
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis of bismuth active pharmaceutical ingredients, bismuth(III) gallate and bismuth citrate†
Daniel
Szczerba
 *a,
Jean-Louis
Do
bc,
Davin
Tan
d,
Hatem M.
Titi
d,
Nicolas
Geoffroy
a,
María del Carmen Marco de
Lucas
a,
Julien
Boudon
a,
Ivan
Halasz
e,
Tomislav
Friščić
*bd and
Simon A. J.
Kimber
*a
*a,
Jean-Louis
Do
bc,
Davin
Tan
d,
Hatem M.
Titi
d,
Nicolas
Geoffroy
a,
María del Carmen Marco de
Lucas
a,
Julien
Boudon
a,
Ivan
Halasz
e,
Tomislav
Friščić
*bd and
Simon A. J.
Kimber
*a
aLaboratoire Interdisciplinaire Carnot de Bourgogne, UMR 6303 CNRS-Université Bourgogne Franche-Comté, 9 Avenue Alain Savary, BP 47870, F-21078 Dijon Cedex, France. E-mail: DanielSzczerba@protonmail.com; sajkimber@protonmail.com
bSchool of Chemistry, University of Birmingham University, Birmingham, B15 2TT, UK. E-mail: t.friscic@bham.ac.uk
cDepartment of Chemistry and Biochemistry, Concordia University, 7141 Sherbrooke St. W. Montréal, QC H4B 1R6, Canada
dDepartment of Chemistry, McGill University, Montréal, QC H3A 0B8, Canada
eRuđer Bošković Institute, Bijenička Cesta 54, 10000 Zagreb, Croatia
First published on 19th April 2024
Abstract
Organobismuth compounds have been known for centuries as substances of medical interest, and continue to be used in medicine today. Bismuth active pharmaceutical ingredients (APIs) are used as digestive aids, including in combating Helicobacter pylori, showing antiviral properties. Here we report the mechanochemical synthesis of bismuth gallate and bismuth citrate. In addition, we revisit the previously reported mechanochemical synthesis of bismuth(III) di- and trisalicylate. In the case of bismuth citrate, which has an unknown structure, we show that hydration induces a reversible transformation to a large (possibly cubic) unit cell.
1 Introduction
Bismuth and bismuth-containing compounds have been used in various treatments since the 16th century, especially those aimed at alleviating digestive issues.1 Indeed, medicinal compounds such as bismuth subsalicylate (Pepto-Bismol®) and bismuth subgallate (Devrom®) are still in widespread use.2,3 The most important condition treated with these drugs is the infection with Helicobacter pylori,4 which is associated with peptic ulcers. Bismuth complexes have also been shown to inhibit the activity of a SARS coronavirus,5 and more recently, bismuth citrate has been shown to suppress replication of the virus responsible for the COVID-19 pandemic.6A recent report of mechanochemical syntheses of bismuth(III) salicylates7 inspired us to explore the possibility of extending this methodology to other bismuth active pharmaceutical ingredients (APIs) (Fig. 1). In this work we present mechanochemical syntheses of bismuth(III) citrate, bismuth(III) gallate, and an improved recipe for selective synthesis of bismuth(III) disalicylate and trisalicylate. Mechanosynthesis as a means for preparing APIs is rapidly developing,8,9 encompassing also various metallodrugs,10 and other hybrid materials, such as metal–organic frameworks.11,12
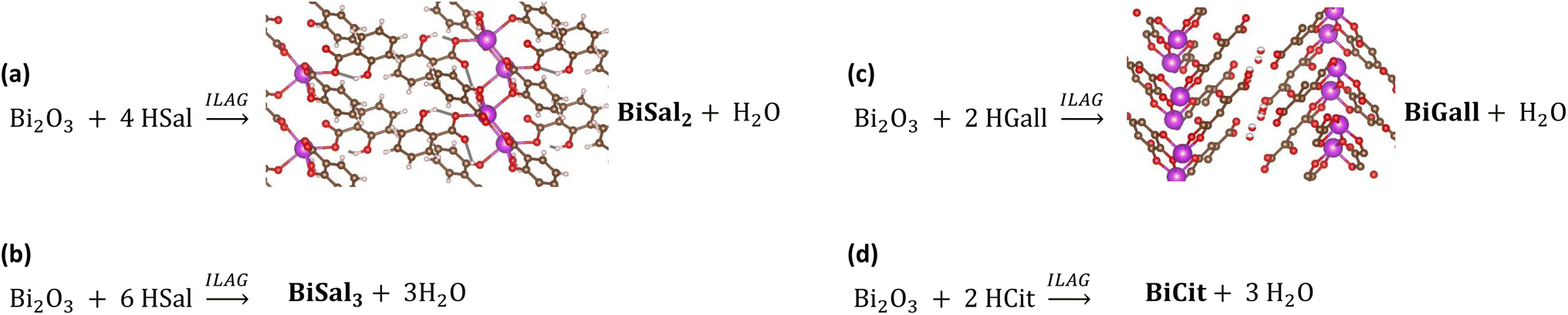 | ||
| Fig. 1 Reaction schemes for synthesized bismuth APIs. (a) The synthesis of bismuth(III) disalicylate with ILAG, showing the known structural model of the compound.7 (b) The synthesis of bismuth(III) trisalicylate. (c) The synthesis of bismuth(III) gallate, showing the known structural model.22 (d) The synthesis of bismuth citrate. | ||
Mechanochemistry is considered to be an environmentally friendly alternative for traditional solvent-based syntheses, a more sustainable solution.8,13–15 Conventionally, large amounts of solvents are used in the synthesis of APIs,16 which is also the case for the bismuth APIs. Methods popular for synthesizing bismuth salicylate,17 gallate,18 citrate and other bismuth APIs use nitric acid and copious amounts of water,19 which is environmentally unfavourable. Additionally, the process requires heating of aqueous solutions for extended time, adding to the carbon footprint of the synthesis. Minimizing the use of solvent is one of the main challenges for “green chemistry”,16 hence working towards discovering solid state synthesis methods is an important goal. Especially for chemical compounds synthesized widely for commercial use, like bismuth APIs are.
Another benefit of mechanochemical over solution-based syntheses is reducing energy use, since mechanochemical reactions are often successful at room temperature.12,20 And even if heating would be required, the energy wasted on heating the bulk of solvent is saved. Other important advantages of mechanochemistry emerge in a shortened time of the reaction, resulting from merging of many reaction steps into a single one, and an improved yield, which is often quantitative. Moreover, limited amount of precursor compounds and intermediate reactions, decreases the amount of side products, which would need to be treated as impurities. In terms of bismuth APIs, the mechanochemical synthesis routes presented in this work employ Bi2O3 and simple organic acids as precursors, which stands in contrast to more elaborate, and potentially expensive metal–organic precursors, and the use of additional acids. Exploring new synthesis routes, with the potential for producing phase pure and crystalline products, is particularly important, given the difficulties in solving the structures of these materials.21
Apart from mechanochemical synthesis having important advantages over solvent-based methods for synthesizing APIs,8,15,23 the chemical compounds synthesized in this work hold their own significance. As indicated, they belong to a group of chemicals widely used of in medicine for centuries, which continues to this day thanks to their low toxicity.24 Bismuth citrate and gallate are directly used as medicine for ulcer treatment, and other intestine issues, and so are derivatives of bismuth salicylates.25 However, these classical uses of those APIs are not the only ones. Medical field is looking for metal–organic compounds capable of forming new structures, that could be used in advanced drug targeting, serving as contrast in medical imaging, and even cancer treatment.26,27 These advanced uses often require the chemicals to be non-toxic to the human organism, and contain a metal element capable of forming interesting for medicine geometry, which makes bismuth compounds so attractive.28 New uses for bismuth(III) salicylates,29 gallate30,31 and bismuth citrate29,32 are often proposed in literature. A green, mechanochemical synthesis of these compounds would fit very well into further experimentation aiming to modify them and test them in new, medical uses.
2 Experimental section
The mechanochemical grinding was done using an InSolido Technologies vibration-type ball mill, IST500, with stainless steel jars (10 and 25 ml) and hardened stainless steel balls weighing 11 and 32 g (7 and 10 mm in diameter). Mechanochemical neat grinding, liquid-assisted grinding (LAG)33–35 and ion- and liquid-assisted grinding (ILAG)33,36 methods were implemented. The latter two methods used distilled water as the default liquid.All chemical ingredients used were purchased from Sigma-Aldrich if not stated otherwise. Bi2O3 (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 999%) was used in all of the synthesis experiments. Salicylic acid (HSal, ≥99.0%), gallic acid (HGall, ≥98.0%, Merck KGaA) and citric acid (HCit, ≥99.5%) were used as the second reaction ingredient, along KNO3 (≥99.0%) and NH4NO3 (≥99.5%) as ionic salt catalysts in the following experiments. For some synthesis citric acid monohydrate (HCit monohydrate, ≥99.0%) was used instead of anhydrous HCit, and for some experiments commercial bismuth citrate (BiCit, 99.999%) was used.
999%) was used in all of the synthesis experiments. Salicylic acid (HSal, ≥99.0%), gallic acid (HGall, ≥98.0%, Merck KGaA) and citric acid (HCit, ≥99.5%) were used as the second reaction ingredient, along KNO3 (≥99.0%) and NH4NO3 (≥99.5%) as ionic salt catalysts in the following experiments. For some synthesis citric acid monohydrate (HCit monohydrate, ≥99.0%) was used instead of anhydrous HCit, and for some experiments commercial bismuth citrate (BiCit, 99.999%) was used.
The Powder X-ray diffraction (PXRD) measurements were done with two diffractometers, a Bruker D2 PHASER LynxEye XE and a Bruker D8 DISCOVER LynxEye XE. Both used Cu Kα radiation, and zero background Si sample holders. Raman spectrum measurements were obtained with a Renishaw inVia spectrometer, using a green (532 nm) laser wavelength. For the TGA measurements, a TA Instruments TGA Discovery with high-temperature Pt sample holders was used.
2.1 Bismuth salicylate
The reaction environment was modified with a series of variables for bismuth(III) disalicylate (BiSal2) and bismuth(III) trisalicylate (BiSal3) to reproducibly obtain full conversion into the desired compounds.The ratios of Bi2O3 to salicylic acid (HSal) were 1:2, 1:4 and 1:6, and the total reactant weights varied from 200 to 300 mg for 10 ml jars, and from 400 to 1000 mg for 25 ml jars. All reagents were weighted on an analytical balance with 0.1 mg precision. The amount of ionic salt (KNO3 or NH4NO3) used in ILAG method varied from 1 to 10 weight % and was used to increase mobility.33 The volume of water added to the reactions varied from 0 to 150 μl per 200 mg of reaction mixture. Stainless steel balls were used for the grinding, with both 7 and 10 mm sizes tested as single balls, and 7 + 7 and 10 + 10 pairs.
Heating of the reaction mixtures to 85 °C for 30 min prior to grinding was tested as a means of enabling the BiSal2 synthesis. While heating, the mixture was kept inside the jars, with the water in the case of LAG and ILAG experiments. Alternatively, the mixture was heated in a holder, before being transferred to the jars with the appropriate amount of water. In the former case, the preheated jars and reaction mixture were rapidly mounted onto the mill. In the case of the latter, the preheated mixture was rapidly transferred to the jars followed by addition of water prior to milling. We also experimented with the cooling fan of the mill, trying syntheses with the fan off and on. The time of grinding varied from 15 to 60 min. The frequency of milling was kept constant at 30 Hz for all reactions, and during the whole reaction period.
Since the mill has two arms, which must be balanced, the samples were synthesized in batches of two at the same time. After synthesis, each sample was transferred into a separate container, for later PXRD pattern measurement.
2.2 Bismuth gallate
The synthesis of bismuth(III) gallate (BiGall) was performed similarly to the salicylates, with changing the reaction environment.The 1:2 and 1:4 ratios of Bi2O3 to gallic acid (HGall) were tested. 200 mg of reaction mixture was used with a 10 ml jar and two 10 mm balls. The addition of 1–2 weight % of NH4NO3 was used for ILAG synthesis. For LAG and ILAG methods, apart from water, acetone, DMF, ethanol, and propanol were tested as the liquid, with 50 μl per 200 mg of reaction mixture. Water and propanol were also used at different volumes, from 30 to 150 μl per 200 mg of reaction mixture, pure and as water–propanol mixtures.
Heating of the reaction mixture inside of the jars for 30 min at 85 °C was implemented for the chosen samples. The time of grinding varied from 30 to 45 min. The cooling fan of the mill was turned off, and the frequency of grinding was set to 30 Hz.
After each synthesis both batches were transferred into separate containers, for later PXRD pattern measurements.
2.3 Bismuth citrate
For the synthesis of bismuth citrate, the mechanochemical route was explored in parallel with synthesis by aging, in a humid environment.Multiple ratios of Bi2O3 to citric acid (HCit) were tested mechanochemically and by aging, these included 2:1, 1:1, 3:4, 1:2, 1:3 and 1:4 ratios. When using the HCit monohydrate, only the 1:2 stoichiometric ratio was used for synthesis.
The aging was performed inside a desiccator, filled with distilled water. The reaction mixture, without additives, was placed in a dish inside the environment. The desiccator was then closed, preventing water from escaping the system, and placed in an oven set to 50–60 °C for 7 days. After that time, each batch of samples was retrieved, dried, ground, and its PXRD pattern measured.
For the mechanochemical synthesis, 300 mg of reaction mixture and 10 ml jars were used. An addition of 2–5 weight % of NH4NO3 or KNO3 was used for ILAG synthesis. Acetone, DMF, ethylene glycol, ethanol, methanol and water were used as the liquid in LAG and ILAG, kept at 75 μl per 300 mg of reaction mixture. For the water, volumes from 75 to 100 μl were tested.
Heating of reaction mixture inside of the jars for 30 min at 85 °C was implemented for most samples. The time of grinding varied from 4 to 50 min. The cooling fan of the mill was turned off, and the frequency of grinding was set to 30 Hz.
Commercial BiCit was ground with LAG method, using water as a liquid, and aged in the described above conditions for testing.
The PXRD measurements of BiCit were done for samples at different steps of drying. Some samples were intentionally mixed with water to create a slurry, which was then slowly dried, while the PXRD pattern was repeatedly measured. Note that these experiments were performed in the compact Bruker D2 PHASER, and that the internal temperature of the diffractometer is typically above room temperature (ca. 35 °C).
:4 O2 and N2 mixture, mimicking air. The rise in temperature was set to 3–5 °C min−1. Different samples were measured at different stages of wetness. Approximately 1 mg of dry material was placed on a sample holder for each measurement.
3 Results
Before beginning a detailed presentation of the results, we briefly summarise the best conditions found for all three bismuth API materials. These can be found in Table 1. Many other details, such as the powder diffraction analysis for the various reactions attempted, can be found in the ESI.†| Product | Precursor A | Precursor B | Molar ratio [A:B] |
Mass [mg] | H2O [μl] | Ionic salt [%] | Heating jars [°C]/[min] | Jar [ml] | Balls no, d[mm] | Time [min] | f [Hz] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Parameters crucial for reaction occurring. | |||||||||||
| BiSal2 | Bi2O3 | HSal | 1:4 |
200 | 75a | 1–2a | 85/30a | 10 | 2, 10a | 30–45 | 30 |
| BiSal3 | Bi2O3 | HSal | 1:6 |
200 | 75a | 1–2 | —a | 10 | 2, 7a | 30 | 30 |
| BiGall | Bi2O3 | HGall | 1:2 |
200 | 100a | 1–2 | 85/30 | 10 | 2, 10 | 45 | 30 |
| BiCit | Bi2O3 | HCit/HCit·H2O | 1:2 |
200 | 75a | — | 85/30a | 10 | 2, 10 | 30 | 30 |
3.1 Bismuth salicylate
Mechanochemical synthesis of pure BiSal2 (Fig. 2a) proved to be a complex process, influenced significantly not only by controlled experimental conditions, but also by external environment. The latter became obvious when experiments conducted based on previous work7 yielded no BiSal2, only BiSal3. At the same time, the exact same reaction environment recreated in other laboratories (in Canada and Croatia) yielded BiSal2 as it should. The unsuccessful attempts to recreate the synthesis were explored, and the challenges identified, improving the reproducibility of the BiSal2 mechanochemical synthesis by tweaking some of the parameters.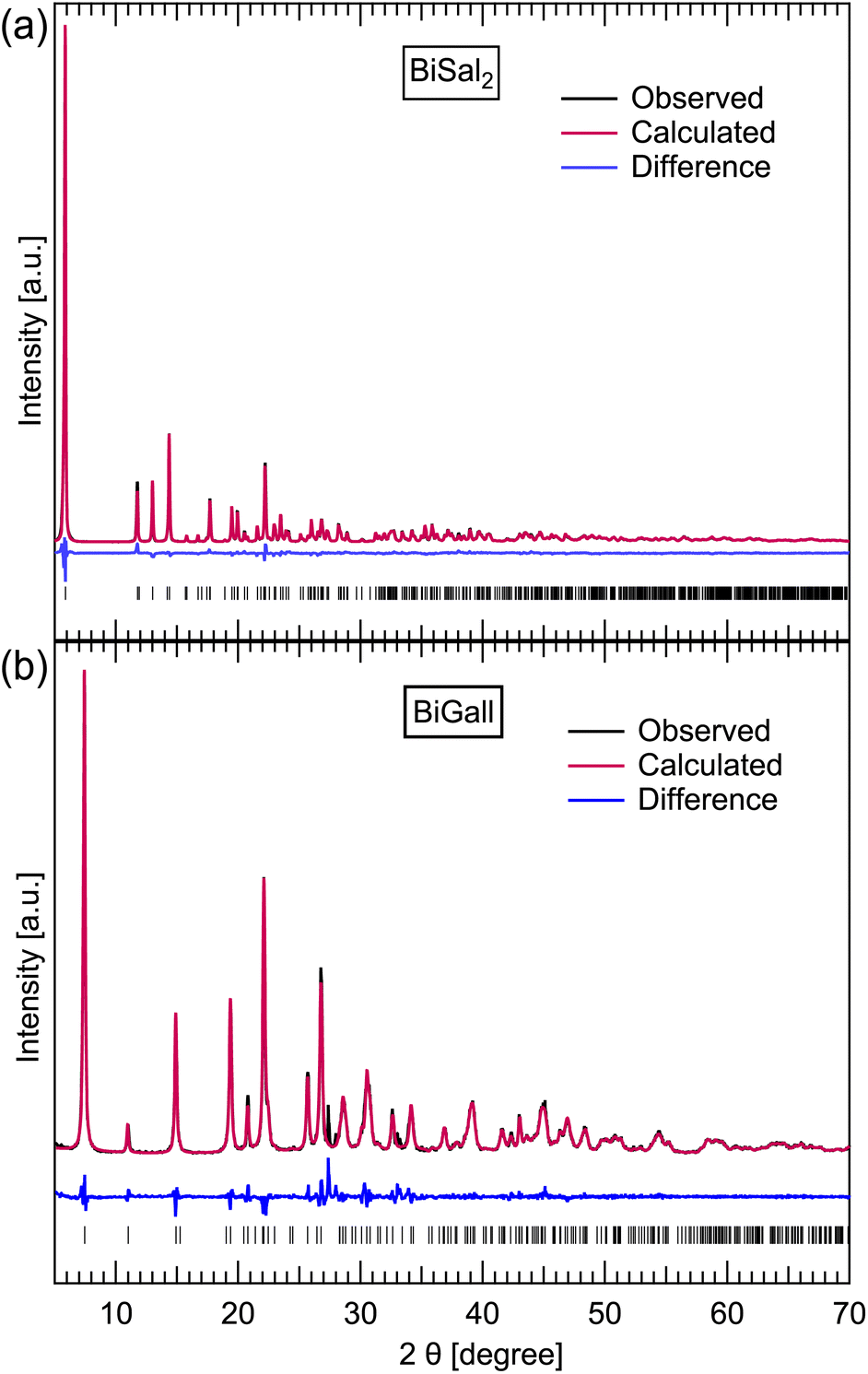 | ||
| Fig. 2 Observed, calculated and difference profiles from the Rietveld fits to the PXRD data the PXRD data for BiSal2 (a) and BiGall (b) at 300 K was fitted using the Rietveld method and the GSAS-II software,37 with wR parameters of 8.68 and 14.96% respectively. The tick marks represent allowed peak positions. The fits show nearly full conversion achieved with mechanochemical synthesis for BiGall, and full conversion for BiSal2. Fitting was done using literature structural data for both BiSal2 (ref. 7) and BiGall.22 | ||
To mechanochemically synthesize high purity BiSal2 the use of ILAG method to improve mobility in the system remains crucial for a full conversion, as stated in previous work.7 Addition of 1–2 mass% of ionic salt (NH4NO3 or KNO3) is enough to have a desired impact. Using water as the liquid is also essential, with no other solvent enabling the reaction. Using 75 μl per 200 mg of reaction mixture provides best results. However, using previously recommended pair of 7 mm steel balls most often yields BiSal3, regardless of the Bi2O3:HSal ratio. Exchanging those for two 10 mm balls, and therefore increasing the force of impact and generated in the grinding process heat, proves to be essential for successful BiSal2 synthesis. Additional step to ensure successful synthesis, is heating the jars with the starting 1:4 (Bi2O3:HSal) reaction mixture and water, at 85 °C for 30 minutes, right before milling. Although heating of the jars was already recommended in previous work,7 doing it with the reaction mixture ensures rapid mounting of the jars. Then 30–45 min of milling is enough to ensure full conversion. If these parameters are not enough to obtain pure BiSal2 yield, turning off the cooling fan of the mill is recommended. For a larger batch, exceeding the amount of reaction mixture over 500 mg for 25 ml milling jars can lead to incomplete conversion. Therefore, the reaction might require further adjustments for larger-scale synthesis. However, below 500 mg the reaction can be reproduced with the same conditions, provided the volume of water is proportionally adjusted.
For the intended synthesis of BiSal3, 7 mm balls and a 1:6 (Bi2O3:HSal) ratio are required. In addition, no heating can be applied to the jars before milling, as a high temperature of reaction mixture favors BiSal2. High room temperature, exceeding 20 °C, may also cause partial conversion to BiSal2, therefore the use of the mill cooling fan is recommended. The volume of water, amount of ionic salt and other parameters may be used as for the BiSal2 synthesis.
Interestingly enough, some commercially available Bi2O3 batches do not convert into BiSal2 under any circumstances, yielding BiSal3 exclusively. Such batches of Bi2O3 can be activated, heating at 600 °C overnight, up to 12 hours. The synthesis of BiSal2 then becomes possible. The heating of such batch is without any mass loss. There is no apparent difference in between batches, according to the labels, including crystalline size. This tends to rule out e.g. carbonate formation, or other reaction with (damp) air, implicating instead the surface crystal structure as the potential culprit for the different behaviour.
The syntheses of BiSal2 and BiSal3 were expanded upon, resulting in a deeper understanding of the differences in reaction conditions required to selectively obtain these compounds. For BiSal2, heating of the reaction mixture before grinding, and maintaining a high temperature during grinding (by turning off a cooling fan) were crucial. In addition, the synthesis worked best using two 10 mm hardened stainless steel balls (rather than 7 mm). In contrast, for synthesizing pure BiSal3, no heating should be applied and the use of 7 mm ball is highly recommended. This indicates that the energy required to obtain BiSal3 through mechanochemical grinding is lower than for BiSal2, and is the key difference apart from using appropriate ratios of Bi2O3 to HSal. The mechanochemically synthesized compounds were pure, with no precursor impurities detected through PXRD analysis nor Rietveld refinement of BiSal2. Important to note is that although the organic acid would typically be ground to an amorphous phase, Bi2O3 from our experience remained crystalline after grinding for up to 1 hour on our setup, remaining easy to qualitatively detect with PXRD. It is also insoluble in water.
3.2 Bismuth gallate
Initially, a mechanochemical synthesis with parameters analogous to the one for BiSal3 was performed for Bi2O3 and HGall mixtures (molar ratios of 1:2 and 1:4), resulting in a pea-green powder. It was determined with PXRD to be BiGall (Fig. 2b). The 1:2 ratio, being stoichiometric, yielded less leftover precursor, and was proceeded with in order to adjust the parameters. The following product's PXRD patterns were compared with each other in terms of ratio of BiGall pattern highest peak to the one of Bi2O3.
Neat grinding with no liquid resulted in most of the Bi2O3 remaining unreacted. In the PXRD pattern this co-exists with broad reflections, that can be tentatively assigned to BiGall formation. Hence, LAG experiments followed. The use of water and propanol as liquid additives improve the reaction significantly. Using ethanol and DMF produces mixtures containing unknown phases. In the case of ethanol, a new peak at larger d-spacing is found, that co-exists with BiGall. A third new phase, with a smaller unit cell is found for DMF (see ESI S3†). Mixtures of water and propanol do not improve the reaction, therefore water was chosen as the most favorable liquid. BiGall crystallizes as a hydrate, therefore it is not surprising that a certain amount of water is important to obtain well-crystalline sample. Adjusting the amount showed us that 100 μl per 200 mg of reaction mixture yields the best results. According to its structure, a minimum of 26.8 μl of water for 200 mg of BiGall is necessary for obtaining a fully crystalline sample, which is less than the volume required for optimising the LAG method. We caution future investigators that all solvents were used directly from the ‘bench’, and that we previously discovered strong water-content effects, investigating solvolysis in the BiSal2 system.38
Finally, using ILAG with 1–2% of NH4NO3 further minimizes the amount of leftover Bi2O3, and repeatably yields good results. We also implemented the heating of the jars with reaction mixture to ensure reproducibility. 45 min of grinding was found to yield better conversion, compared to the 30 min experiments. Based on PXRD pattern Rietveld refinement, mechanochemical synthesis of BiGall was readily achieved with best purity of 94.0%, where the other 4.4% and 1.6% of the mass were leftover Bi2O3 and HGall respectively. Indeed, the literature structure22 could be used to fit our data. To the best of our knowledge, this work is the first report of success by the mechanochemical route.
3.3 Bismuth citrate
In the case of bismuth citrate, the synthesis proves to be less straightforward. However, using Raman and TGA characterization aided in understanding the process.The mechanosynthesis initially resulted in an amorphous product, that could not be readily identified or characterized. Therefore, aging of Bi2O3 mixed with citric acid in wet environment was tested. Afterwards, dried and ground product was measured, revealing that a partial reaction occurred, resulting in PXRD peaks matching commercially available BiCit (2a) (Fig. 3a). Aging in a stoichiometric 1:2 (Bi2O3:HCit) ratio, in a small surface container, results in a nearly full conversion. This aging process is not as reliable as mechanochemical grinding. However, implementing mixing and a container that allows for a better contact in between reaction mixture grains, increases the chemical reaction yield, and even allows for a full conversion. This was proved with two samples, prepared with reaction mixtures in stoichiometric ratios, made using aging. One of them was prepared in a small, porcelain crucible, with a curved bottom. The second reaction was performed in a standard size Petri dish. Both were filled with 1000 mg of reaction mixture. The first one resulted in a nearly full conversion, whereas the second did not, due to the reaction mixture being more scattered on the surface of the dish.
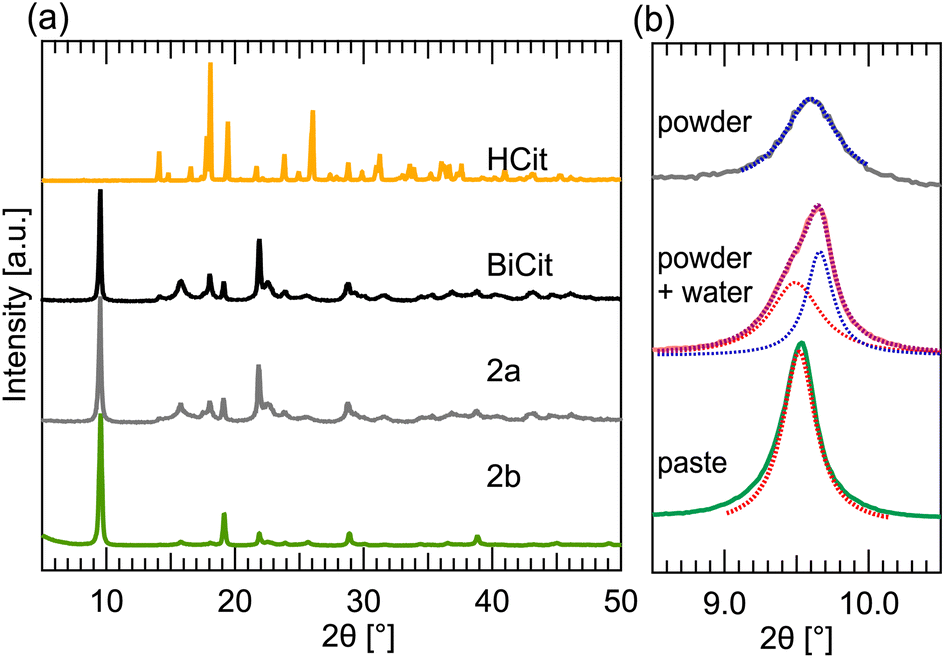 | ||
| Fig. 3 PXRD patterns for bismuth citrate (BiCit) synthesis. (a) Simulation of citric acid (HCit) PXRD pattern, compared to measured PXRD patterns of commercial BiCit, synthesized 2a and synthesized 2b BiCit structures. The 2a and 2b measurements come from the same sample, synthesized by aging 1:1 (Bi2O3:HCit) reaction mixture. 2a was measured as a dry powder, and 2b as a wet paste. (b), PXRD patterns showing transformation of mechanochemically synthesized bismuth citrate, from 2a to 2b structure, from dry powder to wet paste, formed by drying suspension. The graph is focused on the first peak of both structures, showing the change in its position when transforming from 2a to 2b. The peaks were fitted with Lorentzian functions, highlighting two-peak contribution in the intermediate state. | ||
In terms of mechanochemical synthesis, dry grinding experiments yielded an amorphous product. However, the inclusion of water in experiments allowed a well crystalline product to develop. No other liquid yielded successful BiCit synthesis. Assistance of ionic salt (ILAG) proved not to substantially improve the reaction. Therefore, the LAG process is recommended, using 75 μl of water for 200 mg of reaction mixture. The heating of reaction mixture in jars is, unlike the ILAG method, a necessary step for the reaction to occur.
PXRD of a mechanochemical product revealed in some cases an unknown structure (2b) next to the commercial one (2a). All of the PXRD data contained peaks associated with 2a, which led us to believe that the conversion to 2b occurring in some samples was not full. Adjusting the grinding time and Bi2O3:HCit ratio did not yield “pure” 2b, however. No synthesis resulted in obtaining only the new, 2b peaks. However, full conversion of reaction mixture to 2a was achieved at 30 min of grinding, with 1:2 (Bi2O3:HCit) stoichiometric molar ratio.
The yellow reaction mixture, from a dry powder, turns into a white paste after mechanochemical grinding. It was discovered that 2b is present in the product measured right after the reaction, while the consistency of the sample still resembles a wet paste (Fig. 3b). After the sample is completely dry, however, only 2a structure can be seen. In an attempt to rehydrate an already dry sample, it was mixed with water, forming a suspension or a paste. While drying, characteristic peaks of 2b emerge in PXRD datasets. After fully drying, the PXRD patterns contain exclusively 2a structure. Transforming the samples from 2a back to 2b by mixing with water, and the other way when drying proves the change in structure to be reversible.
The same process does not work in the case of commercial BiCit, its structure remaining that of 2a even when exposed to excess of water. However, grinding 200 mg of commercial BiCit with addition of 75 μl of water “activates” the compound. After that, its structure changes from 2a to 2b when exposed to excess of moisture, and back when drying. Aging commercial BiCit in a moist atmosphere does not activate it in the same way.
The Raman spectrum of synthesized BiCit (aged and mechanochemical), commercial BiCit, andnHCit were measured. The spectra were gathered for dry powders, rehydrated paste and drying suspensions, to correspond with the PXRD data (Fig. 4) directly. Comparing the results, it is clear that the synthesized samples have a spectrum identical to commercial BiCit. At the same time, no peaks associated with citric acid are found in the samples, supporting the claim of full conversion. The bands in 1600–1800 cm−1 region, associated with [–COOH] group, are present only in the pure citric acid, due to the [–OH] part of that group releasing a proton to bond with bismuth in bismuth citrate. This further proves the success of the aging and mechanochemical synthesis routes. However, the dry and wet measurements do not differ in terms of peak positions, nor relative intensities. This is unsurprising, given that the Raman spectra in the wavenumber range probed here, are dominated by internal ligand modes.
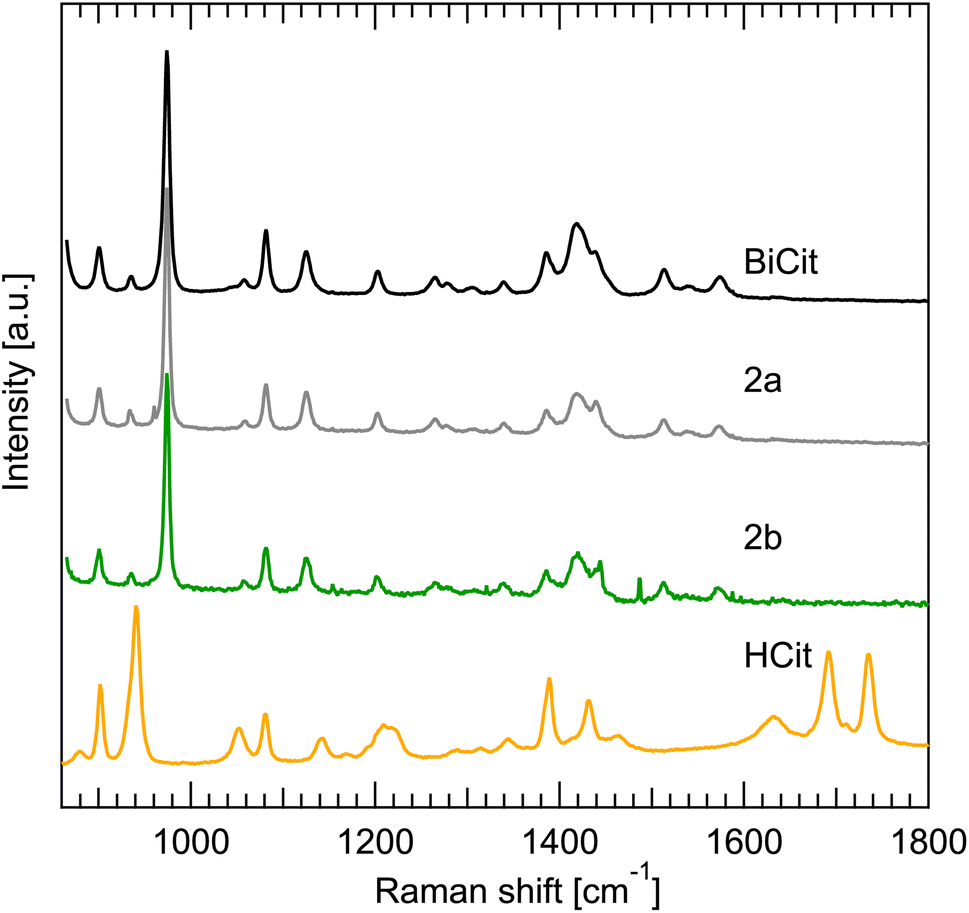 | ||
| Fig. 4 Raman spectrum of commercial bismuth citrate (BiCit) and synthesized 2a and 2b BiCit structures, compared to citric acid (HCit). The BiCit synthesized and commercial BiCit measurements show no apparent difference. The Raman spectrum of 2a and 2b structures also does not differ. HCit peak positions not overlapping with commercial BiCit, are also nowhere to be found in synthesized BiCit. Sharp spikes in the 2b spectrum, not matching peaks from 2a and BiCit measurements, come from the detector background, and are visible due to a longer signal gathering time. | ||
The TGA measurements allowed us to compare the synthesized samples with commercial bismuth citrate (Fig. 5). It was established that the excess of water in each measurement evaporates below 100 °C, and then unbound citric acid decomposes below 200 °C virtually fully. The citric group bound in BiCit (BiC6H5O7) decomposes below 320 °C, to finally leave pure bismuth(III) oxide (Bi2O3) above 420 °C.
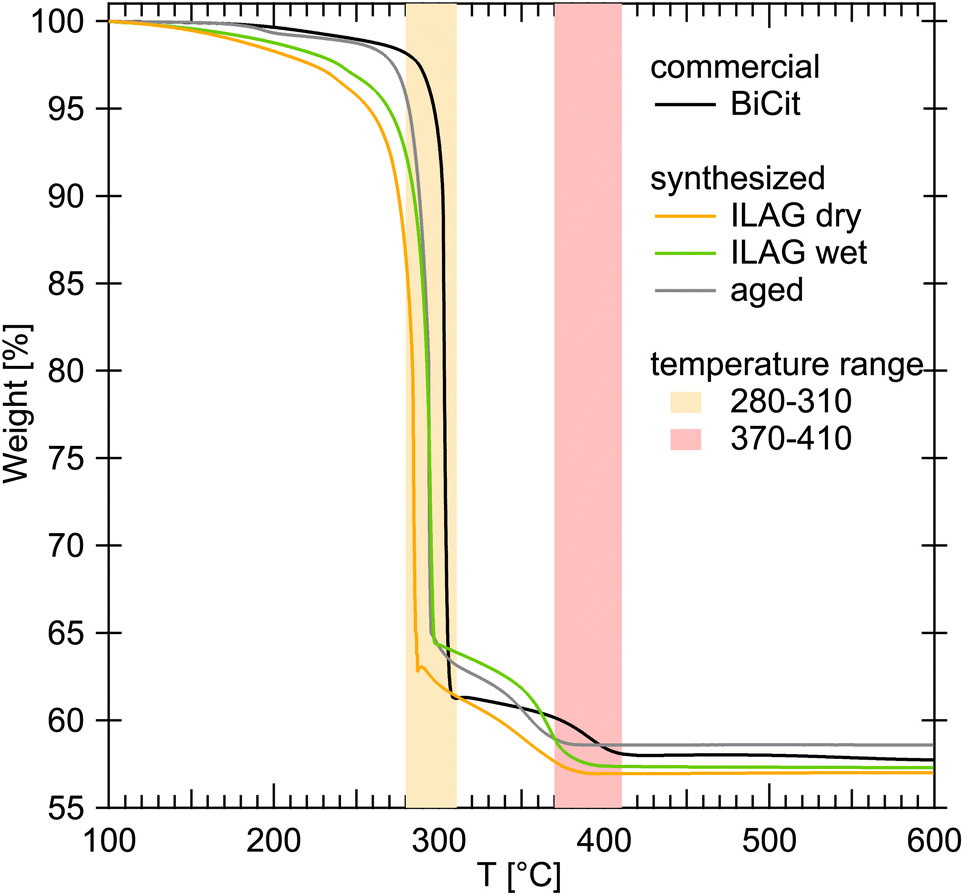 | ||
| Fig. 5 TGA measurements of commercial and synthesized bismuth citrate (BiCit). Temperature ranges for two decomposition stages are highlighted in orange and red. The first one (orange) associated with the combustion of most BiCit to Bi2O3, and partially to species such as bismuth(III) carbonate. The latter (red) is the further reaction of partially combusted BiCit to Bi2O3. No significant traces of leftover citric acid, which would decompose at 200 °C, can be detected in the curves presented for synthesized BiCit. The offsets in temperature, resulting in temperature ranges rather than a single decomposition temperature for each sample, are due to particle size differences. The measurements were normalized to 100% of mass at 100 °C to erase water content bias. | ||
Normalizing the TGA data to 100% of mass at 100 °C for all bismuth citrate samples (Fig. 5), commercial and synthesized, results in comparable mass loss, from 41.4 to 43.0%. This normalization is meant to erase the water evaporation process from all the samples, enabling their direct comparison. The loss of mass in all cases consists of two main steps, first finishing between 280 and 310 °C, likely associated with the following reaction:
| 2BiC6H5O7(s) + 9O2(g) → 9CO2(g) + 5H2O(g) + Bi2(CO3)3(s) | (1) |
And then the second between 370 and 410 °C, likely being a further oxidation into bismuth(III) oxide:
| Bi2(CO3)3(s) → 2CO2(g) + Bi2O2(CO3)(s) | (2) |
| Bi2O2(CO3)(s) → CO2(g) + Bi2O3(s) | (3) |
The small loss of mass at around 200 °C can be attributed to decomposition of excess citric acid that could be present in some of the samples.
Analysis of the difference between 2a and 2b structures using TGA is not a straight forward process, due to the 2b structure being water dependant. Measurements comparing a paste-wet sample and the same sample as dry powder were taken, revealing that both behave similarly, apart from the water evaporation process taking place below 100 °C. Measurements of mechanochemical sample freshly made and rehydrated after drying were also unsuccessfull in capturing any difference.
While the structure of BiCit is currently unknown, our mechanochemical synthesis gave identical diffraction patterns to the commercial bismuth citrate. Raman spectra analysis and TGA experimental data, unambiguously confirm the synthesized BiCit to be no different than commercially available bismuth citrate. A comparison of PXRD data for synthesized and commercial BiCit agrees with that assessment. Moreover, mechanochemically synthesized samples have no traces of the precursor materials, Bi2O3 or HCit, according to all three methods.
We further discovered a reversible structural change in BiCit, from 2a to 2b, which depends upon water exposure. The 2b, widespread peaks indicate high symmetry, most likely of a cubic unit cell. It fits well with a 16.1 Å unit cell in the cubic system. This adds a possible, large unit-cell as another, interesting trait of the structure. We made an attempt to find a solvent, that would allow the 2b to be fully stable. Different solvents, such as acetone, DMF, methanol, ethanol and ethylene glycol, were used in the LAG process. The attempts were unsuccessful in enabling BiCit synthesis, proving water to be the preferred liquid for this reaction.
Identical Raman spectra of 2a and 2b structures paired with the different PXRD patterns, makes it clear that the change in the BiCit structure is structural and not associated with a chemical reaction. One intriguing possibility for the 2a to 2b structural change is the formation of an open framework, as recently seen in the BiGall system.31 Future work will evidently be required to solve both crystal structures, before more progress can be made.
4 Conclusions
We present here detailed protocol for synthesizing bismuth(III) gallate, bismuth citrate, and improved synthesis recipes for bismuth di- and trisalicylate. Of particular interest in the latter case, is that certain commercial batches of Bi2O3 are unreactive, and must be ‘activated’ by calcination overnight.Due to the benefits of the mechanochemical synthesis route, such as its lower environmental impact and sustainability, we believe that these reactions could be implemented in pharmaceutical and chemical industries for an efficient production of bismuth APIs from simple ingredients. The synthesized products were shown to be of high purity, using PXRD data and Rietveld refinements where possible, and TGA and Raman spectroscopy data analysis for bismuth citrate.
Finally, an interesting, water-dependent structural effect was found in bismuth citrate. This effect implies that the water can enter into the 2a structure, which must be porous. Given the known (and complex) hydrolysis behaviour shown by these materials,7,38 as well as the recent discovery of bismuth gallate based frameworks,31 further investigations are merited.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Investments for the Future” program, ISITE-BFC project (contract ANR-15-IDEX-0003), and the EIPHI Graduate School (contract ANR-17-EURE-0002). TF acknowledges the Leverhulme International Professorship and the University of Birmingham for support. We thank Bruno Domenichini for discussions, and Nadine Millot for her support and interest in this work.References
- P. Malfertheiner, M. Nilius and U. Kreusel, Basic and Clinical Aspects of Helicobacter pylori Infection, Berlin, Heidelberg, 1994, pp. 266–279 Search PubMed.
- G. G. Briand and N. Burford, Chem. Rev., 1999, 99, 2601–2658 CrossRef CAS PubMed.
- P. Reynolds, K. Abalos, J. Hopp and M. Williams, Int. J. Clin. Med., 2012, 3, 46–48 CrossRef CAS.
- B. J. Marshall, J. A. Armstrong, G. J. Francis, N. T. Nokes and S. H. Wee, Digestion, 1987, 37(Suppl 2), 16–30 Search PubMed.
- N. Yang, J. A. Tanner, B.-J. Zheng, R. M. Watt, M.-L. He, L.-Y. Lu, J.-Q. Jiang, K.-T. Shum, Y.-P. Lin, K.-L. Wong, M. C. M. Lin, H.-F. Kung, H. Sun and J.-D. Huang, Angew. Chem., Int. Ed., 2007, 46, 6464–6468 CrossRef CAS PubMed.
- S. Yuan, R. Wang, J. F.-W. Chan, A. J. Zhang, T. Cheng, K. K.-H. Chik, Z.-W. Ye, S. Wang, A. C.-Y. Lee, L. Jin, H. Li, D.-Y. Jin, K.-Y. Yuen and H. Sun, Nat. Microbiol., 2020, 5, 1439–1448 CrossRef CAS PubMed.
- V. André, A. Hardeman, I. Halasz, R. S. Stein, G. J. Jackson, D. G. Reid, M. J. Duer, C. Curfs, M. T. Duarte and T. Friščić, Angew. Chem., Int. Ed., 2011, 50, 7858–7861 CrossRef PubMed.
- D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760–7781 RSC.
- D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustain. Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS.
- S. Quaresma, V. André, A. Fernandes and M. T. Duarte, Inorg. Chim. Acta, 2017, 455, 309–318 CrossRef CAS.
- T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 49, 712–715 CrossRef PubMed.
- P. J. Beldon, L. Fábián, R. S. Stein, A. Thirumurugan, A. K. Cheetham and T. Friščić, Angew. Chem., 2010, 122, 9834–9837 CrossRef.
- K. Wieczorek-Ciurowa and K. Gamrat, J. Therm. Anal. Calorim., 2007, 88, 213–217 CrossRef CAS.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- P. Brandão and M. Pineiro, in Sustainable Approaches in Pharmaceutical Sciences, John Wiley & Sons, Ltd, 2023, pp. 255–272 Search PubMed.
- T. Welton, Proc. R. Soc. A, 2015, 471, 20150502 CrossRef PubMed.
- E. V. Timakova, Y. M. Yukhin and T. A. Udalova, Chem. Sustainable Dev., 2009, 305–313 Search PubMed.
- Y. M. Yukhin, O. A. Logutenko, I. A. Vorsina and V. I. Evseenko, Theor. Found. Chem. Eng., 2010, 44, 749–754 CrossRef CAS.
- Y. M. Yukhin, T. V. Daminova, L. I. A. Onina, B. B. Bokhonov, O. A. Logutenko, A. I. Aparnev and K. Y. Mikhailov, Chem. Sustainable Dev., 2004, 395–401 Search PubMed.
- G. Dayaker, D. Tan, N. Biggins, A. Shelam, J.-L. Do, A. D. Katsenis and T. Friščić, ChemSusChem, 2020, 13, 2966–2972 CrossRef CAS PubMed.
- E. Svensson Grape, V. Rooth, M. Nero, T. Willhammar and A. K. Inge, Nat. Commun., 2022, 13, 1984 CrossRef CAS PubMed.
- Y. Wang, S. Takki, O. Cheung, H. Xu, W. Wan, L. Öhrström and A. K. Inge, Chem. Commun., 2017, 53, 7018–7021 RSC.
- M. Solares-Briones, G. Coyote-Dotor, J. C. Páez-Franco, M. R. Zermeño-Ortega, C. M. de la O Contreras, D. Canseco-González, A. Avila-Sorrosa, D. Morales-Morales and J. M. Germán-Acacio, Pharmaceutics, 2021, 13, 790 CrossRef CAS PubMed.
- J. M. Bothwell, S. W. Krabbe and R. S. Mohan, Chem. Soc. Rev., 2011, 40, 4649–4707 RSC.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- M. Kowalik, J. Masternak and B. Barszcz, Curr. Med. Chem., 2019, 26, 729–759 CrossRef CAS PubMed.
- S. A. Shetu, L. M. Sanchez-Palestino, G. Rivera and D. Bandyopadhyay, Tetrahedron, 2022, 129, 133117 CrossRef CAS.
- P. J. Sadler, H. Li and H. Sun, Coord. Chem. Rev., 1999, 185–186, 689–709 CrossRef CAS.
- H. Li and H. Sun, Curr. Opin. Chem. Biol., 2012, 16, 74–83 CrossRef CAS PubMed.
- Z. Wang, Z. Zeng, H. Wang, G. Zeng, P. Xu, R. Xiao, D. Huang, S. Chen, Y. He, C. Zhou, M. Cheng and H. Qin, Coord. Chem. Rev., 2021, 439, 213902 CrossRef CAS.
- E. S. Grape, V. Rooth, S. Smolders, A. Thiriez, S. Takki, D. De Vos, T. Willhammar and A. K. Inge, Dalton Trans., 2022, 51, 14221–14227 RSC.
- R. Wang, T.-P. Lai, P. Gao, H. Zhang, P.-L. Ho, P. C.-Y. Woo, G. Ma, R. Y.-T. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439 CrossRef PubMed.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., 2020, 132, 1030–1041 CrossRef.
- T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546–7550 CrossRef PubMed.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- P. A. Julien, C. Mottillo and T. Friščić, Green Chem., 2017, 19, 2729–2747 RSC.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- D. Szczerba, D. Tan, J.-L. Do, H. M. Titi, S. Mouhtadi, D. Chaumont, M. del Carmen Marco de Lucas, N. Geoffroy, M. Meyer and Y. Rousselin, et al. , J. Am. Chem. Soc., 2021, 143, 16332–16336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Contains experimental details, powder diffraction and Raman analysis. See DOI: https://doi.org/10.1039/d4mr00008k |
| This journal is © The Royal Society of Chemistry 2024 |