
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards tunable exciton delocalization in DNA Holliday junction-templated indodicarbocyanine 5 (Cy5) dye derivative heterodimers†
Gissela
Pascual
 a,
Sebastián A.
Díaz
*b,
Simon K.
Roy
a,
Adam
Meares
bg,
Matthew
Chiriboga
bh,
Kimihiro
Susumu
c,
Divita
Mathur
d,
Paul D.
Cunningham
e,
Igor L.
Medintz
b,
Bernard
Yurke
ai,
William B.
Knowlton
ai,
Joseph S.
Melinger
*e and
Jeunghoon
Lee
*f
a,
Sebastián A.
Díaz
*b,
Simon K.
Roy
a,
Adam
Meares
bg,
Matthew
Chiriboga
bh,
Kimihiro
Susumu
c,
Divita
Mathur
d,
Paul D.
Cunningham
e,
Igor L.
Medintz
b,
Bernard
Yurke
ai,
William B.
Knowlton
ai,
Joseph S.
Melinger
*e and
Jeunghoon
Lee
*f
aMicron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, USA
bCenter for Bio/Molecular Science and Engineering Code 6900, U. S. Naval Research Laboratory, Washington, DC, Virginia 20375, USA. E-mail: sebastian.diaz@nrl.navy.mil
cOptical Sciences Division Code 5600, U.S. Naval Research Laboratory, Washington, DC, USA
dDepartment of Chemistry, Case Western Reserve University, Cleveland, Ohio 44106, USA
eElectronics Science and Technology Division Code 6800, U.S. Naval Research Laboratory, Washington, DC, Virginia 20375, USA. E-mail: joseph.melinger@nrl.navy.mil
fMicron School of Materials Science & Engineering and Department of Chemistry and Biochemistry, Boise State University, Boise, Idaho 83725, USA. E-mail: jeunghoonlee@boisestate.edu
gCollege of Science, George Mason University, Fairfax, Virginia 22030, USA
hVolgenau School of Engineering, George Mason University, Fairfax, Virginia 22030, USA
iDepartment of Electrical & Computer Engineering, Boise State University, Boise, Idaho 83725, USA
First published on 20th September 2024
Abstract
We studied the exciton delocalization of indodicarbocyanine 5 dye derivative (Cy5-R) heterodimers templated by a DNA Holliday junction (HJ), which was quantified by the exciton hopping parameter Jm,n. These dyes were modified at the 5 and 5′ positions of indole rings with substituent (R) H, Cl, tBu, Peg, and hexyloxy (Hex) groups that exhibit different bulkiness and electron-withdrawing/donating capacities. The substituents tune the physical properties of the dyes, such as hydrophobicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) and solvent-accessible surface area (SASA). We tuned the Jm,n of heterodimers by attaching two Cy5-Rs in adjacent and transverse positions along the DNA-HJ. Adjacent heterodimers exhibited smaller Jm,n compared to transverse heterodimers, and some adjacent heterodimers displayed a mixture of H- and J-like aggregates. Most heterodimers exhibited Jm,n values within the ranges of the corresponding homodimers, but some heterodimers displayed synergistic exciton delocalization that resulted in larger Jm,n compared to their homodimers. We then investigated how chemically distinct Cy5-R conjugated to DNA can interact to create delocalized excitons. We determined that heterodimers involving Cy5-H and Cy5-Cl and a dye with larger substituents (bulky substituents and large SASA) such as Cy5-Peg, Cy5-Hex, and Cy5-tBu resulted in larger Jm,n. The combination provides steric hindrance that optimizes co-facial packing (bulky Cy5-R) with a smaller footprint (small SASA) that maximizes proximity. The results of this study lay a groundwork for rationally optimizing the exciton delocalization in dye aggregates for developing next-generation technologies based on optimized exciton transfer efficiency such as quantum information systems and biomedicine.
P) and solvent-accessible surface area (SASA). We tuned the Jm,n of heterodimers by attaching two Cy5-Rs in adjacent and transverse positions along the DNA-HJ. Adjacent heterodimers exhibited smaller Jm,n compared to transverse heterodimers, and some adjacent heterodimers displayed a mixture of H- and J-like aggregates. Most heterodimers exhibited Jm,n values within the ranges of the corresponding homodimers, but some heterodimers displayed synergistic exciton delocalization that resulted in larger Jm,n compared to their homodimers. We then investigated how chemically distinct Cy5-R conjugated to DNA can interact to create delocalized excitons. We determined that heterodimers involving Cy5-H and Cy5-Cl and a dye with larger substituents (bulky substituents and large SASA) such as Cy5-Peg, Cy5-Hex, and Cy5-tBu resulted in larger Jm,n. The combination provides steric hindrance that optimizes co-facial packing (bulky Cy5-R) with a smaller footprint (small SASA) that maximizes proximity. The results of this study lay a groundwork for rationally optimizing the exciton delocalization in dye aggregates for developing next-generation technologies based on optimized exciton transfer efficiency such as quantum information systems and biomedicine.
New conceptsUnderstanding and controlling exciton delocalization in dye aggregates is critical for developing next generation exciton-based materials for quantum computing, light harvesting, and organic optoelectronic applications. We studied a new method to control exciton delocalization between organic dyes templated on DNA nanostructures. By building heterodimers of Cy5 derivatives (Cy5-R) containing a variety of substituents, we were able to obtain dimers that allowed for precise control of exciton delocalization quantified by the exciton hopping parameters (Jm,n) as well as dye dimers that exhibited exciton delocalization that exceeded those of homodimers. Furthermore, through computational modeling based on the exciton theory, we obtained the relative orientation of each dye. These results provided insights into the interactions between the substituents and information on different types of dimer geometries, which could lead to rational design and engineering of molecular quantum materials. |
1. Introduction
There has been growing interest in the use of exciton delocalization in dye aggregates to enhance exciton transfer capabilities for quantum information processing in next-generation technologies.1–4 Dyes can spontaneously self-assemble via noncovalent interactions, such as van der Waals forces and hydrophobic effects, to form x-mer aggregates (e.g., x = 2, dimer and x = 3, trimer). Yet, the self-assembly of dyes in solution is sensitive to solvent properties, such as solubility5 and ionic strength,6 resulting in minimal control of the type of aggregate formation. An alternative approach is the covalent attachment between dyes to form superdyes, examples of which have been obtained using squaraine (SQ),7,8 merocyanine,9 dimethylfluoresceins,10 and perylene bisimide dyes,11 but the synthesis procedure of these superdye molecules requires complex chemical steps. In contrast, engineered DNA structures were identified as an excellent template material to control the aggregation of homo- (m = n) and heteroaggregates (m ≠ n). The hybridization of DNA duplexes by using Watson–Crick–Franklin base pairing between complementary sequences facilitates the engineering of linear, branched, and three-dimensional DNA structures with predictable nanoscale features.12–14 This also enables the assembly of covalently attached dyes along the DNA strands. Dyes are attached to DNA strands for numerous purposes, such as exploring dye properties for new applications in quantum computing and biosensing15,16 and understanding the optical characteristics of dye aggregates.17–33 Two essential features that can be achieved using DNA as templates to assemble dyes are explored in the present study. The first is the ability to form x-mers with desirable dye properties that cannot be achieved without templating (i.e. aggregation of dyes with extreme solubilities, either too high or too low). Some studies have used cyanine (Cys)17–19,24,25 and methyl red17–19 covalently attached to DNA templates to assemble heteroaggregates; others have used DNA templates to promote aggregation of SQs20,30 and Cy5s21 with limited water solubility to investigate exciton delocalization. The second is the capability to control the orientation of transition dipole moment (TDM) of dyes and dye–dye interactions by attaching dyes in adjacent and transverse positions within the 4-arm DNA Holliday junction (DNA-HJ). The position of the dye along the DNA-HJ affects the interdye interaction and optical properties of the aggregate. For example, SQ dimers attached to DNA-HJ templates through a single tether showed a higher exciton delocalization in the adjacent configuration20,30 while Cy5 derivatives attached via two tethers showed a more pronounced exciton delocalization in the transverse configuration.21,23,24Coulombic interaction between dyes enables a collective sharing of electronic excitation energy, leading to quantum delocalized excitation and Frenkel excitons.34 The interaction between dyes m and n enabling the exciton delocalization is quantified directly by the exciton hopping parameter Jm,n. This parameter denotes the exchange energy between the TDM of dyes in the aggregate approximated as a dipole–dipole interaction.35 In the incoherent weak-coupling limit, this exchange energy mediates Förster resonance energy transfer (incoherent hopping); in the strong-coupling regime, it mediates exciton delocalization and coherent transport of energy. Therefore, Jm,n is a key parameter of the Frenkel exciton Hamiltonian and its control is essential for impactful applications of engineered dye aggregates.1 These applications range from biosensing,36,37 and light harvesting,38,39 to quantum computing.4,35,37,40,41
The optical properties of dye aggregates, such as absorption intensity and Jm,n, depend on the relative orientation of the TDM of dyes. According to molecular exciton theory,34,42 strongly coupled TDMs lead to energy level hybridization that splits the excited state levels and redistributes oscillator strength among the new levels into vector sums of the constituent TDMs. An end-to-end TDM orientation forms J-aggregates, where the higher energy transition is out of phase such that, for identical TDMs, the contributions cancel each other and oscillator strength is redistributed to the symmetric lower energy transition, resulting in a red-shifted (i.e. bathochromic shifted) absorption feature relative to the monomer.34,43 In contrast, face-to-face oriented TDMs form H-aggregates redistributing the oscillator strength to the higher energy transition, leading to a blue-shifted absorption spectrum relative to the monomer.34,43 Oblique aggregates have an intermediate configuration between J- and H-aggregates with obliquely oriented TDM, and both energy transitions are allowed34 (Fig. 1). Homodimers20–23,27,30 and heterodimers,7,17–19,24,44 exhibit changes in photophysical behaviors relative to their monomers due to the delocalized excitons.34 In both types of dimers, excited states split into lower and higher energy levels. In homodimers, if we ignore vibronic coupling, the energy splitting is symmetric and determined solely by Jm,n because identical dyes have the same energy state (Fig. 1A). However, in practice, as the peak blueshifts, the vibronic coupling causes oscillator strength to transfer to the higher energy vibronic shoulder such that the splitting between absorption peaks is typically asymmetric.45 In heterodimers, excited state energies split symmetrically about the average of the excited state energies of the constituent dyes, endowing an identical shift to each transition that depends on both Jm,n and the difference in the excited state energies of the monomers. Greater differences in the monomer excited state energies result in smaller shifts. While optical transitions into the higher or lower energy excited state only is allowed in H-aggregate and J-aggregate homodimers respectively, transitions into both the highest and lowest state are allowed in heterodimer H-aggregates due to incomplete cancellation of the nonidentical TDMs7 (Fig. 1B).
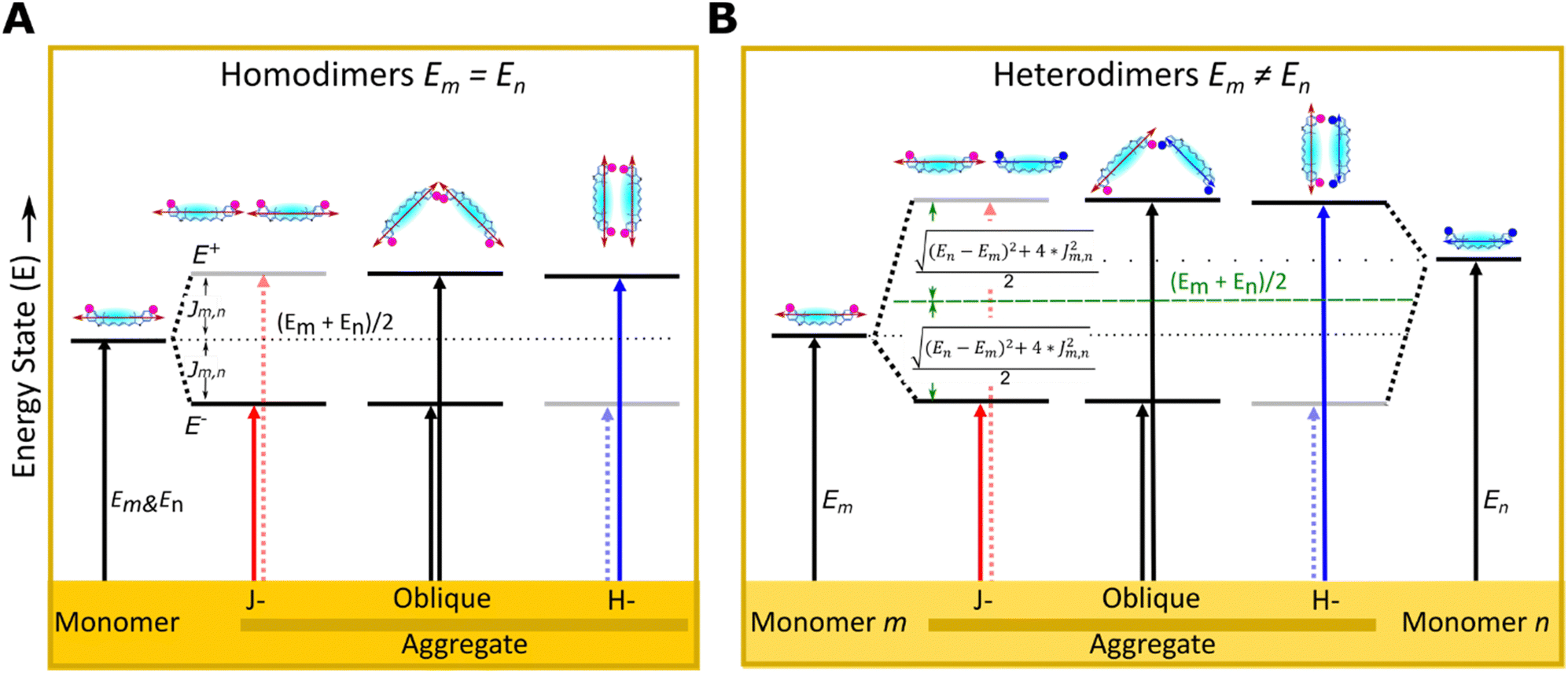 | ||
| Fig. 1 Schematic illustration of electronic transitions of (A) homodimer (m = n) and (B) heterodimers (m ≠ n) relative to their monomers m and n as described by molecular exciton theory.34 The formation of dye aggregates induces exciton delocalization splitting into higher and lower excited energy states (E+ and E−) relative to the corresponding monomers. The transition into a higher- (lower-) energy excitonic state is forbidden for J- (H-) aggregates, respectively (dot arrows). In the dye structure, dots (purple and blue) represent modified substituents of Cy5-R in 5 and 5′ positions of indole rings and the arrow represents the TDM orientation. | ||
Tuning the physical properties of the constituent dyes can alter dye packing in aggregates and is a promising method to tune exciton delocalization. For example, high hydrophilicity (or low hydrophobicity) reduce the aggregation ability of dyes leading to a greater inter-dye distance and resulting in a small Jm,n.20,30 Customized substituents (-R) of dyes, such as hydrogen (H), n-hexyloxy (Hex), triethylene glycol monomethylether (Peg), tert-butyl (tBu), or chloro (Cl), provide the dyes with unique properties (e.g., hydrophobicity)16 that impact the dye aggregation capability. A previous study found that hydrophobicity plays an essential role in Cy5-R aggregation, but the dye orientation and Jm,n are influenced more by the bulkiness of the substituent.21 Homodimer systems provided valuable insights into the understanding of optical properties and the ability to delocalize excitons by tuning the dye properties. However, heterodimer systems may reveal an enhanced ability to tune optical properties and exciton delocalization compared to homodimers. The molecular exciton theory18 supports changes in the absorption spectra feature17,19 that are indicative of a coherent interaction between chemically distinct dyes. Recently, exciton delocalization, tunable electronic structure, and energy transfer were explored by assembling Cy5 and Cy5.5 templated by DNA-HJ.24,25 Heterodimers (Cy5–Cy5.5) exhibited Jm,n (95 meV) between the Jm,n of homodimers Cy5 and Cy5.5 (69–116 meV);24 heterotetramers showed similar results.25
Here, we report a method for systematically tuning the optical properties and exciton delocalization in Cy5-R heterodimers attached to the core of DNA-HJs. There are two major advantages in using the DNA-HJ in this work. First is the chemical flexibility to attach dyes in close proximity at the HJ core in order to characterize and maximize strong excitonic coupling for dyes on DNA. Second is the spatial tunability to place dyes at different sites within the core (e.g., adjacent and transverse dimers), which allows us to explore dye–dye interactions that determine dye packing and orientation. This type of flexibility afforded by the DNA-HJ allows us to study how excitonic coupling from heterodimers might be controlled using different dye types and dye placements, which would likely be impractical using other DNA scaffolds. A significant outcome of the present study is that some Cy5-R heterodimers exhibited greater exciton delocalization ability with a larger Jm,n compared to their corresponding homodimers, highlighting the synergistic interaction of dyes with different chemical properties. Cy5-Rs were chosen for the present study due to the convenient tuning of spectral properties that allow for a detailed characterization of monomers and dimers using steady-state spectroscopy.21 Physical properties of Cy5-R were quantified by the hydrophobicity (logP) and solvent-accessible surface area (SASA), and the substituents were characterized by their bulkiness and electron-withdrawing capacity. Two Cy5-Rs were placed at adjacent (adjacent dimer) or transverse (transverse dimer) positions along the DNA-HJ to tune Jm,n. The KRM model simulation tool based on the Kühn–Renger–May theory45 was used to estimate Jm,n and the orientation of the TDMs of dyes. The tendency observed in some adjacent heterodimer samples to form subpopulations with different Jm,n and packing arrangements led us (i) to investigate all possible dimer configurations in DNA-HJ of Cy5 Peg–Cl, H–Cl, and tBu–H, and (ii) to conduct transient absorption spectroscopy using representative heterodimers to confirm the presence of subpopulations in the adjacent dimer.
2. Materials and methods
2.1 Characterization of Cy5-Rs and synthesis of DNA constructs
The Cy5-R (R: H, Hex, Peg, tBu, or Cl) derivatives, except Cy5-H, are not commercially available. The synthesis and method of attaching these dyes to DNA strands were described by Meares et al.16 Unmodified Cy5 has H atoms in the 5,5′-positions of the indole ring, which were symmetrically substituted by R substituents (Fig. 2A and B). The attachment of customized Cy5 with different substituents into single DNA strands (Fig. 2B and C) via two 3-carbon covalent linkers (labeled DNA) has no significant influence on the DNA optical properties (e.g., absorption spectra). This is because dyes behave independently of the DNA and of the aliphatic linkers.16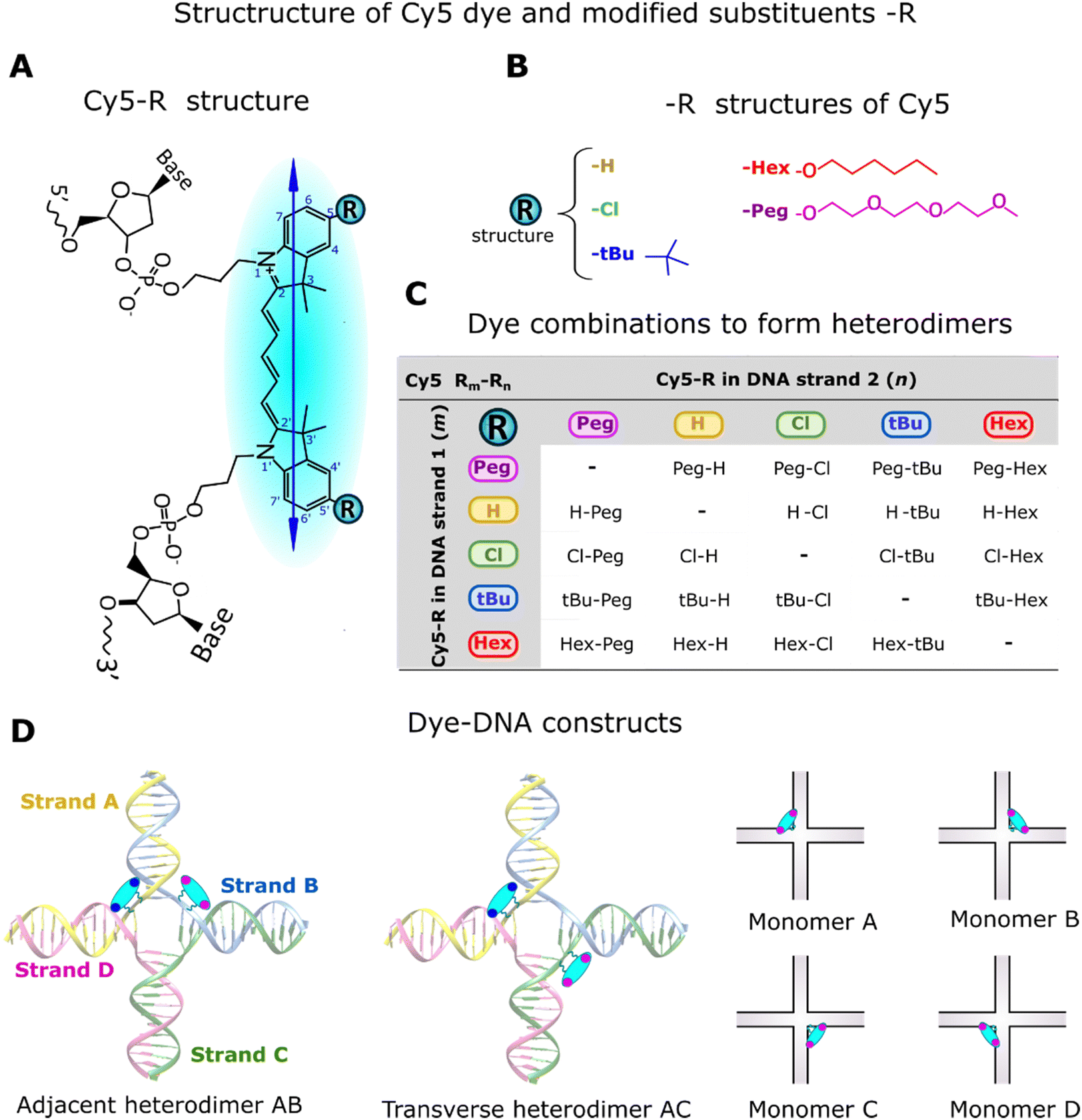 | ||
| Fig. 2 Schemes of the Cy5-R structure with attachments in the DNA Holliday junction (DNA-HJ). (A) Scheme of the chemical structure of Cy5-R, within the DNA backbone. The arrow inside the Cy5-R structure represents the transition dipole moment (TDM), and R indicates the 5- and 5′- positions of the indolenine rings where substituents are placed. (B) Schematic structure of R substituents. (C) Cy5-R dye combinations to form heterodimers (Cy5 Rm–Rn) of Cy5 derivatives. (D) Schematic representation of monomers, and adjacent AB and transverse AC heterodimers templated by DNA Holliday junction (DNA-HJ). | ||
To gain insights into the characteristics of the substituents used to modify Cy5-H, data on local bulkiness (A-value) and electron-withdrawing capacity (Hammett constant, σp) were collected from the available literature (Table 1). A-value represents the energetic preference of the equatorial position imparted by a substituent on cyclohexane; a high A-value indicates a locally bulkier substituent.20,21 The Hammett constant (σp) quantifies the electronic effect of replacing H (σp = 0) with other substituents; electron donating and withdrawing substituents exhibit negative and positive σp, respectively.46 The physical properties of Cy5-Rs were characterized by their hydrophobicity and the area the solvent could access (SASA). The hydrophobicity, represented by the octanol–water partition coefficient (logP), was calculated using Percepta software; the more hydrophobic dyes exhibit a higher logP.16 SASA was calculated using optimized ground state Cy5-R structures in the ChimeraX software package with SASA function.47 A larger SASA indicates a higher accessible surface area by the solvent (buffer solution containing the DNA constructs), which correlates with the size of the substituents.
| Substituents (R) of Cy5s | Characteristics of substituents | Dye Cy5-R | Characteristics of dyes | ||
|---|---|---|---|---|---|
| σ p | A-valueb | logPc |
SASAd | ||
| a Hammet constant in the para position that represents the electron-donating capability of the substituent.21 b Bulkiness of substituents (kcal mol−1).21 c Calculated using Percepta software from Advanced Chemistry Development, Inc. (Toronto, Canada).16 d Solvent accessible surface area (Å2) calculated using Chimera. e Upper range of A-value calculated using a conformational model based on the interconverting conformational enantiomers of bilirubin that assess substituents size from head-to-tail steric comprehension.48 | |||||
| H | 0 | 0 | Cy5-H | 1.73 | 591 |
| Cl | 0.22 | 0.48 | Cy5-Cl | 2.35 | 679 |
| tBu | −0.20 | 1.85e | Cy5-tBu | 3.43 | 670 |
| PEG | −0.26 | ∼0.9 | Cy5-PEG | 1.58 | 1225 |
| Hexyloxy | −0.27 | ∼0.9 | Cy5-Hex | 5.66 | 1033 |
DNA constructs, monomers (Cy5-R) and heterodimers (Cy5 Rm–Rn), were synthesized using equimolar concentrations of labeled and unlabeled (DNA without dye) complementary DNA strands (see DNA strands and nucleotide sequences in Section S1, ESI†). The synthesis of DNA constructs was performed in 1xTAE + 15 mM MgCl2 buffer solution, using a similar approach to that used for homodimers.21 Monomers have one Cy5-R in a DNA-HJ, while adjacent and transverse heterodimers have two different Cy5-Rs (Fig. 2D). Adjacent heterodimers have two Cy5-Rs in two half-complementary DNA strands (e.g., AB), and transverse dimers have two Cy5-Rs in two noncomplementary DNA strands of the DNA-HJ (e.g., AC). The present study includes a total of 76 unique heterodimers. Initially, 40 Cy5 Rm–Rn heterodimers (20 AB/BA adjacent and 20 AC/CA transverse dimers) were synthesized to examine their properties compared to homodimers. However, the observed results led us to synthesize all possible combinations of adjacent (AB/BA, AD/DA, BC/CB, and CD/DC) and transverse (AC/CA and BD/DB) Cy5 Peg–Cl, Cy5 tBu–H, and Cy5 Cl–H heterodimers. All DNA constructs were annealed in an Eppendorf Mastercycler (Eppendorf; Hamburg, Germany) by heating up to 95 °C and then gradually cooling down until to 4 °C (see details in Section S1, ESI†).
2.2 Optical characterization of DNA constructs
Steady-state absorption, circular dichroism (CD), and fluorescence spectra were measured to characterize the optical features of DNA constructs. All measurements were conducted at 20 °C using samples dissolved in 1× TAE with 15 mM MgCl2 buffer.Absorption spectra were measured using a Cary 60 UV-vis spectrophotometer. 2 μM solution of DNA construct (monomer or dimer) was placed in a 10 mm path length quartz spectrometer cell (110 μL), and data were collected from 200 to 900 nm within a 1 nm step interval and 0.1 s integration time.
CD spectra were measured using a JASCO J-1500 CD spectrophotometer. 2 μM solution of DNA construct was filled in a 10 mm path length quartz spectrometer cell (110 μL), and data were collected from 200 to 850 nm with 1 nm step intervals at 100 nm min−1, and 4 s digital integration time.
Fluorescence spectra were measured using a TECAN plate reader exciting from above. 100 μL of 1 μM DNA construct solution was filled in each well of a 96-well black microplate, resulting in a 1.25 mm path length and maximum OD of 0.05 to avoid inner filter effects. The emission intensity was scanned from 630 to 900 nm at a fixed 615 nm excitation wavelength. Fluorescence excitation spectra were measured by scanning the excitation wavelength from 450 to 720 nm with the emission wavelength fixed at 740 nm. In both cases, the data were collected in 1 nm steps.
Ultrafast transient absorption (TA) spectra were recorded using a commercial TA spectrometer (Ultrafast Systems, Helios) driven by a 1 kHz 100 fs Ti:Sapphire amplifier (Coherent, Legend Elite) and a tunable optical parametric amplifier (Coherent, Topas). Dye-labeled DNA solutions were prepared to 6.67 μM in 1× TAE buffer with 15 mM MgCl2 in a 2 mm cuvette. The solution was filtered through 0.2 micron PES filters to remove debris that scatter light. The solution was stirred during illumination to minimize photodegradation. The excitation beam was polarized at the magic angle with respect to the white light probe to eliminate artifacts associated with rotational depolarization. All data were chirp corrected using the ultrafast response of pure buffer. Photoselection of the reddest absorption peak as well as the strongly enhanced 0–1 vibronic shoulder was used to preferentially photoexcite J-like and H-like geometry dimer subpopulations, respectively.
2.3 KRM modeling and data analysis
The orientation of TDMs of dyes and the resulting Jm,n of heterodimers were calculated using the heteroaggregate KRM model simulation tool (version ev1HAv2, 2022). KRM theory,45 which is an extension of molecular exciton theory that includes vibronic coupling rather than only electronic coupling, and it is sometimes called vibronic coupling theory. The theoretical formulation and fitness metrics behind the homoaggregate KRM model simulation tool were described by Roy et al.27 The simulation for heteroaggregates operates similarly to the KRM modeling tool for homoaggregates used in previous studies.20,23,27–30 Both types of KRM model simulation tools simultaneously fit experimental absorption and CD spectra using the key parameters of the different monomers (Table S3, ESI†) to estimate the orientation of TDMs of dyes and their Jm,n in an aggregate. The difference is that the heteroaggregate simulation tool allows input of the characteristics (e.g., energy levels) of each monomer to compute the orientation of TDMs of dyes and Jm,n of the dye heteroaggregate. In contrast, the homoaggregate simulation tool allows only the input of one type of monomers, considering that the same type of dye forms homoaggregates. See more detailed differences between the homoaggregate and heteroaggregate KRM model simulation tools in Section S4 (ESI†).The prepared dye–DNA constructs generally exhibited a single type of dimer in the solution, but some samples exhibited a mixture of dimers (e.g. J- and H-like aggregates). Initially, all samples were modeled as having a single type of dimer using two distinct dye TDMs. However, in some samples, the overlap integral between the experimental and theoretical absorption and CD spectra as a single dimer was low. Therefore, a new approach was modeled using four TDMs of dyes to form two types of dimers, as described by Pascual et al.20 The model was configured such that the two modeled dimers did not interact.
The relationship between Jm,n and properties of the dyes or their substituents was analyzed using linear regression analysis (LRA). The coefficient of determination R-square (R2), scaled from 0 to 1, represents the goodness of fit, and slope (b) measures the changes of Jm,n for each unit change on the independent variable (dye or substituent properties).
3. Results and discussion
3.1 Properties of the Cy5-R monomer templated by DNA-HJ
The absorption and emission spectra of monomers measured in the present study showed similar features to those reported in our previous study of Cy5-R synthesis16 and homodimers.21 The maximum absorption spectra peak of Cy5-R monomers templated by DNA-HJ ranged between 652 and 681 nm, while the emission peaks ranged between 668 and 705 nm (Fig. 3A and B). | ||
| Fig. 3 Optical properties of Cy5-R monomers templated by DNA-HJ. (A) Molar extinction coefficient (ε in M−1 cm−1) of Cy5-R determined using the absorption spectra of monomers. (B) Emission intensity of monomers normalized to absorptance20 at 630 nm excitation wavelength. Measurements were performed in 1xTAE + 15 mM MgCl2 buffer solution. Shadow in (A) and (B) depicts the variation range of spectra intensity exhibited by the monomers A, B, C, and D templated by DNA-HJ, and values indicate the location of the maximum peak wavelength. | ||
The absorption peak of Cy5-R monomers gradually shifted to a longer wavelength (red-shifted) with the increasing electron-donating character of substituents (more negative σp) in the order H < tBu < Peg < Hex (Fig. S1A, ESI†), while Cy5-Cl also showed a red-shift. The absorption peak shifts to longer wavelengths can be explained by the following changes in HOMO/LUMO energy levels imparted by the substituents: An electron-withdrawing group lowers HOMO and LUMO energies but the effect is greater on the LUMO leading to a lower energy transition compared to an H group at the same position. The inverse applies to electron-donating groups, that is, HOMO and LUMO have increased energy but the effect is greater on the HOMO.49 A similar relationship was exhibited between the Cy5-R monomer absorption peaks and bulkiness of substituents (A-value) that increases in the following order: H< Cl < Peg ∼ Hex < tBu (Fig. S1B, ESI†). Nevertheless, the bulkiness of substituents does not necessarily have a direct influence on the absorption and emission peaks, since similar peaks could be expected in dyes with a different bulkiness as observed in other cyanine dyes.50 Regarding the properties of dyes, absorption peaks of Cy5-Rs and logP bear no relationship while a stronger correlation was observed between the Stokes shifts and logP (R2 = 0.98), as well as between SASA and maximum absorption peaks (Fig. S1C–E, ESI†). Interestingly, Cy5-Rs with a larger SASA exhibited a lower emission intensity (Fig. 3B). The causative effect of dye SASA with the absorption peak position and fluorescence emission intensity as well as between logP and Stokes shift is unknown. However, these results confirm that different R substituents provided the Cy5-Rs with new properties that can impact the exciton delocalization in heterodimers.
3.2 Properties of Cy5 Rm–Rn heterodimers templated by DNA-HJ
Changes in absorption, circular dichroism (CD), and fluorescence features of Cy5-R heterodimers templated by DNA-HJ relative to their monomers confirmed the successful formation of aggregates and coherent interaction of two chemically distinct dyes (Fig. 4 and Section S3.2, ESI†). All adjacent and transverse heterodimers exhibited an increase in the oscillator strength of the 0–1 vibronic absorption transition due to vibronic coupling and oscillator strength transfer from the blue-shifted higher energy absorption peak relative to the low-energy peak of monomers (Fig. 4A and D and Sections S3.1 and S3.2, ESI†). As expected, heterodimers exhibited absorption peaks at different wavelengths than homodimers (Fig. S22, ESI†). The energy transition exhibited by heterodimers relative to their monomers confirms the exciton delocalization between two chemically distinct Cy5-Rs. This result agrees with the molecular exciton theory proposed by Kasha34,42 and the vibronic coupling described by Khün.45 Moreover, our findings are supported by studies of other DNA-templated dye aggregates,17–19,24,25,43 and dyes covalently bound between each other7–11 exhibiting different optical features than their respective monomers.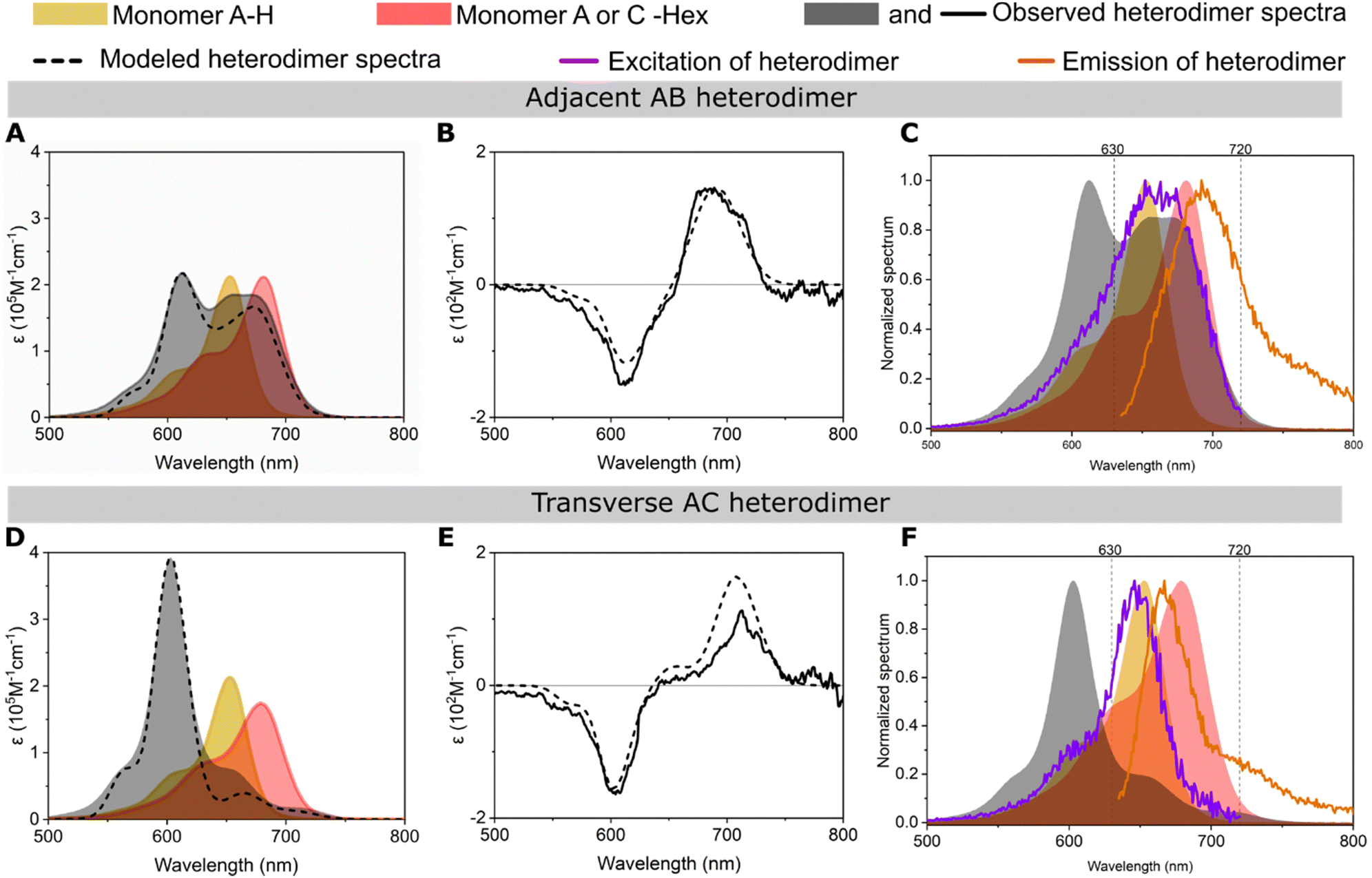 | ||
| Fig. 4 Optical characterization of the representative (A)–(C) adjacent Cy5 H–Hex AB and (D)–(F) transverse Cy5 H–Hex AC heterodimers templated by a DNA Holliday junction (DNA HJ). The experimental (A) and (D) absorption and (B) and (E) circular dichroism (CD) spectra of heterodimers were converted into molar extinction coefficient units (ε M−1 cm−1). The experimental absorption and CD spectra were modeled (dashed line) using a method based on Kühn–Renger–May approach (KRM model). (C) and (F) Plots of excitation and emission spectra of heterodimer (lines) and absorption spectra of monomers and heterodimer (shadow). The excitation and emission spectra were measured at fixed wavelengths of 740 and 615 nm, respectively. See the spectra of all heterodimers of the present study in Section S3 (ESI†). | ||
The adjacent or transverse position of the same pair of dyes in the DNA-HJ template had a remarkable influence on the absorption spectra feature of heterodimers. The features of absorption spectra among transverse heterodimers were similar to each other; they exhibited a predominant peak at a shorter wavelength (high energy level) that indicates a stronger excitonic coupling than adjacent heterodimers. Some adjacent heterodimers showed absorption peaks near the absorption peak of their monomers, especially heterodimers where Cy5-tBu was not involved. Cy5 Hex–H AB is a representative adjacent heterodimer (Fig. 4A) showing an H-aggregate peak at 612 nm resulting from vibronic coupling and oscillator strength transfer between the blue-shifted higher energy 0–0 energy level and the 0–1 vibronic shoulder. This heterodimer exhibited two additional peaks at ∼655 and ∼673 nm, which may represent the presence of either monomers or a secondary dimer subpopulation, as further investigated in Sections 3.3 and 3.4. Other heterodimers with similar behavior to Cy5 Hex–H AB are Cy5 Cl–Hex (AB and BA), Cy5 H–Cl (BA), Cy5 H–Hex (BA) Cy5 H–Peg (BA and AB), and Peg–Cl (AB and BA) (Section S3.2, ESI†). These observations in heterodimers are consistent with the stronger exciton delocalization observed in Cy5 transverse homodimers templated by DNA-HJ compared to adjacent homodimers.21,23,24 The generally different exciton coupling between adjacent and transverse dimers may arise from the interaction of dyes with the neighboring nucleobases51 and the conformation of the DNA structure.52
Fluorescence (emission and excitation) and the absorption spectra of heterodimers indicated the formation of heterodimer aggregates. The heterodimers exhibited fluorescence suppression (FS) relative to the average of the emission of the two monomers in the range of 87 and 99% (Section S3.5, ESI†), which is consistent with the FS that has been observed in strongly coupled Cy5 dimers on DNA.21,24,53 The FS of heterodimers varies depending on the emission intensity of the participant monomers, but there is a general trend of a greater FS in transverse than adjacent heterodimers. This result is consistent with the co-facial arrangement adopted by transverse heterodimers with large Jm,n and short center-to-center dye distance (Rm,n) (Section 3.3), which make quenching of fluorescence from wavefunction overlap more likely. Fluorescence excitation and absorption spectra of the monomers are similar;54,55 however, Cy5-R heterodimers exhibited dissimilar normalized fluorescence excitation and absorption spectra. The excitation spectra of transverse heterodimers were remarkably comparable to the absorption spectrum of one of the monomers, generally to the monomer exhibiting the higher emission intensity (Fig. 4F). This result suggests that the detected emission intensity in heterodimer samples could arise from small subpopulations of monomers that remain despite efforts using equimolar concentrations of labeled and unlabeled DNA strands to synthesize the HJ structures. Samples still can contain traces of monomers regardless of purification during the synthesis of dye–DNA constructs assuring near 100% labeling efficiency.26 Another explanation of this emission intensity of dimers could be the fluctuation of the DNA template that can result in momentaneous separated dyes that behave like monomers.53 On the other hand, some adjacent heterodimers exhibited a dissimilar excitation spectrum to the absorption spectrum of the heterodimer or monomers, suggesting that there may exist a secondary dimer subpopulation (Fig. 4C). These observations led us to investigate the formation of heterodimer subpopulations, namely a mixture of dimers with different Jm,n (Section 3.3), and to confirm our assumption using transient absorption (TA) spectroscopy of representative samples (Section 3.4).
Heterodimers generally exhibited well-defined right-handed bisignate CD spectra (Fig. 4B and E); however, some samples (e.g., transverse Cy5 Peg–H and tBu–Peg) exhibited close-to-zero CD signals. Adjacent and transverse heterodimers exhibited different CD signal features; adjacent heterodimers generally exhibited a stronger signal. The intensity of the CD signal relies on the mutual orientation of the TDM of dyes, the distance between dyes (Rm,n), and the coupling strength of aggregates (Jm,n).27,56 A weak CD signal could be justified by the planar packing arrangement and the suspected presence of dimers with similar packing but opposite chirality.27 The presence of multiple configurations of dimer orientations or dimers with subpopulations (e.g., a mixture of J- and H-aggregates) showing a weak CD is also possible. Nonetheless, a close-to-zero signal does not necessarily signify the absence of exciton delocalization, nor does a high CD signal always correlate with a single population of dimers. A close-to-zero CD signal could indicate the presence of almost equimolar concentrations of chiral dimers where the left- and right-handed CD signals cancel each other. Another possibility is the planar packing arrangement or formation of pure H- or J-dimer. In this study, CD of Cy5 tBu–Hex and Cy5 H–Hex exhibited a single dimer and a mixture of dimer populations, respectively (Sections 3.4 and S3.2, ESI†). These two heterodimers exhibited a strong CD signal, which suggests that a strong CD signal can arise from the presence of a single or a mixture of dimer subpopulations.
Unlike other Cy5-R heterodimers, switching the position of dyes in Cy5 tBu–Cl from AB to BA shifted the CD signal from left handed to right handed, though the BA dimer showed a weakly right-handed CD signal. Both heterodimers exhibited comparable excitonic coupling strength (Section 3.3). This result revealed that the formation of chiral heterodimers is achievable by simply switching the dye position between the A and B strands of the DNA-HJ, where the handedness of the CD signal is favored by the attached position of dyes in the DNA-HJ. A previous study achieved the formation of chiral homodimers by using unequal lengths of the single linker that attaches SQ-Cl2 to the DNA strands.28 In contrast, a change in the length of linkers of Cy5 tBu–Cl was not necessary to form chiral heterodimers in the present study, demonstrated by the CD spectra at different AB:BA heterodimer ratio (Section S3.4, ESI†). Chiral dimers could open research horizons for developing materials that rely on programable system chirality.28
We now discuss the absorption and fluorescence spectra of the heterodimers in Fig. 4 in the context of possible multiple conformations of the DNA-HJ scaffold. It's known that the DNA-HJ can undergo transitions between open and closed structures. However, at the relatively high concentration of Mg2+ (at 15 mM MgCl2) the DNA-HJ exists mainly in the closed form.22,57 We consider absorption and fluorescence spectra anticipated from the two closed form structural isomers, Iso I and Iso II, as shown schematically in Fig. S35 and S36 (ESI†). For transverse heterodimers in Iso I, the dyes are expected to be relatively far apart (dye separation ≳ 30 Å) and weakly coupled. This dimer configuration is expected to produce monomer-like absorption spectra with fluorescence primarily from the lower energy dye. In contrast, for transverse heterodimers in Iso II, the dyes are expected to be closely spaced and strongly coupled. This dye arrangement tends to produce blue-shifted absorption spectra with strongly quenched fluorescence from the strongly coupled heterodimer. In the case of the representative transverse AC heterodimer Cy5 H–Hex, the absorption spectrum is strongly blue-shifted from the constituent monomers. Besides, the fluorescence excitation spectrum overlaps well with the Cy5-H monomer absorption spectrum. These observations support Iso II as the dominant form. If Iso I is present at an equal mole fraction to Iso II then we would expect to observe strong absorption peaks from the Cy5-H and Cy5-Hex monomers. We would also expect strong fluorescence from the Cy5-Hex dye via Förster resonance energy transfer (FRET) from Cy5-H and from direct excitation of Cy5-Hex. However, these signatures are not observed for transverse AC Cy5-H Cy5-Hex or for the other transverse heterodimers studied in this work. We suspect that strong van der Waals attraction between the hydrophobic Cy5 dyes leads to a lower free energy for Iso II.
The situation for adjacent heterodimers is more complex. Fig. S5-2 (ESI†) suggests that the dye separations in Iso I and Iso II are similar. In this situation we would expect Iso I and Iso II to produce similar signatures in their absorption and fluorescence spectra. From these measurements alone, it is not possible to determine whether both structural isomers are present or if one of them is favored.
3.3 Dye packing and exciton delocalization of adjacent and transverse heterodimers
The KRM modeling of heterodimer absorption and CD spectra indicate strong coupling between the two chemically distinct Cy5-Rs and provide the packing arrangement of the dyes (Fig. 5A and B). Using a DNA-HJ template, we achieved the formation of heterodimers with relatively small center-to-center dye distances (Rm,n = 0.34–1.27 nm) leading to strong exciton delocalization (Jm,n = 43–133 meV) (Fig. 5C–H). Marked differences in packing arrangements and exciton delocalization were observed between transverse and adjacent heterodimers templated by the DNA-HJ. All transverse dimers exhibited only one H-like packing (single dimer population). In contrast, some adjacent dimers exhibited two types of packing (dimers with subpopulations) with large and small Jm,n, respectively (Sections S4.1 and S4.2, ESI†). These dimers with subpopulations were observed especially when Cy5-tBu was not part of the dimer. The modeling results confirmed the two types of packing arrangements suspected from the absorption spectra feature in the adjacent heterodimers Cy5 Hex–H (AB and BA), Cy5 Cl–Hex (AB and BA), Cy5 H–Cl (BA), Cy5 H–Peg (BA and AB), and Peg–Cl (AB and BA) (Section 3.2 and Sections S4.1 and S4.2, ESI†). Further insights into the difference in packing arrangements were gained by preparing all possible combinations of adjacent (AB, BC, CD, and AD) and transverse (AC and BD) Cy5 Peg–Cl, Cy5 tBu–H, and Cy5-H–Cl heterodimers (Section S4.3, ESI†). The presence of adjacent dimers with subpopulations was consistent with findings in Cy5-R21 and SQ20 homodimers templated by a DNA-HJ via a double and single linker, respectively. Additionally, selected samples underwent transient absorption spectroscopy measurements which confirmed the presence of dimer subpopulations that exhibited H- and oblique-like packing in adjacent dimers, while transverse dimers showed only one population that exhibited packing of only H-like aggregates (Section 3.4).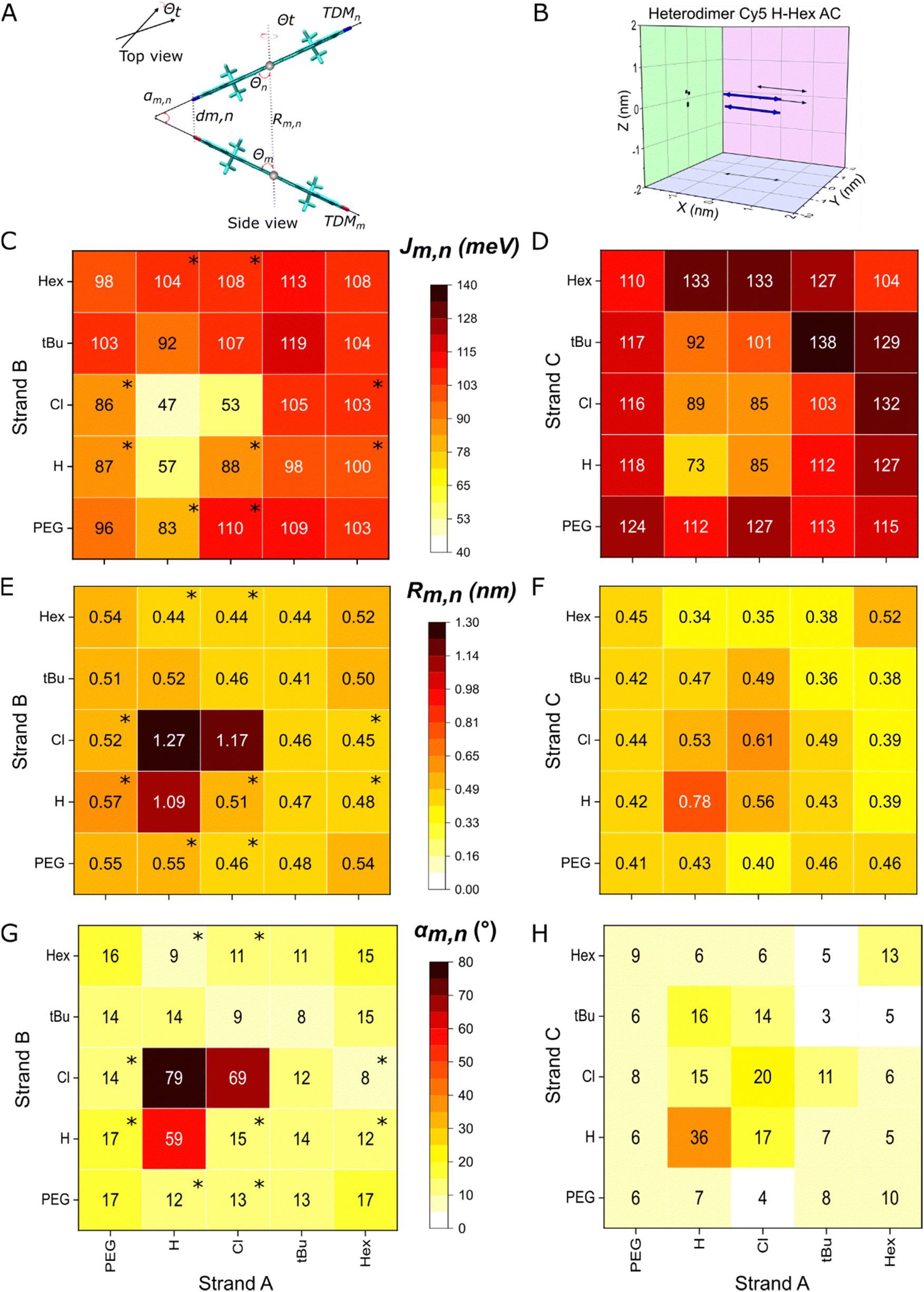 | ||
| Fig. 5 (A) Schematic representation of dyes in dye aggregates (TDM: transition dipole moments of dyes, αm,n: angle between TDM of dyes, dm,n: minimum distance between dyes, Rm,n: center-to-center dye distance Θ: slip angle of dyes m and n, and Θt: twist angle). (B) Representation of a 3D plot of TDM of dyes (blue arrows) projected to XY, YZ, and XZ planes (black arrows) resulting from KRM modeling. (C) Exciton hopping parameter (Jm,n), (E) Rm,n, and (G) αm,n of adjacent heterodimers, and (D) Jm,n, (F) Rm,n, and (H) αm,n of transverse heterodimers resulted from KRM modeling. Data with an asterisk (*) in the heat plot indicate that the heterodimer has subpopulations and the depicted values correspond to the dimer with a larger Jm,n. See details of KRM modeling parameters and results in Section S4 (ESI†). | ||
The formation of two types of dimers with different Jm,n in a sample may arise from the interaction of multiple factors, which requires further investigation. One factor may be the orientation of the Cy5-R relative to the DNA due to its interaction with the neighboring nucleobases,51 influenced by the properties of the dye itself,20,21,30,58 as well as the conformation of the DNA-HJ.52 The DNA-HJ can take on different conformations (Iso I and Iso II)59 potentially affected by the placement of the two Cy5-Rs in adjacent or transverse positions (Section S5, ESI†), as discussed later in this section. Thus, the dye position in the DNA-HJ favors the formation of homogeneous types of aggregates when dyes are attached in transverse positions. In samples exhibiting two types of dimers, the dimer exhibiting the larger Jm,n represents the upper limit on the exciton interaction strength that can be achieved. Therefore, dimers with a larger Jm,n and their respective packing characteristics were used to compare the difference between adjacent and transverse dimers and the influence of dye properties on Jm,n.
The extent of exciton delocalization of heterodimers was influenced by the position of the dyes in the DNA-HJ. Adjacent (AB) dimers exhibited Jm,n between 47 and 119 meV, center-to-center dye distance (Rm,n) between 0.44 and 1.27 nm, and minimum interdye distance (dm,n) between 0.34 and 0.41 nm. Transverse dimers exhibited Jm,n between 73 and 133 meV, Rm,n between 0.34 and 0.56 nm, and oblique angles between TDM of dyes (αm,n) between 4 and 16° (Fig. 5C–H and Sections S4.1 and S4.2, ESI†). The generally large Jm,n and short interdye distances exhibited by transverse dimers compared to adjacent dimers indicated that strong exciton delocalization is favored in transverse dimers in the DNA-HJ. This result is consistent with observations in Cy5 homodimers that have two points of attachment of the dye to the DNA via a 3-carbon linker (Fig. 2A).21,23 However, it differs from observations of SQ homodimers attached to the DNA using a single 5-carbon linker attached to a modified thymine base, which exhibited a larger Jm,n in adjacent than transverse dimers.20,30 The long single linker may provide a greater degree of freedom to SQs for adopting an optimal packing configuration and the two shorter linkers restrict Cy5s as suggested in previous studies.20,21 However, the dye–DNA linker structure could impact conformations of DNA-HJ leading to a different packing configuration when dyes are attached in adjacent and transverse positions within the DNA-HJ.
The structural conformations of DNA-HJ, which depend on the salt concentration for unlabeled DNA-HJ, may explain the stronger exciton delocalization exhibited by transverse Cy5-R heterodimers compared to adjacent heterodimers. A low salt concentration leads to the formation of an X-shape open DNA-HJ structure due to the electrostatic repulsion between the arms,60 whereas high salt concentration (e.g. ≳5 mM Mg2+) leads to the formation of stacked Iso I and Iso II DNA conformers.60–62 The shape of Iso I and Iso II are more likely to be formed in our systems that used 15 mM MgCl2. These two conformers are the same but the adopted structure in each arm relative to the central branch point is opposite (e.g., horizontal H-shaped: Iso I and vertical H-shaped: Iso II). The ratio of these two iso conformers of unlabeled DNA-HJ is base pair sequence dependent,62 but the attachment of Cy5-Rs to the DNA strands in two points may promote one type of DNA-HJ conformer as shown in Fig. S35 (ESI†). Thus, adjacent dimers are likely to adopt oblique-like packing geometry exhibiting longer Rm,n and larger αm,n compared to transverse dimers that adopt face-to-face packing geometry with shorter Rm,n and smaller αm,n as described in Section S4 (ESI†). This approach agrees with the generally smaller Jm,n value in transverse compared to adjacent dimers (Fig. 5C and D).
Successful formation of heterodimers led to higher Jm,n than exhibited by some homodimers, revealing synergistic exciton delocalization of different dyes. The estimated Jm,n values of heterodimers were generally between the Jm,n of homodimers, which is consistent with findings in Cy5–Cy5.5 heterodimers.24 Strikingly, some transverse heterodimers exhibited remarkable dye interaction, exhibiting larger Jm,n values greater than either of their homodimers. Jm,n of transverse AC homodimers Cy5 Cl, Cy5 Hex, and Cy5 H were 85, 104, and 73 meV, respectively.21 In contrast, Jm,n of transverse AC heterodimers Cy5 H–Hex and Cy5 Cl–Hex fall in the range of 127–133 and 132–133 meV, respectively. This result suggests the long hydrophobic tail Hex produces tighter packing of Cy5-Hex (logP = 5.7 and SASA = 1033 Å2) with less hydrophobic Cy5-Rs that have smaller substituents, such as Cy5-H and Cy5-Cl. It seems that the large size and large hydrophobicity of Cy5-Hex enables the spatial accommodation of the relatively small Cy5-Cl in the packing. Additionally, the bulkiness of Hex and Cl orients the TDM of the dyes.21
Physical properties of the dyes played an essential role in the ability to alter Jm,n in heterodimers. The linear regression analysis (LRA), based on the slope (b) and R-squared (R2), revealed a high dependence of Jm,n on the dye pair properties such as hydrophobicity, SASA, and A-value. This dependence was stronger in transverse (higher R2) compared to adjacent Cy5 H–R and Cy5 Cl–R (R: tBu, Peg, Cl, or Hex) heterodimers (Fig. 6 and Section S6, ESI†) possibly influenced by the DNA-HJ conformers as discussed above. In the LRA analysis of Cy5 H–R (Fig. 6A and D), outliers were observed when examining the effect of hydrophobicity and SASA on Jm,n. The reason for the difference in aggregation of Cy5-Peg and Cy5-H, which have comparable hydrophobicity, is due to their molecular sizes and structure. Peg, being an amphiphilic group, can interact with water as H-bond acceptors because of the polyether oxygen atoms, while this interaction is not possible with other R groups. After removing the outliers, it was observed that the transverse Cy5 H–R heterodimers were more dependent on hydrophobicity (R2 = 0.92, b = 27.7) and SASA (R2 = 0.80, b = 0.22) compared to adjacent heterodimer hydrophobicity (R2 = 0.57, b = 20.8) and SASA (R2 = 0.37, b = 0.15). When examining the effect of the bulkiness of substituents on Jm,n, Cy5 H–tBu heterodimers were not included because the local bulkiness of the substituent of the paired dyes arises mainly from tBu. After excluding the outliers, it was found that the transverse Cy5 H–R heterodimers have a higher dependence on the bulkiness of the substituent (R2 = 0.84, b = 124.9) compared to adjacent heterodimers (R2 = 0.53, b = 88.6) (Fig. S37, ESI†). Similarly, examining Cy5 Cl–R, heterodimers with Cy5-Peg and Cy5-tBu were outliers when assessing the influence of hydrophobicity and SASA (Fig. 6B and E and Fig. S38, ESI†), as well as A-value, respectively. After excluding the outliers, transverse heterodimers demonstrated a stronger dependence on the hydrophobicity (R2 = 0.98 b = 24.9), followed by SASA (R2 = 0.91, b = 0.21), and the bulkiness of the substituent (R2 = 0.81, b = 98.9). In contrast, adjacent heterodimers exhibited a weak dependence on hydrophobicity (R2 = 0.49, b = 21.5), followed by the A-value of the substituent (R2 = 0.46, b = 81.5), and SASA (R2 = 0.27, b = 0.14). Additionally, it is noteworthy to highlight that switching the dye position from Cy5 H–Cl AB (Jm,n = 47 meV) to BA (Jm,n = 88 meV) had a great impact on the exciton delocalization. This result supports the potential interaction of dyes with neighboring nucleobases in the DNA-HJ.51
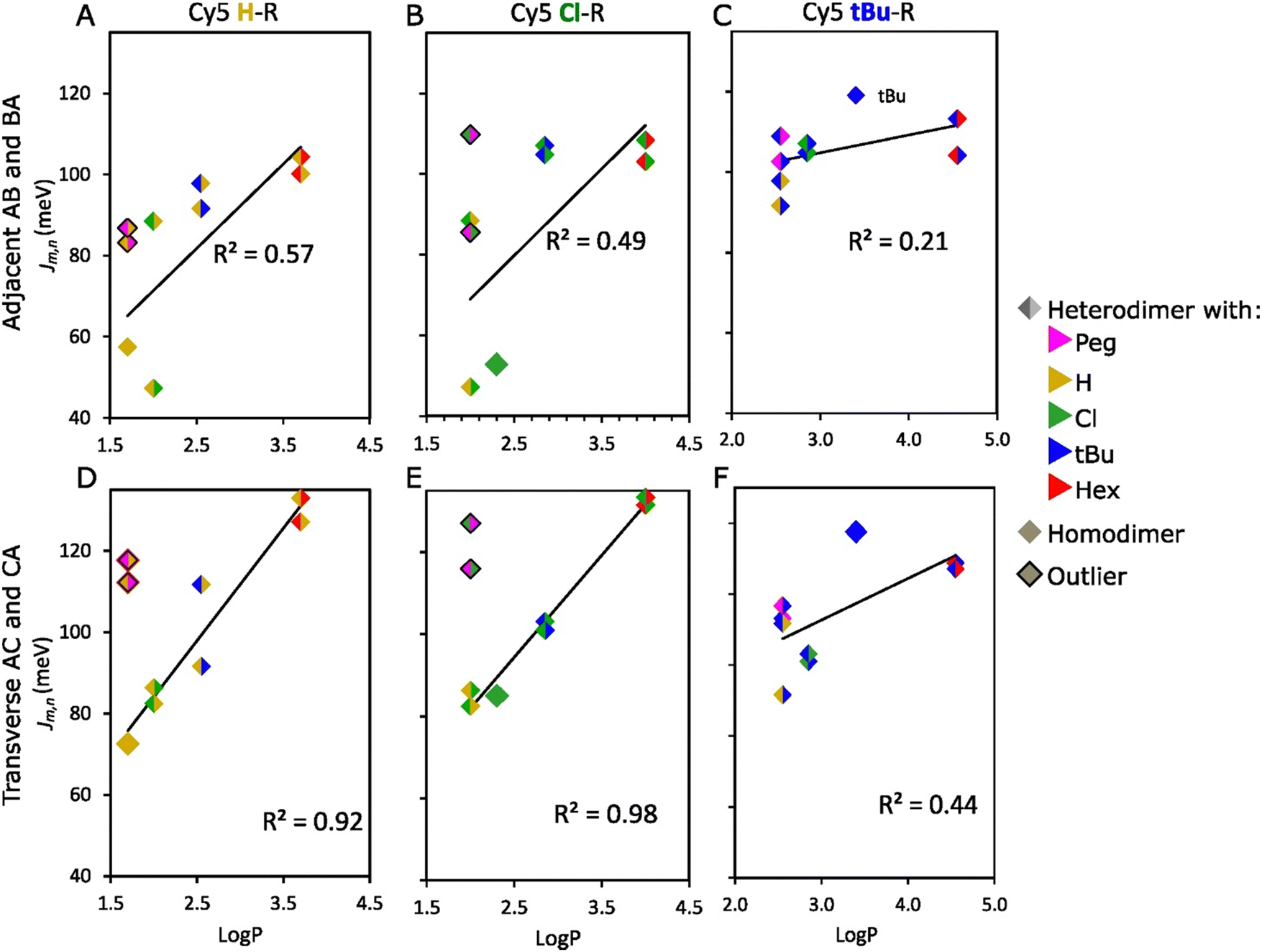 | ||
| Fig. 6 Relationship between hydrophobicity (logP) and exciton hopping parameter (Jm,n) of adjacent (A–C) and transverse (D–F) heterodimers. logP of heterodimers is the average value of the two participant dyes in the aggregates. The outliers in the analysis are describes in Section 3.3 of the main text. See analysis for Cy5 Peg–R, Cy5 Hex–R, and the relationship of Jm,n with other properties of Cy5-R in Section S6 (ESI†). | ||
On the other hand, Jm,n of Cy5 tBu–R (Fig. 6C and F and Fig. S39, ESI†), Cy5 Peg–R, and Cy5 Hex–R were weakly dependent on the dye properties (R2 ≤ 0.61) (Fig. S40 and S41, ESI†). These dyes exhibited large Jm,n for Cy5 tBu, Cy5 Hex, and Cy5 Peg adjacent and transverse homodimers, in the range of 96–119 and 104–138 meV, respectively.21 The large Jm,n of these homodimers was explained by the collective effect of hydrophobicity that increases the propensity of dye interaction and the high A-value that orients the dyes in a co-facial geometry.21 Additionally, the large molecular sizes of tBu, Hex, and Peg (Table 1) enhances the van der Waals forces due to the greater interaction area between dyes and increased electron density around the molecule.63,64 Therefore, heterodimerization between Cy5 tBu, Cy5 Hex, and Cy5 Peg, which are dyes with large and bulky substituents, exhibited generally larger Jm,n values. This is because the characteristics of the substituents promoted the interdye interaction. The Jm,n value of heterodimers falls between the estimated range for homodimers (Fig. 5C and D). This finding is consistent with results for Cy5–Cy5.5 heterodimers.24 Interestingly, when forming heterodimers between dyes with relatively large A-values (tBu, Hex, Peg) and dyes with relatively small A-values (Cl, H), the steric hindrance from the large A-value dye increases the likelihood of forming a co-facial heterodimer with large Jm,n (Section S6, ESI†). This outcome suggests that the large Jm,n mainly arises from dyes that have the large bulkiness of substituents and SASA, which agrees with the result in Cy5-R homodimers that large substituents enforce the H-like packing.21
The LRA revealed no correlation between Jm,n of heterodimers and the electron-withdrawing capacity (σp) of Cy5-R substituents. This result disagrees with a previous finding of enhanced aggregation and dimerization by increasing the electron-withdrawing of substituents.50 This disagreement is explained by the difference between Cy5 structures used in the previous (trimethine bridge and benzothiazole rings) and the present study (pentamethine bridge and indolenine rings). Compared to trimethine-bridged dyes, pentamethine-bridged dyes aggregate more readily.44 Benzothiazole is a sulfur-containing structure that consists of a benzene ring fused to a thiazole ring, while indole consists of a benzene ring fused to a pyrrole ring. The sulfur atom in the thiazole ring confers a negative electrostatic potential region compared to pyrrole rings65 that influences the electron cloud of the dyes, and hence their interdye interaction.
Overall, the results of the present study demonstrated that Jm,n of Cy5 H–R and Cy5 Cl–R heterodimers can be significantly altered by changing the properties of the pairing dye (SASA and hydrophobicity) and the bulkiness (A-value) of their substituents. This result agrees with previous studies which found that dye hydrophobicity enhances the dye packing.20,21,30,58 That is because hydrophobic interactions promote π–π stacking stability of noncovalent interaction.66 However, the bulkiness and size of the dye substituents determine the orientation of the TDM and preference for cofacial interactions and π–π stacking (H-type aggregates) of the dyes.21 Heterodimers formed between Cy5 tBu–R, Cy5 Peg–R, and Cy5 Hex–R were less tunable and they were dependent on their SASA and bulkiness of substituents. Thus, heterodimers formed with dyes that have similar hydrophobicity but different SASA and A-values such as Cy5-Peg and Cy5-H, the larger Jm,n is exhibited by heterodimers formed with dyes that have larger SASA and bulkier substituents (Cy5-Peg). The results of KRM modeling agree with the previous studies that hydrophobicity is one of the main factors bringing dyes into proximity, enhancing the formation of aggregates.20,21,30,58 This result demonstrated that SASA and bulkiness of the substituents are important factors in determining exciton delocalization when hydrophobicity is not in play. It was observed that dyes having comparable bulkiness of their substituents but contrasting hydrophobicity such as Cy5 Peg (logP = 1.7, A-value = ∼0.9) and Cy5 Hex (logP = 5.7, A-value = ∼0.9.) exhibited similar Jm,n values. The overall effect on exciton delocalization of heterodimers depends on the combined properties of dyes and their substituents.
3.4 Transient absorption spectroscopy of selected adjacent and transverse heterodimers
Ultrafast transient absorption (TA) can reveal the presence of multiple subpopulations of dimers in a solution by exploiting differences in their absorption spectra and lifetimes. This method was previously used to distinguish between H-like and J-like aggregates in Cy5-H homodimers,53 which exhibit large and small Jm,n values, respectively. Here we use photoselection to investigate the suspected formation of a mixture of aggregates in transverse and adjacent Cy5-R heterodimers (Fig. 7). Heterodimers Cy5 H–Hex were selected as representative samples because they represent well the ambiguous sample of the dimer with subpopulation (Fig. 4) and the switched position of dyes (e.g., from AB to BA) exhibit similar absorption spectra. For the Cy5 H–Hex AC transverse dimer, photoexcitation at 713 nm selects the reddest peak of the absorption spectra. The resulting TA spectra are characterized by ground state bleach (GSB) spectra that mirror the dimer absorption spectra; the dominant feature is the 0–1 vibronic shoulder of the absorption spectra that gains oscillator strength through vibronic coupling in a H-like co-facial geometry. Photoexcitation at 603 nm directly excites this dominant absorption feature and results in nearly the same spectra. We conclude that the transverse dimer is characterized predominantly by a co-facial H-like geometry.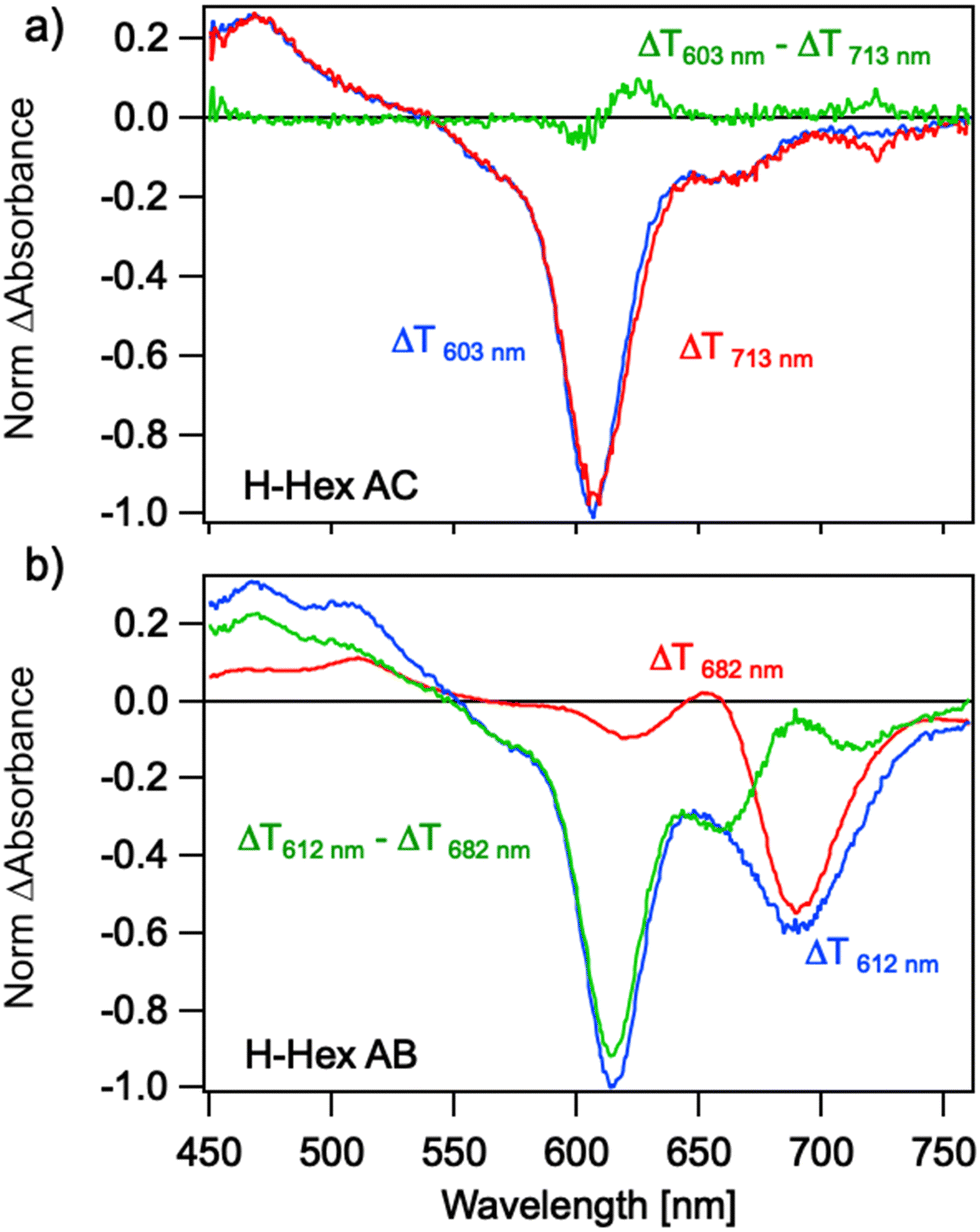 | ||
| Fig. 7 Transient absorption spectra of (a) Cy5 H–Hex AC transverse and (b) Cy5 H–Hex AB adjacent dimer measured 1 ps after photoexcitation at wavelengths chosen to photoselect any H-like or J-like geometry subpopulations. The excitation wavelengths are indicated and color coded. | ||
Conversely, photoselection of the Cy5 H-Hex AB adjacent dimer reveals different TA responses associated with two subpopulations. Photoexcitation at 682 nm selects the reddest peak of the absorption spectra, resulting in TA spectra characterized by a dominant 0–0 GSB peak near 680 nm with an overlapping excited state absorption (ESA) peak near 650 nm. The shape of this response is characteristic of Cy5-R dimers in an end-on-end J-like geometry.53,67 Using molecular exciton theory, the ESA band has been previously assigned to the transition between the symmetric singly excited state, a.k.a molecular exciton, to the lower symmetric delocalized doubly excited state, a.k.a. biexciton.67 However, photoexcitation of the 0–1 vibronic shoulder near 612 nm that dominates the absorption spectra reveals two peaks to the GSB, with the 0–1 peak dominating. This GSB does not mirror the adjacent dimer absorption spectra. Instead, the TA response is a combination of an H-like and J-like dimer response. Subtracting the J-like response measured for 682 nm excitation from the TA spectra measured for 612 nm excitation reveals a TA spectrum that resembles that measured for the transverse dimer. While excitation of Cy5-hex B monomer, which might be present in the sample in small quantities,26 is possible at 682 nm pump wavelength, the associated kinetics (Fig. S42, ESI†) confirm the presence of a subpopulation of J-like dimers. We conclude that the adjacent dimer exhibits two subpopulations: an H-like dimer and a J-like dimer geometry, which have different optical properties. This is qualitatively consistent with the results of KRM modeling of the absorption and CD spectra for these dimers.
4. Conclusions and research outlook
In this study, we demonstrated exciton delocalization (quantified by Jm,n) of heterodimers using five Cy5-R derivatives templated by DNA-HJ to form adjacent and transverse dimer aggregates. Exciton delocalization in Cy5-R heterodimers was evident by the shifts of their absorption spectra relative to their monomers, CD spectra, and significant fluorescence emission suppression. The KRM modeling, used to estimate the orientation of TDMs of the dyes and their Jm,n, suggested that some Cy5-R adjacent dimers form two types of heteroaggregates that have different Jm,n values and packing arrangements while most heterodimers aggregated to form H-aggregates. The mixture of two types of aggregates in samples was confirmed with TA measurements of representative heterodimers.J m,n was tuned using two methods. The first method consisted of using the same pair of dyes templated by the DNA-HJ in adjacent and transverse positions; transverse heterodimers exhibited a higher exciton delocalization compared to adjacent heterodimers. The second method consisted of changing the chemical properties of heterodimers (hydrophobicity, SASA, and bulkiness of the substituents) using five Cy5-Rs with distinct chemical properties. The exciton hopping parameters of heterodimers generally resulted in the range of the exciton hopping parameters exhibited by homodimers. However, some Cy5 Hex–R heterodimers exhibited exceptionally enhanced exciton delocalization energy compared to their homodimers, revealing that chemically distinct dyes can interact synergistically to delocalize excitons. Exciton delocalization of dye with small substituents (Cy5-H and Cy5-Cl) was optimized forming aggregates with another dye with larger substituents (Cy5-Peg, Cy5-Hex, and Cy5-tBu) and hydrophobicity. However, the contrasting results of Cy5 H–R and Cy5 Peg–R indicated that exciton delocalization relies on the bulkiness of the substituent when the dyes have similar hydrophobicity.
Overall, the two major findings of this research are (1) synergistic exciton delocalization of certain dyes when they aggregate, in which heterodimers (e.g., Cy5 H–Hex and Cy5 Cl–Hex) exhibited a larger Jm,n compared to those exhibited by their homodimers and (2) the ability to influence Jm,n using chemically distinct dyes. It offers an expanded framework of exciton delocalization applications in which the formation of J-like and H-like aggregates with different exciton delocalization energies can be achieved. This is possible because the interdye distance can be varied by placing dyes at adjacent or transverse sites in the DNA-HJ, which, in turn, alters the exciton coupling. This may be useful for targeted applications that require tailored photophysical properties. For example, applications such as photothermal therapy benefit from efficient non-radiative decay typically found in strongly coupled H-dimers.68 Alternatively, J-like aggregates are desirable for applications such as artificial photosynthesis, exciton and electron transport, and room-temperature quantum computing.2,4,40 Thus, this work using heterodimers lays the groundwork for engineering molecular quantum materials with broad applications in different fields. The feasibility of tuning the exciton delocalization energy of dye aggregates is demonstrated using dyes with different properties.
Author contributions
AM, MC, KS, DM, SAD, ILM prepared, purified, and characterized the dye-labeled DNA oligos and DNA nanostructures (investigation). GP, SKR, WBK, BY, JL curated the data and analyzed the spectra for KRM modeling (data curation and formal analysis). GP, SAD, PDC, ILM, JSM designed experimental protocols for spectroscopy (methodology) and realized initial analysis (formal analysis). WBK, BY, JL, SAD, ILM, JSM (conceptualization). GP, PDC, ILM, JL, JSM, SAD (original draft). All authors (writing – review & editing).Data availability
The data supporting this article titled “Towards tunable exciton delocalization in DNA Holliday junction-templated indodicarbocyanine 5 (Cy5) dye derivatives heterodimers” by Pascual et al., which include DNA construct design, spectroscopic data of monomers and dimers, input and output parameters for KRM modeling results, 3D plots of transition dipole moments, and transient absorption spectra and kinetics, have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research at the Boise State was supported by the Department of the Navy, Office of Naval Research (ONR) via ONR award no. N00014-22-1-2725. Research at the U.S. Naval Research Laboratory (NRL) was supported by NRL base funding, the NRL Institute for Nanoscience, and ONR award # N0001424WX00409. D. M. was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Number R00EB030013. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- D. Mathur, S. A. Díaz, N. Hildebrandt, R. D. Pensack, B. Yurke, A. Biaggne, L. Li, J. S. Melinger, M. G. Ancona, W. B. Knowlton and I. L. Medintz, Chem. Soc. Rev., 2023, 52, 7848–7948 RSC.
- B. Yurke and W. Kuang, Phys. Rev. A: At., Mol., Opt. Phys., 2010, 81, 033814 CrossRef.
- X. Wang, R. Sha, W. B. Knowlton, N. C. Seeman, J. W. Canary and B. Yurke, ACS Nano, 2022, 16, 1301–1307 CrossRef CAS PubMed.
- M. A. Castellanos, A. Dodin and A. P. Willard, Phys. Chem. Chem. Phys., 2020, 22, 3048–3057 RSC.
- A. K. Singh, M. F. M. Kavungathodi, A. J. Mozer, K. Krishnamoorthy and J. Nithyanandhan, Langmuir, 2022, 38, 14808–14818 CrossRef CAS PubMed.
- W. J. Harrison, D. L. Mateer and G. J. T. Tiddy, J. Phys. Chem., 1996, 100, 2310–2321 CrossRef CAS.
- S. F. Völker, A. Schmiedel, M. Holzapfel, K. Renziehausen, V. Engel and C. Lambert, J. Phys. Chem. C, 2014, 118, 17467–17482 CrossRef.
- H. Ceymann, M. Balkenhohl, A. Schmiedel, M. Holzapfel and C. Lambert, Phys. Chem. Chem. Phys., 2016, 18, 2646–2657 RSC.
- D. Bialas, A. Zitzler-Kunkel, E. Kirchner, D. Schmidt and F. Würthner, Nat. Commun., 2016, 7, 12949 CrossRef CAS PubMed.
- D. Hayes, G. B. Griffin and G. S. Engel, Science, 2013, 21, 1431–1434 CrossRef PubMed.
- S. Ghosh, X. Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills Jr, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- L. I. Markova, V. L. Malinovskii, L. D. Patsenker and R. Häner, Chem. Commun., 2013, 49, 5298–5300 RSC.
- A. Meares, K. Susumu, D. Mathur, S. H. Lee, O. A. Mass, J. Lee, R. D. Pensack, B. Yurke, W. B. Knowlton, J. S. Melinger and I. L. Medintz, ACS Omega, 2022, 7, 11002–11016 CrossRef CAS PubMed.
- H. Kashida, H. Asanuma and M. Komiyama, Angew. Chem., Int. Ed., 2004, 43, 6522–6525 CrossRef CAS PubMed.
- H. Asanuma, T. Fujii, T. Kato and H. Kashida, J. Photochem. Photobiol., C, 2012, 13, 124–135 CrossRef CAS.
- T. Fujii, H. Kashida and H. Asanuma, Chem. – Eur. J., 2009, 15, 10092–10102 CrossRef CAS PubMed.
- G. Pascual, S. K. Roy, G. Barcenas, C. K. Wilson, K. Cervantes-Salguero, O. M. Obukhova, A. I. Krivoshey, E. A. Terpetschnig, A. L. Tatarets, L. Li, B. Yurke, W. B. Knowlton, O. A. Mass, R. D. Pensack and J. Lee, Nanoscale, 2024, 16, 1206–1222 RSC.
- S. A. Díaz, G. Pascual, L. K. Patten, S. K. Roy, A. Meares, M. Chiriboga, K. Susumu, W. B. Knowlton, P. D. Cunningham, D. Mathur, B. Yurke, I. L. Medintz, J. Lee and J. S. Melinger, Nanoscale, 2023, 15, 3284–3299 RSC.
- B. L. Cannon, D. L. Kellis, L. K. Patten, P. H. Davis, J. Lee, E. Graugnard, B. Yurke and W. B. Knowlton, J. Phys. Chem. A, 2017, 17, 6905–6916 CrossRef PubMed.
- B. L. Cannon, L. K. Patten, D. L. Kellis, P. H. Davis, J. Lee, E. Graugnard, B. Yurke and W. B. Knowlton, J. Phys. Chem. A, 2018, 122, 2086–2095 CrossRef CAS PubMed.
- A. U. Chowdhury, S. A. Díaz, J. S. Huff, M. S. Barclay, M. Chiriboga, G. A. Ellis, D. Mathur, L. K. Patten, A. Sup, N. Hallstrom, P. D. Cunningham, J. Lee, P. H. Davis, D. B. Turner, B. Yurke, W. B. Knowlton, I. L. Medintz, J. S. Melinger and R. D. Pensack, J. Phys. Chem. Lett., 2022, 13, 2782–2791 CrossRef CAS PubMed.
- J. S. Huff, S. A. Díaz, M. S. Barclay, A. U. Chowdhury, M. Chiriboga, G. A. Ellis, D. Mathur, L. K. Patten, S. K. Roy, A. Sup, A. Biaggne, B. S. Rolczynski, P. D. Cunningham, L. Li, J. Lee, P. H. Davis, B. Yurke, W. B. Knowlton, I. L. Medintz, D. B. Turner, J. S. Melinger and R. D. Pensack, J. Phys. Chem. C, 2022, 126, 17164–17175 CrossRef CAS PubMed.
- J. S. Huff, P. H. Davis, A. Christy, D. L. Kellis, N. Kandadai, Z. S. D. Toa, G. D. Scholes, B. Yurke, W. B. Knowlton and R. D. Pensack, J. Phys. Chem. Lett., 2019, 10, 2386–2392 CrossRef CAS PubMed.
- S. K. Roy, O. A. Mass, D. L. Kellis, C. K. Wilson, J. A. Hall, B. Yurke and W. B. Knowlton, J. Phys. Chem. B, 2021, 125, 13670–13684 CrossRef CAS PubMed.
- O. A. Mass, S. Basu, L. K. Patten, E. A. Terpetschnig, A. I. Krivoshey, A. L. Tatarets, R. D. Pensack, B. Yurke, W. B. Knowlton and J. Lee, J. Phys. Chem. Lett., 2022, 2022, 10688–10696 CrossRef PubMed.
- O. A. Mass, C. K. Wilson, S. K. Roy, M. S. Barclay, L. K. Patten, E. A. Terpetschnig, J. Lee, R. D. Pensack, B. Yurke and W. B. Knowlton, J. Phys. Chem. B, 2020, 124, 9636–9647 CrossRef CAS PubMed.
- O. A. Mass, C. K. Wilson, G. Barcenas, Ewald A. Terpetschnig, O. M. Obukhova, O. S. Kolosova, A. L. Tatarets, L. Li, B. Yurke, W. B. Knowlton, Ryan D. Pensack and J. Lee, J. Phys. Chem. C, 2022, 126, 3475–3488 CrossRef CAS PubMed.
- S. M. Hart, W. J. Chen, J. L. Banal, R. Hä, M. Bathe, G. S. Schlau-Cohen, W. P. Bricker, A. Dodin, L. Markova, Y. Vyborna and A. P. Willard, Chem, 2020, 7, 752–773 Search PubMed.
- M. Chiriboga, S. A. Diaz, D. Mathur, D. A. Hastman, J. S. Melinger, R. Veneziano and I. L. Medintz, J. Phys. Chem. B, 2022, 126, 110–122 CrossRef CAS PubMed.
- L. Kringle, N. P. D. Sawaya, J. Widom, C. Adams, M. G. Raymer, A. Aspuru-Guzik and A. H. Marcus, J. Chem. Phys., 2018, 148, 85101 CrossRef PubMed.
- M. Kasha, Radiat. Res., 1963, 20, 55–70 CrossRef CAS PubMed.
- B. Yurke, R. Elliott and A. Sup, Phys. Rev. A, 2023, 107, 12603 CrossRef CAS.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC.
- C. Laboda, H. Duschl and C. L. Dwyer, Acc. Chem. Res., 2014, 47, 1816–1824 CrossRef CAS PubMed.
- F. Fassioli, R. Dinshaw, P. C. Arpin and G. D. Scholes, J. R. Soc., Interface, 2014, 11, 20130901 CrossRef PubMed.
- S. Kirstein and S. Daehne, Int. J. Photoenergy, 2006, 1–21 Search PubMed.
- B. Yurke, in Natural Computing Series, ed. Janoska N. and Winfree E., Springer Science and Business Media Deutschland GmbH, Singapore, 2023, vol. Part F821, pp. 125–169 Search PubMed.
- A. M. Childs, D. Gosset and Z. Webb, Science, 2013, 339, 791–794 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- R. A. Garoff, E. A. Litzinger, R. E. Connor, I. Fishman and B. A. Armitage, Langmuir, 2002, 18, 6330–6337 CrossRef CAS.
- O. Kühn, T. Renger and V. May, Chem. Phys., 1996, 204, 99–114 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- S. E. Boiadjiev and D. A. Lightner, J. Am. Chem. Soc., 2000, 122, 11328–11339 CrossRef CAS.
- T. Sutradhar and A. Misra, J. Phys. Chem. A, 2018, 122, 4111–4120 CrossRef CAS PubMed.
- D. Takahashi, H. Oda, T. Izumi and R. Hirohashi, Dyes Pigm., 2005, 66, 1–6 CrossRef CAS.
- K. Cervantes-Salguero, A. Biaggne, J. M. Youngsman, B. M. Ward, Y. C. Kim, L. Li, J. A. Hall, W. B. Knowlton, E. Graugnard and W. Kuang, Int. J. Mol. Sci., 2022, 23, 7690 CrossRef CAS PubMed.
- K. Cervantes-Salguero, M. Kadrmas, B. M. Ward, D. Lysne, A. Wolf, L. Piantanida, G. Pascual and W. B. Knowlton, Langmuir, 2024, 40, 10195–10207 CrossRef CAS PubMed.
- J. S. Huff, D. B. Turner, O. A. Mass, L. K. Patten, C. K. Wilson, S. K. Roy, M. S. Barclay, B. Yurke, W. B. Knowlton, P. H. Davis and R. D. Pensack, J. Phys. Chem. B, 2021, 125, 10240–10259 CrossRef CAS PubMed.
- V. Holde, Molecular Mechanisms of Photosynthesis, 2008, pp. 258–305 Search PubMed.
- V. M. Martínez, F. L. Arbeloa, J. B. Prieto and I. L. Arbeloa, J. Phys. Chem. B, 2005, 109, 7443–7450 CrossRef PubMed.
- F. D. Lewis, L. Zhang, X. Liu, X. Zuo, D. M. Tiede, H. Long and G. C. Schatz, J. Am. Chem. Soc., 2005, 127, 14445–14453 CrossRef CAS PubMed.
- Q. Song, Y. Hu, A. Yin, H. Wang and Q. Yin, Int. J. Mol. Sci., 2022, 23, 9730 CrossRef CAS PubMed.
- A. L. Stadler, B. R. Renikuntla, D. Yaron, A. S. Fang and B. A. Armitage, Langmuir, 2011, 27, 1472–1479 CrossRef CAS PubMed.
- S. Hohng, R. Zhou, M. K. Nahas, J. Yu, K. Schulten, D. M. J. Lilley and T. Ha, Science, 2007, 318, 279–283 CrossRef CAS PubMed.
- R. M. Clegg, A. I. Murchie and D. M. J. Lilleyt, Biophys. J, 1994, 66, 99–109 CrossRef CAS PubMed.
- P. Agarwal, S. H. Kabir and N. Pal, Chem. Phys. Impact., 2023, 7, 100322 CrossRef.
- S. A. McKinney, A. C. Déclais, D. M. J. Lilley and T. Ha, Nat. Struct. Biol., 2003, 10, 93–97 CrossRef CAS PubMed.
- A. D. Buckingham, P. W. Fowler and J. M. Hutson, Chem. Rev., 1988, 88, 963–988 CrossRef CAS.
- J. N. Israelachvili and D. Tabor, Proc. R. Soc. London, Ser. A, 1972, 331, 19–38 CAS.
- T. Sierański, Struct. Chem., 2016, 27, 1107–1120 CrossRef.
- K. M. Guckian, B. A. Schweitzer, R. X.-F. Ren, C. J. Sheils, D. C. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 2000, 122, 2213–2222 CrossRef CAS PubMed.
- P. D. Cunningham, S. A. Díaz, B. Yurke, I. L. Medintz and J. S. Melinger, J. Phys. Chem. B, 2020, 124, 8042–8049 CrossRef CAS PubMed.
- F. Wu, Y. Lu, X. Mu, Z. Chen, S. Liu, X. Zhou, S. Liu and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 32388–32396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00225c |
| This journal is © The Royal Society of Chemistry 2024 |