
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning few-layered graphitic carbons with atomic-sized cobalt hydroxide by harnessing hollow metal–organic frameworks†
Eun Jin
Cho‡
 a,
Bo-Min
Kim‡
a,
WooYeon
Moon
b,
Dong Gyu
Park
c,
Young-Wan
Ju
d,
Won Ho
Choi
*e and
Jeeyoung
Shin
*ac
a,
Bo-Min
Kim‡
a,
WooYeon
Moon
b,
Dong Gyu
Park
c,
Young-Wan
Ju
d,
Won Ho
Choi
*e and
Jeeyoung
Shin
*ac
aDepartment of Mechanical Systems Engineering, Sookmyung Women's University, Seoul 04310, Republic of Korea. E-mail: jshin@sookmyung.ac.kr
bDepartment of Chemical and Biological Engineering, Sookmyung Women's University, Seoul 04310, Republic of Korea
cInstitute of Advanced Materials and Systems, Sookmyung Women's University, Seoul 04310, Republic of Korea
dDepartment of Chemical Engineering, College of Engineering, Wonkwang University, Iksan-si 54538, Republic of Korea
eDepartment of Petrochemical Materials, Chonnam National University, Yeosu-si 59631, Republic of Korea. E-mail: wonhochoi@jnu.ac.kr
First published on 16th September 2024
Abstract
Graphitic carbon exhibits distinctive characteristics that can be modulated by varying the number of carbon layers. Here, we developed a method to control the growth of graphitic carbon layers through pyrolysis of zeolitic imidazolate frameworks (ZIFs). The key is to pyrolyze hollow-structured ZIF-8 containing Co ions to simultaneously obtain an amorphous carbon source for graphitic carbons and Co metal nanoparticles for catalyzing graphitization of amorphous carbons. Owing to sparsely distributed Co ions within ZIF-8, Co nanoparticles are formed, which leads to localized graphitization. The graphitic carbon obtained contained two to five layers, unlike carbonized ZIF-67. The few-layered graphitic carbon was subjected to KOH activation and employed as a support for atomic-sized Co(OH)2 owing to the short routes for Co nanoparticle egress and OH− ion movement. Our strategy does not involve any highly corrosive process for catalyst leaching and can even be used to produce atomic-sized Co(OH)2 with few-layered graphitic carbons.
Introduction
Carbon-based materials, such as graphene and carbon nanotubes, that are chemically stable and lightweight, and have structural versatility have emerged as promising materials in energy-related applications.1 Among these materials, graphitic carbon, predominantly composed of well-ordered sp2 bonded carbon atoms, offers unique advantages such as high electrical and thermal conductivity and remarkable mechanical strength.2,3 These properties are highly influenced by the number of graphitic carbon layers. Therefore, the accurate control of the number of graphitic carbon layers, spanning from a few layers to multiple layers, is of utmost importance as the properties of graphitic carbons can be modulated.Atomic layer deposition (ALD) is a technique used for fabricating graphitic carbons.4,5 It involves gas-phase atom deposition and facilitates the molecular-level control and the production of layer-controlled graphitic carbon.6,7
However, this technique is costly and time-consuming, and these drawbacks limit its feasibility for mass production. Another approach, namely catalyst-assisted graphitization of amorphous carbon, is more efficient and relatively inexpensive, and it provides a high yield. It typically involves multiple steps, including the loading of transition metal catalysts on an amorphous carbon and subsequent high-temperature treatments, and the removal of the residual catalysts.
Structurally well-defined metal–organic frameworks (MOFs), formed by repeated coordination between organic linkers and metal nodes, are an attractive precursory material for catalyst-assisted thermal treatment due to their dual role: they provide a rich carbon source from their organic linkers and act as metallic catalysts through their metal nodes during carbonization.8–10 During the high-temperature treatment, the metallic catalysts play an important role in reducing the activation energy required for the rearrangement of the carbon atoms into a crystalline structure. Nevertheless, the use of carbonized MOFs for producing graphitic carbon has drawbacks: the complete removal of catalyst residues from the produced graphitic carbon is challenging, and the highly corrosive process for removing the metallic catalysts can potentially deform the carbon structure. Furthermore, the number of carbon layers often amounts to several tens of layers, and hence, they obstruct the transportation of ions through the interlayer space during electrochemical operation. In this study, to overcome the limitations, we produced few-layered graphitic carbons through the carbonization of hollow MOFs. The key observation was that the sparse distribution of the catalyst precursor within the hollow MOFs led to the generation of nano-sized catalysts, resulting in localized graphitization on the catalysts.11 These combined attributes—few-layered graphitic carbons and nano-sized catalysts—make our approach allow complete removal of catalysts and stabilization of atomic-sized active materials within graphitic carbon layers.
Results and discussion
We used zeolitic-imidazolate frameworks (ZIFs), which are a subfamily of MOFs. The entire process of producing few-layered graphitic carbons with atomic-sized Co(OH)2 is shown in Fig. 1. During pyrolysis at 900 °C, ZIF-8 composed of Zn ions and 2-methylimidazole transforms into porous amorphous carbon by Zn vaporization and structural rearrangements (Fig. 1a). ZIF-67, composed of Co ions and the same ligands as ZIF-8, promotes the formation of multi-layered graphitic carbons. This is attributed to the catalytic properties of Co metal particles, which facilitate the graphitization process around the large Co metal particles. These results suggest that while ZIF-8 provides a carbon source devoid of heavy metal ions, ZIF-67 contributes catalysts essential for the graphitization of the amorphous carbons derived from ZIF-8. We employed a core–shell structured MOF(ZIF-67@ZIF-8), and then deformed its internal ZIF-67 part, resulting in Co(OH)2 embedded hollow ZIF-8 (hZIF-8). The process used to synthesize hZIF-8 was presented in our previous paper.12 Upon pyrolysis of hZIF-8 (c-hZIF-8) at 900 °C, the sparsely distributed Co(OH)2 were reduced to Co nano-particles, converting amorphous carbon into graphitic carbon. This process produced few-layered graphitic carbon, predominantly in the range of 2–5 layers (Fig. 1b). Subsequent KOH treatment transformed the Co nano-particles into atomic-sized Co(OH)2 while simultaneously removing the Co nano-particles, uniformly distributed within the few-layered graphitic carbon of c-hZIF-8 (Co(OH)2⊂c-hZIF-8).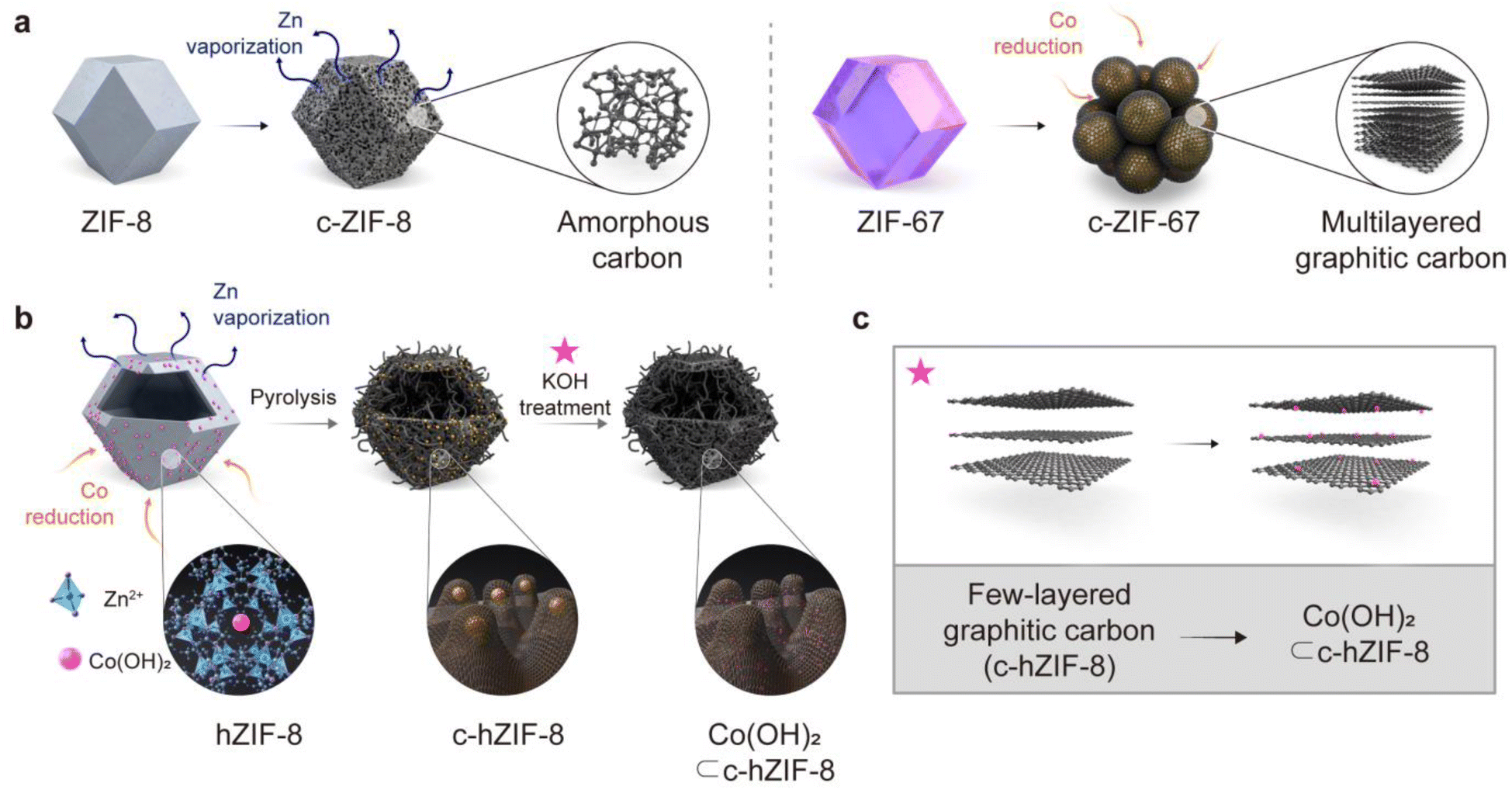 | ||
| Fig. 1 Schematic illustration of the synthesis process of few-layered graphitic carbon with atomic sized Co(OH)2. (a) The structural arrangement of carbon produced after pyrolysis of ZIF-8 and ZIF-67. (b) The entire processes used to produce Co(OH)2⊂c-hZIF-8. (c) After KOH treatment, Co nanoparticles subsequently transformed into atomic-sized Co(OH)2 while uniformly distributing them within the few-layered graphitic carbon of c-hZIF-8 (Co(OH)2⊂c-hZIF-8). | ||
In XRD patterns of carbonized ZIF-67 (c-ZIF-67) heat treated at 900 °C, the distinctive patterns of ZIF-67 vanished and the presence of Co metal (PDF Card no. 00-015-0806) particles was observed (Fig. 2a).13 At a high temperature, 2-methylimidazole undergoes thermal decomposition, breaking down into its fragmented structure. Thus, the crystallinity of ZIF-67 is disrupted. Simultaneously, the large Co particles catalyze the rearrangement of the carbon atoms into a graphitic carbon structure.14,15 Transmission electron microscopy (TEM) and scanning electron microscopy(SEM) images showed the growth of graphitic carbons on the surface of Co metal particles, whose size ranged from 20 to 100 nm, indicating complete structural transformation from ZIF-67 into c-ZIF-67 (Fig. 2b and S1–S4†). However, using pristine ZIF-67 as the precursor has a drawback: its high Co ion concentration leads to the formation of large Co metal particles.16 This is because the Co ions tend to agglomerate because of frequent collisions, resulting in the formation of large Co metal particles.17 The large Co metal particles facilitate the production of graphitic carbons because of their catalytic ability. This chain of events leads to the formation of graphitic carbon shells containing more than 10 layers (Fig. S5†). Consequently, the extraction of the Co metal particles requires the use of strong acids, which inevitably damage the graphitic carbon structure. This results in uncontrollable properties, such as the generation of arbitrary defects and the disruption of electron conduction pathways. We hypothesized that limiting the supply of Co ions could result in the formation of smaller Co metal particles, which would produce few-layered graphitic carbons. Removal of Co metal particles would then be easier, and Co-based active materials could possibly be produced within the graphitic carbons due to the availability of better ion transport pathways.
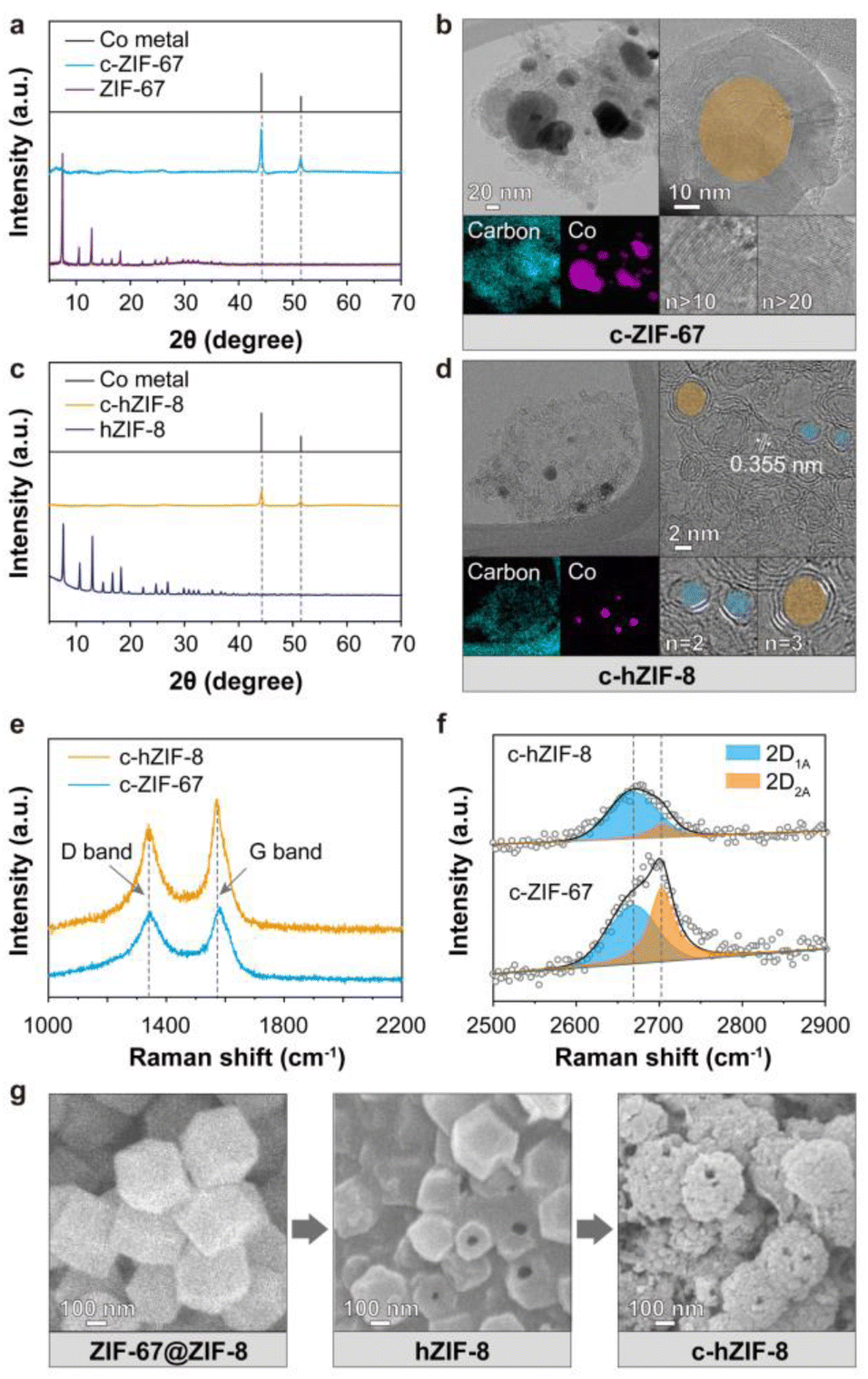 | ||
| Fig. 2 Structural characteristics of ZIF-67, c-ZIF-67, ZIF-67@ZIF-8, hZIF-8, and c-hZIF-8: (a and c) XRD diffraction pattern, (b and d) TEM and EDS images, (e and f) Raman spectra with the position of the G band, D band, and 2D band, and (g) SEM images revealing entire graphitization processes of c-hZIF-8. | ||
Based on the hypothesis, Co(OH)2-embedded hZIF-8 was used since (1) its carbonization provides amorphous carbon without heavy metal particles owing to Zn vaporization at 900 °C and (2) the controllable amount of Co sources supplied from sparse Co(OH)2 governs the growth of Co metal particles. In line with the procedures described in our previous paper,12 selective destruction of the ZIF-67 core resulted in the production of hZIF-8 and the autogenous formation and stabilization of Co(OH)2 within micropores of hZIF-8.
The XRD patterns of hZIF-8 showed high crystallinity, while those of carbonized hZIF-8 (c-hZIF-8) showed the disappearance of the crystallinity of ZIFs and two strong peaks at 44.2° and 51.5° (Fig. 2c). The peaks were attributed to metallic Co, and they indicated that Co metal particles were formed by the reduction of Co(OH)2. Remarkably, a TEM image of c-hZIF-8 showed Co metal nanoparticles surrounded by graphitic carbons containing only a few carbon layers, ranging from 2 to 5 layers, and a lattice spacing of 0.355 nm (Fig. 2d, S6 and S7†).18 Compared with c-ZIF-67, the formation of smaller Co metal particles in the case of c-hZIF-8 resulted in the formation of fewer graphitic carbon layers. SEM images of ZIF-67@ZIF-8, hZIF-8, and c-hZIF-8 showed the entire process used to produce c-hZIF-8, and they highlighted the preserved morphology (evidenced by visible holes on the surfaces in Fig. 2e and S8–S12†). The preserved morphology showed that the graphitization of hZIF-8 occurred under conditions where amorphous carbon was supplied by the thin shell of hZIF-8 and that Co metal nanoparticles catalyzed the graphitization.19 The FT-IR spectrum of hZIF-8 showed that the bonds formed were similar to those observed in ZIF-8, indicating an identical structure (Fig. S13†). However, the c-hZIF-8 spectrum showed distinct peaks below 1500 cm−1, unlike the hZIF-8 spectrum, indicating complete structural transformation from hZIF-8 into c-hZIF-8. A peak at 1637 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonding, suggested that the organic ligands underwent significant structural rearrangement during pyrolysis.20 Raman spectroscopy was employed to assess the degree of graphitization (Fig. 2e) and the number of carbon layers in both c-hZIF-8 and c-ZIF-67 (Fig. 2f). The spectra of the two samples exhibited characteristic peaks at 1580 cm−1 (G band) and 1350 cm−1 (D band).21 The ID/IG signifies the intensity ratio of the D band to the G band.22 Its value for c-hZIF-8 was calculated to be 0.74, while for c-ZIF-67, its value was 0.92. This indicates that c-ZIF-67 had more defects and structural distortions than c-hZIF-8.23 The number of carbon layers was evaluated through the convolution of the ∼2700 cm−1 peak (2D band). The peaks for c-hZIF-8 and c-ZIF-67 show distinct components labeled as 2D1A and 2D2A,24 at 2668 cm−1 and 2703 cm−1, respectively. For c-hZIF-8, the 2DA1 band is more prominent than the 2D2A band. In contrast, c-ZIF-67 exhibits a more prominent 2D2A band at a higher wavenumber. For graphite, the peak at higher wavenumbers is stronger than those at lower wavenumbers (Fig. S14†), suggesting that graphite has more carbon layers than c-hZIF-8 and c-ZIF-67. This result indicates that c-hZIF-8 has fewer carbon layers than c-ZIF-67 and graphite, implying few-layered carbon structures of c-hZIF-8.
C bonding, suggested that the organic ligands underwent significant structural rearrangement during pyrolysis.20 Raman spectroscopy was employed to assess the degree of graphitization (Fig. 2e) and the number of carbon layers in both c-hZIF-8 and c-ZIF-67 (Fig. 2f). The spectra of the two samples exhibited characteristic peaks at 1580 cm−1 (G band) and 1350 cm−1 (D band).21 The ID/IG signifies the intensity ratio of the D band to the G band.22 Its value for c-hZIF-8 was calculated to be 0.74, while for c-ZIF-67, its value was 0.92. This indicates that c-ZIF-67 had more defects and structural distortions than c-hZIF-8.23 The number of carbon layers was evaluated through the convolution of the ∼2700 cm−1 peak (2D band). The peaks for c-hZIF-8 and c-ZIF-67 show distinct components labeled as 2D1A and 2D2A,24 at 2668 cm−1 and 2703 cm−1, respectively. For c-hZIF-8, the 2DA1 band is more prominent than the 2D2A band. In contrast, c-ZIF-67 exhibits a more prominent 2D2A band at a higher wavenumber. For graphite, the peak at higher wavenumbers is stronger than those at lower wavenumbers (Fig. S14†), suggesting that graphite has more carbon layers than c-hZIF-8 and c-ZIF-67. This result indicates that c-hZIF-8 has fewer carbon layers than c-ZIF-67 and graphite, implying few-layered carbon structures of c-hZIF-8.
The N2 physisorption isotherms of ZIF-67 and hZIF-8 showed type I curves, which indicated the presence of only micropores in their crystalline structures (Fig. S15†). By contrast, the isotherms of c-ZIF-67 and c-hZIF-8 exhibited type IV curves characterized by a hysteresis loop. The isotherm of c-hZIF-8 showed distinct adsorption at low relative pressures and a more prominent hysteresis loop than that of c-ZIF-67 (Fig. S16†).25 These different isotherm profiles showed the different pore restructuring processes occurring in the graphitic carbons. The specific surface areas of ZIF-67, c-ZIF-67, hZIF-8, and c-hZIF-8 were estimated to be 1685, 154, 1063, and 197 m2 g−1, respectively (Table S1†). The smaller specific surface areas of c-ZIF-67 and c-hZIF-8 were attributed to the destruction of micropores.26 Although the number of micropores was significantly reduced due to the destruction, the Co metal particles left behind cavities after the sintering process, leading to the formation of more prominent micropores in c-hZIF-8 because of its smaller Co metal particles compared with c-ZIF-67. Furthermore, the multilayered graphitic carbons in c-ZIF-67 hindered the removal of Co metal particles, resulting in a low surface area and pronounced hysteresis being caused by the mesopores (Fig. S17†). The volume ratio of the micropores to mesopores (Vmicro/Vmeso) of c-ZIF-67 and c-hZIF-8 was estimated to 1.02 and 0.94, respectively. This result implied that c-hZIF-8 possesses mesopores derived by Co nano-particles than that of c-ZIF-67 (Fig. S18†). 27
When c-hZIF-8 came into contact with the highly alkaline KOH electrolyte upon electrochemical activation, a corrosive reaction led to the formation of Co(OH)2 (Co(OH)2⊂c-hZIF-8). The few-layered graphitic structure of c-hZIF-8 provided a pathway for the transport of OH− ions and did not hinder the egress of Co metal particles. Upon electrochemical activation, Co metal particles were oxidized and lost electrons, while OH− ions from KOH were reduced and gained electrons; the process culminated in the formation of Co(OH)2. XRD patterns of Co(OH)2⊂c-hZIF-8 showed weak and broad Co(OH)2 peaks (PDF Card no. 00-001-0357), suggesting the production of atomic-sized Co(OH)2 and the absence of Co metal particles (Fig. 3a).28 On the other hand, XRD patterns of Co(OH)2⊂c-ZIF-67 indicated the presence of both Co metal particles and Co(OH)2. The persistence of Co metal particles in Co(OH)2⊂c-ZIF-67 suggests that their extraction from c-ZIF-67 is more challenging compared with their extraction from Co(OH)2⊂c-hZIF-8, owing to the numerous layers of graphitic carbon in c-ZIF-67 obstructing their extraction. TEM and EDS images clearly showed the disappearance of Co metal particles, but indicated the presence of atomic-sized Co(OH)2 in Co(OH)2⊂c-hZIF-8 (Fig. 3b). This was supported by SEM images, which showed the presence of bulk Co(OH)2 in Co(OH)2⊂c-ZIF-67, but not in Co(OH)2⊂c-hZIF-8 (Fig. S19 and S20†). The TEM image confirms the preservation of the few-layered graphitic carbons after KOH treatment (Fig. S21†). It shows the formation of atomic-sized Co(OH)2 without any residual Co metal particles within the few-layered graphitic carbons in c-hZIF-8.
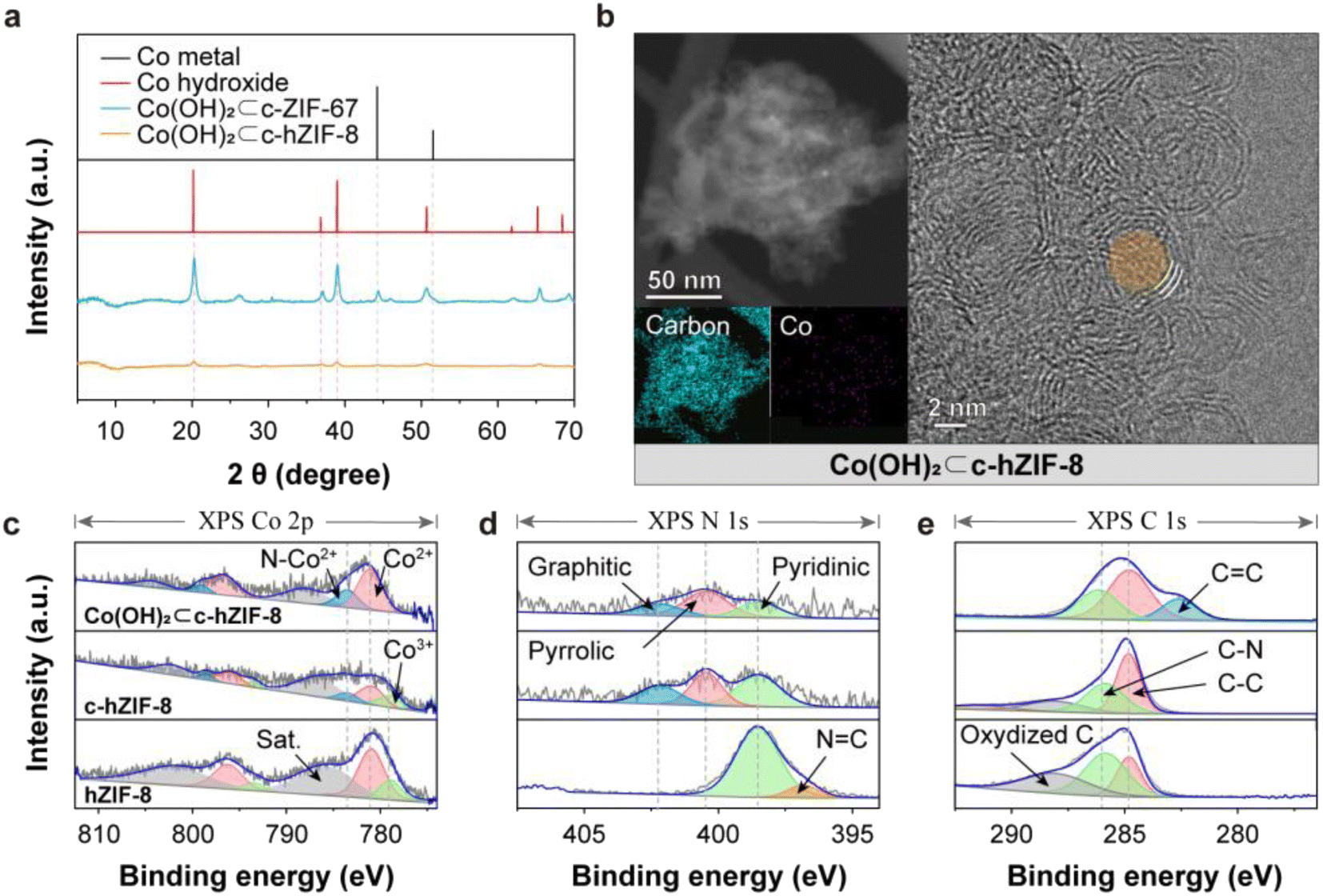 | ||
| Fig. 3 Chemical characteristics of hZIF-8, c-hZIF-8, and Co(OH)2⊂c-hZIF-8. (a) Comparison of XRD patterns of Co metal and Co(OH)2 for Co(OH)2⊂c-ZIF-67 and Co(OH)2⊂c-hZIF-8. (b) STEM and EDS images of Co(OH)2⊂c-hZIF-8. (c–e) Co 2p, N 1s, and C 1s XPS core level spectra of the as-synthesized hZIF-8, c-hZIF-8, and Co(OH)2⊂c-hZIF-8. | ||
XPS deconvolutions elucidated the transition from Co metal particles to Co(OH)2 (Fig. S22–S24†). The Co 2p XPS spectrum (Fig. 3c) of hZIF-8 indicated the Co2+/Co3+ ratio to be 3.05; thus, Co2+ was predominant, and the amount of Co3+ was very small (because of the presence of stabilized Co(OH)2 in micropores).12 In the Co 2p XPS spectrum of c-hZIF-8, the ratio was significantly lower (1.88) because of the transformation of Co2+ ions into larger Co metal particles. The Co metal particles were easily oxidized upon exposure to air, and the oxidized surface of Co metal particles, along with Co ions adsorbed on graphitic carbons, appeared as Co2+, Co3+, and N–Co2+, instead of metallic Co. This result differs from the XRD patterns, which indicate the presence of Co metal particles.29,30 In the spectrum of Co(OH)2⊂c-hZIF-8, the Co2+ peaks were predominant consistent with the Co2+ peak of pristine Co(OH)2. The Co3+ peak disappeared, suggesting that Co3+ species were transformed into Co(OH)2. Compared with the N 1s XPS spectrum of hZIF-8, the N 1s XPS spectra of c-hZIF-8 and Co(OH)2⊂c-hZIF-8 showed three types of N, namely graphitic, pyrrolic, and pyridinic N (Fig. 3d).31 The strong N peak at 397 eV corresponded to the 2-methylimidazole organic linkers, but the presence of graphitic and pyrrolic N showed the structural transformation of N-doped carbon structures.32 The similar ratio of the three N types in the spectra of c-hZIF-8 and Co(OH)2⊂c-hZIF-8 indicated that N in graphitic carbons was not affected by KOH. The C 1s XPS spectrum of Co(OH)2⊂c-hZIF-8 showed the disappearance of the oxidized C (C–O, CO, or O–CO) peak, and the appearance of a distinct peak corresponding to graphitic carbon (CC) (Fig. 3e).33 The disappearance of the oxygenated carbon peak suggested that KOH removed the oxygen-containing functional groups from the carbon surface. The emergence of the CC peak, which was also apparent in the FT-IR spectrum, suggested that some carbons in c-hZIF-8 were reordered to form a graphitic CC structure (Fig. S25†). These results show that KOH treatment produced Co(OH)2 on few-layered graphitic carbons while removing oxygenated carbons and absorbed Co ions, apart from promoting additional graphitization of the remaining amorphous carbons.
The electrochemical performance of Co(OH)2⊂c-hZIF-8 and Co(OH)2⊂c-ZIF-67 was investigated using a three-electrode cell and 1 M KOH electrolyte. Co(OH)2 provides advantageous properties including improved redox activity, superior chemical stability, rapid ion transport, and cost efficiency.34,35 During the long-term cycling test, the specific capacitance initially increased as a result of the KOH activation process, reached a peak, and then decreased (Fig. S26†). At the beginning, the corrosion reaction transformed Co metal nanoparticles into Co(OH)2, leading to a temporary increase in the specific capacitance up to 60 cycles. As evident from the KOH activations, the CV curves of Co(OH)2⊂c-hZIF-8 showed an oxidation reaction occurring at around 0.28 V, and the conversion of Co metal nanoparticles to Co2+ was observed, which involved the reaction Co + 2 OH− → Co(OH)2 + 2 e− (Fig. S27†).36 After the complete conversion of Co metal nanoparticles into Co(OH)2, only a single redox reaction occurred in Co(OH)2⊂c-hZIF-8.37 Compared with the case of Co(OH)2⊂c-ZIF-67, the shape of the CV curve remained largely unchanged, with only slight peak shifts being observed (Fig. S28 and S29†). The specific capacitance retention plot showed that Co(OH)2⊂c-hZIF-8 retained 80.1% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, which was much higher than the value of 38.2% for Co(OH)2⊂c-ZIF-67 (Fig. 4a). The enhanced stability implies that the integrated structure of Co(OH)2⊂c-hZIF-8 remained intact for 10000 cycles, without any deformation or detachment of Co(OH)2. The CV curves of Co(OH)2⊂c-hZIF-8 (Fig. 4b) show the redox behavior after every 2000 cycles (Co(OH)2⊂c-ZIF-67 is shown in Fig. S30†). The oxidation of Co2+ to Co3+ occurred at a potential of 0.51 V, and the reduction reaction occurred at 0.39 V. At 10000 cycles, redox reactions occurred at 0.53 and 0.38 V. The relatively constant potential difference indicates the high reversibility of the electrochemical reactions. As the scan rate increased, the redox peak showed a slight potential shift (Fig. S31 and S32†), but the shape of the curve remained similar to that observed at lower scan rates. These results showed that the redox profiles were consistent irrespective of the scan rate, indicating high rate performance.38 The results also indicated that Co(OH)2⊂c-hZIF-8 not only exhibited stable performance but also showed good reversibility over 10000 cycles in the potential window of 0–0.6 V. The galvanic discharge profiles of c-hZIF-8 at different current densities showed a voltage plateau at approximately 0.39 V, as shown in CV curves (Fig. 4c, S33 and S34†). In the CV curves, Co(OH)2⊂c-hZIF-8 shows minimal changes in the position and shape of the oxidation-reduction peaks over 10000 cycles, whereas Co(OH)2⊂c-ZIF-67 exhibits significant changes in both the position and shape of the peaks after just 1000 cycles. These contrasting results indicate that Co(OH)2⊂c-hZIF-8 undergoes minimal structural change. This behavior indicated pseudocapacitive characteristics associated with the presence of Co(OH)2 (ref. 39) in Co(OH)2⊂c-hZIF-8 and Co(OH)2⊂c-ZIF-67. Although the specific capacitance decreased as the current density increased, Co(OH)2⊂c-hZIF-8 exhibited a higher specific capacitance than Co(OH)2⊂c-ZIF-67 (Fig. 4d, Tables S2 and S3†). Tables S2 and S3† show a high capacitance of 257.2 F g−1 at 1 A g−1 for Co(OH)2⊂c-hZIF-8; this is superior to the capacitance of 169.2 F g−1 at 1 A g−1 for Co(OH)2⊂c-ZIF-67 by about 65.8%. This is attributed to the structural features of Co(OH)2⊂c-hZIF-8, such as Co(OH)2 being atomic sized, the presence of few-layered graphitic carbon, and both micropores and mesopores being present, that resulted in enhanced performance.
000 cycles, which was much higher than the value of 38.2% for Co(OH)2⊂c-ZIF-67 (Fig. 4a). The enhanced stability implies that the integrated structure of Co(OH)2⊂c-hZIF-8 remained intact for 10000 cycles, without any deformation or detachment of Co(OH)2. The CV curves of Co(OH)2⊂c-hZIF-8 (Fig. 4b) show the redox behavior after every 2000 cycles (Co(OH)2⊂c-ZIF-67 is shown in Fig. S30†). The oxidation of Co2+ to Co3+ occurred at a potential of 0.51 V, and the reduction reaction occurred at 0.39 V. At 10000 cycles, redox reactions occurred at 0.53 and 0.38 V. The relatively constant potential difference indicates the high reversibility of the electrochemical reactions. As the scan rate increased, the redox peak showed a slight potential shift (Fig. S31 and S32†), but the shape of the curve remained similar to that observed at lower scan rates. These results showed that the redox profiles were consistent irrespective of the scan rate, indicating high rate performance.38 The results also indicated that Co(OH)2⊂c-hZIF-8 not only exhibited stable performance but also showed good reversibility over 10000 cycles in the potential window of 0–0.6 V. The galvanic discharge profiles of c-hZIF-8 at different current densities showed a voltage plateau at approximately 0.39 V, as shown in CV curves (Fig. 4c, S33 and S34†). In the CV curves, Co(OH)2⊂c-hZIF-8 shows minimal changes in the position and shape of the oxidation-reduction peaks over 10000 cycles, whereas Co(OH)2⊂c-ZIF-67 exhibits significant changes in both the position and shape of the peaks after just 1000 cycles. These contrasting results indicate that Co(OH)2⊂c-hZIF-8 undergoes minimal structural change. This behavior indicated pseudocapacitive characteristics associated with the presence of Co(OH)2 (ref. 39) in Co(OH)2⊂c-hZIF-8 and Co(OH)2⊂c-ZIF-67. Although the specific capacitance decreased as the current density increased, Co(OH)2⊂c-hZIF-8 exhibited a higher specific capacitance than Co(OH)2⊂c-ZIF-67 (Fig. 4d, Tables S2 and S3†). Tables S2 and S3† show a high capacitance of 257.2 F g−1 at 1 A g−1 for Co(OH)2⊂c-hZIF-8; this is superior to the capacitance of 169.2 F g−1 at 1 A g−1 for Co(OH)2⊂c-ZIF-67 by about 65.8%. This is attributed to the structural features of Co(OH)2⊂c-hZIF-8, such as Co(OH)2 being atomic sized, the presence of few-layered graphitic carbon, and both micropores and mesopores being present, that resulted in enhanced performance.
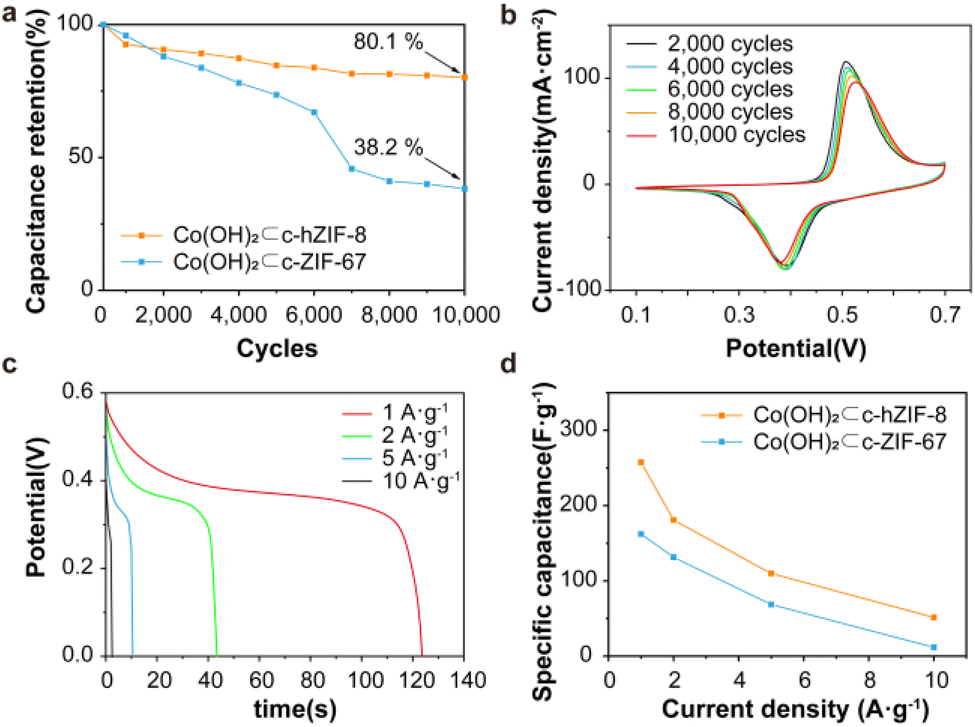 | ||
| Fig. 4 Electrochemical characterization of Co(OH)2⊂c-hZIF-8. (a) The specific capacitance retention rate of Co(OH)2⊂c-hZIF-8 and Co(OH)2⊂c-ZIF-67 after a 10000-cycle test at 50 mV s−1. (b) Cyclic voltammetry curves. The change in the CV curve shape was monitored every 2000 cycles. (c) Galvanostatic discharge profiles. (d) The variation of the specific capacitance with the current density for Co(OH)2⊂c-hZIF-8 and Co(OH)2⊂c-ZIF-67. | ||
Conclusions
In this study, we synthesized few-layered graphitic carbons and subsequently produced and stabilized atomic-sized Co(OH)2 within the few-layered graphitic carbons. For this, we used Co ion embedded hollow ZIF-8 as the precursor material, which served as a source of Co metal nanoparticles and amorphous carbons. Upon carbonization at 900 °C, the chemical structure of hZIF-8 was initially amorphized and Zn ions were vaporized. Subsequently, Co metal nanoparticles were produced from the sparsely distributed Co precursors, and the amorphous carbons were graphitized in the vicinity of the Co nanoparticles. Notably, the resulting graphitic carbon layers were few in number—in the range of 2–5 layers—owing to the use of nanosized Co catalysts; the number of layers was significantly lower than that in multilayered graphitic carbons produced through pristine ZIF-67. After graphitization, the remaining Co nanoparticles were completely converted to Co(OH)2via electrochemical KOH activation. These findings show that limiting the supply of the catalyst precursor promotes the formation of nanosized metallic catalysts, which facilitate the formation of few-layered graphitic carbon structures. In particular, our strategy can be used to simultaneously produce and stabilize atomic-sized active materials in few-layered graphitic carbons. The production of atomic-sized Co(OH)2 in few-layered graphitic carbons enhances capacitance retention by 80.1% after 10000 cycles and results in a high specific capacitance of 257.2 F g−1 at 1 A g−1. This study unveils a potential strategy to manipulate the number of graphitic carbon layers used as supporting materials and to facilitate the production of atomic-sized materials used as active materials.
Experimental
Materials
Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 98+%), zinc(II) nitrate hexahydrate (Zn(NO3)2·6H2O, 98+%), 2-methylimidazole (2-mim, 99%), polyvinylpyrrolidone (PVP, mol. wt. 10000), methanol (99.9+%), ethanol (99.5+%), poly(tetrafluoroethylene) (PTFE, 1 μm), and Ni foam were purchased from Sigma-Aldrich. Ethylene glycol (EG, 99%) was purchased from Daejung Chemicals and Metals Co. Ltd, deionized (DI) water was obtained from a water purifying system, and Ketjen Black (KB, EC 300 JD) was purchased from Pyunghwa Co. Ltd
Preparation of the electrode
The catalyst ink preparation method was used, and it is as follows: c-hZIF-8, KB, and PTFE in an 8:1:1 ratio were mixed and used as the electrode sample. The mixing was achieved using ethanol, and the resulting ink was uniformly coated onto a nickel foam substrate during the electrode fabrication process. The electrode was dried in a vacuum oven at 75 °C for 12 h, and it was washed once with acetone. Subsequently, it was dried in a vacuum oven at 75 °C for 24 h.
Structural characterization
X-ray diffraction (XRD) patterns were obtained using a Bruker D8 Advance diffractometer at 40 kV and 40 mA, with Cu Kα radiation. TEM images were obtained using a JEM-2100F (JEOL) that was operated at an acceleration voltage of 200 kV. SEM images were obtained using a JSM-7600F (JEOL). FT-IR spectra were measured using a Nicolet IS50 FTIR spectrometer (Thermo Fisher Scientific) with powder-pressed KBr pellets in the wave number range of 4000 to 400 cm−1. The BET specific surface area and pore size distribution were measured using a BELSORP MINI X (MicrotracBEL), and XPS measurement was performed with a K-Alpha XPS system (Thermo Fisher Scientific). Raman measurements were conducted using an XperRAM-S (NANOBASE) and a laser (λ = 532 nm) excitation source.Electrochemical characterization
To evaluate the supercapacitor characteristics of the prepared electrodes, we performed CV and GCD tests with a three-electrode cell configuration by using a potentiostat (VSP, BioLogic). The fabricated electrodes were used as working electrodes. A platinum wire, a Hg/HgO electrode (saturated KOH), and 1 M KOH were used as the counter electrode, reference electrode, and electrolyte, respectively. The CV and GCD experiments were performed in a voltage window between 0 and 0.6 V. CV was performed at scan rates of 0.5 to 50 mV s−1, and GCD tests were performed at current densities from 1 to 10 A g−1. Additionally, CV for examining capacitance retention was performed in a voltage window between 0.1 and 0.7 V at a scan rate of 50 mV s−1.The specific capacitance (F g−1) was calculated from the GCD curves by using the following equation. 42
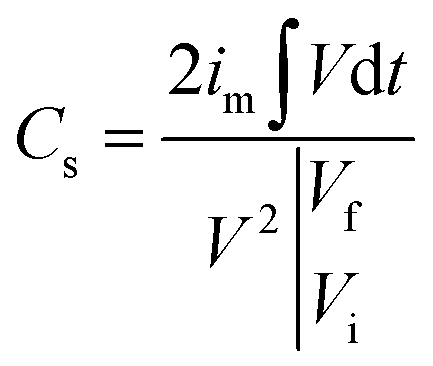
Data availability
Please contact the corresponding author for all data requests.Author contributions
W. H. C and J. S conceptualized the idea. E. J. C conducted the material measurements/analysis and Y. W. J helped to conduct XPS measurements. W. Y. M and D. G. P helped to conduct BET measurements. E. J. C and B.-M. K conducted the electrochemical measurements/analysis. W. H. C and J. S supervised the project and wrote the manuscript with E. J. C and B.-M. K. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. RS-2023-00218255 & No. 2021R1C1C2012825)References
- Y. Wang, L. Liu, T. Ma, Y. Zhang and H. Huang, Adv. Funct. Mater., 2021, 31, 2102540 CrossRef CAS.
- C. Lu and X. Chen, ACS Nano, 2021, 15, 18777–18793 CrossRef CAS PubMed.
- Y. Luo, Y. Yan, S. Zheng, H. Xue and H. Pang, J. Mater. Chem. A, 2019, 7, 901–924 RSC.
- D. Guo, J. Wang, L. Zhang, X. Chen, Z. Wan and B. Xi, Small, 2020, 16, 2002432 CrossRef CAS.
- C. Marichy and N. Pinna, Coord. Chem. Rev., 2013, 257, 3232–3253 CrossRef CAS.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS.
- A. S. Asundi, J. A. Raiford and S. F. Bent, ACS Energy Lett., 2019, 4, 908–925 CrossRef CAS.
- M. I. Anwar, M. Asad, L. Ma, W. Zhang, A. Abbas, M. Y. Khan, M. Zeeshan, A. Khatoon, R. Gao, S. Manzoor, M. Naeem Ashiq, S. Hussain, M. Shahid and G. Yang, Coord. Chem. Rev., 2023, 478, 214967 CrossRef CAS.
- Z. Liang, R. Zhao, T. Qiu, R. Zou and Q. Xu, EnergyChem, 2019, 1, 100001 CrossRef.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS.
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 RSC.
- W. H. Choi, B. C. Moon, D. G. Park, J. W. Choi, K. Kim, J. Shin, M. G. Kim, K. M. Choi and J. K. Kang, Advanced Science, 2020, 7, 2000283 CrossRef CAS.
- S. You, X. Gong, W. Wang, D. Qi, X. Wang, X. Chen and N. Ren, Adv. Energy Mater., 2016, 6, 1501497 CrossRef.
- T. Y. Chen, L. Y. Lin, D. S. Geng and P. Y. Lee, Electrochim. Acta, 2021, 376, 137986 CrossRef CAS.
- F. Marpaung, M. Kim, J. H. Khan, K. Konstantinov, Y. Yamauchi, M. S. A. Hossain, J. Na and J. Kim, Chem.–Asian J., 2019, 14, 1331–1343 CrossRef CAS.
- Y. Zhang, Y. Lin, H. Jiang, C. Wu, H. Liu, C. Wang, S. Chen, T. Duan and L. Song, Small, 2018, 14, 1702074 CrossRef.
- A. K. Díaz-Duran and F. Roncaroli, Eur. J. Inorg. Chem., 2021, 2021, 2830–2839 CrossRef.
- C. Young, J. Kim, Y. V. Kaneti and Y. Yamauchi, Appl. Energy Mater., 2018, 1, 2007–2015 CrossRef CAS.
- M. Zhu, Q. Chen, J. Kan, J. Tang, W. Wei, J. Lin and S. Li, Energy Technol., 2019, 7, 1800963 CrossRef.
- Y. Pan, S. Yan, Y. Liu, Z. Tian, D. Li, Y. Chen, L. Guo and Y. Wang, Electrochim. Acta, 2022, 422, 140560 CrossRef CAS.
- J. Wang, Y. Song, C. Zuo, R. Li, Y. Zhou, Y. Zhang and B. Wu, J. Colloid Interface Sci., 2022, 625, 722–733 CrossRef CAS PubMed.
- J. Yan, Y. Huang, Y. Yan, X. Zhao and P. Liu, Composites, Part A, 2020, 139, 106107 CrossRef CAS.
- D. B. Schuepfer, F. Badaczewski, J. M. Guerra-Castro, D. M. Hofmann, C. Heiliger, B. Smarsly and P. J. Klar, Carbon, 2020, 161, 359–372 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- F. Ambroz, T. J. Macdonald, V. Martis and I. P. Parkin, Small Methods, 2018, 2, 1800173 CrossRef.
- Z. Zhao, Z. Zhang, Y. Zhao, J. Liu, C. Liu, Z. Wang, G. Zheng, G. Huang and Y. Mei, Adv. Funct. Mater., 2019, 29, 1906365 CrossRef CAS.
- A. Galarneau, F. Villemot, J. Rodriguez, F. Fajula and B. Coasne, Langmuir, 2014, 30, 13266–13274 CrossRef CAS PubMed.
- H. Tabassum, A. Mahmood, Q. Wang, W. Xia, Z. Liang, B. Qiu, R. Zhao and R. Zou, Sci. Rep., 2017, 7, 43084 CrossRef PubMed.
- I. Kone, Z. Ahmad, A. Xie, Y. Tang, Y. Sun, Y. Chen, X. Yang and P. Wan, Energy Technol., 2020, 8, 2000409 CrossRef CAS.
- Z. Wang, Y. Lu, Y. Yan, T. Y. P. Larissa, X. Zhang, D. Wuu, H. Zhang, Y. Yang and X. Wang, Nano Energy, 2016, 30, 368–378 CrossRef CAS.
- Y. Xue, Y. Guo, Q. Zhang, Z. Xie, J. Wei and Z. Zhou, Nano-Micro Lett., 2022, 14, 162 CrossRef CAS.
- X. R. Wang, J. Y. Liu, Z. W. Liu, W. C. Wang, J. Luo, X. P. Han, X. W. Du, S. Z. Qiao and J. Yang, Adv. Mater., 2018, 30, 1800005 CrossRef.
- K. Jayaramulu, D. P. Dubal, B. Nagar, V. Ranc, O. Tomanec, M. Petr, K. K. R. Datta, R. Zboril, P. Gómez-Romero and R. A. Fischer, Adv. Mater., 2018, 30, 1705789 CrossRef PubMed.
- L. Cao, F. Xu, Y. -Y. Liang and H. -L. Li, Adv. Mater., 2004, 16, 1853–1857 CrossRef CAS.
- C. Yuan, L. Hou, L. Shen, D. Li, F. Zhang, C. Fan, J. Li and X. Zhang, Electrochim. Acta, 2010, 56, 115–121 CrossRef CAS.
- G. Samuel. An Introduction to Electrochemistry, Maurice Press, 2008 Search PubMed.
- T. Nguyen and M. de F. Montemor, Adv. Sci., 2019, 6, 1801797 CrossRef PubMed.
- A. Gupta, C. A. Allison, M. E. Ellis, J. Choi, A. Davis, R. Srivastava, F. M. de Souza, D. Neupane, S. R. Mishra, F. Perez, A. Kumar, R. K. Gupta and T. Dawsey, Int. J. Hydrogen Energy, 2023, 48, 9551–9564 CrossRef CAS.
- C. H. Wu, Y. F. Wu, P. Y. Lee, S. Yougbaré and L. Y. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 43180–43194 CrossRef CAS PubMed.
- Q. Lai, Y. Zhao, Y. Liang, J. He and J. Chen, Adv. Funct. Mater., 2016, 26, 8334–8344 CrossRef CAS.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS.
- L. Q. Mai, A. Minhas-Khan, X. Tian, K. M. Hercule, Y. L. Zhao, X. Lin and X. Xu, Nat. Commun., 2013, 4, 2923 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00203b |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |