/MAS solid-state NMR spectra of sample 1a(i) obtained by ball milling (blue, A), RAM (red, B), and the second stoichiomorph 1a(ii) obtained by RAM (green, C). Inset: overlay of peak for the C covalently bonded to N in these three products to illustrate the identical 13C chemical shifts for the ball milled and RAM products for 1a(i), but different chemical shift for the second stoichiomorph 1a(ii). | ||
Fig. 7 and 8 depict the 13C CP/MAS NMR spectra of XB acceptor moieties alongside all cocrystals formed with the two XB donors, 1 and 2. Notable changes in chemical shifts are evident upon the formation of XB cocrystals, with their corresponding δiso values listed in Table 4. Strikingly, these NMR data also provide clear evidence that products generated via RAM and via ball milling are isomorphic; this is consistent with the PXRD data presented above and in the ESI.† In some cases, ball milling gives broader NMR lines which could be indicative of partial amorphization of the product (see ESI†). This effect could possibly be reduced by using shorter milling times, but these experiments were not pursued presently.
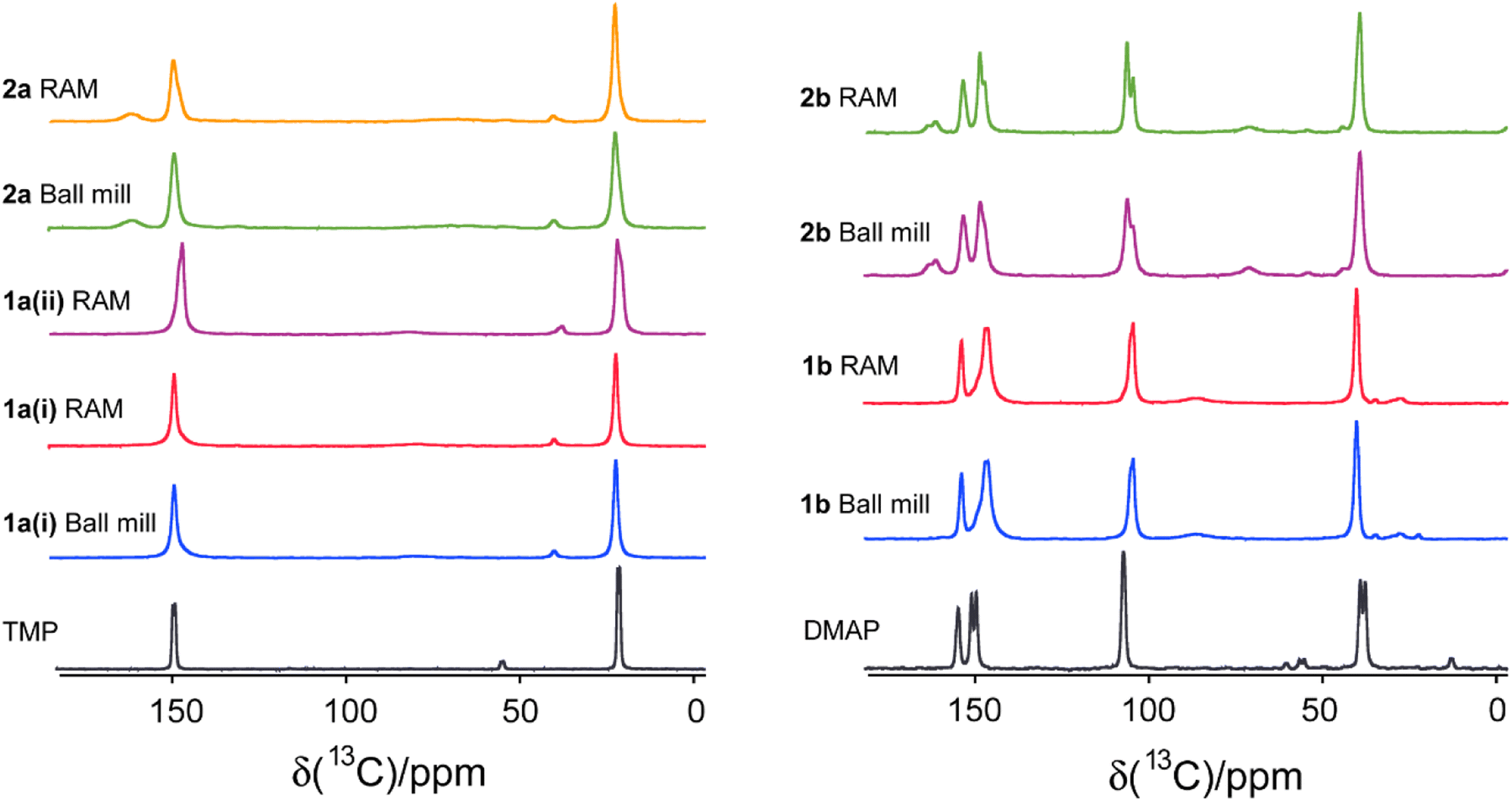 | ||
| Fig. 7 Comparison of 13C CP/MAS NMR spectra for monotopic XB acceptors (TMP and DMAP) and their respective cocrystals produced via ball milling and RAM. The corresponding chemical shifts are listed in Table 4. | ||
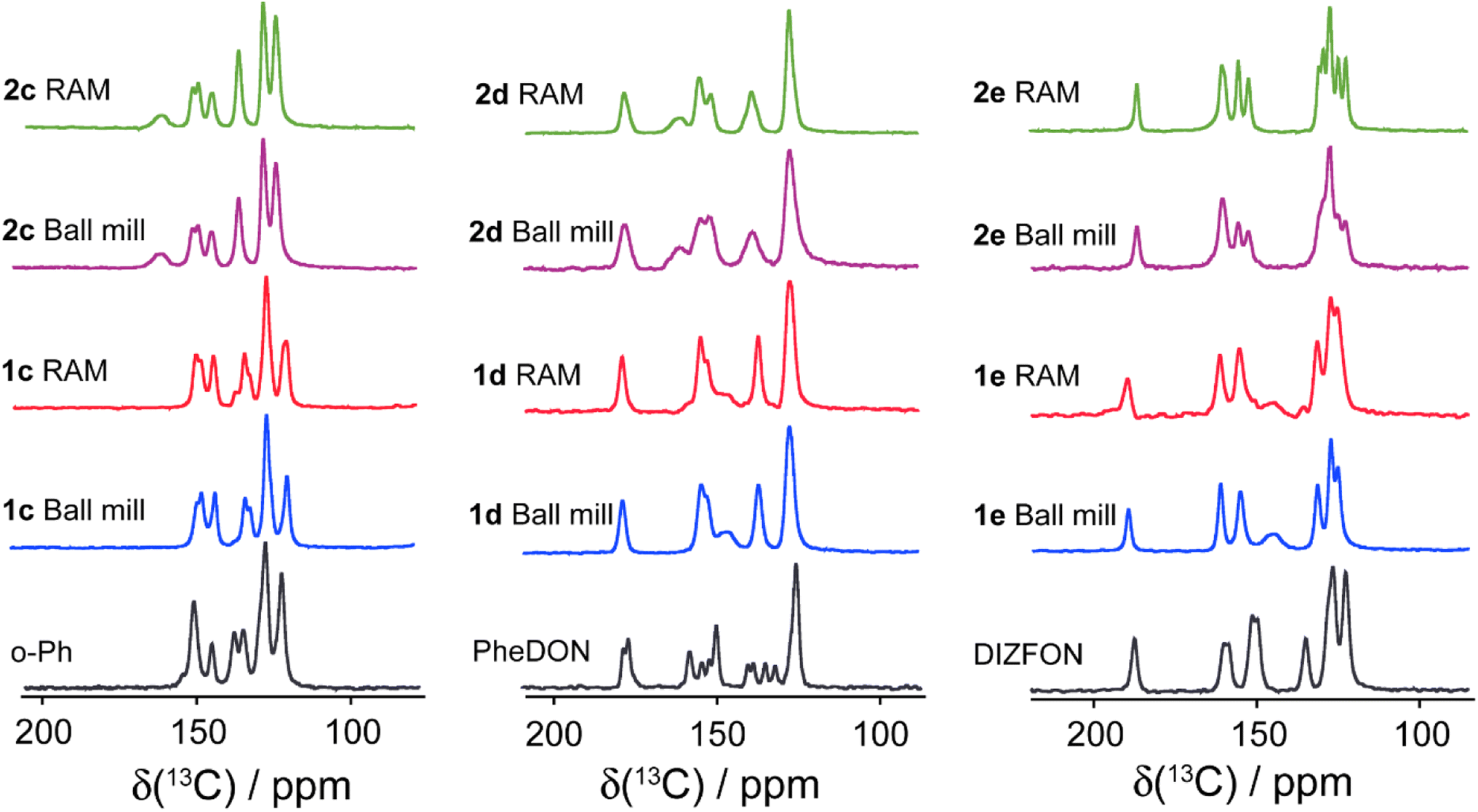 | ||
| Fig. 8 Comparison of 13C CP/MAS NMR spectra of ditopic XB acceptors (o-Ph, PheDON, and DIZFON) and their cocrystals produced via ball milling and RAM. The chemical shifts are detailed in Tables 4 and 5. | ||
| Cocrystal | XB acceptor | δ iso/ppm XB acceptor | δ iso/ppm cocrystal |
|---|---|---|---|
| a Chemical shifts of donor molecules are also evident; these are marked on spectra shown in the ESI. b Errors in chemical shift measurements are attributed to the lack of certainty regarding the quantitative impact of residual 13C–14N dipolar coupling on peak positions. | |||
| 1a(i) | TMP | 142.7 & 149.1 ± 0.1 | 149.3 ± 0.3b |
| 1a(ii) | TMP | 142.7 & 149.1 ± 0.1 | 147.1 ± 0.5 |
| 2a | TMP | 142.7 & 149.1 ± 0.1 | 149.5 ± 0.6 |
| 1b | DMAP | 150.9 & 149.5 ± 0.1 | 146.6 ± 0.6 |
| 2b | DMAP | 150.9 & 149.5 ± 0.1 | 147.8 ± 0.7 |
| 1c | o-Ph | 150.8 & 145.1 ± 0.3 | 149.8 & 148.4 |
| & 144.0 ± 0.3 | |||
| 2c | o-Ph | 150.8 & 145.1 ± 0.3 | 151.2 & 149.5 |
| & 145.1 ± 0.4 | |||
| 1d | PheDON | 158.4 & 154.7 | 154.8 & 153.2 |
| 152.5 & 150.3 ± 0.4 | & 147.1 ± 0.6 | ||
| 2d | PheDON | 158.4 & 154.7 | 161.4 & 155.5 |
| 152.5 & 150.3 ± 0.4 | & 152.0 ± 0.5 | ||
| 1e | DIZFON | 160.7 & 152.0 ± 0.4 | 162.5 & 156.4 ± 0.4 |
| 2e | DIZFON | 160.7 & 152.0 ± 0.4 | 162.1 & 157.1 |
| & 154.0 ± 0.4 | |||
In a statistical and computational analysis conducted by Allen et al. on NO2–X synthons involving both monocoordinate and bifurcated XB, it was observed that as the interaction distance decreased, the XB angles tended to become more directional and linear.69 This observation suggests that bifurcated halogen bonding interactions, where one halogen atom interacts with two Lewis bases, will correlate with smaller C–X⋯O angles. Conversely, smaller angles correlate with weaker interactions when compared to more linear monocoordinated XB interactions. In the cocrystals involving acceptor molecules c and d with donors 1 and 2, which engage in bifurcated halogen-bonded interactions, the contact lengths between I and N atoms are relatively long, corresponding to weaker XB interactions. Consequently, the C–N covalent bond in the acceptor molecule will be weakened less than it is in the presence of a stronger monocoordinated XB, resulting in a lower 13C chemical shift for the C atom covalently bonded to the N atom within the acceptor moiety. More generally, the data in Table 4 show that the 13C chemical shifts of the acceptor molecules can increase or decrease upon cocrystallization; increases are generally noted for TMP and DIZFON, while decreases are noted for DMAP and more subtle changes are noted for o-Ph and PheDON.
In another study conducted by our group,70 it was shown that an increase in the C–I bond length of halogen bond donor 1 as a result of XB formation corresponded to an increase in the 13C chemical shift. The δiso values for 13C covalently bonded to iodine and fluorine in the XB donor moieties are listed in Table 5. Consistent with previous work, increases in the C–I chemical shifts relative to that for pure donor 1 (76.50(0.50) ppm)70 and for pure donor 2 (67.6 ± 2.4 ppm)9 are generally observed. Interestingly, exceptions are seen in both cases for cocrystals formed with acceptor d, which forms bifurcated halogen bonds; such exceptions could therefore possibly be NMR signatures of bifurcated XB, but more data are needed to confirm the generality of this finding.
| Cocrystal | XB donor | δ iso/ppm C–I | δ iso/ppm C–F |
|---|---|---|---|
| a In 2e, the resonance frequency of the C atom bonded to F is obscured by the intense and distinct signals from the acceptor moieties' carbon. Therefore, we are unable to report the chemical shift of the C–F carbon. | |||
| 1a(i) | p-DITFB | 78.7 ± 0.8 | — |
| 1a(ii) | p-DITFB | 82.3 ± 0.8 | — |
| 2a | sym-TFTIB | 68.6 ± 2.2 | 161.9 ± 0.9 |
| 1b | p-DITFB | 86.7 ± 1.4 | — |
| 2b | sym-TFTIB | 71.2 ± 0.9 | 163.4 ± 0.6 |
| 1c | p-DITFB | 77.4 ± 1.4 | — |
| 2c | sym-TFTIB | 67.1 ± 1.2 | 161.5 ± 0.8 |
| 1d | p-DITFB | 73.8 ± 1.2 | — |
| 2d | sym-TFTIB | 63.2 ± 1.4 | 161.6 ± 0.6 |
| 1e | p-DITFB | 76.8 ± 1.2 | — |
| 2e | sym-TFTIB | 68.6 ± 1.4 | —a |
Fluorine-19 SSNMR spectroscopy is a powerful technique that has found extensive utility in characterizing halogen-bonded compounds due to the high receptivity of fluorine nuclei, high natural abundance, and the wide chemical shift range.9,71Fig. 9 illustrates the 19F MAS SSNMR spectra acquired for all cocrystals and the perfluorinated XB donor starting materials. Importantly, changes in the 19F chemical shift upon the formation of halogen-bonded cocrystals serve as valuable independent indicators of cocrystallization events. These spectra also provide an additional independent perspective that the products obtained via RAM and via ball milling are identical.
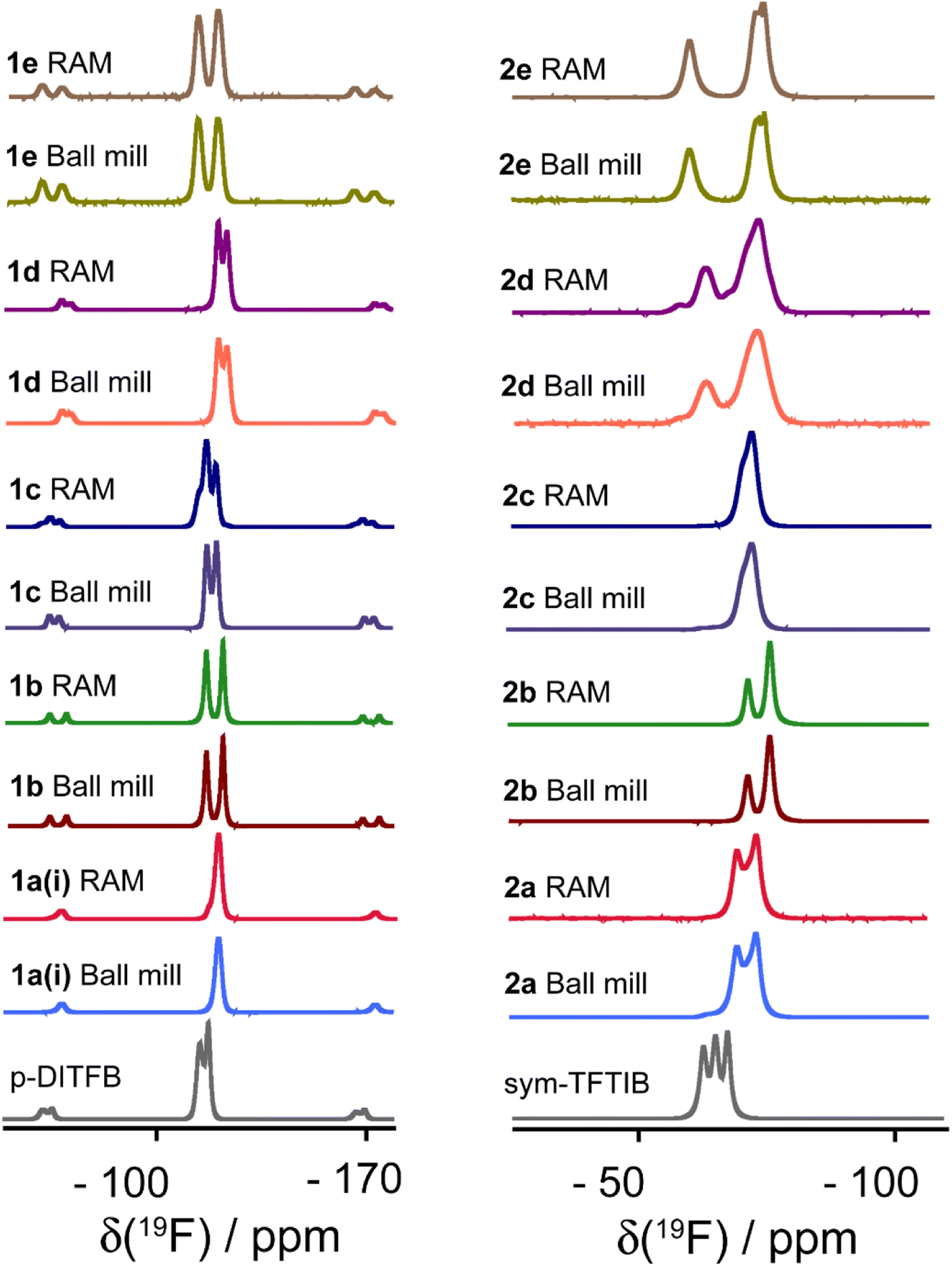 | ||
| Fig. 9 Comparison of 19F MAS SSNMR spectra of XB donors (p-DITFB and sym-TFTIB) and their cocrystals produced via ball milling and RAM. The chemical shifts are detailed in Table 6. | ||
The 19F chemical shifts are listed in Table 6, and the results demonstrate a consistent and notable decrease in the 19F chemical shift for all samples upon the formation of halogen-bonded cocrystals. Since the crystallographic and electronic environment encountered by 19F nuclei are highly sensitive to XB and certain short interactions involving 19F nuclei with I or H atoms, as revealed by XRD, the extent of chemical shift changes exhibits variability across the compounds. Shifts on the order of up to −8 ppm are observed as a result of cocrystallization. These findings highlight the sensitivity of 19F SSNMR as a probe to detect structural changes associated with XB cocrystallization. It is noted that while the 19F NMR resonances for cocrystal 2d, which features a large number of crystallographically non-equivalent molecules, are somewhat broader than for the other cocrystals, there is no clear spectral resolution of these multiple sites.
| Cocrystal | δ iso/ppm cocrystal | XB donor | δ iso/ppm XB donor |
|---|---|---|---|
| 1a(i) | −118.9 ± 0.5 | p-DITFB | −112.6 & −115.5 ± 0.6 |
| 1b | −114.8 & −120.4 ± 0.3 | ||
| 1c | −115.0 & −118.3 ± 0.6 | ||
| 1d | −118.8 & −121.7 ± 0.6 | ||
| 1e | −121.2 & −118.8 ± 0.7 | ||
| 2a | −68.3 & −72.1 ± 0.7 | sym-TFTIB | −61.8 & −64.1 & −66.5 ± 0.3 |
| 2b | −70.4 & −74.8 ± 0.3 | ||
| 2c | −71.2 ± 0.8 | ||
| 2d | −62.2 & −72.6 ± 0.7 | ||
| 2e | −59.1 & −72.0 & −73.4 ± 0.4 |
Conclusions
We have demonstrated that resonant acoustic mixing can consistently promote halogen-bond-induced cocrystallization of donor and acceptor molecules. Eleven halogen-bonded cocrystals have been prepared via RAM, including two novel structures which were characterized by single-crystal X-ray diffraction and one novel stoichiomorphic product which is inaccessible via ball milling. One of the notable advantages of RAM-based cocrystallization is its capacity to transform reactants into halogen-bonded interaction products through direct interactions, both under neat mixing conditions and when aided by the addition of small amounts of polar or moderately polar solvents. For most cocrystals studied herein, the inclusion of even a minimal quantity of these solvents serves to significantly enhance the cocrystallization process and increase the completeness of the transformation, further highlighting the potential of RAM in optimizing cocrystal synthesis. Powder X-ray diffraction and 13C CP/MAS and 19F MAS NMR spectroscopy have provided compelling evidence for the phase purity of samples obtained from RAM experiments and for the degree of polymorphic control available when small volumes of liquid are employed in mechanochemical reactions. Overall, this work demonstrates the potential of RAM for the production of known and novel cocrystalline assemblies, including polymorphic and stoichiomorphic structures.Data availability
Data are available upon reasonable request from the authors.Author contributions
A. N. prepared samples, carried out PXRD and SSNMR experiments, analysed all data, and wrote the first draft of the manuscript, and edited the manuscript. J. S. O. solved the single-crystal X-ray structures. D. L. B. directed research, secured funding, analysed data, and wrote and edited the manuscript.Conflicts of interest
The authors declare they have no conflicts of interest.Acknowledgements
D. L. B. is grateful to the Natural Sciences and Engineering Research Council of Canada for funding. The authors thank the solid-state NMR group at the University of Ottawa for helpful comments, and Dr Patrick Szell and Dr Tristan Georges for technical assistance.References
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Prot. Sci., 2013, 22, 139–152 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- P. M. J. Szell, S. A. Gabriel, R. D. D. Gill, S. Y. H. Wan, B. Gabidullin and D. L. Bryce, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 157–167 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- P. S. Ho, Future Med. Chem., 2017, 9, 637–640 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- A. Ravi, A. S. Oshchepkov, K. E. German, G. A. Kirakosyan, A. V. Safonov, V. N. Khrustalev and E. A. Kataev, Chem. Commun., 2018, 54, 4826–4829 RSC.
- L. Catalano, S. Perez-Estrada, H.-H. Wang, A. J.-L. Ayitou, S. I. Khan, G. Terraneo, P. Metrangolo, S. Brown and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139, 843–848 CrossRef CAS PubMed.
- J.-P. Gliese, S. H. Jungbauer and S. M. Huber, Chem. Commun., 2017, 53, 12052–12055 RSC.
- L. Carreras, M. Serrano-Torné, P. W. N. M. van Leeuwen and A. Vidal-Ferran, Chem. Sci., 2018, 9, 3644–3648 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- K. Horie, M. Barón, R. B. Fox, J. He, M. Hess, J. Kahovec, T. Kitayama, P. Kubisa, E. Maréchal, W. Mormann, R. F. T. Stepto, D. Tabak, J. Vohlídal, E. S. Wilks and W. J. Work, Pure Appl. Chem., 2004, 76, 889–906 CrossRef CAS.
- L. Takacs, JOM, 2000, 52, 12–13 CrossRef CAS.
- L. Takacs, J. Therm. Anal. Calorim., 2007, 90, 81–84 CrossRef CAS.
- L. Takacs, J. Mater. Sci., 2004, 39, 4987–4993 CrossRef CAS.
- Ball Milling towards Green Synthesis: Applications, Projects, Challenges, ed. A. Stolle and B. Ranu, Royal Society of Chemistry, Cambridge, UK, RSC Green Chemistry Series, No. 31, 2015, p. 303 Search PubMed.
- I. Huskić, C. B. Lennox and T. Friščić, Green Chem., 2020, 22, 5881–5901 RSC.
- M. Solares-Briones, G. Coyote-Dotor, J. C. Páez-Franco, M. R. Zermeño-Ortega, C. M. de la O Contreras, D. Canseco-González, A. Avila-Sorrosa, D. Morales-Morales and J. M. Germán-Acacio, Pharmaceutics, 2021, 13, 790 CrossRef CAS PubMed.
- D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustain. Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS.
- P. Cerreia Vioglio, M. R. Chierotti and R. Gobetto, Adv. Drug Delivery Rev., 2017, 117, 86–110 CrossRef CAS PubMed.
- D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002–4011 CrossRef CAS PubMed.
- T. Stolar, A. Prašnikar, V. Martinez, B. Karadeniz, A. Bjelić, G. Mali, T. Friščić, B. Likozar and K. Užarević, ACS Appl. Mater. Interfaces, 2021, 13, 3070–3077 CrossRef CAS PubMed.
- L. Gonnet, C. B. Lennox, J.-L. Do, I. Malvestiti, S. G. Koenig, K. Nagapudi and T. Friščić, Angew. Chem., Int. Ed., 2022, 61, e202115030 CrossRef CAS PubMed.
- F. Effaty, L. Gonnet, S. G. Koenig, K. Nagapudi, X. Ottenwaelder and T. Friščić, Chem. Commun., 2023, 59, 1010–1013 RSC.
- C. B. Lennox, T. H. Borchers, L. Gonnet, C. J. Barrett, S. G. Koenig, K. Nagapudi and T. Friščić, Chem. Sci., 2023, 14, 7475–7481 RSC.
- L. Gonnet, T. H. Borchers, C. B. Lennox, J. Vainauskas, Y. Teoh, H. M. Titi, C. J. Barrett, S. G. Koenig, K. Nagapudi and T. Friščić, Faraday Discuss., 2023, 241, 128–149 RSC.
- A. Pichon, A. Lazuen-Garay and S. L. James, CrystEngComm, 2006, 8, 211–214 RSC.
- K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS PubMed.
- F. Toda, K. Tanaka and A. Sekikawa, J. Chem. Soc., Chem. Commun., 1987, 279–280 RSC.
- G. Kaupp, in Organic Solid State Reactions, ed. F. Toda, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 95–183 Search PubMed.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed.
- G. Kaupp, in Encyclopedia of Physical Organic Chemistry, John Wiley & Sons, Inc., 2017, pp. 1–80 Search PubMed.
- A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter, Chem. Commun., 2005, 880–882 RSC.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890–891 RSC.
- A. O. Patil, D. Y. Curtin and I. C. Paul, J. Am. Chem. Soc., 1984, 106, 348–353 CrossRef CAS.
- M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS.
- M. C. Etter and D. A. Adsmond, J. Chem. Soc., Chem. Commun., 1990, 589–591 RSC.
- V. R. Pedireddi, W. Jones, A. P. Chorlton and R. Docherty, Chem. Commun., 1996, 987–988 RSC.
- K. Lisac, S. Cepić, M. Herak and D. Cinčić, Chem.: Methods, 2022, 2, e202100088 CAS.
- X. Ding, A. W. Crawford, W. P. Derrick, D. K. Unruh, R. H. Groeneman and K. M. Hutchins, Chem.–Eur. J., 2021, 27, 16329–16333 CrossRef CAS PubMed.
- C. Schumacher, K.-N. Truong, J. S. Ward, R. Puttreddy, A. Rajala, E. Lassila, C. Bolm and K. Rissanen, Org. Chem. Front., 2024 10.1039/D3QO01512B.
- D. Braga, G. Palladino, M. Polito, K. Rubini, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem.–Eur. J., 2008, 14, 10149–10159 CrossRef CAS PubMed.
- A. Pichon and S. L. James, CrystEngComm, 2008, 10, 1839–1847 RSC.
- H. M. Titi, J.-L. Do, A. J. Howarth, K. Nagapudi and T. Friščić, Chem. Sci., 2020, 11, 7578–7584 RSC.
- D. J. am Ende, S. R. Anderson and J. S. Salan, Org. Process Res. Dev., 2014, 18, 331–341 CrossRef CAS.
- K. Nagapudi, E. Y. Umanzor and C. Masui, Int. J. Pharm., 2017, 521, 337–345 CrossRef CAS PubMed.
- R. Tanaka, S. Osotprasit, J. Peerapattana, K. Ashizawa, Y. Hattori and M. Otsuka, Pharmaceutics, 2021, 13, 56 CrossRef CAS PubMed.
- M. Bui, P. Chakravarty and K. Nagapudi, Faraday Discuss., 2022, 241, 357–366 RSC.
- S. R. Anderson, P. Dubé, M. Krawiec, J. S. Salan, D. J. am Ende and P. Samuels, Propellants, Explos., Pyrotech., 2016, 41, 783–788 CrossRef CAS.
- S. R. Anderson, D. J. am Ende, J. S. Salan and P. Samuels, Propellants, Explos., Pyrotech., 2014, 39, 637–640 CrossRef CAS.
- M. Michalczyk, W. Zierkiewicz and S. Scheiner, Cryst. Growth Des., 2022, 22, 6521–6530 CrossRef CAS PubMed.
- J. L. Syssa-Magale, K. Boubekeur, P. Palvadeau, A. Meerschaut and B. Schöllhorn, CrystEngComm, 2005, 7, 302–308 RSC.
- P. M. J. Szell, S. A. Gabriel, E. Caron-Poulin, O. Jeannin, M. Fourmigué and D. L. Bryce, Cryst. Growth Des., 2018, 18, 6227–6238 CrossRef CAS.
- R. Wang, D. Hartnick and U. Englert, Z. Kristallogr., 2018, 233, 733–744 CAS.
- B. Ji, W. Wang, D. Deng and Y. Zhang, Cryst. Growth Des., 2011, 11, 3622–3628 CrossRef CAS.
- L. C. Roper, C. Präsang, V. N. Kozhevnikov, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, Cryst. Growth Des., 2010, 10, 3710–3720 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2005, 5, 1013–1021 CrossRef CAS.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- F. H. Allen, J. P. M. Lommerse, V. J. Hoy, J. A. K. Howard and G. R. Desiraju, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 1006–1016 CrossRef.
- J. Viger-Gravel, S. Leclerc, I. Korobkov and D. L. Bryce, CrystEngComm, 2013, 15, 3168–3177 RSC.
- C. M. Widdifield, G. Cavallo, G. A. Facey, T. Pilati, J. Lin, P. Metrangolo, G. Resnati and D. L. Bryce, Chem.–Eur. J., 2013, 19, 11949–11962 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further synthetic details; additional NMR spectra; powder X-ray diffractograms. Crystallographic information files for compounds 2d and 2e have been deposited with the CCDC with cif numbers 2305189 and 2305188. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3mr00028a |
| This journal is © The Royal Society of Chemistry 2024 |