
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA non-invasive osteopontin-targeted phase changeable fluorescent nanoprobe for molecular imaging of myocardial fibrosis†
Xueli
Zhao
a,
Yuze
Qin
a,
Bo
Wang
a,
Jiao
Liu
a,
Yueyue
Wang
a,
Kun
Chen
c,
Jia
Zhao
a,
Lanlan
Zhang
a,
Yuanming
Wu
*b and
Liwen
Liu
 *a
*a
aDepartment of Ultrasound, Xijing Hypertrophic Cardiomyopathy Center, Xijing Hospital, Fourth Military Medical University, Xi'an, Shaanxi 710032, China. E-mail: liuliwen@fmmu.edu.cn
bDepartment of Biochemistry and Molecular Biology, Shaanxi Provincial Key Laboratory of Clinical Genetics, School of Basic Medicine, Fourth Military Medical University, Xi'an, Shaanxi 710032, China. E-mail: wuym@fmmu.edu.cn
cDepartment of Anatomy, Histology and Embryology and K.K. Leung Brain Research Centre, School of Basic Medicine, Fourth Military Medical University, Xi'an, Shaanxi 710032, China
First published on 3rd June 2024
Abstract
Due to the elevated fatality rate of cardiovascular diseases, myocardial fibrosis emerges as a prominent pathological alteration in the majority of heart ailments and their associated pathologies, thereby augmenting the likelihood of sudden cardiac death. Consequently, the prompt and obligatory identification of myocardial fibrosis assumes paramount importance in averting malignant incidents among patients afflicted with cardiac disorders. Herein, with higher expression osteopontin (OPN) found in cardiac fibrosis tissue, we have developed a dual-modality imaging probe, namely OPN targeted nanoparticles (OPN@PFP-DiR NPs), which loaded perfluoropentane (PFP) for ultrasound (US) and 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) for near-infrared fluorescence (NIR) of molecular imaging, to investigate the molecular features of cardiac fibrosis using US and NIR imaging. Subsequently, the OPN@PFP-DiR NPs were administered intravenously to a mouse model of myocardial infarction (MI). The US and NIR molecular imaging techniques were employed to visualize the accumulation of the nanoparticles in the fibrotic myocardium. Hence, this research presents a valuable noninvasive, cost-effective, and real-time imaging method for evaluating cardiac fibrosis, with promising clinical applications.
Introduction
Cardiac fibrosis, characterized by the excessive accretion of extracellular matrix proteins in the myocardium, is a noteworthy pathological modification observed in diverse heart diseases and associated conditions, including myocardial ischemia, inherited cardiomyopathies, and myocarditis.1–3 Furthermore, cardiac fibrosis has been closely linked to the progress of malignant arrhythmias, heart failure, and sudden death.4,5 Consequently, it is of paramount significance to actualize early and precise detection of myocardial fibrosis through application of proactive theragnostic tools, thereby circumventing the high risks of unfavorable outcomes associated with cardiovascular disease.Currently, computed tomography (CT), ultrasound (US), and magnetic resonance imaging (MRI) are the most routine methods for clinical diagnosis of myocardial fibrosis.6–8 However, these conventional methods possess inherent limitations and drawbacks although they do have various advantages. For example, positron emission tomography (PET) and single-photon emission computed tomography (SPECT) have lower spatial resolution compared to other modalities, require daily probe production, and involve the use of ionizing radiation, rendering them less suitable for longitudinal patient monitoring.9–11 Despite being regarded as the gold standard for the noninvasive identification of myocardial fibrosis, late gadolinium enhancement cardiac magnetic resonance (LGE-CMR) is hindered by its inadequate spatial resolution and vulnerability to metallic implants.12–14 Traditional US contrast agents are confined to the vascular space, while optical imaging is constrained to superficial or semi-invasive applications due to its reliance on light penetration into the tissue.6,15 Significantly, these conventional imaging techniques exhibit reduced sensitivity in detecting disease at its early stages and cannot distinguish between active fibrogenesis and stable scars. Moreover, these methods primarily emphasize the morphological alterations in fibrotic myocardium.16,17 Biopsy, although utilized, is an imperfect method for identifying activated molecular pathways and carries inherent risks.18,19 Therefore, it is imperative to create new sophisticated imaging strategies, which are economical, non-ionizing, extremely sensitive, and capable of providing high-resolution images to achieve timely detection of myocardial fibrosis and its early warning signs.
Molecular imaging technology represents a novel avenue for the prompt identification of myocardial fibrosis.20 Unlike traditional imaging methods, molecular imaging technology stands out for its capacity to detect initial indications of the condition21,22 and holds the potential for precise early diagnosis of myocardial fibrosis. Particularly noteworthy is the emergence of US molecular imaging as a viable clinical technique, given its simplicity, precise targeting, and real-time visualization of lesion structure. This technique shows promise surmounting the limitations associated with existing PET/PECT, CT, and MRI imaging methods.23,24 Nevertheless, its main drawback lies in its inadequate sensitivity.25 Near-infrared fluorescence imaging (NIR) has emerged as a prevalent technique for assessing pathophysiological microenvironments, owing to its noninvasive detection, easy operation, and real-time monitoring capabilities,26–29 whereas albeit with the disadvantage of limited penetration depth, while this limitation can be mitigated by integrating with US molecular imaging, thereby effectively compensating for the restricted penetration depth. Hence, the incorporation of NIR with US molecular imaging through the development of a US/NIR multimodal molecular probe can provide facilitating rapid, high-resolution imaging with a deep penetration depth, therefore enabling the effectively evaluation of myocardial fibrosis. In addition, targeted delivery is of utmost importance in improving the specificity and sensitivity of molecular probes. Osteopontin (OPN), a phosphorylated glycoprotein, demonstrates significant expression in fibrotic myocardium, while its presence in normal tissues is either absent or minimal.21,30 This unique attribute renders OPN an ideal contender for the early detection of myocardial fibrosis. Leveraging the abundant expression of OPN in myocardial fibrosis, along with the advantageous features of deep tissue penetration in US molecular imaging and high sensitivity of NIR. Phase change material perfluoropentane (PFP) allows ultrasonic molecular imaging without the large particle size and short half-life of conventional microbubble contrast agents.31,32 As for NIR imaging, DiR (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide) is a common photosensitizer. DiR's water-insoluble nature and rapid elimination from the bloodstream following systemic administration, however, severely restrict its use in imaging and therapeutic procedures.33,34 Therefore, the development of effective tools for improving the delivery of PFP and DiR is essential for the successful US/NIR imaging.
Our research aims to design a dual modal imaging system utilizing US/NIR technology to specifically target OPN with PFP coated phase change nanoparticles as the carrier and DiR as the fluorescent agent, to enable early detection and assessment of myocardial fibrosis at the molecular level (Fig. 1), thereby facilitating prevention of sudden death and identification of potential drug targets.
 | ||
| Fig. 1 (A) Synthetic procedures for OPN-targeted nanoparticles (OPN@PFP-DiR NPs) which loaded perfluoropentane (PFP) for ultrasound (US) and 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) for near-infrared fluorescence (NIR) of molecular imaging, via two step emulsion method and carbodiimide method. (B) Enhanced dual-modal imaging performed by OPN@PFP-DiR NPs via intravenous injection. | ||
Materials and methods
Reagents and materials
PLGA-COOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, MW = 10000) was obtained from Jinan Daigang Biomaterial Co., Ltd (China). Perfluoropentane (PFP), 2-(N-morpholino) ethane sulfonic acid (MES), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and various fluorescent dyes, including 4′,6-diamidino-2-phenylindole (DAPI) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 1,10-dioctadecyl-3,3,30,30-tetramethylindotricarbocyanine iodide (DiR), were all obtained from Sigma-Aldrich Chemical Co. (USA). Osteopontin (OPN) and FITC-OPN peptide were purchased From QYAOBAO Biochem Ltd in China. Except where otherwise stated, all reagents and solvents were analytical grade and bought from Chengdu Kelong Chemical Reagent Factory in Sichuan, China.
:50, MW = 10000) was obtained from Jinan Daigang Biomaterial Co., Ltd (China). Perfluoropentane (PFP), 2-(N-morpholino) ethane sulfonic acid (MES), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and various fluorescent dyes, including 4′,6-diamidino-2-phenylindole (DAPI) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), 1,10-dioctadecyl-3,3,30,30-tetramethylindotricarbocyanine iodide (DiR), were all obtained from Sigma-Aldrich Chemical Co. (USA). Osteopontin (OPN) and FITC-OPN peptide were purchased From QYAOBAO Biochem Ltd in China. Except where otherwise stated, all reagents and solvents were analytical grade and bought from Chengdu Kelong Chemical Reagent Factory in Sichuan, China.
Preparation of OPN-targeted and DiR loaded PFP nanoparticles (OPN@PFP-DiR NPs)
Targeting OPN@PFP-DiR NPs were synthesized using a previously established method with minor modifications.35,36 To begin, a solution consisting of 50 mg PLGA-COOH and 1 mg DiR fluorescent dye in 2 mL dichloromethane (CH2Cl2) was prepared. And subsequently 200 μL PFP was gradually introduced to the above solution, which was then subjected to emulsification using an ultrasonic probe operating at 100 W for a duration of 2 min (with a vibration cycle of 5 s on/5 s off to prevent phase transition). The emulsified solution described above was then added to a total volume of 20 mL, consisting of 19 mL PVA (2% w/v) solution and 1 mL 2% didodecyl dimethylammonium bromide (DDAB) solution, and then homogenized (FJ300-SH, Shanghai, China) for 5 min in preparation for another emulsion. The emulsion, containing 15 mL of a 2% (w/v) isopropyl alcohol solution, was stirred using a magnetic stirrer (HJ-1, Ronghua, China) for 6 h to remove CH2Cl2 and eliminate foam. Following this, the prepared solution was centrifuged at a speed of 10000 rpm for 5 min at 4 °C. The supernatant was thrown away, and the precipitate was washed three times to acquire non-targeting PFP-DiR NPs.
OPN peptides were conjugated to the surface of PFP-DiR NPs using the typical carbodiimide method. The prepared PFP-DiR NPs were dispersed in 5 mL of MES buffer (0.1 mol L−1, pH = 5.5) along with 46 mg EDC and 23 mg NHS for agitation and incubated for a duration of 30 min. After undergoing centrifugation three times for 5 min at 10000 rpm, and the supernatant was discarded each time and 1 mL fresh MES buffer (0.1 mol L−1, pH = 5.5) solution was added in order to extract the residual EDC and NHS. The aforementioned solution was subsequently supplemented with 1 mL OPN peptides solution (1 mg mL−1), subsequently subjected to an incubation period of 2 h with continuous shaking. Furthermore, following three consecutive centrifugations for 5 min at 10000 rpm, and the supernatant was disposed of and 1 mL fresh MES buffer (0.1 mol L−1, pH = 8.0) solution was added each time to extract the remaining OPN peptides. OPN@PFP-DiR NPs were harvested, and all of the procedures mentioned above were done at a temperature of 4 °C.
Characterization of OPN@PFP-DiR NPs
To gain a deeper apprehension of the characterization of prepared nanoparticles, the morphology was detected using the transmission electron microscope (TEM, Hitachi H7600, Japan) and scanning electron microscopy (SEM, Hitachi S4800, Japan). A laser particle size analyzer (NanoBook90Plus PALS, Brookhaven, USA) was utilized to measure the particle size and zeta potential of the nanodroplets at a temperature of 4 °C. The efficiency of OPN conjugation with non-targeted PFP-DiR NPs was ascertained by measuring the fluorescence of FITC-labeled OPN and DiR-labeled PFP-DiR NPs using an inverted fluorescence microscope (CKX41, Olympus, Japan).The phase transition behavior of OPN@PFP-DiR NPs was investigated using both heating and focused ultrasound irradiation to induce the phase transition. Initially, OPN@PFP-DiR NPs were subjected to heating at temperatures of 25 °C, 40 °C, 45 °C, and 50 °C for 4 min utilizing a heater with a constant temperature. Furthermore, the in vitro generation of phase transition of OPN@PFP-DiR NPs was achieved through the application of low-intensity focused ultrasound (LIFU, LMSC051 ACA; Chongqing Medical University, China) at varying frequencies. The process of phase transition of the nano-contrast agents was observed using an inverted fluorescence microscope.
Evaluation of NIR and US molecular imaging ability in vitro
The in vitro NIR molecular imaging was conducted using the Xenogen IVIS Spectrum imaging system (PerkinElmer, USA) with a 745 nm excitation wavelength. The samples were diluted to varying concentrations of DiR, ranging from 8 μg mL−1 to 80 μg mL−1, and subsequently introduced into the 96-well plates using air-free water as a control. The mean fluorescence intensities were quantified.In order to assess the US molecular imaging capability of OPN@PFP-DiR NPs in vitro, the samples were subjected to different LIFU frequencies (1 W cm−2, 2 W cm−2, 3 W cm−2, and 4 W cm−2) and durations (1 min, 2 min, 3 min, and 4 min). The US images were obtained using the ultrasonography instrument Vevo 2100 Imaging System, (Fuji Film Visual Sonics, Canada) and the mean echo intensities within the region of interest (ROI) have been quantified.
In vitro cell viability
To assess the cytotoxicity in vitro, H9C2 and rat primary cardiac fibroblasts (CFs) cells were cultured in 96-well plates at a density of 5000 cells per well for a duration of 24 h. Subsequently, 100 μL of OPN@PFP-DiR NPs with varying concentrations were incubated for an additional 24 h. The viability of the cells was measured using the cell counting kit-8 (CCK-8) kit from Beyotime Biotechnology, China.Establishment of MI model mice and evaluation of cardiac fibrosis
Myocardial infarction (MI) was induced by permanent left coronary artery ligation in C57BL/6 mice using an established protocol,37 all animal experiments used in this institute strictly follow “The NIH Guidelines On The Use Of Laboratory Animals” and are approved by the Animal Welfare and Ethics Committee of the Laboratory Animal Centre of the Air Force Medical University (License number: IACUC-20240006). All animals underwent anesthesia using 2% isoflurane and were induced by ligating the left anterior descending (LAD) coronary artery with 8–0 silk. Electrocardiogram (ECG) and echocardiography were employed for the evaluation of the model establishment. Myocardial tissue from the infarct location was removed for pathological analysis two weeks following surgery, validating the presence of the formation hallmarks of cardiac fibrosis.Ex vivo localization of FITC-PN@PFP-DiR NPs
Frozen sections (10 μm) of both infarcted and normal myocardium (n = 5 for each group) were utilized for the ex vivo localization of FITC-OPN@PFP-DiR NPs. The localization of the nucleus and fibrosis tissue was achieved through the use of DAPI and OPN-FITC, respectively. A 10 μL volume of OPN-FITC(1 mg mL−1) was applied to the sections, followed by a 15 min incubation at room temperature and subsequent washing with PBS three times to eliminate any unbound OPN-FITC. The sections were then examined with a confocal laser scanning microscope (CLSM, Nikon A1R, Japan). After being sealed with an antifluorescent quencher and covered with a coverslip. Following OPN-FITC imaging, the same sections were stained with Picrosirius red (PSR), Masson staining and Hematoxylin & Eosin (H&E) staining, and the resulting fluorescent pictures were compared. The fibrosis content was calculated using the Image-Pro Plus (IPP) 6.0 imaging software.Evaluation of NIR and US molecular imaging ability in vivo
For in vivo NIR molecular imaging, mice were randomly assigned to three groups and administered intravenous injections of 100 μL saline, non-targeted PFP-DiR NPs and targeted OPN@PFP-DiR NPs (with a concentration of 100 μg mL−1 DiR) respectively. The major organs, including the heart, liver, spleen, lung, and kidney, were extracted following the sacrificial procedure of the mice after 24 h following the injection. The fluorescence signal was then quantified using the Living Image Software (PerkinElmer, USA).With the same group as NIR molecular imaging, MI mice were administered non-targeted PFP-DiR NPs or targeted OPN@PFP-DiR NPs intravenously via the tail vein for in vivo US molecular imaging. Subsequently, a 3 W cm−2 LIFU irradiation was applied for 10 min. Following LIFU sonication, US images were recorded and compared.
In vivo biocompatibility evaluation
So as to ascertain the biosafety and biocompatibility of the OPN@PFP-DiR NPs in vivo, all normal mice were randomly divided into test and control groups. An intravenous administration of OPN@PFP-DiR NPs was received by the test group, whereas the control group was injected with saline. After a period of 48 h, the major organs (heart, liver, kidney, spleen, and lung) were afterward harvested for histopathological examination.Statistical analysis
Quantitative data were presented as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism 9.5 (GraphPad, Inc., San Diego, CA, USA). All experiments were performed using n ≥ 3 biological replicates. Student's t-test was performed to assess differences between the two groups. One-way ANOVA or two-way ANOVA was used for multigroup comparisons. P values < 0.05 were considered statistically significant (NS, no significance, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).Results and discussion
Synthesis and characterization of OPN@PFP-DiR NPs
Using two-step emulsification and carbodiimide technique, we effectively constructed OPN-targeted phase changeable fluorescent nanoparticles (OPN@PFP-DiR NPs). The emulsion formed by OPN@PFP-DiR NPs exhibited light blue (Fig. 2A). The OPN@PFP-DiR NPs had uniform diameters, regular and spherical forms, and no discernible aggregation or adhesion, as shown by TEM and SEM images (Fig. 2B and C). As we known, larger particles are more probable to be engulfed by the reticuloendothelial systems, which shortens the time that blood circulates and causes particle accumulation.38,39 Smaller particles, on the other hand, have the benefit of more readily accessing the interior region of the target, but if the particles get too small, phase transitions become more challenging.40 Considering these factors, we developed OPN@PFP-DiR NPs with dimensions of 187.7 ± 9.5 nm (Fig. 2D), exhibiting high water monodispersity and homogeneity (PDI = 0.08 ± 0.02), and a zeta potential of 3.4 ± 1.9 mV (Fig. 2E). Furthermore, it was observed that the size of OPN@PFP-DiR NPs did not exhibit significant variation following 30 days storage period at 4 °C, as depicted in Fig. 2F. This exceptional stability of OPN@PFP-DiR NPs during storage ensures their suitability for future experimental applications.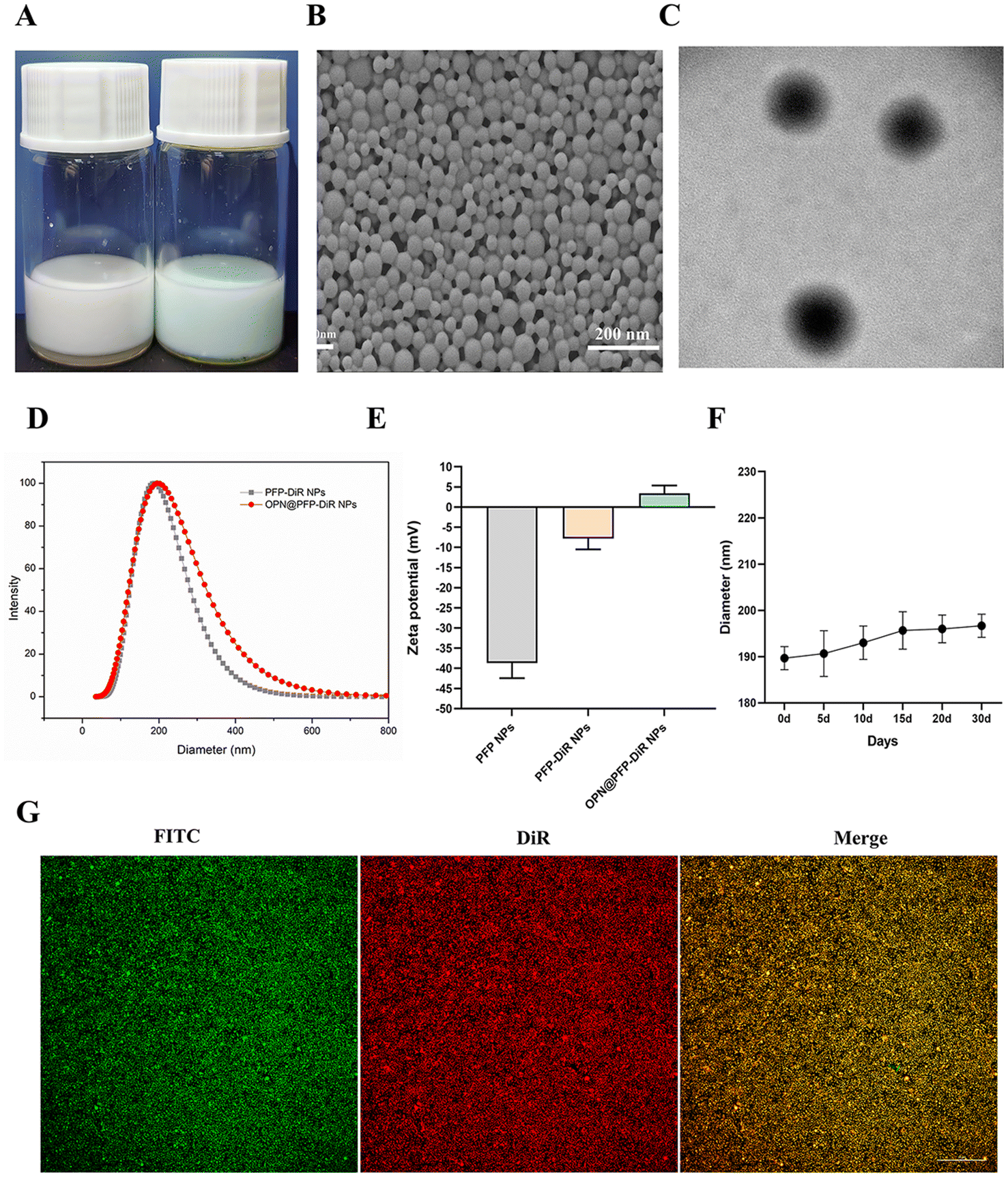 | ||
| Fig. 2 Characteristics of the OPN@PFP-DiR NPs. (A) Macroscopic views of free DiR and OPN@PFP-DiR NPs at the same PLGA concentration in PBS representative SEM images (B) and TEM images (C) of OPN@PFP-DiR NPs. (D) The size distribution of the different PLGA NPs. (E) Size changes of OPN@PFP-DiR NPs at 4 °C after long-term storage. (F) Zeta potential of different PLGA NPs (G) Fluorescence images showed preferential connection between FITC-labeled OPN peptide and DiR-labeled PFP-DiR NPs. | ||
Considering that PFP-DiR NPs lack inherent targeting capabilities of cardiac fibrosis tissue, their accumulation at the heart is solely facilitated through enhanced permeability. To address this limitation, OPN peptide, which possesses a high binding affinity to cardiac fibrosis locations, was conjugated onto the surface of the PFP-DiR NPs. The evaluation of OPN conjugation efficiency entailed an investigation into the correlation between FITC-labeled OPN peptides and DiR-labeled PFP-DiR NPs. This association was visually validated by the amalgamation of orange images showcased in Fig. 2G, which depicted a seamless connection between PFP-DiR NPs nanoprobes (red) and OPN peptides (green). These findings indicated that the NHS/EDC linker played a role in the covalent attachment that demonstrated a high degree of controllability for the immobilization of biomolecules on the surface and offered enough stability for the surface active agent. Another piece of evidence indicating the successful conjugation is that the surface zeta potential of nanoparticles exhibited a noticeable change following the conjugation reaction, from −7.7 ± 2.7 mV to 3.4 ± 1.9 mV (Fig. 2E). The aforementioned properties of prepared nanoparticles demonstrated that OPN@PFP-DiR NPs were successfully fabricated.
Further investigation is warranted to investigate the thermal effect and LIFU irradiation as pivotal factors in inducing phase transition of as-synthesized OPN@PFP-DiR NPs, with the aim of enhancing the comprehension of this phenomenon. Due to their small size, the initial OPN@PFP-DiR NPs posed challenges in differentiation into microbubbles. However, significant microbubbles observed in Fig. 3A and B and video S1† indicated that elevated temperature and LIFU frequency facilitated the transition from liquid NPs to gaseous microbubbles. At an elevated temperature of 50 °C, a significant proportion of the particles demonstrated a gradual expansion, concomitant with the emergence of larger bubbles. This observation provides empirical support for the influence of external temperature on the phase change process of OPN@PFP-DiR NPs. Likewise, an escalation in LIFU frequency led to a proportional increase in the production of bubbles. This occurrence can be attributed to the rising temperature caused by LIFU, which triggers a gaseous transformation of the enclosed PFP component within the particular nanoagents, thus illustrating the temperature-responsive characteristic of US imaging.32 It is noteworthy that, consistent with previous investigations, adjacent bubbles exhibited a tendency to amalgamate and generate larger entities during the microbubble formation process.41
 | ||
| Fig. 3 (A) Optical microscopic image of OPN@PFP-DiR NPs phase transformation by heating from 25 to 50 °C. (B) Optical microscope graphics of phase-transformation after LIFU-irradiated treatment of OPN@PFP-DiR NPs at different frequency (1 W cm−2, 2 W cm−2, 3 W cm−2 and 4 W cm−2) for 3 min. (C and D) NIR images and intensities of OPN@PFP-DiR NPs with different concentrations of DiR. (E and G) US molecular imaging of OPN@PFP-DiR NPs after irradiation by LIFU for 3 min under different condition (1 W cm−2, 2 W cm−2, 3 W cm−2 and 4 W cm−2). (F and H) Gray-scale values of CEUS-mode imaging of OPN@PFP-DiR NPs after LIFU irradiation under different frequency and duration time, as shown in the corresponding figure. | ||
NIR and US imaging of the NPs in vitro
In vitro NIR and US imaging characteristics of OPN@PFP-DiR NPs were examined. DiR is a widely used fluorescent dye.33,42 The fluorescence of OPN@PFP-DiR NPs with varying DiR concentrations was examined to describe their fluorescent characteristics. Self-quenching was found when the DiR concentration above 40 μg mL−1, which was consistent with the other NIR dyes43 (Fig. 3C and D). Additionally, the potential of OPN@PFP-DiR NPs for US imaging was investigated by quantifying the ultrasound values derived from PFP. The impetus of LIFU at different power densities and durations resulted in a progressive amplification of echo intensity ranging from 1 to 3 W cm−2 for 3 min. After reaching its peak at 3 W cm−2 and then decreasing at the 4W cm−2 (Fig. 3E and F). In the 3 W cm−2 group, the echo intensity demonstrated a linear signal enhancement over time, reaching its maximum after a 4 minutes exposure to LIFU (Fig. 3G and H). These findings offer substantiation for the effectiveness of LIFU in stimulating the phase transition of PFP-based NPs, thereby establishing the groundwork for ultrasound imaging of cardiac fibrosis.Cardiac fibrosis after myocardial infarction
The ligation of anterior descending coronary artery was performed to successfully establish a model of myocardial infarction. Following two weeks, the presence of ST-segment elevation in the ECG confirmed the successful establishment of the MI model (Fig. 4A). Furthermore, echocardiography was conducted to assess the cardiac function of MI mice (Fig. 4B). There was a notable increase in the left ventricular internal diameter in diastole (LVIDd) (Fig. 4C). Additionally, the results revealed a significant reduction in both the ejection fraction (EF) and fractional shortening (FS)(Fig. 4D and E). All the above-mentioned discoveries suggested the successful construction of MI-model mice for in vivo cardiac fibrosis. | ||
| Fig. 4 MI model mice were successfully constructed. (A) ECG before (left) and after (right) modeling. (B) Echocardiograms before (left) and after (right) modeling. Cardiac function indicators including left ventricular internal dimension in diastole (LVIDd) (C), ejection fraction (EF%) (D) and fractional shortening (FS%) (E) were determined by echocardiography. | ||
Ex vivo OPN-FITC targeted ability and quantitative analysis
H&E (Fig. 5Aa), Masson (Fig. 5Ab) and PSR (Fig. 5Ac) staining, were performed on cardiac tissue slices, among them, hematoxylin stains for cell nuclei, eosin stains for extracellular matrix and cytoplasm, Masson staining for keratin and muscle fibers, collagen and bone, and cell nuclei, and PSR staining for collagen. The results showed that the model group had markedly worsened cardiac fibrosis and myocardial cell injury compared with control group. Meanwhile, frozen slices of the mouse myocardium with and without myocardial fibrosis were used in the ex vivo experiment. FITC labeled OPN exhibited more green fluorescence signal under CLSM while the myocardium of the normal mouse demonstrated a very minor quantity of green fluorescence (Fig. 5B) and then the semiquantitative analysis of Masson and PSR staining conducted by each group yielded results that were consistent with the characterization of OPN expression obtained through CLSM (Fig. 5C). Through this ex vivo investigation, we were able to validate the accumulation of OPN at the site of myocardial fibrosis. Conversely, no notable accumulation was observed in the nonfibrotic region, establishing a foundation for forthcoming in vivo investigations. | ||
| Fig. 5 The staining outcomes of myocardial fibrosis in myocardial infarction and normal group, myocardial tissues were assessed using various techniques, including (A) HE staining (Aa), Masson staining (Ab), and Sirius red staining (Ac). Notably, the infarcted myocardium exhibited significant collagen fibrosis. (B) FITC-OPN staining was employed to compare myocardial tissue between the myocardial infarction and normal groups. (C) A semiquantitative analysis was conducted to evaluate the staining results of Masson staining, PSR staining, and FITC-OPN staining, comparing the control group with the experimental group. | ||
NIR and US imaging of the OPN@PFP-DiR NPs in vivo
Subsequently, dual-modal imaging characteristics of NIR/US of the OPN@PFP-DiR NPs were assessed in vivo. All procedures were performed under anesthesia using 2% isoflurane. MI mice were intravenously administered with saline, PFP-DiR NPs and OPN@PFP-DiR NPs, each with an equivalent dosage of DiR. After 24 h, the mice were autopsied to collect major organs for ex vivo fluorescent imaging. The infarcted hearts that received OPN@PFP-DiR NPs exhibited a stronger fluorescent signal compared to those that received PFP-DiR NPs consistent with quantitative results, respectively (Fig. 6A and B), which validated the notable heart targeting ability of OPN@PFP-DiR NPs. In contrast, a higher level of nanoparticle accumulation was observed in the livers of animals administered with PFP-DiR NPs in comparison to other organs, indicating a notable elimination of nanoparticles by the liver macrophages. Because the liver has a far more abundant aggregation of nanomaterials than the myocardial site, NIR imaging at the heart site is therefore not very visible. This also explains why, similar to the earlier study,34,44ex vivo imaging is a common technique for fluorescence imaging.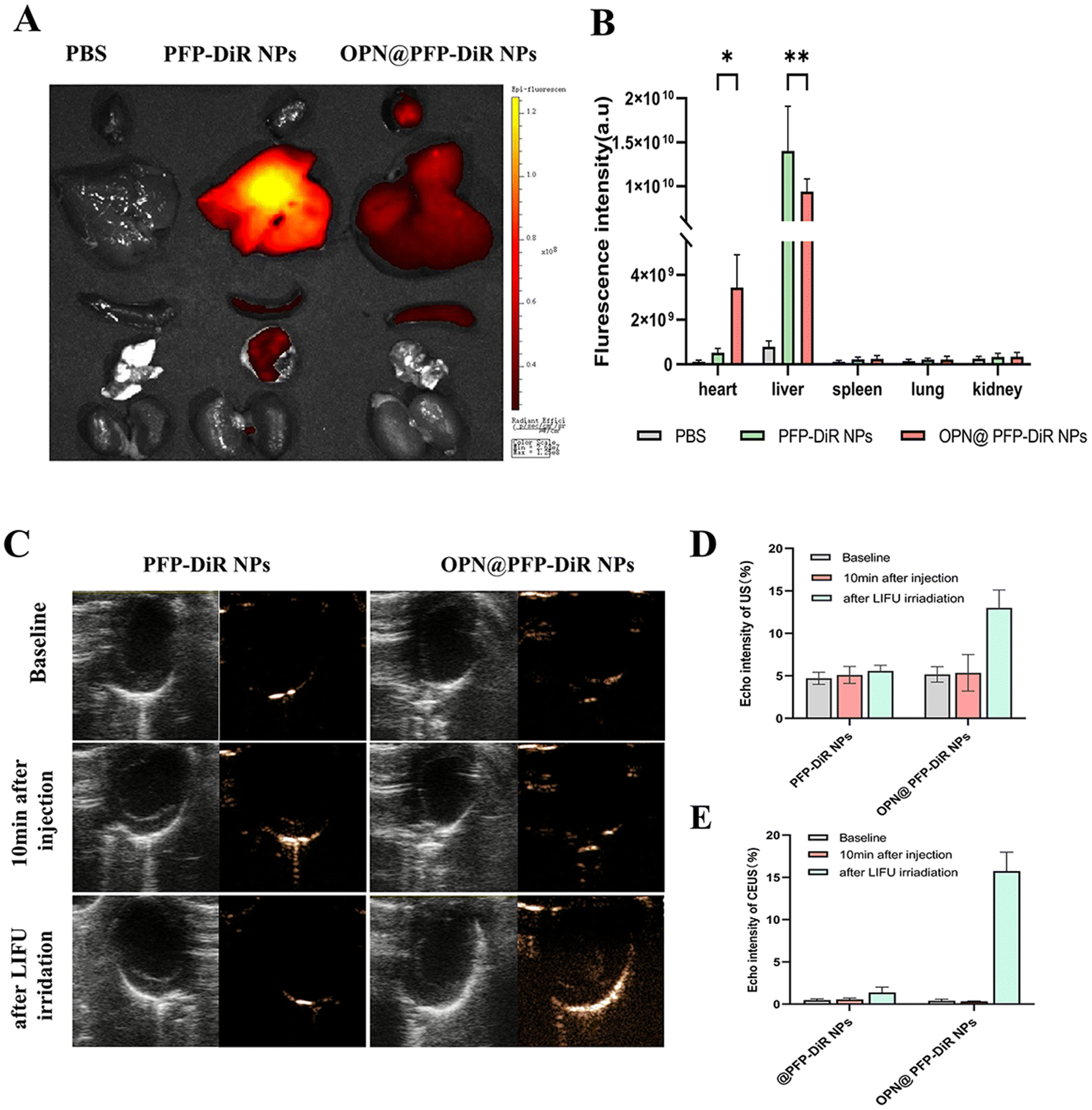 | ||
| Fig. 6 The performance of in vivo NIR and US imaging is evaluated. (A) NIR images are obtained. Representative ex vivo fluorescent imaging of mouse organs (heart, lung, liver, kidney, and spleen) at 24 h post-intravenous injections of OPN@PFP-DiR NPs and PFP-DiR NPs. (B) Quantitative analysis of fluorescent intensities (n = 3 per group). The utilization of B-Mode and CEUS imaging (C), as well as (D and E) the assessment of echo intensities before and after LIFU irradiation (3 W cm−2, 10 min) for both PFP-DiR NPs and OPN@PFP-DiR NPs. | ||
Through intravenous injection of targeting OPN@PFP-DiR NPs and non-targeting PFP-DiR NPs in MI model, the in vivo confirmation of the cardiac fibrosis-targeting US imaging capacity of OPN@PFP-DiR NPs was accomplished considering its exceptional performance during in vitro US imaging. As shown in Fig. 6C, echo intensity analysis demonstrated that the acquired images had superior gray scale intensity at a frequency of 3 W cm−2. This finding provides evidence that the frequency of LIFU is critical in boosting phase transition. Therefore, a frequency of 3 W cm−2 was chosen for subsequent research due to its suitability for ultrasonography with OPN@PFP-DiR NPs and its ability to prevent thermal injury to the skin. The post-injection imaging performance of all groups with LIFU impetus was researched. Prior to LIFU irradiation, no ultrasound enhancement was observed in the PFP-DiR NPs and OPN@PFP-DiR NPs group, indicating that these nanoagents had limited ability to spontaneously undergo a phase transition in vivo. However, LIFU-triggered OPN@PFP-DiR NPs exhibited significantly enhanced capability for US molecular imaging. Furthermore, it was observed that the echo intensity values were considerably higher in the OPN@PFP-DiR NPs + LIFU group compared to the PFP-DiR NPs + LIFU group (Fig. 6D), indicating successful cardiac accumulation. These experimental outcomes confirm that OPN@PFP-DiR NPs have the ability to traverse the vascular endothelium, specifically bind to myocardial fibrosis, and enhance US imaging signals. Conversely, nanoparticles lacking OPN modification were unable to target the fibrotic myocardium, resulting in the aforementioned challenges. Meanwhile, PFP has a boiling point of 29 °C, theoretically making it liquid at room temperature but gaseous at body temperature.42,45 However, the temperature at which the nanoparticles undergo phase change was increased upon their encapsulation by PLGA shell. Previous research has demonstrated that smaller particle sizes necessitate higher temperatures for phase transition.46 Consequently, the encapsulated PFP does not undergo phase transformation at the average body temperature of approximately 37 °C. Additional investigation is necessary to optimize the nanodroplets in order to decrease their vaporization threshold within living organisms. This optimization would specifically enhance the vaporization of nanodroplets in targeted tissues, as their current imaging enhancement is comparatively inferior to that of microbubbles, despite the administration of higher quantities of nanodroplets.
Toxicity of and safety evaluation of NPs in vitro and vivo
Even though numerous studies have shown that PLGA-based nanoparticles with adequate surface modification have low cytotoxicity.7,47 Till, careful evaluations are required because cytotoxicity is not only controlled by the chemical composition of the particle, but also by the surface physicochemical features, which control how nanoparticles interact with cells.48 As depicted in Fig. 7A, no significant cytotoxicity was detected in all concentrations of OPN@PFP-DiR NPs following 24 h incubation. Meanwhile, no damage was seen in any of the organs after 48 h after OPN@PFP-DiR NPs injection (Fig. 7B), confirming the comparatively great biocompatibility of as-prepared nanoprobes. Taken together, these findings clearly provide significant promise for enabling low-dose, very effective targeted delivery to myocardial fibrosis. Further research is necessary to determine the exact mechanism underlying the preferential binding and internalization of OPN peptide in cardiac fibrosis. Furthermore, we will plan additional experiments to investigate the dual mode of imaging features of OPN@PFP-DiR NPs in larger animals. The goal of this work is to break through the current barrier in the clinical and experimental translation of targeted therapy for pathological cardiac diseases. | ||
| Fig. 7 The in vitro and in vivo toxicity and safety assessment of nanoparticles (OPN@PFP-DiR NPs). (A) Morphology of normal H9C2 cells and cardiac fibroblasts (CFs) after co-incubation with OPN@PFP-DiR NPs. Scale bars, 200 μm. (B) The cytotoxicity of NPs on cells was evaluated after a 24 hours incubation period. (C) The main organs, namely the heart, lung, liver, kidney, and spleen, were subjected to HE staining under a light microscope 48 h following the injection of OPN@PFP-DiR NPs. No observable damage was detected in any of the organs. | ||
Conclusion
In conclusion, an OPN targeting nanoparticles (OPN@PFP-DiR NPs) has been successfully developed to enable dual-modality imaging capabilities, facilitating the direct visual diagnosis of cardiac fibrosis at the molecular level. Drawing inspiration from that OPN is prominently expressed in cardiac fibrosis tissue, OPN-modified nanoprobes exhibited exceptional targeting abilities specifically for cardiac fibrosis in a MI model. The encapsulated PFP can undergo a transformation into microbubbles due to LIFU-induced thermal effect, thereby endowing the nanoprobes with significantly enhanced US molecular imaging capacity for cardiac fibrosis imaging. Through in vitro and in vivo experiments, we further validated that nanoprobes presented to a fluorescence imaging potential, enabling real-time NIR and US molecular imaging monitoring and allowing precise detection of early-stage cardiac fibrosis. Together, this work addresses the obstacles related to the detection of cardiac fibrosis in clinical practice, and render the OPN@PFP-DiR NPs highly promising for molecular diagnosis of cardiac fibrosis and noninvasive evaluation of early clinical cardiovascular events, thereby advancing the field of precision medicine.Conflicts of interest
The authors declare no financial or otherwise that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the financial support through the National Natural Science Foundation of China (82001831, 82271893, 82071932, 82371974, 82202169), Shaanxi Province Qin Chuangyuan “scientist + engineer” team construction (2022KXJ-123).References
- T. Senra, B. M. Ianni, A. C. P. Costa, C. Mady, M. Martinelli-Filho, R. Kalil-Filho and C. E. Rochitte, J. Am. Coll. Cardiol., 2018, 72, 2577–2587 CrossRef PubMed.
- H. Kurose, Cells, 2021, 10, 1716 CrossRef CAS PubMed.
- S. Neuber, M. R. Ermer, M. Y. Emmert and T. Z. Nazari-Shafti, Int. J. Mol. Sci., 2023, 24, 10480 CrossRef CAS PubMed.
- J. G. Travers, F. A. Kamal, J. Robbins, K. E. Yutzey and B. C. Blaxall, Circ. Res., 2016, 118, 1021–1040 CrossRef CAS PubMed.
- N. G. Frangogiannis, Cardiovasc. Res., 2021, 117, 1450–1488 CrossRef CAS PubMed.
- A. Sonaglioni, G. L. Nicolosi, E. Rigamonti, M. Lombardo and L. La Sala, Int. J. Mol. Sci., 2022, 23, 10944 CrossRef CAS PubMed.
- A. K. Barton, E. Tzolos, R. Bing, T. Singh, W. Weber, M. Schwaiger, Z. Varasteh, R. Slart, D. E. Newby and M. R. Dweck, Eur. Heart J. Cardiovasc. Imaging, 2023, 24, 261–275 CrossRef PubMed.
- F. P. de Carvalho and C. F. Azevedo, Radiographics, 2020, 40, 336–353 CrossRef PubMed.
- W. Chen and J. Jeudy, Curr. Cardiol. Rep., 2019, 21, 76 CrossRef PubMed.
- H. Bulluck, S. K. White, G. M. Frohlich, S. G. Casson, C. O'Meara, A. Newton, J. Nicholas, P. Weale, S. M. Wan, A. Sirker, J. C. Moon, D. M. Yellon, A. Groves, L. Menezes and D. J. Hausenloy, Circ. Cardiovasc. Imaging, 2016, 9, e003900 Search PubMed.
- X. Zhang, Q. Zhao, J. Yang, T. Wang, F. Chen and K. Zhang, Coord. Chem. Rev., 2023, 484, 215115 CrossRef CAS.
- Z. Weng, J. Yao, R. H. Chan, J. He, X. Yang, Y. Zhou and Y. He, JACC Cardiovasc. Imaging, 2016, 9, 1392–1402 CrossRef PubMed.
- K. G. Ordovas and C. B. Higgins, Radiology, 2011, 261, 2359–2374 Search PubMed.
- T. Wang, X. Zhang, Y. Xu, Y. Xu, Y. Zhang and K. Zhang, J. Mater. Chem. B, 2022, 10, 7361–7383 RSC.
- T. T. Le, W. Huang, G. K. Singh, D. F. Toh, S. H. Ewe, H. C. Tang, G. Loo, J. A. Bryant, B. Ang, E. L. Tay, W. M. Soo, J. W. Yip, Y. Y. Oon, L. Gong, J. B. Lunaria, Q. W. Yong, E. M. Lee, P. S. D. Yeo, S. C. Chai, P. P. Goh, L. F. Ling, H. Y. Ong, A. M. Richards, V. Delgado, J. J. Bax, Z. P. Ding, L. H. Ling and C. W. L. Chin, Front. cardiovasc. med., 2021, 8, 750016 CrossRef CAS PubMed.
- G. Shiha, A. Ibrahim, A. Helmy, S. K. Sarin, M. Omata, A. Kumar, D. Bernstien, H. Maruyama, V. Saraswat, Y. Chawla, S. Hamid, Z. Abbas, P. Bedossa, P. Sakhuja, M. Elmahatab, S. G. Lim, L. Lesmana, J. Sollano, J. D. Jia, B. Abbas, A. Omar, B. Sharma, D. Payawal, A. Abdallah, A. Serwah, A. Hamed, A. Elsayed, A. AbdelMaqsod, T. Hassanein, A. Ihab, G. H. H, N. Zein and M. Kumar, Hepatol. Int., 2017, 11, 1–30 CrossRef PubMed.
- S. B. Montesi, P. Desogere, B. C. Fuchs and P. Caravan, J. Clin. Invest., 2019, 129, 24–33 CrossRef PubMed.
- A. Pucci, A. Aimo, V. Musetti, X. Barison, G. Vergaro, D. Genovesi, A. Giorgetti, S. Masotti, C. Arzilli, C. Prontera, L. E. Pastormerlo, M. A. Coceani, M. Ciardetti, N. Martini, C. Palmieri, C. Passino, C. Rapezzi and M. Emdin, J. Am. Heart Assoc., 2021, 10, e020358 CrossRef CAS PubMed.
- J. Xu, E. Reznik, H. J. Lee, G. Gundem, P. Jonsson, J. Sarungbam, A. Bialik, F. Sanchez-Vega, C. J. Creighton, J. Hoekstra, L. Zhang, P. Sajjakulnukit, D. Kremer, Z. Tolstyka, J. Casuscelli, S. Stirdivant, J. Tang, N. Schultz, P. Jeng, Y. Dong, W. Su, E. H. Cheng, P. Russo, J. A. Coleman, E. Papaemmanuil, Y. B. Chen, V. E. Reuter, C. Sander, S. R. Kennedy, J. J. Hsieh, C. A. Lyssiotis, S. K. Tickoo and A. A. Hakimi, Elife, 2019, 8, e38986 CrossRef PubMed.
- P. Desogere, S. B. Montesi and P. Caravan, Chemistry, 2019, 25, 1128–1141 CrossRef CAS PubMed.
- K. H. Jung and K. H. Lee, J. Pathol. Transl. Med., 2015, 49, 5–12 CrossRef PubMed.
- X. Zhang, Z. Zeng, H. Liu, L. Xu, X. Sun, J. Xu and G. Song, Nanoscale, 2022, 14, 3306–3323 RSC.
- D. T. Fetzer, Radiology, 2023, 307, e222735 CrossRef PubMed.
- H. He, X. Zhang, L. Du, M. Ye, Y. Lu, J. Xue, J. Wu and X. Shuai, Adv. Drug Deliv. Rev., 2022, 186, 114320 CrossRef CAS PubMed.
- K. Tang, C. Niu, Y. Xu, Y. Zhu, S. Tang, M. Zhang and Q. Zhou, RSC Adv., 2018, 8, 5407–5419 RSC.
- Y. Dai, X. Sha, X. Song, X. Zhang, M. Xing, S. Liu, K. Xu and J. Li, Int. J. Nanomed., 2022, 17, 5413–5429 CrossRef CAS PubMed.
- F. Zeng, Z. Fan, S. Li, L. Li, T. Sun, Y. Qiu, L. Nie and G. Huang, ACS Nano, 2023, 17, 19753–19766 CrossRef CAS PubMed.
- G. Selvaggio and S. Kruss, Nanoscale, 2022, 14, 9553–9575 RSC.
- R. Jiao, X. Lin, Q. Zhang, Y. Zhang, W. Qin, Q. Yang, C. Xu, F. Chen and K. Zhang, Prog. Mater. Sci., 2024, 142, 101230 CrossRef CAS.
- I. Abdelaziz Mohamed, A. P. Gadeau, A. Hasan, N. Abdulrahman and F. Mraiche, Cells, 2019, 8, 1558 CrossRef PubMed.
- F. Li, L. Chen, S. Zhong, J. Chen, Y. Cao, H. Yu, H. Ran, Y. Yin, C. Reutelingsperger, S. Shu and Z. Ling, ACS Nano, 2024, 18, 4886–4902 CrossRef CAS PubMed.
- Q. Zhou, Y. Zeng, Q. Xiong, S. Zhong, P. Li, H. Ran, Y. Yin, C. Reutelingsperger, F. W. Prinze and Z. Ling, ACS Appl. Mater. Interfaces, 2019, 11, 23006–23017 CrossRef CAS PubMed.
- J. Guan, Y. Wu, H. Wang, H. Zeng, Z. Li and X. Yang, Nanoscale, 2021, 13, 19399–19411 RSC.
- J. E. Lemaster, F. Chen, T. Kim, A. Hariri and J. V. Jokerst, ACS Appl. Nano Mater., 2018, 1, 1321–1331 CrossRef CAS PubMed.
- Y. Zhong, M. Ye, L. Huang, L. Hu, F. Li, Q. Ni, J. Zhong, H. Wu, F. Xu, J. Xu, X. He, Z. Wang, H. Ran, Y. Wu, D. Guo and X. J. Liang, Adv. Mater., 2022, 34, e2109955 CrossRef PubMed.
- Y. Zhong, Y. Zhang, J. Xu, J. Zhou, J. Liu, M. Ye, L. Zhang, B. Qiao, Z. G. Wang, H. T. Ran and D. Guo, ACS Nano, 2019, 13, 3387–3403 CrossRef CAS PubMed.
- S. McLaughlin, V. Sedlakova, Q. Zhang, B. McNeill, D. Smyth, R. Seymour, D. R. Davis, M. Ruel, M. Brand, E. I. Alarcon and E. J. Suuronen, Adv. Funct. Mater., 2022, 32, 2204 CrossRef.
- J. Dolai, K. Mandal and N. R. Jana, ACS Appl. Nano Mater., 2021, 4, 6471–6496 CrossRef CAS.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed.
- J. Xu, J. Zhou, Y. Zhong, Y. Zhang, J. Liu, Y. Chen, L. Deng, D. Sheng, Z. Wang, H. Ran and D. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 42525–42535 CrossRef CAS PubMed.
- X. Zhao, W. Luo, J. Hu, L. Zuo, J. Wang, R. Hu, B. Wang, L. Xu, J. Li, M. Wu, P. Li and L. Liu, J. Nanobiotechnol., 2018, 16(36), 1–4 Search PubMed.
- Y. Li, Y. Wu, J. Chen, J. Wan, C. Xiao, J. Guan, X. Song, S. Li, M. Zhang, H. Cui, T. Li, X. Yang, Z. Li and X. Yang, Nano Lett., 2019, 19, 5806–5817 CrossRef CAS PubMed.
- D. Sheng, T. Liu, L. Deng, L. Zhang, X. Li, J. Xu, L. Hao, P. Li, H. Ran, H. Chen and Z. Wang, Biomaterials, 2018, 165, 1–13 CrossRef CAS PubMed.
- X. Liu, B. Chen, J. Chen, X. Wang, X. Dai, Y. Li, H. Zhou, L. M. Wu, Z. Liu and Y. Yang, Adv. Mater., 2024, 36, e2308477 CrossRef PubMed.
- L. Bin, Y. Du, Y. Zhang, Q. Xiao, X. Chen, Z. Liu and Z. Du, J. Drug Delivery Sci. Technol., 2022, 78, 103989 CrossRef CAS.
- N. Rapoport, D. A. Christensen, A. M. Kennedy and K. H. Nam, Ultrasound Med. Biol., 2010, 36, 419–429 CrossRef PubMed.
- X. Zhao, X. Wang, J. Wang, J. Yuan, J. Zhang, X. Zhu, C. Lei, Q. Yang, B. Wang, F. Cao and L. Liu, Int. J. Nanomed., 2020, 15, 1321–1333 CrossRef CAS PubMed.
- R. Qiao, H. Qiao, Y. Zhang, Y. Wang, C. Chi, J. Tian, L. Zhang, F. Cao and M. Gao, ACS Nano, 2017, 11, 1816–1825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00042k |
| This journal is © The Royal Society of Chemistry 2024 |