
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContrast enhanced photoacoustic detection of fibrillar collagen in the near infrared region-I†
Inna
Solomonov‡
a,
Irene
Locatelli‡
 b,
Silvia
Tortorella‡
c,
Manu
Unni
a,
Shay-Lee
Aharoni
a,
Elisa
Alchera
b,
Erica
Locatelli
c,
Mirko
Maturi
c,
Chiara
Venegoni
b,
Roberta
Lucianò
d,
Andrea
Salonia
be,
Angelo
Corti
ef,
Flavio
Curnis
f,
Valeria
Grasso
gh,
Gayathri
Malamal
g,
Jithin
Jose
g,
Mauro
Comes Franchini
*c,
Irit
Sagi
*a and
Massimo
Alfano
*b
b,
Silvia
Tortorella‡
c,
Manu
Unni
a,
Shay-Lee
Aharoni
a,
Elisa
Alchera
b,
Erica
Locatelli
c,
Mirko
Maturi
c,
Chiara
Venegoni
b,
Roberta
Lucianò
d,
Andrea
Salonia
be,
Angelo
Corti
ef,
Flavio
Curnis
f,
Valeria
Grasso
gh,
Gayathri
Malamal
g,
Jithin
Jose
g,
Mauro
Comes Franchini
*c,
Irit
Sagi
*a and
Massimo
Alfano
*b
aDepartment of Immunology and Regenerative Biology, Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: irit.sagin@weizmann.ac.il
bDivision of Experimental Oncology/Unit of Urology, URI, IRCCS Ospedale San Raffaele, Milan, Italy. E-mail: alfano.massimo@hsr.it
cDepartment of Industrial Chemistry “Toso Montanari”, University of Bologna, Via P. Gobetti 85, 40129 Bologna, Italy. E-mail: mauro.comesfranchini@unibo.it
dDepartment of Pathology, IRCCS San Raffaele Hospital and Scientific Institute, Milan, Italy
eVita-Salute San Raffaele University, Milan, Italy
fDivision of Experimental Oncology, IRCCS San Raffaele Scientific Institute, Milan, Italy
gFUJIFILM Visualsonics Inc., Amsterdam, the Netherlands
hFaculty of Engineering, Institute for Materials Science, Christian-Albrecht University of Kiel, Kiel, Germany
First published on 7th June 2024
Abstract
Fibrillar collagen accumulation emerges as a promising biomarker in several diseases, such as desmoplastic tumors and unstable atherosclerotic plaque. Gold nanorods (GNRs) hold great potential as contrast agents in high-resolution, biomedically safe, and non-invasive photoacoustic imaging (PAI). This study presents the design and characterization of a specialized imaging tool which exploits GNR assisted targeted photoacoustic imaging that is tailored for the identification of fibrillar collagen. In addition to the photoacoustic characterization of collagen in the NIR 1 and 2 regions, we demonstrate the detailed steps of conjugating a decoy to GNRs. This study serves as a proof of concept, that demonstrates that conjugated collagenase-1 (MMP-1) generates a distinct and collagen-specific photoacoustic signal, facilitating real-time visualization in the wavelength range of 700–970 nm (NIR I). As most of the reported studies utilized the endogenous contrast of collagen in the NIR II wavelength that has major limitations to perform in vivo deep tissue imaging, the approach that we are proposing is unique and it highlights the promise of MMP-1 decoy-functionalized GNRs as novel contrast agents for photoacoustic imaging of collagen in the NIR 1 region. To our knowledge this is the first time functionalized GNRs are optimized for the detection of fibrillar collagen and utilized in the field of non-invasive photoacoustic imaging that can facilitate a better prognosis of desmoplastic tumors and broken atherosclerotic plaques.
Introduction
Many malignancies are distinguished by profound alterations in the extracellular matrix (ECM).1 Pathological remodeling of the ECM leads to the development of a desmoplastic microenvironment characterized by the deposition of fibrillar collagens and other structural proteins, including fibronectin, hyaluronan and tenascin C, among others.2–5 Notably, fibrillar collagens generate structurally specific complex filaments or bundles called fibers, contrasting with non-fibrillar collagens and other ECM proteins that form networks within the ECM with less pronounced features. This process triggers drastic changes in the biophysical and biochemical properties of the tissue, altering cell signaling, migration and proliferation, as well as promoting tumor growth and metastasis.1 Consequently, the detection of desmoplasia, either preceding tumor formation or present in the early stages of desmoplastic tumors, holds immense potential for enhancing patient outcomes and optimizing treatment efficacy. Notably, the excessive deposition of fibrillar collagen is a hallmark feature of the desmoplastic microenvironment in various malignancies, such as bladder cancer, breast cancer, and gastric cancer.2,6,7 Other than oncological diseases, exposure of collagen by atherosclerotic plaques is an indicator of plaque progression and instability.8 With this understanding, the exploration of desmoplastic microenvironments for early prognostic indicators was deemed imperative, particularly leveraging fibrillar collagens as a disease biomarker.Over the past decade, photoacoustic imaging (PAI) has risen as a rapid, non-invasive, and safe biomedical imaging modality, synergizing the advantages of optical absorption and ultrasound resolution. Operating on the principle of pulsed-laser light absorption by endogenous or exogenous contrast agents within tissues, PAI generates an acoustic wave collected using a transducer, thereby furnishing real-time functional and molecular insights with exceptional resolution and tissue-penetration capabilities.9 The first window of the near infrared wavelength region (NIR I, 700–1000 nm) is ideal for deep tissue in vivo PA imaging as it has compromised optical absorption and scattering that can enable the detection of oxy–deoxy hemoglobin. In addition to hemoglobin, other prominent chromophores in this window that can be visualized are melanin and lipids.10,11 To date, most of the reported studies utilized the second window of NIR (NIR-II, 1100 to 2000 nm) for the detection of collagen. While collagen has very distinct characteristics in the NIR II window, and intravascular PA imaging has demonstrated visualization possibilities,12 this wavelength range is not ideal for in vivo deep tissue imaging due to the limited energy of the commonly used light sources for PA imaging in this window. However, this limitation can be mitigated by utilizing contrast agents. Recent study demonstrated the feasibility of deeper tissue imaging in the NIR-II region by utilizing specially designed responsive optical probes as contrast agents and imaging deep up to 116 mm.13
Targeted exogenous contrast agents offer the dual advantage of amplifying the PAI response and selectively targeting regions of interest, thus expanding PAI's scope to molecular imaging.14–17 Several contrast agents have been developed, but the majority of these are based on small molecules that are difficult to adapt for the real targeting of specific tissues or lesions. Nanoparticles, and in particular gold nanoparticles, are of interest as promising contrast agents for their unique optical properties, possibility to tune the light absorption wavelength and safety profile due to their inert nature.18 Several types of gold nanoparticles have been designed and explored for this purpose, and among them, gold nanorods (GNRs) have shown the highest extinction coefficient and a good PA conversion efficiency. Furthermore, by modifying their aspect ratio (ratio between length and width) it is possible to tune the wavelength of maximum absorption of light, thereby enabling a perfect match between contrast agent properties and PAI application.19 In particular, the near-infrared region (NIR I), which is placed above 750 nm, is to be preferred for PAI because it leverages the optical window that allows for deeper tissue penetration and overcomes the interference from different endogenous contrast molecules present in tissues:20 GNRs with the maximum absorption wavelength peaking at 800 nm represent the best candidates for this scope.21
In this study we aimed to demonstrate that the steps for conjugating a decoy to GNRs and thus functionalized GNRs can be used to detect collagen in the NIR-I wavelength range. In addition to the ex vivo characterization of collagen in both NIR-I and NIR-II regions, here, we demonstrated the feasibility of an innovative approach and visualization strategy for the non-invasive detection of the desmoplastic ECM, by exploiting fibrillar collagens as prime candidates of desmoplastic areas and the utilization of GNRs as plasmonic nanoantennas for PAI.
To facilitate GNR localization to fibrillar collagens, these nanoparticles were functionalized with a highly specific and selective decoy receptor. We needed to engineer a molecule that would recognize fibrillar collagens with high affinity and specificity, without affecting their structure or functions and without exhibiting proteolytic activity. This collagen decoy receptor was engineered by inactivating human collagenase-1, i.e., matrix metalloproteinase 1 (MMP-1), which demonstrates remarkable affinity for fibrillar collagens, primarily degrading collagen types I and III with high specificity.22,23 Additionally, MMP-1 specifically cleaves interstitial collagen types I, II, and III within the triple-helical collagen chain, yielding classic ¾ and ¼ fragments.24,25
In vitro experiments confirmed the binding of the MMP-1 decoy to collagen type I from both human and rat sources. Subsequently, the localization of GNRs conjugated with the MMP-1 decoy was evaluated ex vivo using a rat model of bladder carcinoma, demonstrating the maintenance of MMP-1 decoy receptor conformation and its ability to mediate targeted delivery of GNRs to collagen-rich sites within the neoplastic ECM. Finally, we validated the decoy's ability to facilitate GNR-assisted photoacoustic signal generation specifically for fibrillary collagens. To the best of our knowledge, the presented study is pioneering in demonstrating the capability of detecting collagen in the NIR-I range. Given the widespread utilization of the NIR-I range for the in vivo imaging of various tissue chromophores such as oxy/deoxy hemoglobin, lipids, and melanin, our study offers an additional value for comprehensive tissue characterization as it integrates collagen detection by utilizing specifically tailored GNRs as a contrast agent.
Results
Expression, purification and characterization of the MMP1-decoy
To develop a specific and selective receptor decoy for human fibrillary collagens, the scaffold of one of the major enzymes capable of degrading this protein, such as MMP-1, was chosen. Previous reports have shown that both the catalytic and hemopexin domains of MMP-1 are required for strong MMP-1-collagen interactions.26–29 Thus, the zymogen form of MMP-1 was inactivated due to known mutation E200A30 and expressed in HEK293-6E cells, allowing the generation of soluble secreted proteins with >95% and a yield of 21 ± 4 mg L−1 (Fig. 1A).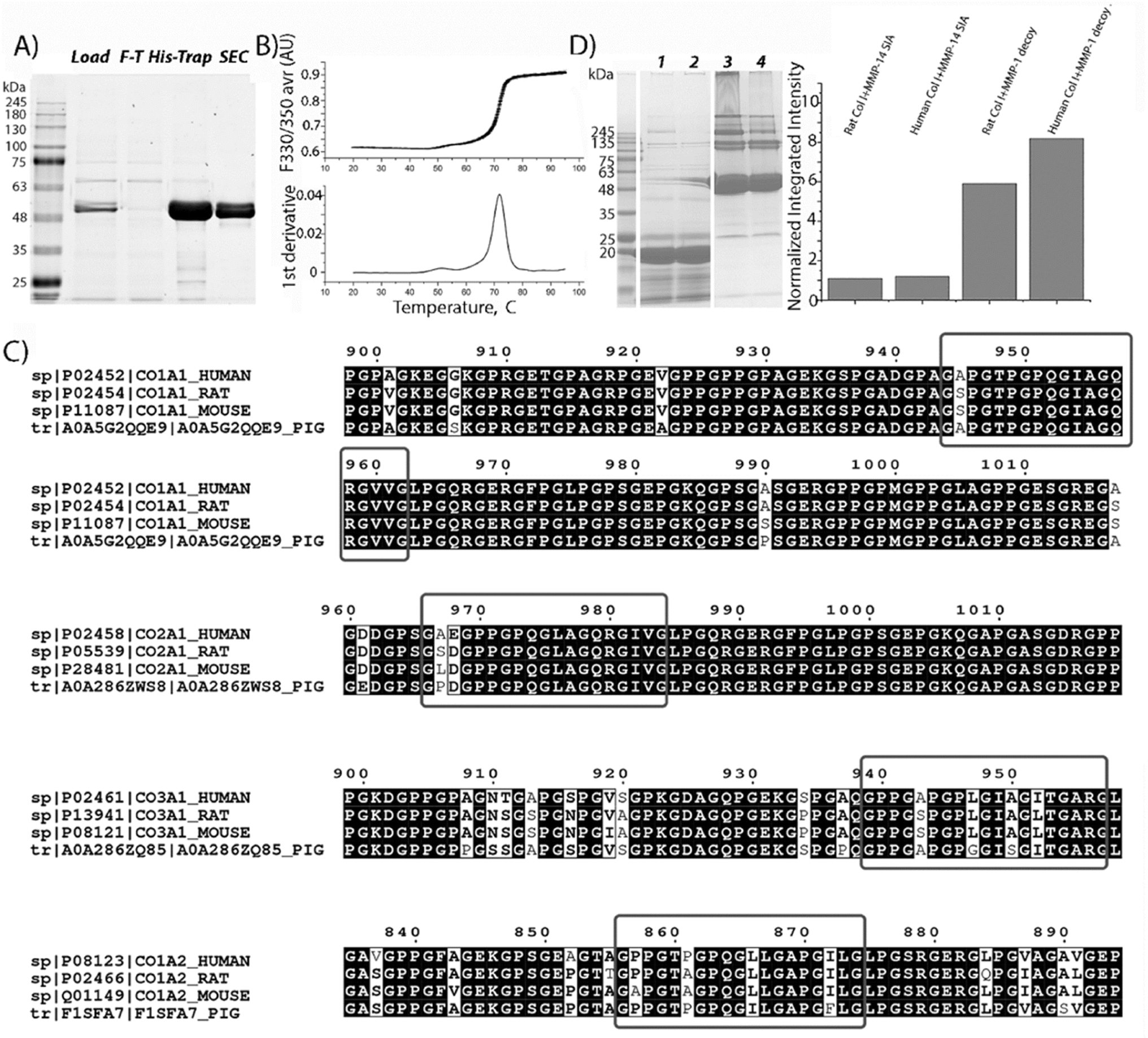 | ||
| Fig. 1 Characterization of the MMP-1 decoy. (A) SDS-PAGE analysis of MMP-1 decoy purification. Lane labels: molecular mass standards; L, load; F-T, flow-through; His-Trap, protein eluted using imidazole from the Ni column; SEC, size exclusion chromatography using a Superdex 75. (B) MMP-1 decoy thermostability by DSF, showing the melting curve and derivative plot used to determine the Tm of the decoy. (C) BLAST search results; red rectangles show the alignment of collagen fragments from different origins with human collagen MMP interaction domains identified33 or supposed32 to be MMP interaction domains. (D) Direct specific binding of the MMP1-decoy to collagen type I from different species revealed by pull down assay. Left: the SDS-PAGE image of the decoy pulling down different collagens. lanes 1 and 2: negative control and beads immobilized with the MMP14 based decoy (MMP-14 SIA). Lanes 3 and 4: the MMP-1 decoy binds to reconstituted collagen type I from rats and humans, respectively. Right: the densitometry analysis of total pulled down collagen (all bands above 139 kDa). The bars represent the normalized integrated density of collagen compared to that of the decoy. | ||
The thermostability analysis of the MMP-1 decoy was conducted using differential scanning fluorimetry (DSF). The melting temperature (Tm) of the MMP-1 decoy was quantified to be 71.7 ± 0.03 °C (Fig. 1B). Notably, this Tm value closely resembles that of active enzyme zymogens such as MMP-2 (71.3 °C) and MMP-8 (74.8 °C),31 indicating a high level of protein stability for the MMP-1 decoy.
To predict the recognition and binding of the MMP-1 decoy to collagens from other origins, the sequence similarities of collagen fragments around cleavage sites, as defined in human fibrillar collagens,32,33 were compared with those from various species, using basic local alignment search tool BLAST and Clustal Omega (Fig. 1C).34 A high degree of sequence similarity and sequence identity in the MMP interaction domain was revealed for collagen type I from different origins, for both the α1 and α2 chains. However, a single substitution of Ala with Ser was observed at position 945 in the α1 chain of rat and mouse collagen, differing from human collagen type I. Additionally, one substitution (P856A) and two substitutions (L866I and I872F) were identified in the α2 chain of collagen type I in mice and pigs respectively. These differences indicate that the MMP-1 decoy will interact with rat collagen type I, but its binding ability has to be checked for that of mice and pigs, which harbor two substitutions in the collagen interaction domain. Furthermore, only one substitution (P861A) is observed in rat collagen type III compared to that of humans, suggesting the interaction between the decoy and collagen type III in rats. Finally, two substitutions are detected in the MMP interaction domain of collagen type II in rats (A967S and E968D), mice (A867L and E968D) and pigs (A967P and E968D) and in collagen type III in mice (P857A and P861A) and pigs (L866I and I872F) indicating weakened interactions with the decoy molecule.
To confirm the predicted binding of the MMP-1 decoy to collagen type I from different origins, a pull-down assay with a His-tagged MMP-1 decoy and reconstituted collagens from humans and rats, was conducted. The eluted MMP-1-decoy complexes were analyzed by SDS-PAGE, followed by densitometry analysis (Fig. 1D). As predicted, the MMP-1 decoy binds both rat and human collagen type I. Remarkably, in comparison to published data,27 we observed the binding of the latent form of inactivated collagenase to collagen type I obtained from different origins. The data strongly suggest that the decoy binding occurs due to the interaction of the hemopexin domain with collagens, since the MMP-1 decoy exists in zymogen form. The analysis of collagen bands (>139 kDa) obtained as a result of the pull-down procedure shows that the integrated density value for human collagen is higher than that of rat collagen. These results confirm BLAST prediction, pointing to a stronger binding of the decoy to human collagen type I than to rat collagen. Remarkably, the inactive variant mutant of the MMP14 catalytic domain,35 which is non-specific to fibrillar collagens, does not bind to human or rat collagen type I, further indicating the specific binding of the MMP-1 decoy to collagen I.
Synthesis and characterization of GNRs@Chit-Dec ex vivo
With the aim to provide a biocompatible system for future in vivo applications, we opted to use GNRs coated with chitosan, whose biocompatibility of the entire nanosystem (GNRs@Chit) or of the single components of the nanosystem has been previously demonstrated.21,36–38The presence of a N-term 8xHis-tag in the MMP1-decoy sequence, exploited for performing decoy purification, was also utilized in a conjugation approach based on “Ni-NTA labelling” for coupling to GNRs. Chitosan-coated GNRs (GNRs@Chit) were prepared from CTAB-coated GNRs and characterized as we recently reported.21 A summary of the materials characterization from previously published data is available (Fig. S1 in the ESI†). The nanosystem displayed free amino groups on chitosan chains, which were exploited for binding a bifunctional linker able to couple to the biopolymeric matrix with the targeting decoy protein. For this purpose (N-[5-(4-isothiocyanatobenzyl)amido-1-carboxypentyl]iminodiacetic acid (isothiocyanobenzyl-NTA)) was selected. The isothiocyanate group is known to readily react with free amino groups forming stable thiourea linkages, which have been widely used to couple biomolecules, dyes, and nanostructures with a click chemistry approach.39,40 On the other hand, the nitrilotriacetic acid (NTA) terminus of the linker can be activated for proteins containing six adjacent histidine residues (His-tagged proteins), by complexation of Ni2+ ions, as previously reported (Fig. 2).41,42
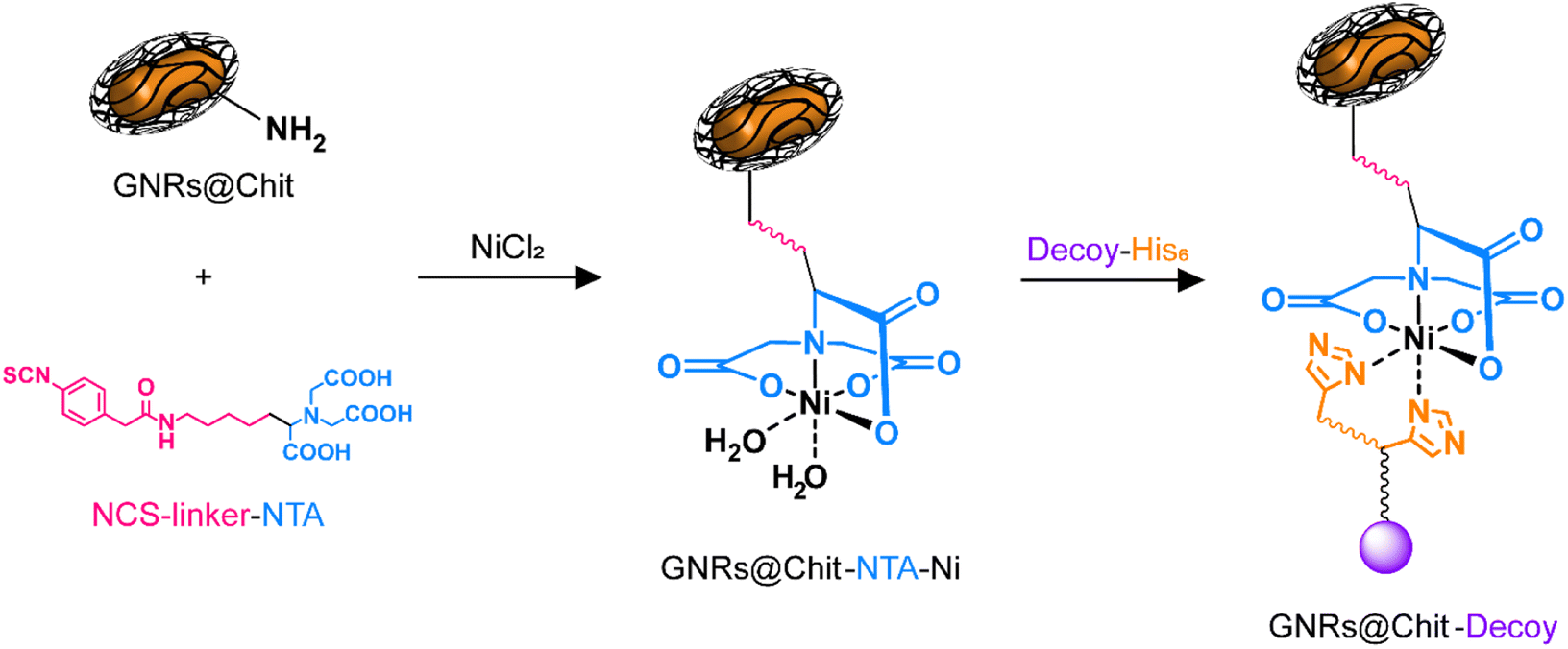 | ||
| Fig. 2 Coupling of the His-tagged decoy to chitosan-coated GNRs. First, the free amino groups of GNRs@Chit are reacted with the isothiocyanate terminus of the bifunctional linker. Then, the NTA terminus is exploited for Ni2+ complexation, thus forming a Ni-NTA complex able to bind His-tagged proteins. | ||
His-tagged green fluorescent protein (GFP) was used as a model protein to verify the effectiveness of the developed GNRs@Chit functionalization approach. Fluorescence emission spectra were recorded on purified GNRs@Chit-GFP and on the wastewaters collected during purification, revealing the quantitative binding of the His-tagged protein to the nanosystem (Fig. 3A) and no fluorescence signal arising from the wastewaters. Therefore, this approach was adopted for the His-tagged decoy molecule whose conjugation to the GNRs did not induce aggregation or reshaping of the nanoparticles (Fig. 3B), neither modify the properties of the metallic core of the nanosystem (Fig. 3C and D). The functionalization with the MMP1-decoy did not significantly affect the ζ-potential, which shifted from +40 ± 6 mV for GNRs@Chit to +37 ± 5 mV for GNRs@Chit-Dec, and there was a small shift in the position of the longitudinal surface plasmon resonance band from 800 nm for GNRs@Chit to 803 nm for GNRs@Chit-Dec, with no appreciable reduction of peak intensity or increase in the values of full width at half-maximum (Table 1).
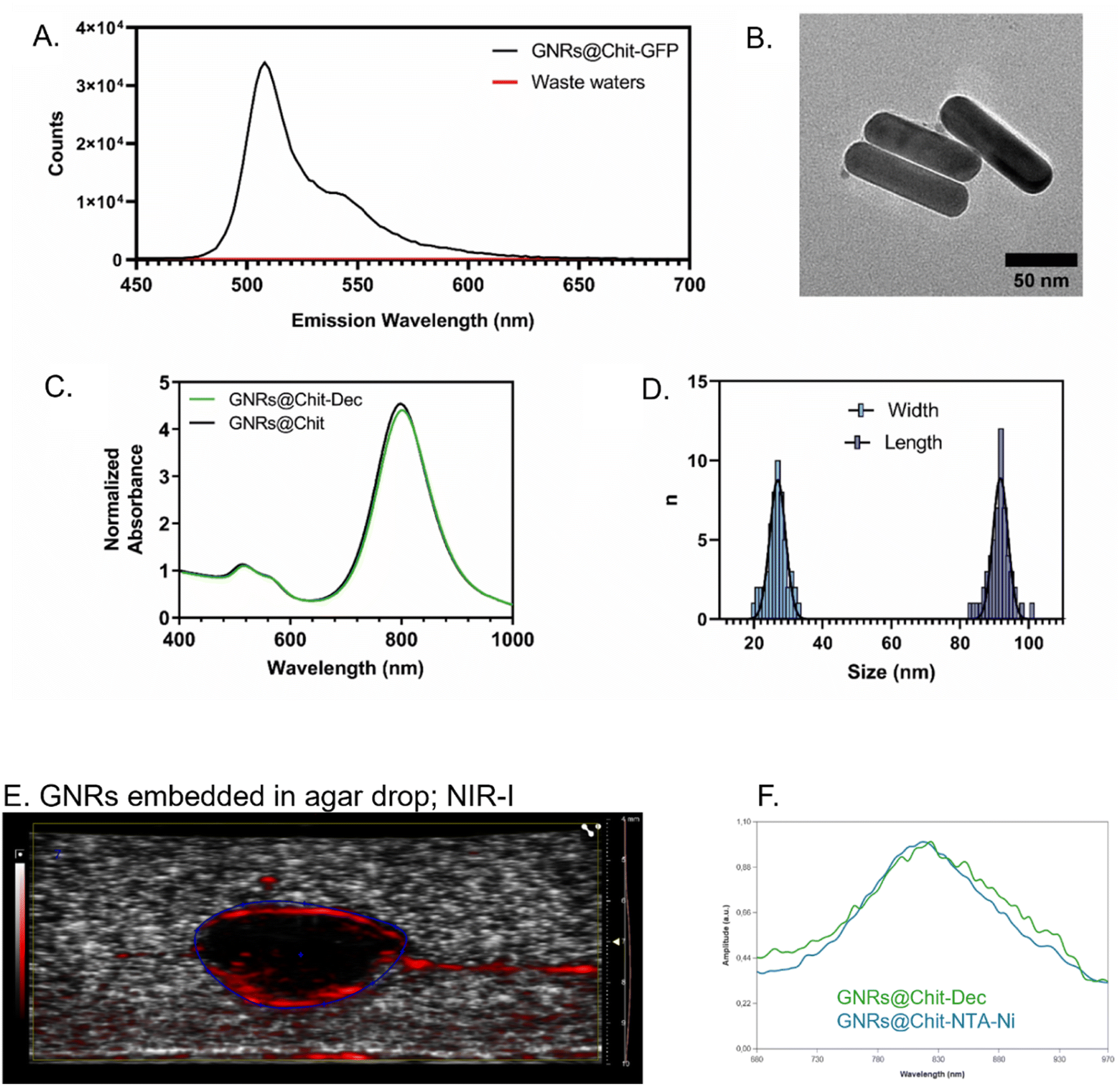 | ||
| Fig. 3 Effectiveness of the conjugation approach and characterization of GNRs@Chit-Dec. (A) Fluorescence emission spectrum of GNRs@Chit-GFP compared to the wastewaters collected during its purification, revealing quantitative attachment of the His-tagged protein to the Ni-activated nanosystem. (B) Representative TEM image of GNRs@Chit-Dec. (C) Normalized Uv-vis spectrum of GNRs@Chit-Dec compared to GNRs@Chit. (D) Distribution of widths and lengths of n = 55 GNRs@Chit-Dec measured by TEM. The solid lines represent the Gaussian fit for both dimensions. Photoacoustic imaging of an agar drop containing GNR@Chit-NTA-Ni (6 nmol Au) (E) and its associated PA spectra in NIR-I (680–970 nm), followed by the overlay with the PA spectra of GNR@Chit-Dec (6 nmol Au) (F). | ||
| GNRs@CTAB | GNRs@Chit | GNRs@Chit-Dec | |
|---|---|---|---|
| a Data for GNRs@CTAB and GNRs@Chit are from ref. 21. b Data expressed as mean ± SD. | |||
| VIS-NIR | |||
| λ max (nm) | 798 | 800 | 803 |
| FWHM (nm) | — | 113 | 112 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| TEM | |||
| GNRs lengthb (nm) | — | 88.2 ± 6.4 | 91.8 ± 2.2 |
| GNRs widthb (nm) | — | 25.7 ± 2.0 | 26.9 ± 2.3 |
| Aspect ratiob | — | 3.4 ± 0.5 | 3.4 ± 0.4 |
| Zeta potential (mV) | +29.8 | +40 ± 6 | +37.0 ± 5 |
We also assessed whether the conjugation of the decoy to the GNRs could affect the PA spectra of GNRs@Chit, which occurs in NIR-I. The PA spectra of GNRs in the absence or presence of the MMP-1 decoy were obtained, by embedding GNRs in agar drops. The GNRs were identified in B mode and the PA spectra were acquired in NIR-I, demonstrating that the conjugation of the decoy to the GNRs has not modified their expected PA spectra that peaked at 810 nm (Fig. 3E and F), as previously reported for the conjugation with a small peptide.21
To visualize the targeting ability of the MMP-1 decoy, a rat bladder carcinoma model was first established. Among the various bladder cancer models available, the orthotopic bladder cancer model mediated by the bladder-specific carcinogen nitrosamine (N-(4-hydroxybutyl)nitrosamine: BBN) was opted. Employing this compound allows all stages of bladder cancer development and progression to be monitored, thus mimicking the pathological processes that happen in humans,43,44 which include the formation of desmoplastic stroma (Fig. 4a and b).
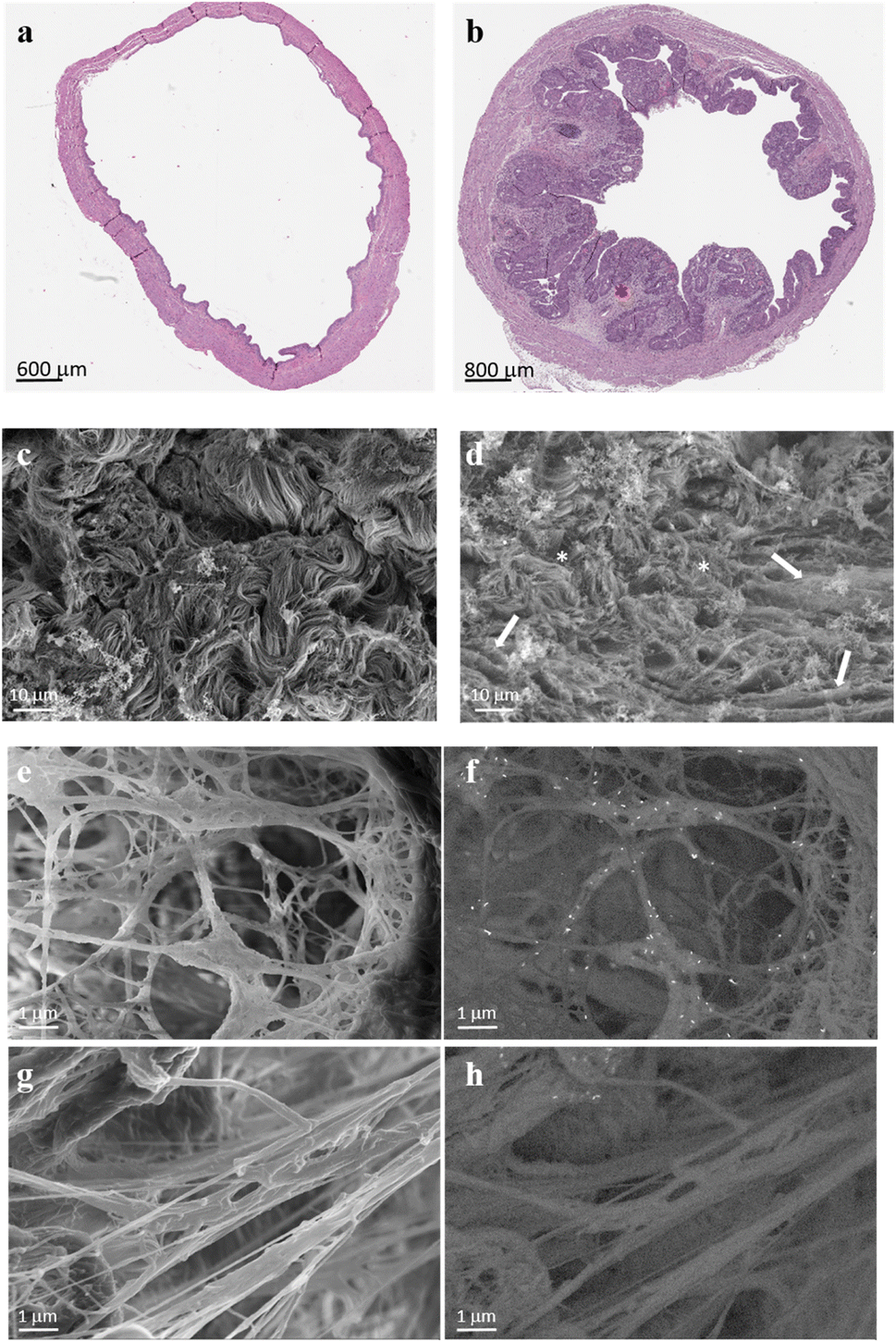 | ||
| Fig. 4 Images illustrating the progression of bladder carcinoma in rats and the interactions of GNRs with collagen fibrils enriched in ECM scaffolds derived from bladder carcinoma tissues. Representative H&E section bladder from an 8-month rat (a) and rat bladder carcinoma after 6 months of BBN treatment (b), used for the isolation of the ECM; the red asterisk shows a typical desmoplastic stroma, which applies to all tumors present in that bladder. Representative SEM images of the ECM isolated from healthy (c) and carcinoma rat bladder (d) tissues; ECM scaffolds from healthy tissues consist of tightly packed collagen fibers with similar diameters, creating a wavy pattern. In contrast, bladder carcinoma scaffolds exhibit disrupted collagen packing, characterized by a disorganized composition of fibrils (thinnest fibrils indicated by asterisks) and fibers with varying diameters. Linearization of thick collagen fibers, as expected in solid tumors, is also observed (indicated by the arrow). The images of at least 7 fields in different locations within the desmoplastic region of each decellularized bladder from three rats were captured. Topography of the tumor ECM captured in the secondary electron mode (e) and acquired in the backscattered electron mode (f) to highlight the binding of GNRs@Chit-Dec to collagen fibrils (indicated by white dots). Panels (g) and (h) refer to the same experiment shown in panels (e) and (f), but with the ECM incubated with the control nanoparticles GNRs@Chit-NTA-Ni. | ||
To assess the specificity of GNRs@Chit-Dec to recognize fibrillar collagen we used electron microscopy, because this technique allows the visualization of both collagen fibers and GNRs at the same time and could be used to demonstrate the colocalization of gold with the collagen. The ECM scaffolds obtained by decellularization of rat bladder and rat bladder cancer enriched with desmoplastic areas were subjected to high resolution scanning electron microscopy (Fig. 4c and d), confirming the desmoplastic modification that was detected using hematoxylin eosin stain (Fig. 4a). In particular, distinctive features of the bladder cancer ECM were the thickening and linearization of collagen fibers, in agreement with previous information reported for human bladder cancer.45
We investigated the MMP1-decoy's targeting activity for delivering GNRs to collagen fibrils using ECM scaffolds derived from rat bladder cancer, as they maintain the structure and morphology of the tumor microenvironment (Fig. 4c and d). Scanning electron microscopy images captured after incubation of GNRs@Chit-Dec with the ECM from bladder cancer showed direct adsorption of the decoy onto the fibrils. Conversely, the control nanoparticles lacking the decoy (GNRs@Chit-NTA-Ni) did not bind to the fibrils (Fig. 4e–h).
Identification of the PA spectra of collagen I in NIR-II
To evaluate the recognition of fibrillar collagens, mainly type I collagen, by PAI of GNRs@Chit-Dec, we set up in vitro and ex vivo experimental conditions. A sterile, neutral purified porcine gelatin absorbable sponge made of collagen I, Spongostan™, was used to identify the PA spectra of collagen I in the NIR I and NIR-II regions. B-mode US imaging was used to locate Spongostan™ below the US gel (Fig. 5A) and to identify its PA spectra (Fig. 5B). The specific shape of the PA spectra of collagen was identified by superimposing the spectra of a water-based US gel on the spectra of Spongostan™ (Fig. 5B). For collagen type I we ascribed a characteristic shape of the spectra in NIR-II (1200–1350 nm wavelengths) and two peaks at 1450 and 1920 nm that we ascribed to the ultrasound gel. Rat tail tendons (Fig. 5C) were used to establish the in vivo spectra of collagen I in NIR-II (Fig. 5D), which was superimposable to that of Spongostan™ (Fig. 5D). This information provided the reference spectra of collagen I in the NIR-II range, with a characteristic three-peak shape of the PA spectra of collagen, with peaks at 1210/1250/1280 nm wavelengths.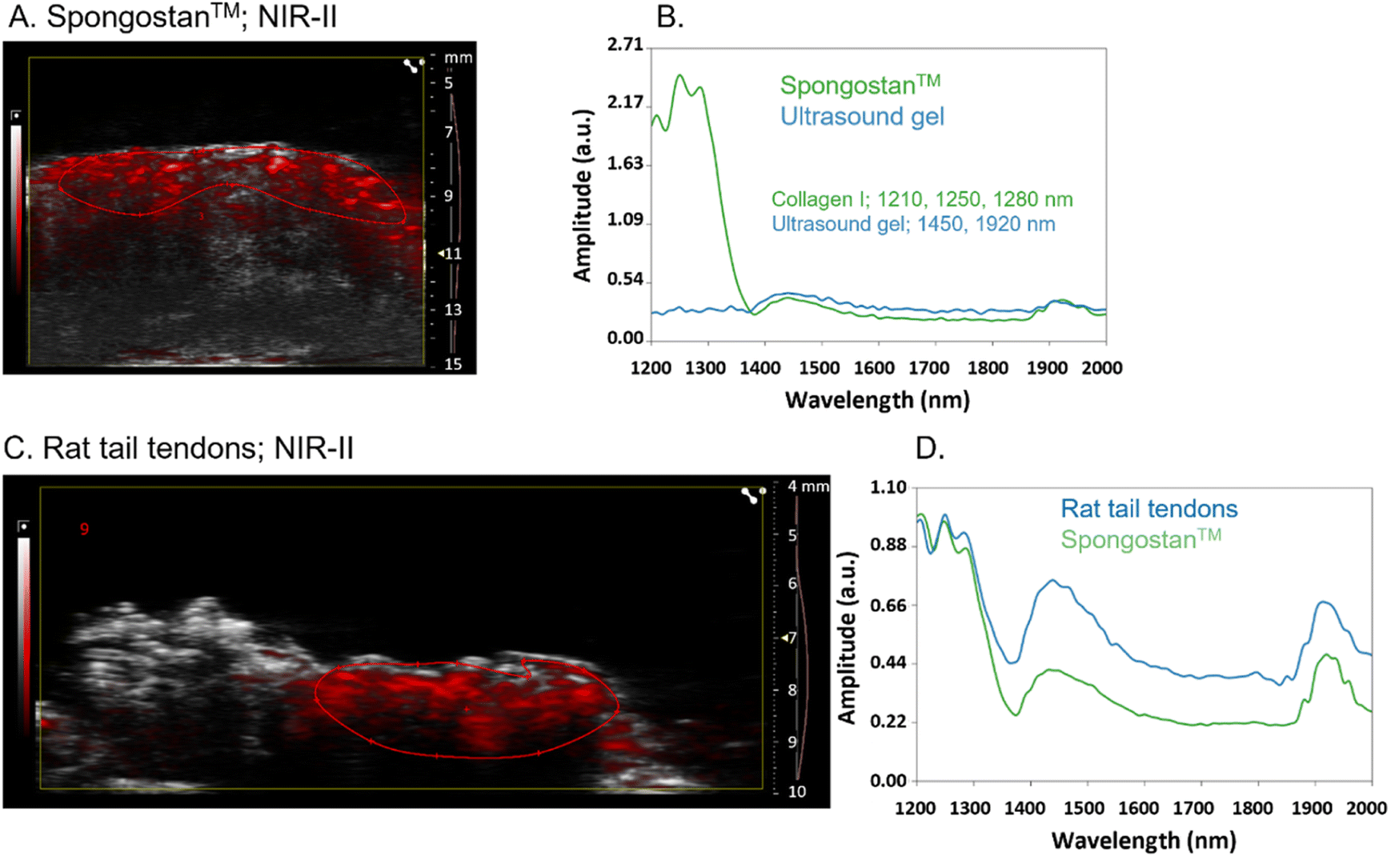 | ||
| Fig. 5 PA spectra of collagen I in NIR-II. Photoacoustic imaging (overlay of B mode in grayscale and photoacoustic signal in red scale) of Spongostan™ (A) and its associated PA spectra in NIR-II (1200–2000 nm) followed by the overlay with the PA spectra of the ultrasound gel (B); the wavelength of the three peaks of the collagen spectra and of the two peaks of the ultrasound gel are reported. Photoacoustic imaging of rat tail tendons (C) and their associated PA spectra in NIR-II, followed by the overlay with the PA spectra of Spongostan™ (D); the PA spectra of rat tail tendons and Spongostan™ were normalized to highlight the similarities. The right axis of US imaging shows (i) the acquisition depth in millimeters (mm), (ii) the yellow arrow the focus of the US imaging, and (iii) the red line the time gain compensation. | ||
The MMP-1 decoy delivers a GNR-assisted photoacoustic signal to collagen in NIR-I
We then investigated the PA spectra of the rat tail tendons and GNRs@Chit-Dec in NIR-I. The rat tail tendons were characterized by an unspecific spectrum and low absorbance in NIR-I, compared with the specific and intense bell-shape spectra that were acquired for GNRs@Chit-Dec in NIR-I (Fig. 6A).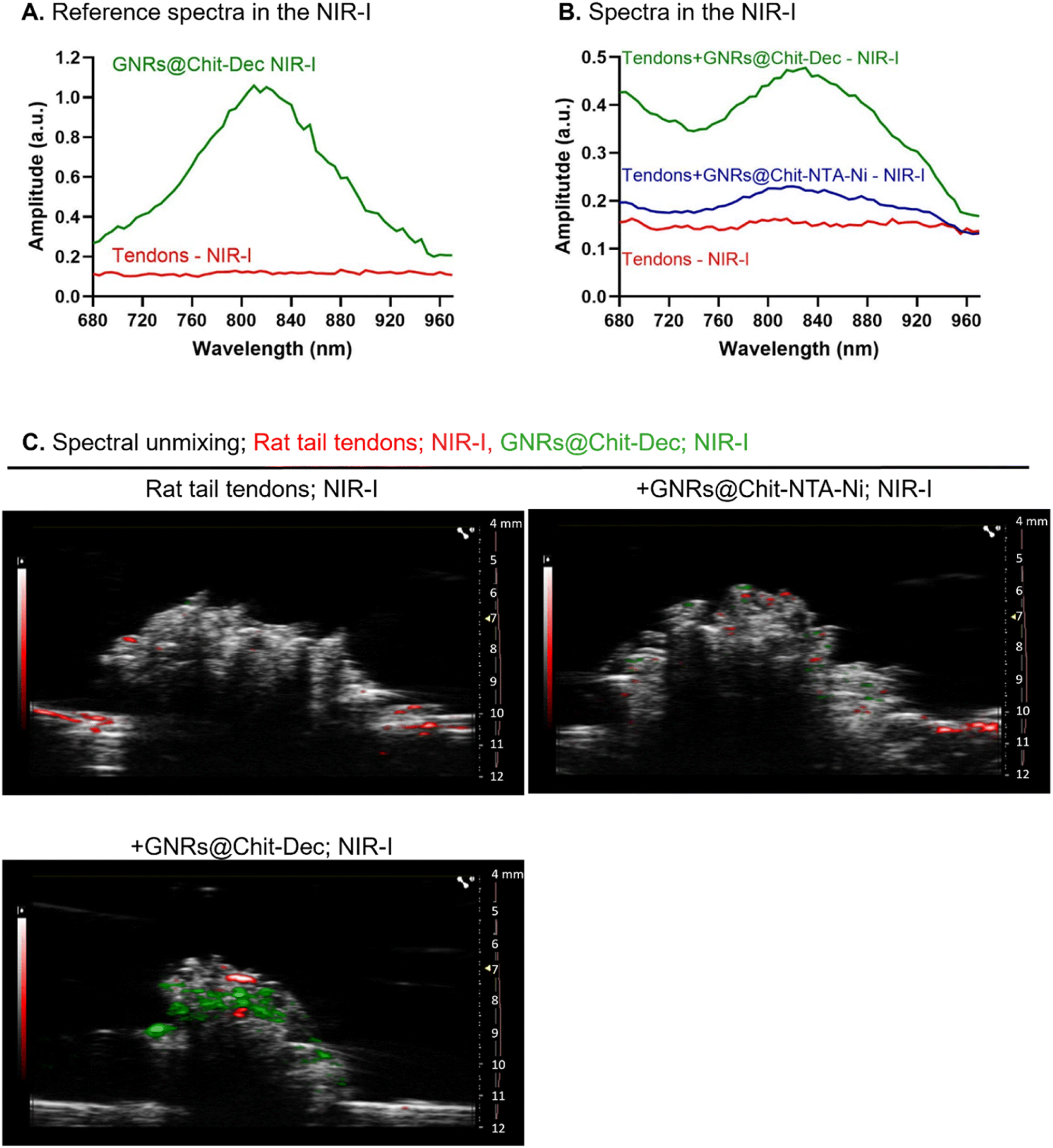 | ||
| Fig. 6 GNRs@Chit-Dec allows photoacoustic detection of collagen in NIR-I. Reference spectra of GNRs@Chit-Dec and rat tail tendons in NIR-I (A). PA spectra or rat tail tendons alone, incubated with GNRs@Chit-NTA-Ni, or GNRs@Chit-Dec (B). PA imaging of rat tail tendons alone, incubated with GNRs@Chit-NTA-Ni (100 nmol Au) or GNRs@Chit-Dec (100 nmol Au), after spectral unmixing using the reference spectra of rat tail tendons and GNRs@Chit-Dec acquired in NIR-I (C); the right axis of US imaging shows (i) the acquisition depth in millimeters (mm), (ii) the yellow arrow the focus of the US imaging, and (iii) the red line the time gain compensation. | ||
The PA spectra in NIR-I was then acquired for the rat tail tendons alone, incubated with either GNRs@Chit-NTA-Ni or GNRs@Chit-Dec. The tendons did not show specific PA spectra, while the PA spectra of GNRs were detected in the samples incubated with either GNRs@Chit-NTA-Ni or GNRs@Chit-Dec (Fig. 6B). Indeed, an increased PA signal of the GNRs was acquired from tendons incubated with GNRs@Chit-Dec vs. GNRs@Chit-NTA-Ni (Fig. 6B).
The PA reference spectra of rat tail tendons and GNRs@Chit-Dec were then used to unmix the PA spectra in NIR-I (within 680–970 nm) of rat tail tendons alone, incubated with GNRs@Chit-NTA-Ni or GNRs@Chit-Dec. After spectral unmixing collagen visualization was poor in rat tail tendons alone or incubated with GNRs@Chit-NTA-Ni, while the incubation with GNRs@Chit-Dec allowed better visualization of collagen (Fig. 6C). This information demonstrated the specificity of the decoy to collagen I in its natural microenvironment. This feasibility test also proved that the Ni-NTA chemical approach, applied for decoy conjugation through histidines, maintains the MMP-1 decoy conformation, allowing GNR delivery to fibrillar collagens and giving the PA signal. GNRs@Chit-Dec show the potential to be used as contrast agents for photoacoustic imaging of collagen I in NIR-I.
Discussion
This study demonstrated the feasibility of detecting fibrillar collagen in NIR-I, using a novel contrast agent for PAI represented by GNRs@Chit conjugated with an MMP-1 derived decoy (GNRs@Chit-Dec).We investigated the feasibility of this innovative visualization strategy for the non-invasive detection of the desmoplastic ECM, by assessing fibrillar collagens as prime candidates of desmoplastic areas and the utilization of GNRs as plasmonic nanoantennas for PAI. We also demonstrated that the steps for conjugating the decoy to GNRs@Chit did not affect the properties of neither GNRs nor the decoy. Worth noting is that the long-term storage of GNRs@Chit-Dec was ensured by the lyophilization step, as previously demonstrated by us in a related study.21 In fact, in this study, the photoacoustic properties and the collagen-binding capabilities of GNRs@Chit-Dec have been assessed after lyophilization and prolonged storage of the materials (up to 12 months).
This study presents a proof of concept for an innovative non-invasive approach aimed at early detecting extracellular matrix proteins within desmoplastic microenvironments, which are a distinctive pathological feature in many cancers. Our investigation demonstrates the successful targeting of collagen in both homeostatic and desmoplastic microenvironments through the conjugation of the MMP-1 decoy with innovative GNRs. Effective targeting is achieved through the distinctive structure of collagenase MMP-1 that allows recognition and binding to the collagen binding site of the hemopexin domain, which is distant from cross-linking sites.33 This capability allows the MMP-1 decoy to deliver GNRs to collagens with varying degrees of cross-linking, including linearized collagen fibrils in the desmoplastic region.
GNRs@Chit-Dec function effectively as photoacoustic antennas, generating GNR-assisted photoacoustic signals specific to collagen I, which were visualized in real-time. To investigate the feasibility of GNRs@Chit-Dec as contrast agents for PAI, the acquisition was performed in NIR-I. In general, PA imaging in NIR I normally facilitates the detection of endogenous tissue chromophores such as oxy/deoxygenated hemoglobin, while the detection of collagen is limited as it has a modest optical contrast in this wavelength range.46 With the help of functionalized GNRs this limitation can be vanquished and the collagen can be visualized in the NIR I range. As the utilization of NIR-II wavelength has energy impairment for in vivo deep tissue imaging the use of GNRs@Chit-Dec for PAI in NIR-I can serve as a potential candidate for the exploration of desmoplastic microenvironments.
In addition to imaging in the NIR-I range, we also report a characteristic shape of the PA spectra of collagen in the range of 1200–1300 nm wavelength. This range of wavelengths was identified using a pure gelatin sponge, which allows the separation of the PA contribution of the water and, potentially, other contrast agents that could be present in the acellular dermal matrix recently used to derive the PA spectra of collagen.47 Furthermore, the use of a dry sponge allows the shape of the spectra of collagen vs. the shape of the spectra of water present in the ultrasound gel to be magnified, avoiding interference by the use of acetic acid commonly used to dissolve collagen.48 The range of 1200–1300 nm wavelength for the PA spectra of collagen, and that of the water in NIR-II, is in agreement with previous reports.48–52 What is relevant in the PA spectra of collagen identified in our study is the characteristic three-peak shape in the range of 1200–1300 nm wavelengths, a resolution that was not shown in the above mentioned studies. Indeed, the shape of PA spectra is at the basis of the spectral unmix approach used to differentiate specific tissue components from the overall PA signal of the tissue and thus to provide detailed molecular imaging.53 The characteristic three-peak shape of the PA spectra of collagen, with peaks at 1210/1250/1280 nm wavelengths, provides a more accurate identification of collagen-rich tissue area. The spectrum of collagen was reported to share many features similar to that of another structural protein that is elastin, although the peak of elastin spectra is at 1275 nm.54 The three-peak shape of the PA spectra of collagen in NIR-II lays the groundwork for better discrimination of the two structural components in vivo. Although conventional spectral unmixing facilitates the detection of specific chromophores by leveraging user input spectral profiles, the presence of highly absorbing chromophores such as oxy/deoxy hemoglobin can hamper the sensitivity of lower absorbing chromophore visualizations. However, a recent study showcased the possibility of detecting prominent and non-prominent absorbers through data-driven spectral unmixing.53 Incorporating such artificial intelligence (AI) based approaches can further enhance the detection of collagen in the NIR-I region.
The versatility of GNRs extends beyond their current application, as they can be functionalized with other highly specific and selectively designed decoy molecules, enabling recognition of various extracellular matrix biomarkers within the desmoplastic microenvironment. These molecular probes bear paramount significance in the development of non-invasive, non-ionizing, and highly sensitive contrast agents tailored for precise photoacoustic imaging. This approach holds immense promise for the prognosis of a multitude of solid tumors, including bladder carcinoma, pancreatic cancer, colon cancer, breast cancer, and prostate cancer. Other than desmoplastic areas, another potential application of GNRs@Chit-Dec is for the detection of disrupted atherosclerotic plaque that by exposing the fibrillary collagen ignites the formation of coronary thrombosis presenting as an acute coronary syndrome, a life-threatening disease.55 The potential applications of decoy receptors conjugated with GNRs may be extended into multiple medical applications. These constructs may find utility in specific drug delivery, photothermal therapy, photodynamic therapy, and radiation therapy.56–59 Moreover, their implications in diagnostic imaging continue to hold immense prospects.
The successful conjugation of the MMP-1 decoy and GNRs offers a powerful tool for non-invasive detection and targeted intervention within the desmoplastic microenvironment. This approach may be easily adapted to other ECM biomarkers and its potential integration into a spectrum of medical applications underscores its significance in both the diagnostic and therapeutic realms.
Experimental
Materials and methods
:3 to form a DNA–PEI complex. After adding the DNA–PEI complex to the cells, the culture was incubated on an orbital shaker at 37 °C in a humidified incubator containing 5% CO2 for 5 days. Tryptone N1 was introduced to the cells at a final concentration of 0.5% 24 h post-transfection.
The expressed cells were harvested and centrifuged at 300g for 20 min at 4 °C. The supernatant was collected, filtered through a 0.22 μm membrane, and loaded onto a His-Trap 5 mL column in an AKTA purification system. The column was equilibrated with an equilibration buffer (TNC buffer: 50 mM Tris–HCl (pH 7.5), 150 mM NaCl and 10 mM CaCl2) with 20 mM imidazole and eluted using an elution buffer (TNC buffer with 250 mM imidazole). The folded proteins were concentrated to ∼10 mL using a 10 kDa cut-off Amicon (Merck) and further purified through size-exclusion chromatography (SEC) using a Superdex 75 pre-equilibrated with TNC buffer. The fraction eluted at 120–140 mL of the SEC column was concentrated to approximately 1 mg mL−1 using a 10 kDa cut-off Amicon (Merck), filtered through a 0.22 μm PVDF membrane, and stored at −80 °C. The purification protocol yielded 21 ± 4 mg L−1 of purity >95%.
The sequence analysis with BLAST was performed using the NCBI BLAST web interface (https://blast.ncbi.nlm.nih.gov/Blast.cgi). This search involved comparing the sequence of human collagen type I (genes COL1A and COL1A2 with accession numbers P02452 and P08123, respectively) to collagens of different types from different origins in the public NCBI database. Accession numbers for fibrillar collagens examined are: Homo sapiens COL2A1, P02458; Homo sapiens COL3A1, P02461; Mus musculus COL1A1, P11087.4; Mus musculus COL1A2, Q01149; Mus musculus COL2A1, P28481; Mus musculus COL3A1, P08121; Rattus norvegicus COL1A1, P02454; Rattus norvegicus COL1A2, P02466; Rattus norvegicus COL2A1, P05539; Rattus norvegicus COL3A1, P13941; Sus scrofa (pig) COL1A1, A0A5G2QQE9; Sus scrofa (pig) COL1A2, F1SFA7; Sus scrofa (pig) COL2A1, A0A286ZWS8; Sus scrofa (pig) COL3A1, A0A286ZQ85. Alignment of the genes of interest was performed using Clustal Omega,62 and the figure was generated using ESPript 3.0.63
To assess the delivery activity of the MMP-1 decoy the ECM scaffolds were incubated with a fresh solution of GNRs@Chit-Dec for 24 h at 4 °C. Subsequently, the scaffolds were gently washed with DDW three times for 30 min each at 4 °C. The samples were then fixed in a 0.1 M cacodylate buffer solution (pH 7.4) containing 2.5% paraformaldehyde and 2.5% glutaraldehyde (pH 7.2) for 60 min at room temperature followed by three washes in the same buffer. The samples were dehydrated through a series of ascending ethanol concentrations up to 100% ethanol. The dehydrated samples were observed using a Zeiss FEG Ultra55 SEM operating at 5 kV.
High-resolution ultrasound (US) and photoacoustic (PA) imaging have been performed using the Vevo LAZR-X platform (FUJIFILM VisualSonics, Inc., Toronto, ON, Canada). The imaging platform includes a high frequency US system (Vevo 3100) combined with an Nd:YAG nano-second pulsed laser with a repetition rate of 20 Hz. The linear US transducer array MX550D consists of 256 elements with a nominal center frequency of 40 MHz (25–55 MHz bandwidth) and a spatial resolution of 40 μm with a maximum imaging depth of 15 mm. Light from the laser is delivered to the tissue through optical fibers mounted on either side of the transducer. The photoacoustic spectra have been acquired between 680 nm and 970 nm (NIR-I) or between 1200 and 2000 nm (NIR-II) with a step size of 5 nm. The parameters used for the B-mode were: 2D power 100% and 2D gain 13 dB and for the PA-mode were: PA power 100% and PA gain 44 dB.
Author contributions
Writing the original draft of the manuscript: MA. Review and editing: IS, EL, MM, AS, VG, GM, JJ, MCF, IS, and MA. Conceptualization: MCF, IS, and MA. Data curation and data analysis: IS, IL, ST, MU, SLA, EA, EL, MM, CV, RL, FC, VG, GM, JJ, MCF, IS, and MA. Investigation: IS, IL, ST, MU, SLA, EA, EL, MM, CV, and FC. Methodology; IS, IL, ST, MU, SLA, EA, EL, MM, CV, FC, VG, GM, and JJ. Funding acquisition: MA.Conflicts of interest
The authors declare no competing financial interests; GM and JJ are employees of FujiFilm VisualSonics.Acknowledgements
We thank Dr Y. Fridmann Sirkis and the Protein Analysis Unit for suggestions. The EM studies were conducted at the Moskowitz Center for Nano and Bio-Nano Imaging at the Weizmann Institute of Science. This study has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 801126 (https://cordis.europa.eu/project/id/801126) and from the Horizon Europe framework programme under grant agreement no. 101113193 (https://cordis.europa.eu/project/id/101113193). The funding source had no role in the design of this study, data interpretation and writing of the report.References
- J. Winkler, A. Abisoye-Ogunniyan, K. J. Metcalf and Z. Werb, Nat. Commun., 2020, 11, 5120 CrossRef CAS PubMed.
- Z. H. Zhou, C. D. Ji, H. L. Xiao, H. B. Zhao, Y. H. Cui and X. W. Bian, J. Cancer, 2017, 8, 1466–1476 CrossRef PubMed.
- C. J. Whatcott, C. H. Diep, P. Jiang, A. Watanabe, J. LoBello, C. Sima, G. Hostetter, H. M. Shepard, D. D. Von Hoff and H. Han, Clin. Cancer Res., 2015, 21, 3561–3568 CrossRef CAS PubMed.
- C. A. Iacobuzio-Donahue, P. Argani, P. M. Hempen, J. Jones and S. E. Kern, Cancer Res., 2002, 62, 5351–5357 CAS.
- M. B. Amin, Mod. Pathol., 2009, 22(Suppl 2), S96–S118 CrossRef PubMed.
- S. Kauppila, F. Stenback, J. Risteli, A. Jukkola and L. Risteli, J. Pathol., 1998, 186, 262–268 CrossRef CAS PubMed.
- M. Fang, J. Yuan, C. Peng and Y. Li, Tumor Biol., 2014, 35, 2871–2882 CrossRef CAS PubMed.
- S. K. Nadkarni, B. E. Bouma, J. de Boer and G. J. Tearney, Laser Med. Sci., 2009, 24, 439–445 CrossRef PubMed.
- M. J. C. Lee and C. Kim, in Application of Nanoscience in Photomedicine, ed. M. R. Hamblin and P. Avci, Elsevier, 2015, ch. 3, pp. 31–47 Search PubMed.
- S. W. Cho, T. T. V. Phan, V. T. Nguyen, S. M. Park, H. Lee, J. Oh and C. S. Kim, Photoacoustics, 2023, 29, 100456 CrossRef PubMed.
- V. Grasso, H. W. Hassan, P. Mirtaheri, R. Willumeit-Römer and J. Jose, Front. Signal Process., 2022, 2, 1–17 Search PubMed.
- J. Leng, J. Zhang, C. Li, C. Shu, B. Wang, R. Lin, Y. Liang, K. Wang, L. Shen, K. H. Lam, Z. Xie, X. Gong, J. Ge and L. Song, Biomed. Opt Express, 2021, 12, 1934–1946 CrossRef PubMed.
- F. Ding, J. Feng, X. Zhang, J. Sun, C. Fan and Z. Ge, Adv. Drug Deliv. Rev., 2021, 173, 141–163 CrossRef CAS PubMed.
- M. H. Y. Cheng, Y. Mo and G. Zheng, Adv. Healthcare Mater., 2021, 10, e2001549 CrossRef PubMed.
- S. L. P. Aggarwal and F. A. Papay, Exp. Dermatol., 2022, 31, 1128–1135 Search PubMed.
- M. Maturi, E. Locatelli, I. Monaco and M. Comes Franchini, Biomater. Sci., 2019, 7, 1746–1775 RSC.
- W. Choi, B. Park, S. Choi, D. Oh, J. Kim and C. Kim, Chem. Rev., 2023, 123, 7379–7419 CrossRef CAS PubMed.
- W. Li and X. Chen, Nanomedicine, 2015, 10, 299–320 CrossRef CAS PubMed.
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS PubMed.
- A. Sun, H. Guo, Q. Gan, L. Yang, Q. Liu and L. Xi, Opt. Express, 2020, 28, 9002–9013 CrossRef PubMed.
- E. Alchera, M. Monieri, M. Maturi, I. Locatelli, E. Locatelli, S. Tortorella, A. Sacchi, A. Corti, M. Nebuloni, R. Luciano, F. Pederzoli, F. Montorsi, A. Salonia, S. Meyer, J. Jose, P. Giustetto, M. C. Franchini, F. Curnis and M. Alfano, Photoacoustics, 2022, 28, 100400 CrossRef PubMed.
- R. Visse and H. Nagase, Circ. Res., 2003, 92, 827–839 CrossRef CAS PubMed.
- A. Pardo and M. Selman, Int. J. Biochem. Cell Biol., 2005, 37, 283–288 CrossRef CAS PubMed.
- G. B. Fields, J. Biol. Chem., 2013, 288, 8785–8793 CrossRef CAS PubMed.
- I. Solomonov, E. Zehorai, D. Talmi-Frank, S. G. Wolf, A. Shainskaya, A. Zhuravlev, E. Kartvelishvily, R. Visse, Y. Levin, N. Kampf, D. A. Jaitin, E. David, I. Amit, H. Nagase and I. Sagi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10884–10889 CrossRef CAS PubMed.
- I. M. Clark and T. E. Cawston, Biochem. J., 1989, 263, 201–206 CrossRef CAS PubMed.
- J. A. Allan, R. M. Hembry, S. Angal, J. J. Reynolds and G. Murphy, J. Cell Sci., 1991, 99(Pt 4), 789–795 CrossRef CAS PubMed.
- G. Murphy, J. A. Allan, F. Willenbrock, M. I. Cockett, J. P. O'Connell and A. J. Docherty, J. Biol. Chem., 1992, 267, 9612–9618 CrossRef CAS PubMed.
- M. L. Patterson, S. J. Atkinson, V. Knauper and G. Murphy, FEBS Lett., 2001, 503, 158–162 CrossRef CAS PubMed.
- L. Chung, D. Dinakarpandian, N. Yoshida, J. L. Lauer-Fields, G. B. Fields, R. Visse and H. Nagase, EMBO J., 2004, 23, 3020–3030 CrossRef CAS PubMed.
- N. Meraz-Cruz, F. Vadillo-Ortega, A. M. Jimenez-Garduno and A. Ortega, Heliyon, 2020, 6, e03865 CrossRef PubMed.
- R. C. Billinghurst, L. Dahlberg, M. Ionescu, A. Reiner, R. Bourne, C. Rorabeck, P. Mitchell, J. Hambor, O. Diekmann, H. Tschesche, J. Chen, H. Van Wart and A. R. Poole, J. Clin. Invest., 1997, 99, 1534–1545 CrossRef CAS PubMed.
- S. M. Sweeney, J. P. Orgel, A. Fertala, J. D. McAuliffe, K. R. Turner, G. A. Di Lullo, S. Chen, O. Antipova, S. Perumal, L. Ala-Kokko, A. Forlino, W. A. Cabral, A. M. Barnes, J. C. Marini and J. D. San Antonio, J. Biol. Chem., 2008, 283, 21187–21197 CrossRef CAS PubMed.
- F. Madeira, M. Pearce, A. R. N. Tivey, P. Basutkar, J. Lee, O. Edbali, N. Madhusoodanan, A. Kolesnikov and R. Lopez, Nucleic Acids Res., 2022, 50, W276–W279 CrossRef CAS PubMed.
- E. M. Adams, S. Pezzotti, J. Ahlers, M. Ruttermann, M. Levin, A. Goldenzweig, Y. Peleg, S. J. Fleishman, I. Sagi and M. Havenith, JACS Au, 2021, 1, 1076–1085 CrossRef CAS PubMed.
- S. Rodrigues, M. Dionisio, C. R. Lopez and A. Grenha, J. Funct. Biomater., 2012, 3, 615–641 CrossRef CAS PubMed.
- A. Madni, R. Kousar, N. Naeem and F. Wahid, J. Bioresour. Bioprod., 2021, 6, 11–25 CrossRef CAS.
- J. K. Kim, H. P. Kim, J. D. Park, K. Ahn, W. Y. Kim, M. Gulumian, G. Oberdorster and I. J. Yu, Part. Fibre Toxicol., 2021, 18, 5 CrossRef CAS PubMed.
- I. Monaco, P. Armanetti, E. Locatelli, A. Flori, M. Maturi, S. Del Turco, L. Menichetti and M. Comes Franchini, J. Mater. Chem. B, 2018, 6, 2993–2999 RSC.
- G. Martinez-Edo, M. C. Llinas, S. Borros and D. Sanchez-Garcia, Nanomaterials, 2019, 9, 1–16 CrossRef PubMed.
- J. F. Hainfeld, W. Liu, C. M. Halsey, P. Freimuth and R. D. Powell, J. Struct. Biol., 1999, 127, 185–198 CrossRef CAS PubMed.
- S. Knecht, D. Ricklin, A. N. Eberle and B. Ernst, J. Mol. Recognit., 2009, 22, 270–279 CrossRef CAS PubMed.
- C. Vasconcelos-Nobrega, A. Colaco, C. Lopes and P. A. Oliveira, In Vivo, 2012, 26, 727–739 CAS.
- L. Martinez-Vidal, M. Chighizola, M. Berardi, E. Alchera, I. Locatelli, F. Pederzoli, C. Venegoni, R. Luciano, P. Milani, K. Bielawski, A. Salonia, A. Podesta and M. Alfano, Commun. Biol., 2023, 6, 217 CrossRef PubMed.
- M. Alfano, M. Nebuloni, R. Allevi, P. Zerbi, E. Longhi, R. Luciano, I. Locatelli, A. Pecoraro, M. Indrieri, C. Speziali, C. Doglioni, P. Milani, F. Montorsi and A. Salonia, Sci. Rep., 2016, 6, 36128 CrossRef CAS PubMed.
- A. P. Regensburger, L. M. Fonteyne, J. Jungert, A. L. Wagner, T. Gerhalter, A. M. Nagel, R. Heiss, F. Flenkenthaler, M. Qurashi, M. F. Neurath, N. Klymiuk, E. Kemter, T. Frohlich, M. Uder, J. Woelfle, W. Rascher, R. Trollmann, E. Wolf, M. J. Waldner and F. Knieling, Nat. Med., 2019, 25, 1905–1915 CrossRef CAS PubMed.
- E. Park, Y. J. Lee, C. Lee and T. J. Eom, J. Biomed. Opt., 2020, 25, 1–8 Search PubMed.
- B. Ferraro, P. Giustetto, O. Schengel, L. T. Weckbach, L. Maegdefessel and O. Soehnlein, Thromb. Haemostasis, 2023, 123, 545–554 CrossRef PubMed.
- R. Nachabe, D. J. Evers, B. H. Hendriks, G. W. Lucassen, M. van der Voort, E. J. Rutgers, M. J. Peeters, J. A. Van der Hage, H. S. Oldenburg, J. Wesseling and T. J. Ruers, J. Biomed. Opt., 2011, 16, 087010 CrossRef PubMed.
- Y. Yan, N. Gomez-Lopez, M. Basij, A. V. Shahvari, F. Vadillo-Ortega, E. Hernandez-Andrade, S. S. Hassan, R. Romero and M. MehrMohammadi, Biomed. Opt Express, 2019, 10, 4643–4655 CrossRef CAS PubMed.
- Y. Zhu, L. A. Johnson, Z. Huang, J. M. Rubin, J. Yuan, H. Lei, J. Ni, X. Wang, P. D. R. Higgins and G. Xu, Biomed. Opt Express, 2018, 9, 1590–1600 CrossRef CAS PubMed.
- S. K. Sekar, I. Bargigia, A. D. Mora, P. Taroni, A. Ruggeri, A. Tosi, A. Pifferi and A. Farina, J. Biomed. Opt., 2017, 22, 15006 CrossRef PubMed.
- V. Grasso, R. Willumeit-Römer and J. Jose, Photoacoustics, 2022, 26, 100367 CrossRef PubMed.
- S. Konugolu Venkata Sekar, J. S. Beh, A. Farina, A. Dalla Mora, A. Pifferi and P. Taroni, Biophys. Chem., 2017, 229, 130–134 CrossRef CAS PubMed.
- J. F. Bentzon, F. Otsuka, R. Virmani and E. Falk, Circ. Res., 2014, 114, 1852–1866 CrossRef CAS PubMed.
- A. Jahangiri-Manesh, M. Mousazadeh, S. Taji, A. Bahmani, A. Zarepour, A. Zarrabi, E. Sharifi and M. Azimzadeh, Pharmaceutics, 2022, 14, 1–27 CrossRef PubMed.
- L. Xie, X. Zhang, C. Chu, Y. Dong, T. Zhang, X. Li, G. Liu, W. Cai and S. Han, J. Nanobiotechnol., 2021, 19, 454 CrossRef CAS PubMed.
- J. K. Cheong, E. H. Ooi, Y. S. Chiew, L. Menichetti, P. Armanetti, M. C. Franchini, E. Alchera, I. Locatelli, T. Canu, M. Maturi, V. Popov and M. Alfano, Comput. Methods Progr. Biomed., 2023, 230, 107363 CrossRef PubMed.
- M. Nejabat, A. Samie, M. Ramezani, M. Alibolandi, K. Abnous and S. M. Taghdisi, J. Controlled Release, 2023, 354, 221–242 CrossRef CAS PubMed.
- R. C. Billinghurst, L. Dahlberg, M. Ionescu, A. Reiner, R. Bourne, C. Rorabeck, P. Mitchell, J. Hambor, O. Diekmann and H. Tschesche, J. Clin. Investig., 1997, 99, 1534–1545 CrossRef CAS PubMed.
- S. M. Sweeney, J. P. Orgel, A. Fertala, J. D. McAuliffe, K. R. Turner, G. A. Di Lullo, S. Chen, O. Antipova, S. Perumal and L. Ala-Kokko, J. Biol. Chem., 2008, 283, 21187–21197 CrossRef CAS PubMed.
- F. Madeira, M. Pearce, A. R. Tivey, P. Basutkar, J. Lee, O. Edbali, N. Madhusoodanan, A. Kolesnikov and R. Lopez, Nucleic Acids Res., 2022, 50, W276–W279 CrossRef CAS PubMed.
- X. Robert and P. Gouet, Nucleic Acids Res., 2014, 42, W320–W324 CrossRef CAS PubMed.
- E. M. Adams, S. Pezzotti, J. Ahlers, M. Rüttermann, M. Levin, A. Goldenzweig, Y. Peleg, S. J. Fleishman, I. Sagi and M. Havenith, JACS Au, 2021, 1, 1076–1085 CrossRef CAS PubMed.
- G. P. Luke, S. Y. Nam and S. Y. Emelianov, Photoacoustics, 2013, 1, 36–42 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00204k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |