
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of a ruthenium complex covalently bonded to multilayer graphene and its evaluation as a photocatalyst†‡
Lesly V.
Rodríguez-Flórez§
ab,
María
de Gracia Retamosa
ab,
Miriam
Navlani-García¶
c,
Diego
Cazorla-Amorós
 *c,
Carmen
Nájera
b,
Miguel
Yus
b and
José M.
Sansano
*a
*c,
Carmen
Nájera
b,
Miguel
Yus
b and
José M.
Sansano
*a
aDepartamento de Química Orgánica e Instituto de Síntesis Orgánica (ISO), University of Alicante, E-03080 Alicante, Spain. E-mail: jmsansano@ua.es
bCentro de Innovación en Química Avanzada (ORFEO-CINQA), University of Alicante, E-03080 Alicante, Spain
cDepartamento de Química Inorgánica and Instituto Universitario de Materiales, University of Alicante, E-03080 Alicante, Spain. E-mail: cazorla@ua.es
First published on 9th February 2024
Abstract
Multilayer graphene (MLG), obtained by mild sonication of graphite in NMP, was functionalised via 1,3-dipolar cycloaddition with azomethine ylides generated by thermal 1,2-prototropy from various imino esters. The microwave-assisted functionalisation took place in five hours at 100 °C. The resulting MLG, containing substituted proline-based amine functional groups, was characterized using XPS and showed a nitrogen loading three times that obtained for the same transformation performed for five days using convection-assisted heating. The preparation of the imino ester containing a bipyridine unit at the arylidene position allowed for the preparation of the corresponding functionalised MLG, which incorporated the ruthenium atom to achieve a heterogeneous MLG-Ru complex. This supported complex was tested, as a proof of concept, as a photocatalyst of the aerobic oxidative hydroxylation of 4-methoxyphenylboronic acid.
The association of transition metal catalysis with photocatalysis (known as metallaphotocatalysis)1,2 gives access to unique very reactive intermediate species (impossible to generate using other protocols) under mild conditions. Versatility, modulation, scope and tolerance are also important features of these catalysed transformations. In addition, these processes are complementary to those traditionally used in the field of transition metal catalysis. The new frontiers of this area have been made to involve, among other features, ready access to robust and recoverable photocatalysts3 in order to minimize costs and operational work, which would be very attractive from the industrial point of view.4 Support materials to covalently bond the metal have been sought, and carbon materials constitute one of the preferred types of such materials nowadays. In this regard, graphene is a very useful and robust atomic-scale scaffold for the design of new nanomaterials. Due to the electrical, optical and mechanical properties of graphene,5–10 functionalisation of these planar structures can produce many useful synergies.11 Its covalent functionalisation, however, is not very simple because of its low reactivity. So, harsh reaction conditions (pressure, temperature, etc.) are usually required for this purpose. It is well known that the reactivity also depends on the type of graphene-based material used as a support,12–14 and on the number of graphene layers.15–17
In most of the examples reported in the literature, multilayer graphene (MLG)18 can be used as 4pi or 2pi components of [4 + 2] cycloadditions19,20 in the presence of very reactive dienophiles or dienes, respectively. However, only [3 + 2] cycloadditions involving fleeting azomethine ylides, generated in situ via the decarboxylation route (also called the Prato reaction),21–23 have been found to allow the introduction of a secondary amine as a functional group.24–32 This type of functionalisation takes place on the basal plane of the sheets and not on the edges, thus allowing a more complete exploitation of the support surface.21 The synthesis of the graphene-based material often requires ultrasound-mediated dispersion (liquid exfoliation) of graphite in appropriate organic solvents such as pyridine30 or N-methylpyrrolidone (NMP).33,34 The reaction with a large excess of N-alkylglycines and the corresponding aldehyde in a multicomponent sequence at 130–180 °C for 3–5 days affords the prolinate-functionalised graphene. Our group reported the first thermal 1,2-prototropy of imino esters to MLG, achieved at 90 °C for 5 days and in a totally atom-economical process as no decarboxylation occurred during the generation of the intermediate azomethine ylide (Scheme 1a).35
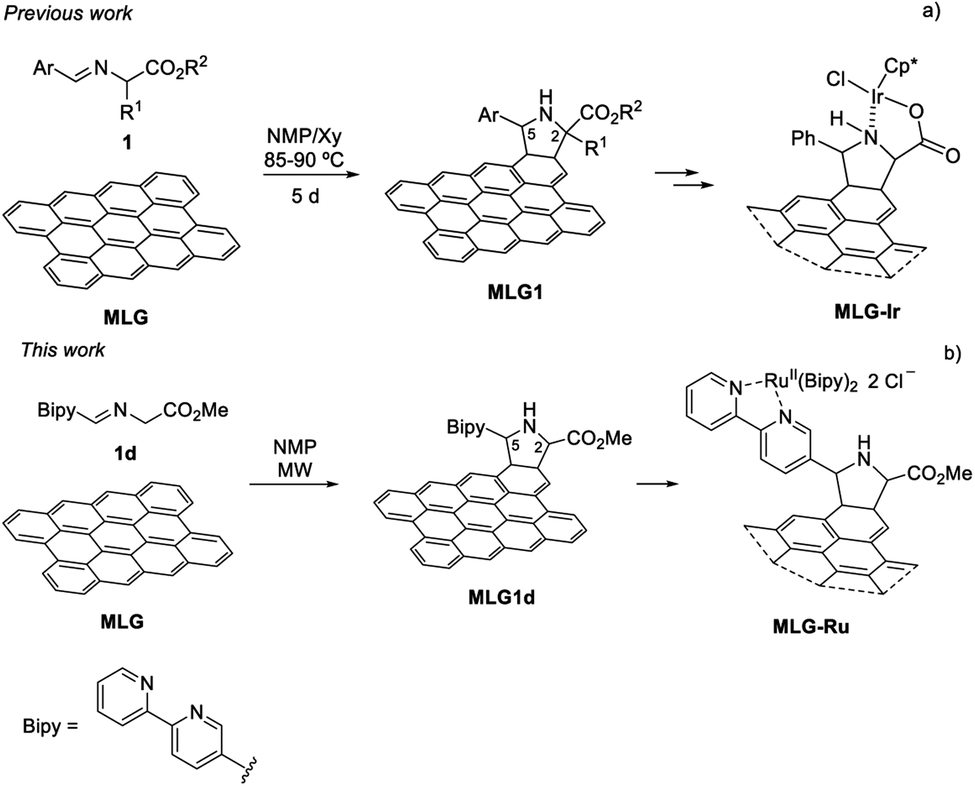 | ||
| Scheme 1 Synthesis of functionalised MLG from imino esters 1. | ||
In this contribution, the functionalisation of MLG using a 1,3-dipolar cycloaddition (1,3-DC) of azomethine ylides under microwave (MW)-assisted heating was studied and compared with the analogous process carried out using convection-assisted heating. With this strategy, it was possible to anchor a 2,2′-bipyridine unit to the ruthenium atom (Scheme 1b). The final goal of this metal-graphene-supported material was its application as a photocatalyst in a proof of concept.
The overall incorporation of nitrogen into pristine MLG,36 under conventional heating (90 °C) in NMP after 5 days was 0.37% (ref. 35 and 37) (Table 1, entry 1). With this reference we started a microwave-assisted heating survey of reactions carried out for 5 h at the same temperature (Table 1, entry 2). Here, a notable increase in the functionalisation was observed (1.00% nitrogen content, with all such contents determined using thermogravimetric analysis) when only using 120 mg of the starting imino ester 1, instead of 100 mg used per day in the mentioned convection-assisted heating. This value encouraged us to study the limit of the functionalisation of MLG under these conditions.38,39
|
|
||||||
|---|---|---|---|---|---|---|
| Ent. | 1 | Heating | T (°C) | t (h) | ML1 | N (%) |
| a A mass of 15 mg of graphene (in 50 mL of NMP), addition of imino ester 120 mg. b Determined from TG analysis. c CH = convection-assisted heating. Addition of 120 mg of imino ester per day. d A mass of 100 mg of imino ester 1a was added. e A mass of 150 mg of imino ester 1a was added. | ||||||
| 1 | 1a | CHc | 90 | 120 | MLG1a | 0.37 (ref. 35) |
| 2 | 1a | MW | 90 | 5 | MLG1a | 1.00 |
| 3 | 1a | MW | 100 | 5 | MLG1a | 1.45 |
| 4 | 1a | MW | 100 | 6 | MLG1a | 1.32 |
| 5 | 1a | MW | 100 | 8 | MLG1a | 0.52 |
| 6 | 1a | MW | 100 | 1 | MLG1a | 0.32 |
| 7 | 1a | MW | 100 | 3 | MLG1a | 0.69 |
| 8 | 1a | MW | 100 | 5 | MLG1a | 1.38 |
| 9 | 1a | MW | 100 | 5 | MLG1a | 1.44 |
| 10 | 1a | MW | 110 | 5 | MLG1a | 1.43 |
| 11 | 1a | MW | 120 | 5 | MLG1a | 1.23 |
| 12 | 1b | MW | 100 | 5 | MLG1b | 2.28 |
| 13 | 1c | MW | 100 | 5 | MLG1c | 1.35 |
When increasing the temperature to 100 °C, and keeping the reaction time at 5 h, the incorporation of nitrogen was 1.45% (Table 1, entry 3). After heating the reaction mixture for 6 or 8 h, a mass loss was detected (Table 1, entries 3 and 4). It seemed that the MW energy could induce some degradation of the organic material covalently bonded to the MLG. This result was observed in our previous research when allowing the reaction to proceed for more than 5 d. These reactions performed for 6 and 8 h were repeated three times and the results were essentially identical. Immediately, the reactions carried out for 1 and 3 h were analysed. In both cases, the extent of the organic functionalization was lower than that when the reaction was run for 5 h. So, neither longer reaction times (6 and 8 h) nor a shorter one (3 h) increased the nitrogen content during the functionalisation of MLG (Table 1, entries 4–7). Adding 100 mg of imino ester 1a did not increase the functionalisation level (Table 1, entry 8). In addition, with 150 mg of imino ester 1a, the incorporation of the nitrogen atoms was very similar to the transformation run using 100 mg of 1a (Table 1, entry 9). Increasing the temperature to 120 °C, while keeping the reaction time at 5 h, was not beneficial for the reaction of graphene with imino ester 1a (Table 1, entry 11). So, submitting a suspension of graphene in NMP (50 mL, 0.1 mg mL−1), containing 120 mg of imino ester 1a, to microwave irradiation (reaching 100 °C) for 5 h constituted the optimal set of reaction parameters to achieve a very good functionalisation of graphene.
These conditions were applied to another two imino esters just to test the reproducibility of this procedure. Specifically, the 2-pyridyl-substituted glycine-derived imino ester 1b and its alanine analog 1c were tested as azomethine ylide precursors in this 1,3-DC (Table 1, entries 12 and 13). For the reaction of substrate 1b, the nitrogen content (2.28%) observed was less than double that observed for the same transformation run with imino ester 1a (compare entries 3 and 12 of Table 1). The alaninate 1c afforded a slightly lower functionalisation (1.35%) in MLG1c (Table 1, entry 13), possibly due to a higher steric hinderance.
Once the microwave-assisted functionalisation was optimized, the synthesis of the 2,2′-bipyriridine-derived imino ester 1d was undertaken. The chemical yield of aldehyde precursor 4 was extremely low,40 so a modification of it was designed in order to obtain this aldehyde in large quantities and higher yields. For this purpose, reactions from several references were selected and combined, as shown in Scheme 2. 2-(Iodoacetyl)pyridine40 was not used due to the waste material generated in this step. 2-(Bromoacetyl)pyridine41 was used instead, affording cleanly the corresponding pyridinium salt 2, in 82% yield, after treatment with pyridine at rt.41 Bipyridine intermediate 3 was isolated in a 38% yield by reacting 2 with crotonaldehyde in the presence of ammonium acetate.42 Aldehyde 4 was generated (65%) by subjecting 3 to a benzylic oxidation with selenium oxide, following the published procedure (Scheme 2).43 Imino ester 1d was formed quantitatively, after generation of the free base of Gly-OMe, in dichloromethane at rt44 (Scheme 2).
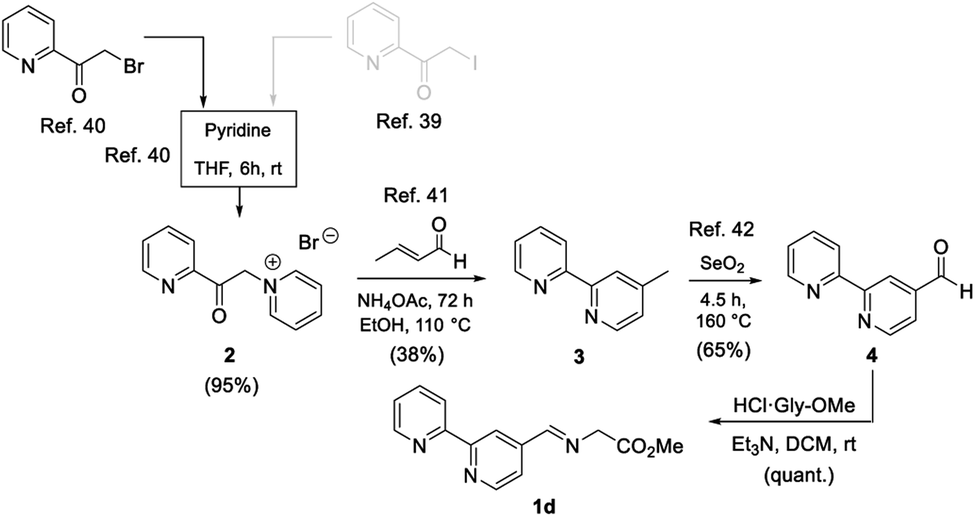 | ||
| Scheme 2 Synthesis of imino ester 1d. | ||
The synthesis of the ruthenium-supported graphene photocatalyst (MLG-Ru) was performed by achieving a thermal 1,2-prototropy-mediated 1,3-DC between pristine graphene and 1d under standard conditions, giving rise specifically to MLG1d, which incorporated 4.01% nitrogen atoms. In the second step, MLG1d was allowed to react with Ru(Bpy)2Cl2·2H2O complex in tetrachloroethane (40 mg of MLG1d in 20 mL) under reflux for 48 h45 (Scheme 3). The amount of ruthenium, determined from an ICP analysis, was 0.80%, which revealed that approximately 60% of ruthenium atoms were coordinated by a bipyridine unit covalently bonded to MLG.
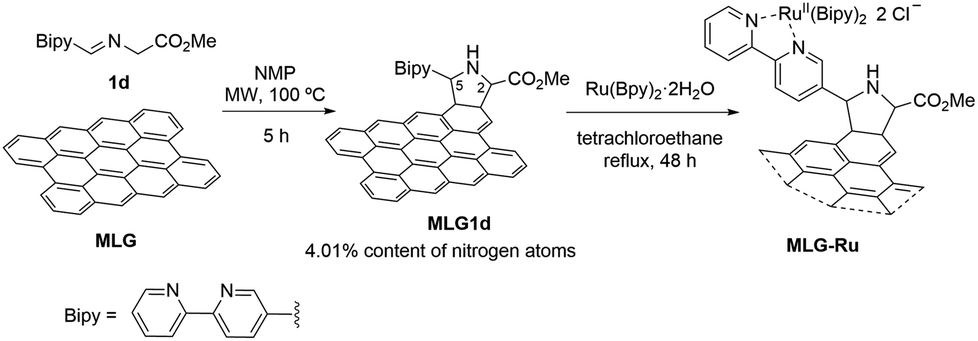 | ||
| Scheme 3 Synthesis of functionalised MLG-Ru from MLG1d. | ||
As a proof of concept, an evaluation of the catalytic activity of this heterogeneous MLG-Ru complex in the aerobic oxidative hydroxylation of arylboronic acids46 was carried out. For this purpose, 4-methoxyphenylboronic acid (5, 10 mg, 0.07 mmol) was selected as the starting material to undergo this oxidation to the corresponding 4-methoxyphenol (6) with 20 mg of MLG-Ru complex (0.80%, 1.5 × 10−3 mmol, 2 mol% Ru), under an air atmosphere and irradiation with white LED light (14 W), and using diisopropylethylamine (DIPEA, 0.14 mmol, 2 equiv.) and DMF as solvent. Initially, very disappointing results were obtained when concentrated solutions of 5 (0.70 and 0.14 M) and MLG-Ru were employed (Table 2, entries 1 and 2). More diluted suspensions in DMF (0.07 and 0.04 M of 5) afforded partial conversions at 36 h (Table 2, entries 3 and 4).47 Perhaps, the low transparency of the most concentrated dense black suspensions impeded an efficient transmission of light to the reactive site. For this reason, the reaction was more sluggish than the published same reaction with Ru(Bpy)3Cl2·2H2O where an almost complete conversion at room temperature was observed at 72 h (Table 2, entry 5). The 0.07 M concentration was selected rather than 0.04 M because smaller amounts of DMF were required. Several control experiments were designed to demonstrate the necessity of all of the components involved. The absence of MLG-Ru catalyst (Table 2, entries 6–8), air (Table 2, entry 9), triethylamine (Table 2, entry 10), and white LED light (Table 2, entries 11 and 12) in each case yielded no reaction. As shown in the last entry of Table 2, we also tested the recycling of the heterogeneous catalyst under the standard conditions described in entry 5 of Table 2. The reaction was reproducible and the structure of the catalyst remained intact according to microscopy, XPS and ICP mass spectrometry analyses (see ESI‡).
|
|
||||||
|---|---|---|---|---|---|---|
| Ent. | DMF | [5]b | t (h) | C. E.c | 6 (%) | 6 (%) |
| a A mass of 20 mg of MLG-Ru, in DMF, 5 (0.07 mmol), DIPEA (0.14 mmol), white LED light (14 W), and air atmosphere at room temperature. nr = no reaction. b In mol L−1. c C. E. = control experiments. d Conversions determined from analysis of crude 1H NMR spectra. e Isolated yield after flash chromatography. f MLG was used instead of MLG-Ru. g MLG-1d was used instead of MLG-Ru. h Recovered MLG-Ru from entry 5 was reused in a new reaction. | ||||||
| 1 | 0.1 mL | 0.70 | 36 | — | nr | — |
| 2 | 0.5 mL | 0.14 | 36 | — | nr | — |
| 3 | 1.0 mL | 0.07 | 36 | — | 45 | 40 |
| 4 | 1.5 mL | 0.04 | 36 | — | 48 | 41 |
| 5 | 1.0 mL | 0.07 | 72 | — | 94 | 88 |
| 6 | 1.0 mL | 0.07 | 72 | MLG | nr | — |
| 7 | 1.0 mL | 0.07 | 72 | MLG1d | nr | — |
| 8 | 1.0 mL | 0.07 | 72 | No MLG-Ru | nr | — |
| 9 | 1.0 mL | 0.07 | 72 | Inert atm. | nr | — |
| 10 | 1.0 mL | 0.07 | 72 | No DIPEA | nr | — |
| 11 | 1.0 mL | 0.07 | 72 | Blue light | nr | — |
| 12 | 1.0 mL | 0.07 | 72 | Darkness | nr | — |
| 13 | 1.0 mLh | 0.07 | 72 | — | 95 | 88 |

In conclusion, the 1,3-DC of dispersed MLG and stabilized azomethine ylides 1, originated from thermal 1,2-prototropy of imino esters, occurred under microwave irradiation in shorter reaction times (5 h versus 5 d) and with a final MLG1 three times more functionalised than for the already published work.35 To the best of our knowledge, this was the first time that this dipolar cycloaddition occurred with a total atom economy under microwave irradiation and it was the highest level of functionalisation of MLG achieved. The bipyridine unit was covalently bonded to the graphene and allowed for a complex to form with the ruthenium atom. The resulting heterogeneous catalyst acted as a photocatalyst in the aerobic oxidative hydroxylation of 4-methoxyphenylboronic acid and proceeded with a shorter reaction time than that needed for the reaction described in the original contribution.46 This supported ruthenium catalyst was found (using TG, and TEM) to be robust and could be recycled in a second reaction batch. The catalytic efficiency of this heterogeneous supported complex offered slightly lower chemical yields for the mentioned oxidative hydroxylation than did the homogeneous reaction reported in the literature46 using longer reaction times. The current reaction system showed an opacity to light, a drawback in our proof-of-concept experiment, but showed the advantage of the ability to separate and recycle the catalyst.
Author contributions
The manuscript was written and corrected through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Spanish Ministerio de Ciencia, Innovación y Universidades (project RED2018-102387-T), the Spanish Ministerio de Economía, Industria y Competitividad, Agencia Estatal de Investigación (AEI) and Fondo Europeo de Desarrollo Regional (FEDER, EU) (projects CTQ2017-82935-P and PID2019-107268GB-I00), the Generalitat Valenciana (IDIFEDER/2021/013, GVA-COVID19/2021/079 and CIDEGENT/2020/058), Medalchemy S. L. (Medalchemy-22T) and the University of Alicante (VIGROB-068, UAUSTI21-05). LVR-F thanks Generalitat Valenciana for Grisolía's fellowship (GRISOLIAP/2020/111). This work is also part of the R+D+I project PID2021-123079OB-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”. MNG is grateful for the grant RYC2021-034199-I funded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future”.Notes and references
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem, 2017, 1, 1–19 CrossRef.
- L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS PubMed.
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang, P. Huo and X. Li, Chin. J. Catal., 2022, 43, 178–214 CrossRef CAS.
- L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS PubMed.
- A. D. Ghuge, A. R. Shirode and V. J. Kadam, Curr. Drug Targets, 2017, 18, 724–733 CrossRef CAS PubMed.
- Introduction to Graphene: Chemical and Biochemical Applications, ed. A. Pattammattel and C. V. Kumar, Elsevier, Oxford, 2017 Search PubMed.
- V. G. Shevchenko, P. M. Nedorezova, and A. N. Ozerin, in Graphene Science Handbook: Electrical and Optical Properties, ed. M. Aliofkhazraei, N. Ali, W. I. Milne, C. S. Ozkan, S. Mitura and J. L. Gervasoni, CRC press, Boca Ratón, 2016, ch. 34, p. 553 Search PubMed.
- G. M. Vlasceanu, R.-M. Amarandi, M. Ionita, T. Tite, H. Iovu, L. Pilan and J. S. Burns, Biosens. Bioelectron., 2018, 117, 283–302 CrossRef CAS PubMed.
- K. C. Wasalathilake, G. Ayoko, and C. Yan, in Recent Advances in Graphene Research, ed. P. K. Nayak, InTech Open, London, 2016, ch. 9, pp. 195–213 Search PubMed.
- A. R. Urade, I. Lahiri and K. S. Suresh, JOM, 2023, 75, 614–630 CrossRef CAS PubMed.
- Results indicated that it is possible to fine tune the band gap of graphene by varying the number of azomethine groups attached. P. A. Denis and F. Iribarne, Chem. Phys. Lett., 2012, 550, 111–117 CrossRef CAS.
- D. D. Chronopoulos, A. Bakandritsos, P. Lazar, M. Pykal, K. Cepe, R. Zboril and M. Otyepka, Chem. Mater., 2017, 29, 926–930 CrossRef CAS PubMed.
- Q. H. Wang, Z. Jin, K. K. Kim, A. J. Hilmer, G. L. C. Paulus, C.-J. Shih, M.-H. Ham, J. D. Sánchez-Yamagishi, K. Watanabe, T. Taniguchi, J. Kong, P. Jarillo-Herrero and M. S. Strano, Nat. Chem., 2012, 4, 724–732 CrossRef CAS PubMed.
- X. Fan, R. Nouchi and K. Tanigaki, J. Phys. Chem. C, 2011, 115, 12960–12964 CrossRef CAS.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398–405 CrossRef CAS PubMed.
- F. M. Koehler, A. Jacobsen, K. Ensslin, C. Stampfer and W. J. Stark, Small, 2010, 6, 1125–1130 CrossRef CAS PubMed.
- G. Diankov, M. Neumann and D. Goldhaber-Gordon, ACS Nano, 2013, 7, 1324–1332 CrossRef CAS PubMed.
- For a revised nomenclature of graphene family, see: A. Bianco, H. M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- P. A. Denis, Chem. Phys. Lett., 2017, 684, 79–85 CrossRef CAS.
- J. C. Yuan, G. H. Chen, W. G. Weng and Y. Z. Xu, J. Mater. Chem., 2012, 22, 7929–7936 RSC.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS PubMed.
- G. Bottari, M. A. Herranz, L. Wibmer, M. Volland, L. Rodríguez-Pérez, D. M. Guldi, A. Hirsch, N. Martín, F. D'Souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464–4500 RSC.
- L. Rodríguez-Pérez, M. Á. Herranz and N. Martín, Chem. Commun., 2013, 49, 3721–3735 RSC.
- V. Bekiari, A. Karakassides, S. Georgitsopoulou, A. Kouloumpis, D. Gournis and V. Georgakilas, J. Mater. Sci., 2018, 53, 11167–11175 CrossRef CAS.
- J. Dai, Y. Li, Z. Huang and X. Huang, New J. Chem., 2015, 39, 9586–9595 RSC.
- S. Zhao, H. Wang, L. Xin, J. Cui and Y. Yan, Chem.–Asian J., 2015, 10, 1177–1183 CrossRef CAS PubMed.
- J. Huang, Y. Wu, D. Wang, Y. Ma, Z. Yue, Y. Lu, M. Zhang, Z. Zhang and P. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 3732–3741 CrossRef CAS PubMed.
- B. Xiao, X. Wang, H. Huang, M. Zhu, P. Yang, Y. Wang and Y. Du, J. Phys. Chem. C, 2013, 117, 21303–21311 CrossRef CAS.
- G. L. Liu, C. L. Yu, C. C. Chen, W. H. Ma, H. W. Ji and J. C. Zhao, Sci. China: Chem., 2011, 54, 1622–1626 CrossRef CAS.
- V. Georgakilas, A. B. Bourlinos, R. Zboril, T. A. Steriotis, P. Dallas, A. K. Stubos and C. Trapalis, Chem. Commun., 2010, 46, 1766–1768 RSC.
- P. Mehra and A. Paul, J. Phys. Chem. C, 2022, 126, 6135–6146 CrossRef CAS.
- L. Basta, A. Moscardini, F. Fabbri, L. Bellucci, V. Tozzini, S. Rubini, A. Griesi, M. Gemmi, S. Heun and S. Veronesi, Nanoscale Adv., 2021, 3, 5841–5852 RSC.
- Y. Hernández, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. GunKo, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef PubMed.
- U. Khan, A. O'Neill, M. Loyta, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- M. Ferrándiz-Saperas, A. Ghisolfi, D. Cazorla-Amorós, C. Nájera and J. M. Sansano, Chem. Commun., 2019, 55, 7462–7465 RSC.
- The starting graphite was prepared as in ESI‡ of ref. 35.
- This percentage means% by gained mass compared to the entire MLG structure.
- To the best of our knowledge, this is the first time that a functionalisation of multilayer graphene using a freshly generated azomethine ylide from imino esters is carried out using a microwave-assisted process.
- For other microwave-assisted functionalisations of graphene, see: (a) M. V. Sulleiro, S. Quiroga, D. Peña, D. Pérez, E. Guitián, A. Criado and M. Prato, Chem. Commun., 2018, 54, 2086–2089 RSC; (b) E. Váquez, F. Giacalone and M. Prato, Chem. Soc. Rev., 2014, 43, 58–60 RSC.
- E. C. Constable, E. Figgemeier, C. E. Housecroft, J. Olsson and Y. C. Zimmermann, Dalton Trans., 2004, 1918–1927 RSC.
- Y. Katoh, Y. Tsujimoto, C. Yamamoto, T. Ikai, M. Kamigaito and Y. Okamoto, Polym. J., 2011, 43, 84–90 CrossRef CAS.
- Y. Huang, Q. Lin, J. Wu and N. Fu, Dyes Pigm., 2013, 99, 699e704 Search PubMed.
- P. Beer, F. Szemes, P. Passaniti and M. Maestri, Inorg. Chem., 2004, 43, 3965–3975 CrossRef CAS PubMed.
- G. Caleffi, O. Larrañaga, M. Ferrándiz-Saperas, P. Costa, C. Nájera, A. de Cózar, F. P. Cossío and J. M. Sansano, J. Org. Chem., 2019, 84, 10593–10605 CrossRef CAS PubMed.
- K. Kodama, A. Kobayashi and T. Hirose, Tetrahedron Lett., 2013, 54, 5514–5517 CrossRef CAS.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- In the corresponding crude 1H NMR spectra the adduct boronic acid water DMF was observed as referred in the literature: T. D. James, K. R. A. Samankumara-Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1910–1922 CrossRef.
Footnotes |
| † Dedicated to Professor Miquel Angel Pericàs Brondó on the occasion of his retirement from the Institute of Chemical Research of Catalonia, ICIQ. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00075g |
| § This author contributed throughout the experimental work. |
| ¶ This author contributed to the preparation of the pristine multilayer graphene suspensions. |
| This journal is © The Royal Society of Chemistry 2024 |