
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble-layered protein nanoparticles conjugated with truncated flagellin induce improved mucosal and systemic immune responses in mice†
Joo Kyung
Kim
 ,
Wandi
Zhu
,
Chunhong
Dong
,
Lai
Wei
,
Yao
Ma
,
Timothy
Denning
,
Sang-Moo
Kang
and
Bao-Zhong
Wang
*
,
Wandi
Zhu
,
Chunhong
Dong
,
Lai
Wei
,
Yao
Ma
,
Timothy
Denning
,
Sang-Moo
Kang
and
Bao-Zhong
Wang
*
Center for Inflammation, Immunity & Infection, Georgia State University Institute for Biomedical Sciences, 100 Piedmont Ave SE, Atlanta, Georgia 30303, USA. E-mail: bwang23@gsu.edu; Tel: +1 404-413-3580
First published on 6th September 2024
Abstract
Influenza viral infection poses a severe risk to global public health. Considering the suboptimal protection provided by current influenza vaccines against circulating influenza A viruses, it is imperative to develop novel vaccine formulations to combat respiratory infections. Here, we report the development of an intranasally-administered, self-adjuvanted double-layered protein nanoparticle consisting of influenza nucleoprotein (NP) cores coated with hemagglutinin (HA) and a truncated form of bacterial flagellin (tFliC). Intranasal vaccination of these nanoparticles notably amplified both antigen-specific humoral and cellular immune responses in the systematic compartments. Elevated antigen-specific IgA and IgG levels in mucosal washes, along with increased lung-resident memory B cell populations, were observed in the respiratory system of the immunized mice. Furthermore, intranasal vaccination of tFliC-adjuvanted nanoparticles enhanced survival rates against homologous and heterologous H3N2 viral challenges. Intriguingly, mucosal slow delivery of the prime dose (by splitting the dose into 5 applications over 8 days) significantly enhanced germinal center reactions and effector T-cell populations in lung draining lymph nodes, therefore promoting the protective efficacy against heterologous influenza viral challenges compared to single-prime immunization. These findings highlight the potential of intranasal immunization with tFliC-adjuvanted protein nanoparticles to bolster mucosal and systemic immune responses, with a slow-delivery strategy offering a promising approach for combating influenza epidemics.
New conceptsOur study demonstrates a novel approach in vaccine design through the creation of self-adjuvanted double-layered protein nanoparticles conjugated with truncated flagellin (tFliC). This innovation is distinct from existing research by incorporating both influenza nucleoprotein (NP) cores and hemagglutinin (HA) coatings within a single nanoparticle, coupled with the adjuvant tFliC for enhanced immune activation. Unlike traditional influenza vaccines, which often fall short in providing comprehensive mucosal immunity, our nanoparticles stimulate robust systemic and mucosal immune responses, notably elevating IgA and IgG levels and promoting lung-resident memory B cell populations. The additional insight provided by our work is the significant enhancement of germinal center reactions and effector T-cell populations through a slow-delivery vaccination strategy, which greatly improves protective efficacy against heterologous influenza challenges. This concept advances materials science by showcasing how nanoparticle-based platforms can be optimized for targeted delivery and controlled release, achieving potent and durable immune responses and addressing critical gaps in current influenza vaccine efficacy. |
Introduction
The transmission of influenza viruses and subsequent infections result in 3 to 5 million severe illnesses and 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–650000 deaths globally each year.1 Currently, the primary measure for preventing and controlling the spread of seasonal influenza is the administration of seasonal flu vaccines.2 The Food and Drug Administration (FDA) has approved three primary types of annual influenza vaccines: inactivated quadrivalent, live-attenuated, and subunit.3 These vaccines employ hemagglutinin (HA) proteins as the immunodominant antigens, eliciting a protective antibody immune response.3,4 Nonetheless, due to constant antigenic mutations and mismatch between circulating and vaccine strains, these vaccines offer suboptimal protection against circulating influenza A viruses.5,6 Furthermore, seasonal flu vaccines have no protection against new emerging influenza pandemics.7 Additionally, traditional intramuscular vaccines fail to stimulate strong mucosal immunity at the respiratory tract surfaces, the portal of virus entry.8 While live attenuated vaccines can induce mucosal secretory IgA (sIgA) and tissue-resident memory T cells (TRM) responses, which are significant in anti-viral functions within the pulmonary region, the mechanisms to enhance the longevity of mucosal vaccine-induced TRM remain understudied.9 Safe but effective mucosal influenza vaccine formulations that can provide cross-protection are urgently needed to address the challenges posed by influenza epidemics and pandemics and alleviate the global burden of influenza.
000–650000 deaths globally each year.1 Currently, the primary measure for preventing and controlling the spread of seasonal influenza is the administration of seasonal flu vaccines.2 The Food and Drug Administration (FDA) has approved three primary types of annual influenza vaccines: inactivated quadrivalent, live-attenuated, and subunit.3 These vaccines employ hemagglutinin (HA) proteins as the immunodominant antigens, eliciting a protective antibody immune response.3,4 Nonetheless, due to constant antigenic mutations and mismatch between circulating and vaccine strains, these vaccines offer suboptimal protection against circulating influenza A viruses.5,6 Furthermore, seasonal flu vaccines have no protection against new emerging influenza pandemics.7 Additionally, traditional intramuscular vaccines fail to stimulate strong mucosal immunity at the respiratory tract surfaces, the portal of virus entry.8 While live attenuated vaccines can induce mucosal secretory IgA (sIgA) and tissue-resident memory T cells (TRM) responses, which are significant in anti-viral functions within the pulmonary region, the mechanisms to enhance the longevity of mucosal vaccine-induced TRM remain understudied.9 Safe but effective mucosal influenza vaccine formulations that can provide cross-protection are urgently needed to address the challenges posed by influenza epidemics and pandemics and alleviate the global burden of influenza.
In the past few years, the research community has been deeply engaged in developing universal influenza vaccines.10 Significant progress has been achieved in identifying epitopes that induce broadly neutralizing antibodies and elucidating their roles in inducing comprehensive and durable protective immunity against influenza virus infections.11 Specifically, conserved regions of the influenza virus haemagglutinin (HA) stalk, shared among different viral strains, have been targeted due to their slower mutation rate, offering cross-reactive protection against various influenza virus strains.12 Additionally, the conserved protective epitopes on the HA head are attractive targets of universal influenza vaccine due to their strong immunogenicity to induce the antibodies with more potent reactivities.13,14 Previous studies have also demonstrated that the conserved influenza T cell epitopes in influenza internal proteins, such as influenza nucleoprotein (NP), trigger cross-protective immune responses and provide protection against antigenically drifted strains, indicating the potential of NP as a T-cell immunogen for universal vaccines.15–17 Compared to the current influenza vaccines, which primarily target the HA head domain, vaccines that induce antibodies directed toward various antigens could enhance cross-protection against divergent influenza virus strains.18
Apart from the antigen selection, developing suitable adjuvants and vaccine platforms with minimal unnecessary inflammation to enhance the antigen-stimulated immune responses is especially critical for mucosal vaccine design. While various FDA-approved adjuvants exist, none have been approved for human mucosal vaccines, necessitating further exploration of novel biocompatible mucosal adjuvants. Particulate vaccine platforms demonstrate superiority in enhancing antigen immunogenicity and vaccine potency.19,20 Nanoparticle vaccines can protect antigens from rapid degradation, facilitate controlled release, and prolong exposure to antigen-presenting cells (APCs), enhancing antigen presentation.21,22 Nanoparticle platform conjugating both adjuvant and antigen could offer significant advantages for activating APCs and eliciting adaptive immune responses.23 Co-localizing antigens and adjuvants within nanoparticles improves the delivery efficiency to APCs and ensures more targeted and effective immune responses.24,25 Consequently, intranasal vaccination with well-designed antigen/adjuvant-co-incorporated vaccines can stimulate innate immune cell activation in the respiratory tract and foster robust systemic and mucosal immune responses crucial for defending against influenza viral infection.26–28
Slow delivery immunization (splitting a dose into multiple sequential applications) offers another promising approach to enhance antigen immunogenicity, particularly against challenging pathogens like influenza. Studies have demonstrated that the slow delivery enhances T follicular helper (TFH) cell responses and germinal center (GC) B cell populations, leading to significantly higher titers of autologous neutralizing antibodies (nAbs) in rhesus monkeys (RMs). Traditional immunization methods often struggle to induce broadly neutralizing antibody (bnAb) responses due to various immunological barriers, such as B cell immunodominance and the quality and quantity of GCs.29 By altering the B cell repertoire, the slow delivery redirects the immune system's focus from immunodominant non-neutralizing epitopes to a more diverse range of epitopes, potentially facilitating the development of potent and broad neutralizing antibody responses. This approach mirrors the kinetics of natural infection, providing sustained antigen presentation and promoting antibody affinity maturation, thereby offering a promising strategy for combating infectious diseases where conventional methods have fallen short.29,30
In this study, we incorporated a truncated version of flagellin (tFliC) onto double-layered protein nanoparticles containing influenza NP as the core and HA as the coating antigen to enhance the immunogenicity and protection following intranasal vaccination. tFliC, generated by removing the central variable region of the full-length flagellin to address its safety concerns, retained the ability to stimulate toll-like receptor 5 (TLR5) signaling pathways, thus facilitating robust adaptive immune responses.31–33 Alongside the increased humoral and cellular immune responses in systematic compartments, intranasal vaccination with tFliC-conjugated protein nanoparticles elicited robust mucosal immune responses, particularly in the respiratory system. Importantly, we pioneered the slow-delivery immunization strategy during mucosal vaccination to significantly enhance local germinal center responses, thereby providing better protection against heterologous influenza viral infection. These findings suggest the potential of conjugated tFliC as a promising mucosal adjuvant.
Results
Fabrication and characterization of double-layered SDAD protein nanoparticles
Protein nanoparticles were successfully fabricated using the SDAD crosslinker to generate double-layered HA3/NP (coating/core, HN) and HA3-tFliC/NP (THN) protein nanoparticles.34 The synthesis process involved the generation of an influenza NP nanoparticle core through ethanol desolvation, followed by conjugation with SDAD to form an SDAD-conjugated NP core. Subsequently, HA3 alone or combined with tFliC was conjugated onto the SDAD-conjugated NP core through an ultraviolet-activated reaction at 350 nm (Fig. 1A). Coomassie blue staining confirmed the composition of the synthesized HN and THN protein nanoparticles, including NP, HA3, and tFliC (Fig. 1B).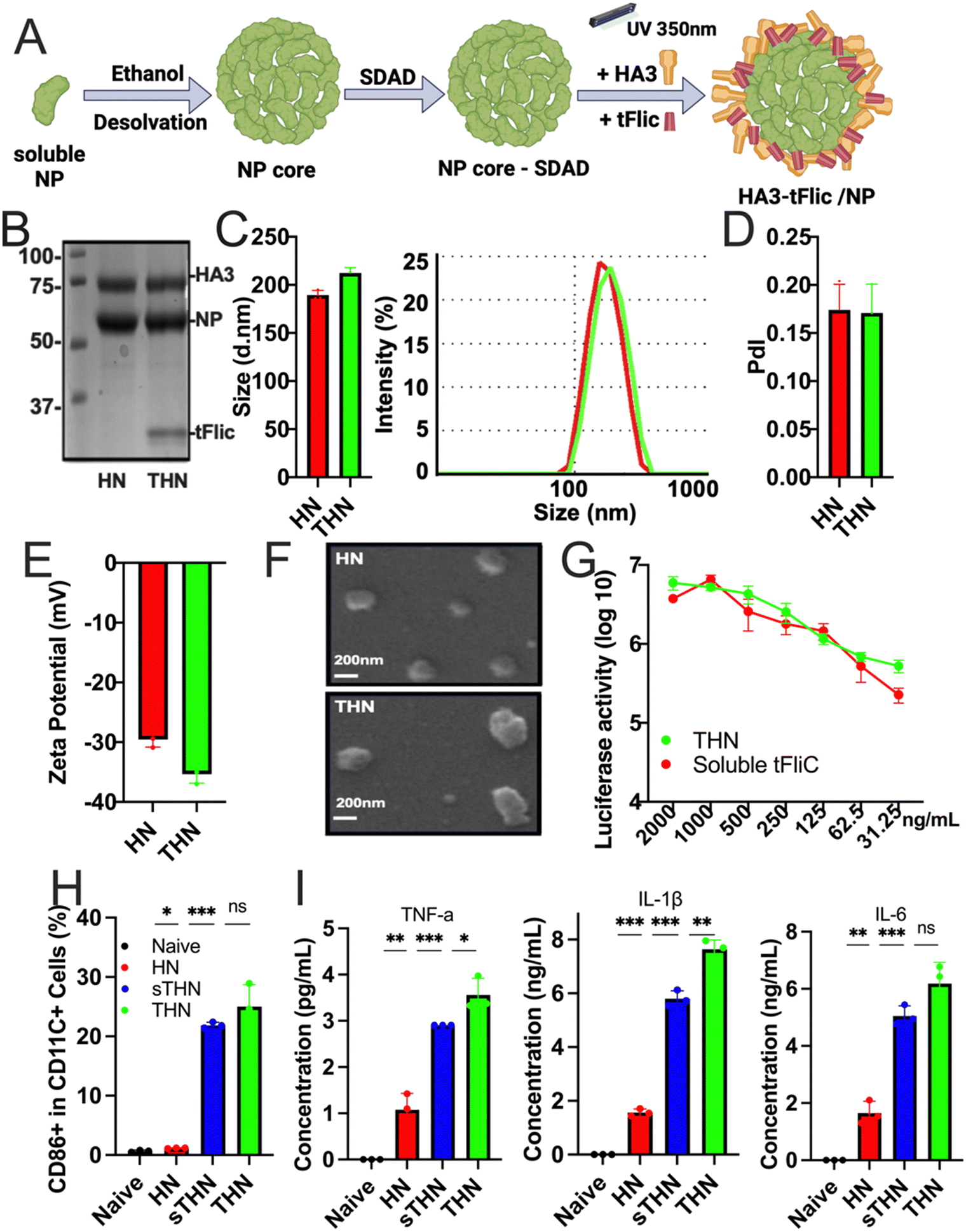 | ||
| Fig. 1 Fabrication and characterization of double-layered HN and THN protein nanoparticles. (A) Diagram of SDAD protein nanoparticle generation. (B) Coomassie blue staining analysis of the fabricated HN and THN nanoparticles. (C) Size distribution of protein nanoparticles. (D) Polydispersity index (PDI) of the nanoparticles. (E) Zeta potential of nanoparticles. (F) SEM images of the layered HN and THN nanoparticles. (G) TLR5-specific bioactivity of HA3/tFliC-NP nano and soluble tFliC. (H) Bone marrow-derived dendritic cells (BMDCs) were stimulated with protein nanoparticles with and without tFliC adjuvant. The expression of CD86 on CD11c+ BMDCs. (I) TNF-α, IL-1β, and IL-6 secretions from BMDCs cultured supernatant after stimulations with protein nanoparticles. The histograms were presented as mean ± SEM. Statistical significance was analyzed using the F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
Dynamic light scattering (DLS) analysis revealed the sizes of the synthesized double-layered protein nanoparticles: HN at 173.2 ± 19.0 nm and THN at 206.1 ± 22.4 nm (Fig. 1C). The polydispersity index (PDI) of the two nanoparticle groups was less than 0.2 (Fig. 1D). Additionally, the zeta potentials of all the double-layered protein nanoparticles were negative: HN at −29.57 ± 1.02 mV and HNT at −35.34 ± 1.45 mV (Fig. 1E). Scanning electron microscopy (SEM) further characterized their roughly spherical morphology (Fig. 1F). Compared to the NP core nanoparticles, we observed an increased size of HA-layered NP protein nanoparticles (HN) (Fig. S1, ESI†).
Additionally, the biological activity of tFliC was assessed by its ability to stimulate the TLR5 innate signal in 293 T cells transfected with plasmids of TLR5 and NF-KB-luciferase reporter genes. THN nanoparticles and soluble tFliC-supplemented HN nanoparticles demonstrated comparable levels of luciferase activities, indicating the retention of the TLR5-ligand activity of tFliC in THN (Fig. 1G). These data demonstrate the successful fabrication, characterization, and functional validation of double-layered HN and THN protein nanoparticles, confirming their composition, size, morphology, and retention of TLR5-ligand activity of tFliC in THN. Meanwhile, we found that intranasally administered fluorescently labeled SDAD protein nanoparticles locally accumulated in lung tissue for over 48 hours (Fig. S2, ESI†). It is important to further characterize the protein nanoparticle induced innate and adaptive immune responses in both systematic compartment and respiratory region.
Enhanced dendritic cell uptake, maturation, and cytokine secretion
We analyzed cellular uptake using JAWS II cells (Fig. S3, ESI†). The mean fluorescence intensity for protein nanoparticles (PNP) was significantly higher than that of soluble proteins, indicating greater cellular uptake. The DPBS control consistently showed minimal uptake, serving as a baseline reference. The protein nanoparticles were also evaluated for their ability to stimulate bone marrow-derived dendritic cell (BMDC) maturation. These innate cells' maturation and cytokine secretion have been recognized as potential indicators of antigen presentation and subsequent antigen-specific immune activation after immunization.34 Both THN and HN nanoparticles mixed with tFliC significantly enhanced the expression of BMDC maturation markers CD40, CD80, and CD86 on CD11c+ BMDCs compared to controls (Fig. 1H and Fig. S4, ESI†). Moreover, the secretion of TNF-α, IL-1β, and IL-6 from BMDCs after nanoparticle treatments was significantly elevated (Fig. 1I), suggesting the potential role of protein nanoparticles in DC maturation and pro-inflammatory cytokine production. The in vitro activation of BMDCs by the nanoparticles indicated the potential for inducing effective immune responses in vivo.Humoral immune responses and cross-reactivity of immune sera
We evaluated the immunogenicity of HA3/NP nanoparticles (HN), soluble tFliC with HA3/NP nanoparticles (sTHN), and HA3-tFliC/NP nanoparticles with the tFliC conjugated on their surface (THN) in mice. Mice sera were collected four weeks after the intranasally prime and boost vaccinations to examine the antigen-specific antibody responses using enzyme-linked immunosorbent assay (ELISA) assays. Following boost immunization in mice, THN and sTHN formulations led to elevated HA3 and Aichi-specific IgG, IgG1, and IgG2a levels compared to HN or the naïve mice. Although the tFliC-supplemented nanoparticle stimulated increased IgG isotypes, the immunity exhibited a Th2-leaning antibody response with a dominant IgG1 subtype (Fig. 2A and B).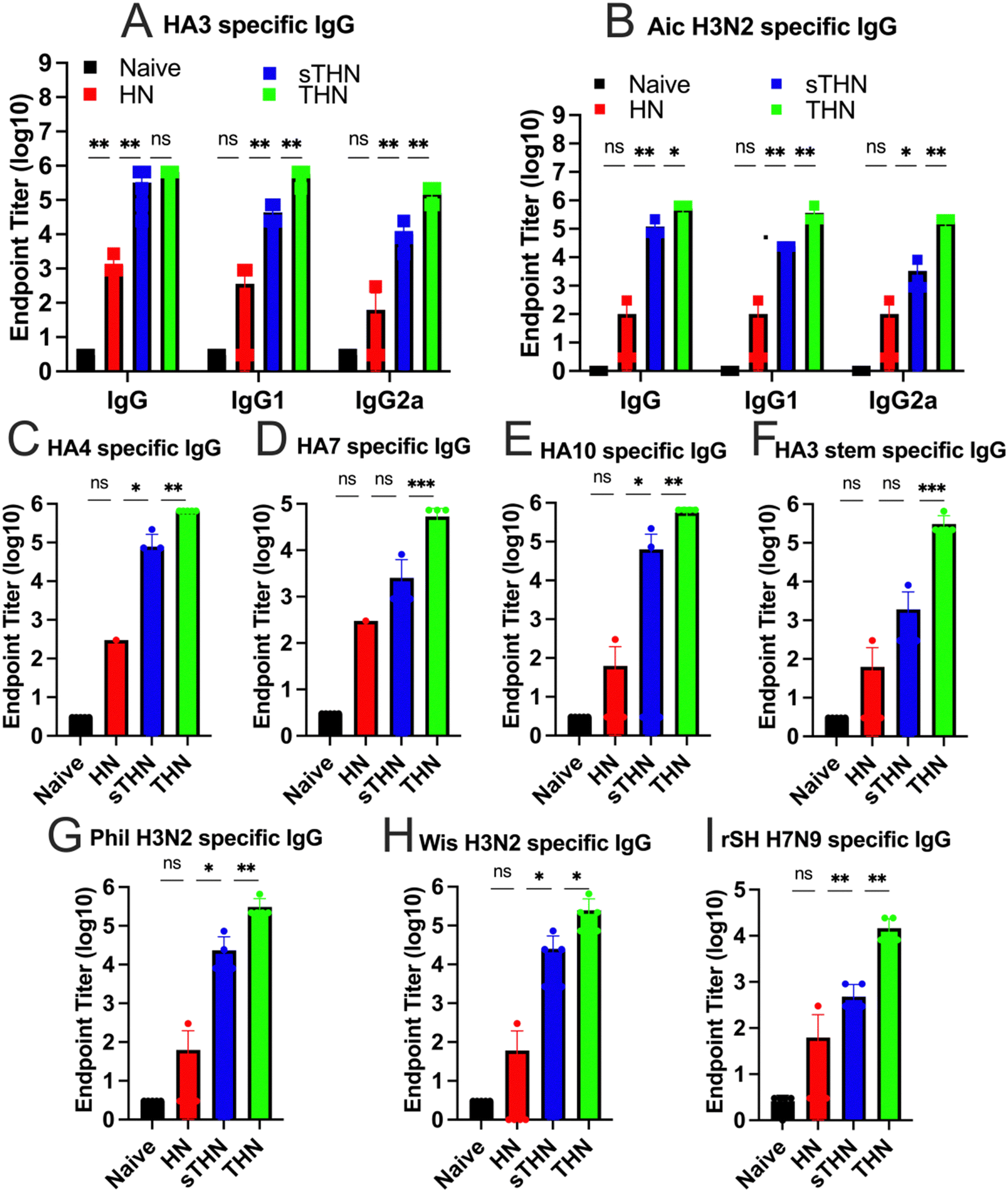 | ||
| Fig. 2 Humoral immune responses and cross-reactivities of immune sera. BALB/c mice (n = 5 per group) were intranasally immunized twice at a 4-week interval with protein nanoparticles. Antigen-specific antibodies in the immune sera were analyzed four weeks post-boost immunization. (A) HA3-specific IgG and IgG isotypes. (B) Aichi-specific IgG, IgG1, and IgG2a levels after boost. (C)–(I) Cross-reactivities of immune sera with various proteins (HA4, HA7, HA10, and HA3 stem) and viruses (Phi, Wis, and rSH) were measured. The histograms were presented as mean ± SEM. Statistical significance was analyzed using the F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
Furthermore, we investigated the serum cross-reactivities with various influenza HA proteins and viruses from group 2. As depicted in Fig. 2C–E, the groups immunized with HN nanoparticles alone exhibited low levels of antibodies. Although additional soluble tFliC (sTHN) induced improved HA4 and HA10 specific IgG levels, mice administered with tFliC conjugated THN displayed significantly higher cross-reactive antibodies against all selected HA proteins, including HA4, HA10, and HA7. Specifically, significantly higher levels of HA3 stem-specific antibodies were induced in the THN group when the tFliC adjuvant was crosslinked on the nanoparticle surface compared to the other groups (Fig. 2F). Similar results were observed using various strains, including A/Philippine/2/82 (Phil, H3N2), A/Wisconsin/2/2013 (Wis, H3N2), and reassortant A/Shanghai/2/2013(rSH, H7N9), as coating antigens. The THN nanoparticle immunization significantly boosted Phil-, Wis-, and rSH-specific IgG antibody levels (Fig. 2G and H). Overall, the self-adjuvanted THN nanoparticles elicited enhanced cross-reactive antibodies against various HA proteins and the HA3 stem domain, suggesting potential for broader protection.
Cellular immune responses after IN immunization
The systemic cellular immune responses in spleens and bone marrow were assessed using an enzyme-linked immunosorbent spot (ELISpot) assay four weeks post-boost immunization. Results indicated a notable increase in HA3 and HA4-specific IgG-secreting B cells in the bone marrow (Fig. 3A and B) and splenocytes (Fig. 3C and D) of the THN group compared to other groups. Furthermore, supplementation with soluble tFliC in protein nanoparticles (sTHN) induced a moderate increase in cytokine-secreting splenocytes (Fig. 3E–H). tFliC-conjugated nanoparticles (THN) significantly augmented the secretion of IL-4 (Fig. 3E and F) and IFN-γ (Fig. 3G and H) by splenocytes following HA3 and HA4 stimulation. These observations imply that adding tFliC adjuvant to the surface of nanoparticles enhances the systemic immune responses after intranasal immunization.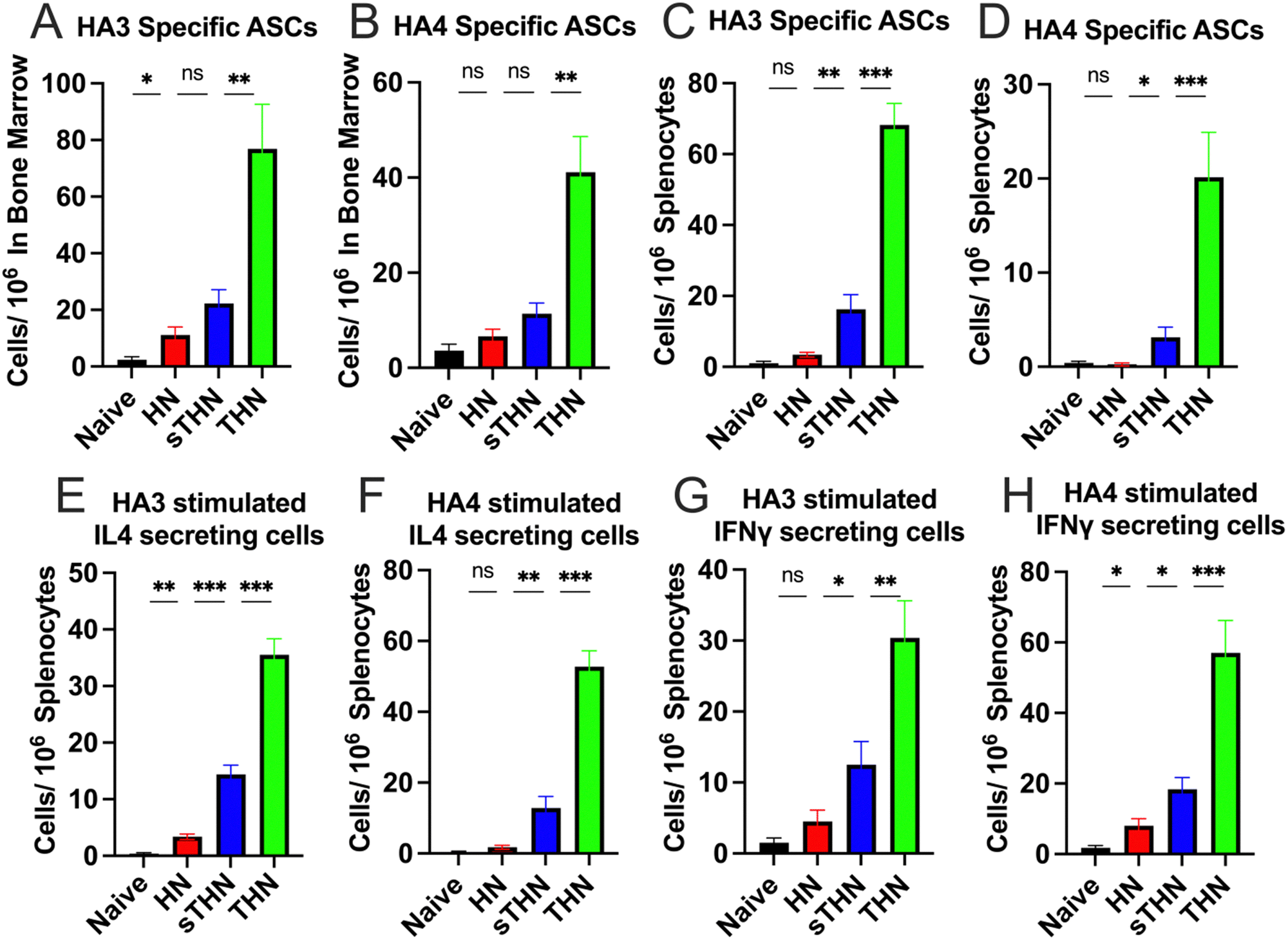 | ||
| Fig. 3 Cellular immune responses after nanoparticles intranasal immunization. Mice were intranasally immunized twice at a 4-week interval with different protein nanoparticles. Spleens and bone marrow were collected and analyzed by ELISpot assays four weeks post-boost immunization. HA3-and HA4-specific antibody-secreting cells in the (A) and (B) bone marrows and (C) and (D) splenocytes were measured. (E) and (F) IL-4 and (G) and (H) IFN-γ and secreting splenocytes were measured after HA3 and HA4 protein stimulation. The histograms were presented as mean ± SEM. Statistical significance was analyzed using the F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
Protective efficacy against influenza viral challenge
We assessed the protective efficacy of SDAD protein nanoparticles against both homologous and heterologous viral infections four weeks post-boosting vaccination. Mice were challenged with 5× LD50 of Aic (Fig. 4A) or 3 × LD50 of Phil (Fig. 4B), and their body weights were monitored over 14 days after infection.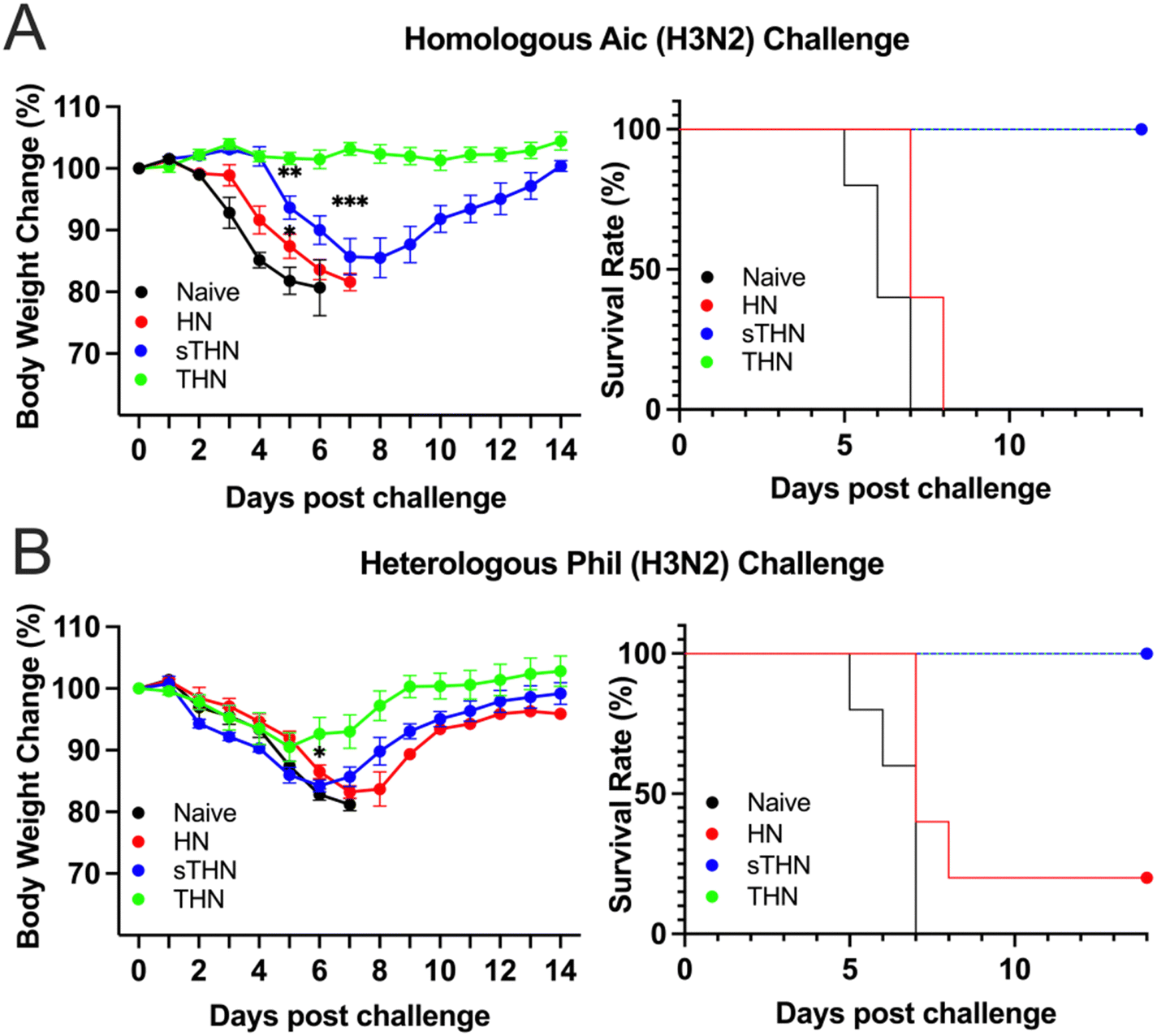 | ||
| Fig. 4 Protective efficacy against influenza viral challenge. Four weeks after the boost immunization, groups of mice were challenged with the (A) homologous Aic (H3N2) and (B) heterosubtypic Phil (H3N2) influenza viruses. Body weight changes were monitored for 14 days after the viral challenge, and the survival rates were calculated. The data were presented as mean ± SEM. | ||
For the Aic challenge, mice receiving THN immunizations displayed no body weight loss and maintained a 100% survival rate throughout the viral infections. Although mice receiving sTHN immunization also had a 100% survival rate, they experienced substantial body weight loss between days 4 and 8. In contrast, HN-immunized and naïve mice experienced rapid body weight loss and succumbed to the infection between days 5 and 8 post-challenge.
Regarding the heterologous Phil challenge, mice receiving tFliC-adjuvanted immunization (either sTHN or THN) demonstrated a 100% survival rate despite experiencing body weight loss during the initial 5 days post-challenge (∼8% of the original weight on day 5). The THN group displayed milder symptoms and a quicker recovery in body weight from day 6 onwards, regaining their original body weight by day 14. In contrast, the sTHN group exhibited significant weight loss and other symptoms, such as reduced activity, ruffled fur, and hutched posture from days 3 to 9 (Fig. 4B). Mice immunized with HN also experienced substantial body weight loss and had only a 20% survival rate against the Phil challenge. At the same time, naïve mice succumbed to the infection seven days after the challenge.
These results highlighted that the tFliC crosslinked on the nanoparticle's surface significantly enhanced the protective efficacy after intranasal immunization against both homologous and heterologous influenza viral infections, as evidenced by improved survival rates and reduced body weight loss.
Mucosal immune responses in pulmonary tissue after intranasal vaccinations
In addition to the systemic immune responses, mucosal vaccines promote the production of secretory IgA (sIgA), thereby reducing the severity and, in certain instances, the fatality rates of numerous prevalent respiratory and gastrointestinal diseases.35,36 Therefore, we evaluated whether the tFliC-adjuvanted nanoparticles could induce mucosal immune responses by measuring the antigen-specific antibody levels in the mucosa and the tissue-resident memory B cell populations (BRM) in the lung four weeks after the booster vaccination.Antibody analysis in bronchoalveolar lavage fluid (BALF) revealed significantly elevated levels of Aic- and HA3-specific IgG and IgA in the tFliC-conjugated THN group (Fig. 5A–D). In contrast, the HN group did not elicit any responses, and the soluble tFliC adjuvanted HN nanoparticles (sTHN) merely induced increased Aic- and HA3-specific IgG in BALF compared to the naïve mice (Fig. 5A and C). Antibody responses were also evaluated in nasal wash samples, where only the THN group exhibited heightened virus and HA3-specific IgG and IgA levels compared to the HN, sTHN, and naïve groups (Fig. 5E–H).
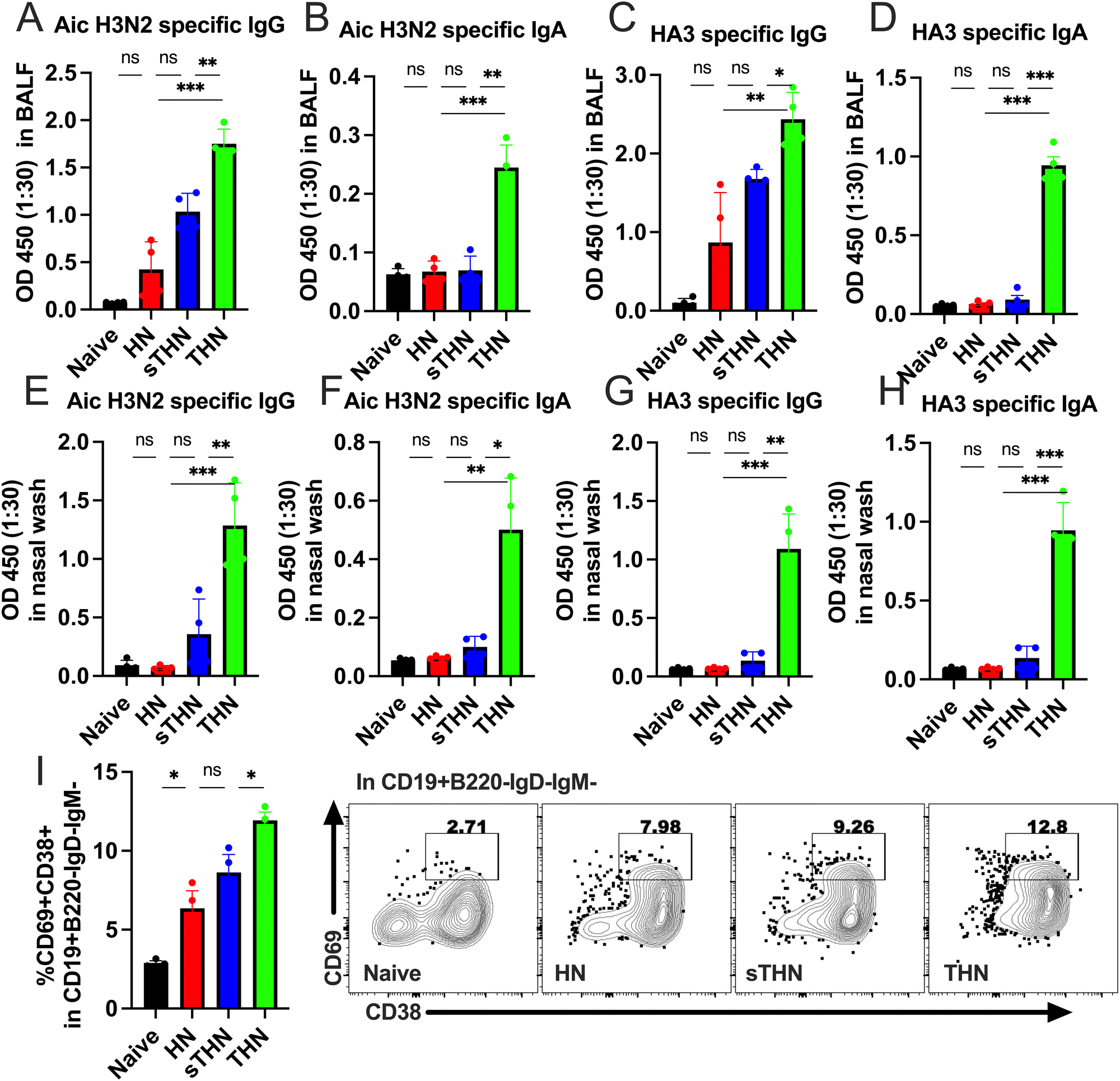 | ||
| Fig. 5 Mucosal immune responses and cell populations in pulmonary tissue. Mice were intranasally immunized twice at a 4-week interval with protein nanoparticles with and without tFliC adjuvant. The nasal and BALF washes were collected one month post-boost immunization to determine the IgA and IgG levels by ELISA. (A) and (B) Aichi-specific IgG and IgA levels in BALF. (C) and (D) HA3-specific IgG and IgA levels in BALFs. (E) and (F) Aichi-specific IgG and IgA levels in nasal washes. (G) and (H) HA3-specific IgG and IgA in nasal washes. (I) Cell populations in localized pulmonary tissue were analyzed one month post-boosting intranasal immunization. The study focused on the frequencies of CD38+CD69+ populations within CD19+B220−IgD−IgM− cells in the lungs. The data were presented as mean ± SEM. Statistical significance was analyzed using the F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
Moreover, we investigated the impact of vaccines on the induction of the BRM population in the lungs. We defined the BRM as CD19+B220−IgD−IgM−CD38+CD69+ populations in the lungs,34 with the gating strategy displayed in Fig. S3 (ESI†). Mice immunized with HN nanoparticles, with or without soluble tFliC adjuvant, demonstrated increased percentages of CD38+CD69+ BRM in the lungs compared to the naïve group. Notably, tFliC-conjugated THN immunization stimulated significantly enhanced percentages of BRM in the lungs compared to the HN and sTHN groups (Fig. 5I). These findings suggest that the intranasal vaccination of tFliC-displayed protein nanoparticles enhances mucosal antibody responses and promotes increased BRM populations in local tissues, indicating a potential strategy for improving mucosal immunity.
Intranasally sequential-prime immunization enhanced the protective efficacy against influenza viral challenge
To further improve immune reactions, we investigated whether extended exposure to antigen and adjuvant could elicit increased protective immunity after vaccination. We split the prime dose into 5 sequential administrations given every other day to create a sequential prime (slow delivery) process, extending antigen exposure in priming vaccination. The dosing schedules are depicted in Fig. 6A. We intranasally immunized one group of mice with five consecutive doses of 3 μg of THN every other day (designated as THN-S). On the day of the last administration, another group of mice received a single-prime immunization of 15 μg of THN (designated as THN). Four weeks after the primary immunization, mice in the THN and THN-S groups received a secondary vaccination with 15 μg of THN through the intranasal route.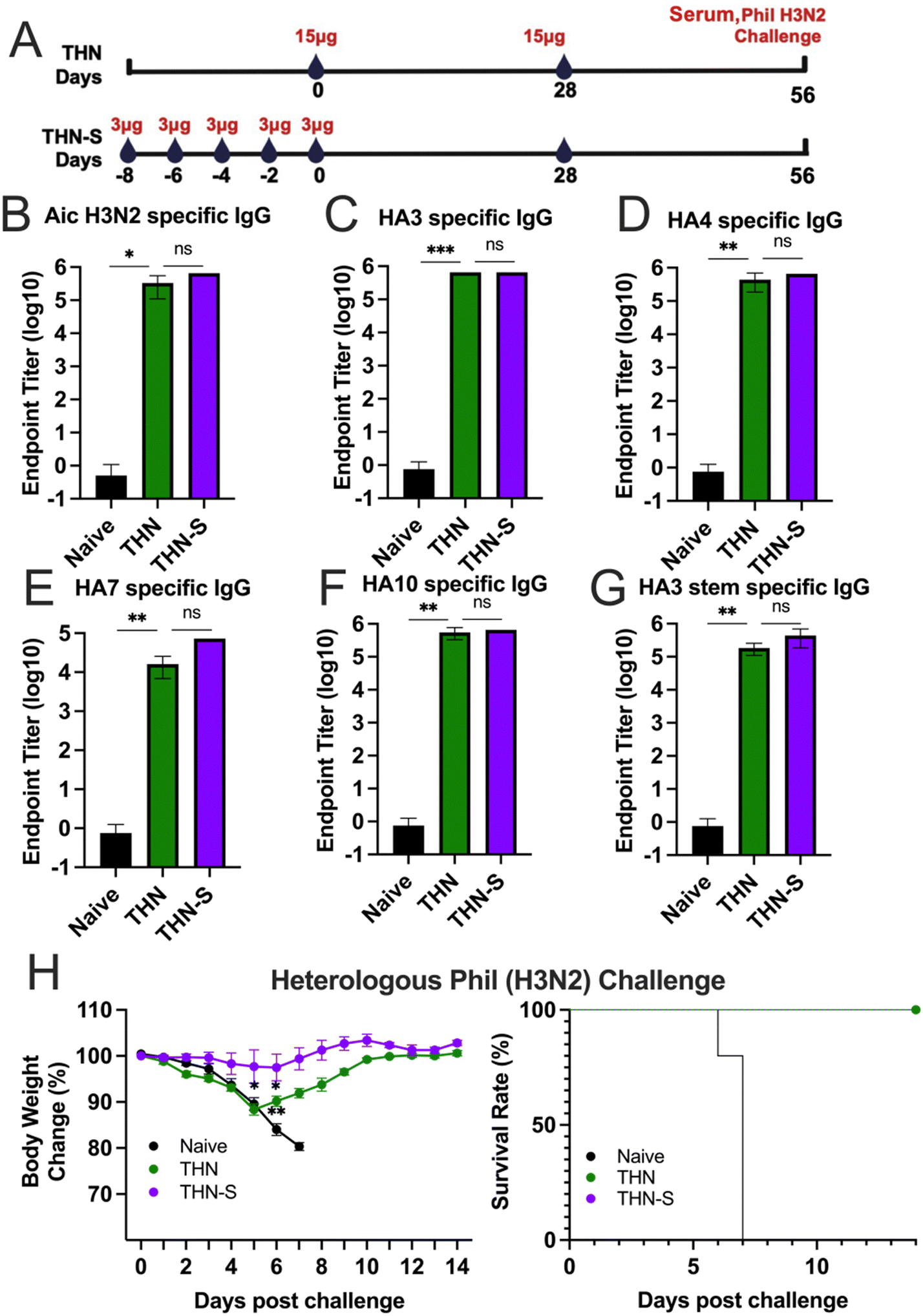 | ||
| Fig. 6 Sequential-prime immunization enhances the protective efficacy against influenza challenge. (A) Experimental schematic. Groups of mice (n = 3) were primarily immunized with a total of 15 μg of tFliC conjugated protein nanoparticles at one time (THN) or with 3 μg of vaccine in 8 consecutive days (THN-S), respectively. The mice were boosted four weeks after the primary immunization. (B)–(G) Cross-reactivities of immune sera with Aichi virus and various proteins (HA3, HA4, HA7, HA10, and HA3 stem) were measured. (H) The body weight changes and the mice survival rates after heterologous A/Phil (H3N2) influenza viral challenge. The histograms were presented as mean ± SEM. Statistical significance was analyzed using the F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
Mice sera were collected four weeks after the prime and boost vaccinations to evaluate the variance in inducing immune responses between single- and sequential-prime immunizations. The results demonstrated that both the single- (THN) and sequential- (THN-S) prime immunization significantly increased the Aic and HA3-specific IgG levels compared to the naïve group (Fig. 6B and C). However, no significant difference was observed in the antibody levels between the two THN-immunized groups. Additionally, both mouse groups immunized with tFliC-adjuvanted nanoparticles displayed robust and comparable cross-reactive antibody titers against various HA proteins from HA phylogenetic group 2 (HA4, HA7, and HA10) (Fig. 6D–F) and the conserved HA3 stem domain in sera (Fig. 6G).
The protective efficacy against viral infection was subsequently assessed. Mice were challenged with 3 × LD50 of A/Phil (H3N2) influenza virus at 4 weeks after the boosting immunizations, and changes in body weight were monitored for 14 days (Fig. 6H–I). As expected, the naïve mice experienced significant body weight loss and succumbed to the infection 7 days after the challenge. In contrast, the THN group that received a single-prime immunization exhibited moderate body weight loss during the first five days following the challenge but began to recover from day 6 onwards with a 100% survival rate. Remarkably, the sequential-prime immunization (THN-S) conferred complete protection against the heterologous A/Phil viral challenge without losing body weight over 14 days.
In conclusion, while both single- and sequential-priming immunizations systematically induced similar antibody levels, the sequential-priming strategy significantly strengthened the protective immunity of tFliC-adjuvanted protein nanoparticles against heterologous influenza viral infection.
Extended antigen exposure in mucosa enhances the germinal center reactions in localized draining lymph nodes
To better understand the underlying mechanisms of the enhanced protection in the sequential prime, we intranasally immunized mice with THN via sequential prime immunization (THN-S), as previously described, and further examined the secreted IgA (sIgA) levels in mucosal washes and GC reactions in the mediastinal lymph nodes (mLNs) 2 weeks after immunization. A group of mice immunized with 15 μg of THN was included as a control, and the experiment outline is shown in Fig. 7A. The THN group showed increased Aic- and HA3-specific sIgA in BALFs and nasal washes, consistent with the results in Fig. 5. Notably, the slow delivery of THN in the mucosal surface significantly augmented the antigen-specific sIgA levels in mucosal washes compared to conventional single immunization (Fig. 7B).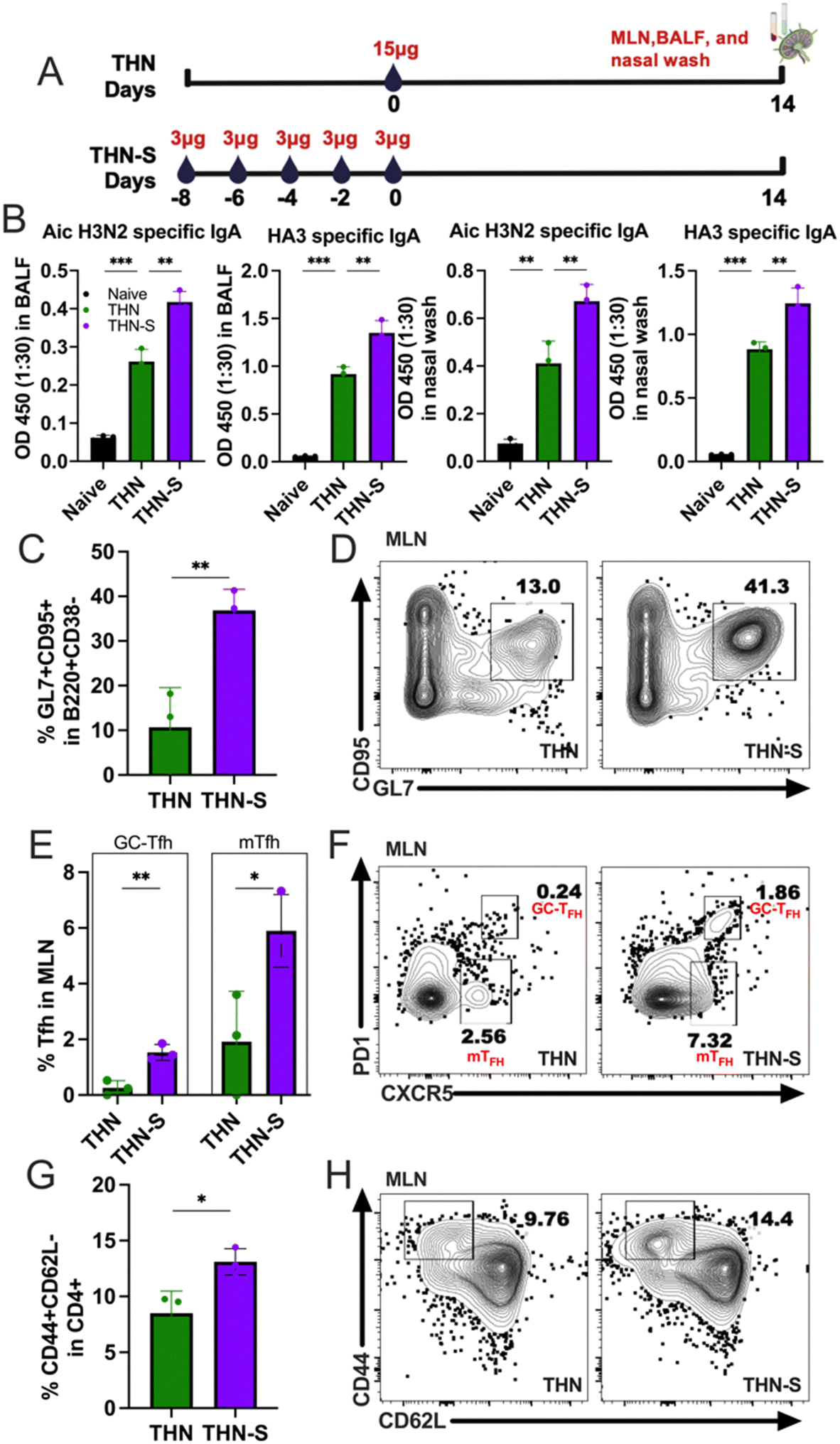 | ||
| Fig. 7 Sequential immunization leads to increased mucosal immune responses and germinal center reactions. (A) Groups of mice (n = 3) were immunized with 15 μg of HA3/tFliC-NP nanoparticle according to the dosing schedules shown above. Mice received internasal prime immunization with a single dose (×1) of 15 μg or five doses of (×5) 3 μg tFliC conjugated protein nanoparticles. BALF and nasal wash were collected and analyzed 14 days post-intranasal immunization. (B) Aichi- and HA3-specific IgA levels in BALFs and nasal washes were examined. Flow cytometry analysis and the statistics of (C) and (D) GC B cell (CD95+GL7+), (E) and (F) Tfh (CXCR5+PD1+) cell populations, and (G) and (H) effector T cell (CD44+CD62L−) were examined in mediastinal lymph nodes (MLN). The histograms were presented as mean ± SEM. Statistical significance was analyzed using the F-test and two-tailed t-test. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
The mLNs were collected from the THN-S and THN immunized groups and processed to analyze GC B cells and CD4+ T follicular helper (Tfh) cells. Remarkably, the slow delivery of tFliC-conjugated protein nanoparticles induced significantly higher percentages of GL7 and CD95 double-positive B cell populations than single-dose immunization (Fig. 7C and D). Additionally, the slow delivery of THN stimulated an increase in CD4+ Tfh cells, with significantly increased percentages of CXCR5+PD-1+CD4+ populations in mLNs compared to the conventional single-dose immunization (Fig. 7E and F). Interestingly, a CXCR5+PD-1− population, which might represent the memory Tfh (mTfh),29 was observed in both THN and THN-S groups but was higher in the mice that received the antigen slow delivery strategy (Fig. 7E and F). Furthermore, THN slow delivery resulted in increased populations of CD4+ effector T cells (CD4+CD44+CD62L−) compared to single-dose immunization (Fig. 7G and H).
In conclusion, our findings revealed a significant increase in GC B and Tfh cell percentages following sequential prime immunization in mLNs. These results suggest that extended mucosal antigen exposure might enhance the establishment and development of GC responses in the lung-draining lymph nodes, which could produce more antibodies in the respiratory system upon encountering secondary antigen exposure or viral infection in the airway.
Discussion
The development of mucosal vaccines presents a unique opportunity to trigger immune responses at primary infection sites. However, their efficacy is often hindered by challenges related to vaccine delivery and antigen uptake. Nanoparticle platforms with high immunogenicity, targeted delivery, and controlled release have garnered significant attention in developing a universal influenza vaccine.19,37 This study explored double-layered protein nanoparticles, which exhibit desirable properties such as optimal size, high antigen load with minimal crosslinker presence, safety, and controlled disassembly.38Nanoparticles within the 20–200 nm range can efficiently traverse lymphatic endothelial cell junctions, whereas larger particles struggle to enter lymphatic vessels. Our nanoparticles, approximately 200 nm with a narrow size distribution (PDI less than 0.3), were well-suited for effective antigen trafficking. Intranasal administration of protein nanoparticles alone has been shown to elicit weak immune responses, necessitating the optimization of nanoparticle sizes and the incorporation of mucosal adjuvants to enhance antigen uptake and processing.34,36 In this study, we aimed to design and evaluate potent mucosal influenza vaccine candidates utilizing protein nanoparticles adjuvanted with truncated flagellin (tFliC), offering a novel approach to vaccine development.
Flagellin, particularly its full-length form (FliC), has demonstrated promising adjuvanticity and tolerance in human clinical trials.39,40 Nevertheless, concerns regarding FliC-induced immunity, particularly related to the hypervariable regions, have limited its clinical utilization.32 We employed a truncated version of FliC (tFliC) to mitigate these concerns, eliminating the previously identified hyperimmunogenic central variable region sequence.31 Our investigation sought to determine whether incorporating tFliC as an adjuvant could enhance the immunogenicity of THN protein nanoparticles, thereby promoting systemic and mucosal antibody responses. The tFliC-conjugated nanoparticles maintained TLR5 innate signaling activity of flagellin, promoting dendritic cell maturation and cytokine secretion, crucial for initiating and regulating immune responses. Notably, intranasal administration of soluble tFliC or tFliC-conjugated protein nanoparticles exhibited no adverse effects, such as significant body weight loss (Fig. S5, ESI†), reduced activity, ruffled fur, or hutched posture in mice post-immunization, indicating a favorable safety profile as an adjuvant.
Moreover, our study provides insights into the immunogenicity and protective efficacy of double-layered HN (HA3/NP) and THN (HA3-tFlic/NP) SDAD protein nanoparticles across the systemic and mucosal compartments. We observed that protein nanoparticles adjuvanted with tFliC elicited potent mucosal immune responses, underscoring their potential as mucosal vaccines. Evaluating humoral immune responses revealed a substantial enhancement in antigen-specific antibody production with the addition of tFliC compared to nanoparticle-alone formulations. The immune sera demonstrated cross-reactivity against various influenza strains and targeted the conserved HA3 stem domain, suggesting broad protective potential. Given the association of HA stalk-specific antibodies with enhanced clearance of infected cells and viral particles, these findings hold promise for providing broader and more durable immunity against influenza viruses, potentially mitigating the burden of seasonal epidemics and future pandemics.41
Furthermore, assessment of cellular immune responses demonstrated increased IL-4 and IFN-γ secreting lymphocytes in tFliC-adjuvanted nanoparticle-immunized mice. Incorporating tFliC also synergistically enhanced cross-protection against homologous and heterologous influenza viral challenges, as evidenced by the reduced body weight loss and improved survival rates compared to control groups. Including tFliC in protein nanoparticles might enhance their immunogenicity and protective efficacy through several mechanisms. Firstly, it activates TLR5 signaling pathways, inducing the production of pro-inflammatory cytokines and promoting the maturation of APCs, leading to enhanced antigen presentation and T cell activation. Moreover, conjugation of the adjuvant to nanoparticles ensures controlled and stable presentation of tFliC on the nanoparticle surface, sustained interaction with immune cells, and prolonged immune stimulation.42 This sustained presentation enhances the efficacy of tFliC as an adjuvant, promoting durable immune responses. Additionally, conjugation improves targeting specific immune cells or tissues, minimizing systemic side effects and enhancing bioavailability.43 Overall, incorporating tFliC into protein nanoparticles harnesses its adjuvant properties and the advantages of nanoparticle delivery, thereby enhancing immunogenicity and protection. Future research could further explore the application of this vaccine platform to elucidate the underlying mechanisms of tFliC's adjuvant activity.
Vaccine slow delivery over 7–14 days, such as using osmotic pumps or repeated small-dose injections, has enhanced immune responses relative to traditional immunization approaches.44,45 Studies on HIV vaccines have indicated that subcutaneous slow delivery results in augmented humoral and GC responses in non-human primates.29 Similarly, research on whole inactivated virus (WIV) influenza vaccines in mice has revealed that prolonged antigen exposure over 28 days leads to elevated levels of high-avidity antibodies, induces class switching, and broadens binding epitopes.46 This study is particularly significant as it examined sequential prime via the mucosal route for the first time, which enhanced the vaccine-induced immune responses. This approach induced enhanced immune responses in the respiratory system, including increased antigen-specific IgA levels in mucosal samples and enhanced GC reactions in lung-draining lymph nodes compared to conventional single-priming immunization. Sequential prime notably enhanced GC-Tfh cells, primarily located within the germinal center, crucial for mediating interactions with B cells during germinal center reactions. Consequently, sequential prime facilitated B-cell antibody class switching and contributed to the enhanced production of mucosal antibodies. Our data also suggest that sequential prime can augment the levels of mTfh cells, typically found outside germinal centers in peripheral tissues or circulation, supporting long-term memory responses to pathogens upon re-exposure to antigens.47 This enhanced immunogenicity translated into superior protection against heterologous influenza viral challenges. Sequentially primed mice demonstrated complete protection and rapid recovery from heterologous viral infection. These findings indicate that sequential prime could be a promising strategy to enhance immune responses and improve vaccine efficacy against influenza and other infectious diseases.
In conclusion, our findings highlight the potential of tFliC-adjuvanted protein nanoparticles as effective influenza vaccine candidates. The results emphasize the critical role of suitable adjuvants in enhancing vaccine immunogenicity and mucosal immune responses during intranasal immunizations. A slow-delivery strategy maintaining an elongated vaccine release significantly boosts IgA antibody levels in mucosal samples and enhances the germinal center reaction in mLNs, which are integral to respiratory mucosal immunity. Furthermore, the study demonstrates the effectiveness of sequential prime to strengthen mucosal immune responses against influenza viruses, thereby improving protection against heterologous viral infections.
Conclusion
In conclusion, our research demonstrates the potential of intranasal immunization using self-adjuvanted double-layered protein nanoparticles to enhance both systemic and mucosal immune responses against influenza. These nanoparticles, consisting of an influenza nucleoprotein core coated with hemagglutinin and a truncated form of bacterial flagellin (tFliC), significantly amplified antigen-specific humoral and cellular responses. Immunized mice exhibited elevated antigen-specific IgA and IgG levels in serum and mucosal washes, along with increased lung-resident memory B cell populations. Furthermore, the tFliC-adjuvanted nanoparticles improved survival rates following homologous and heterologous H3N2 viral challenges. The study also highlighted the efficacy of slow delivery of the prime dose, which significantly enhanced germinal center reactions, effector T-cell populations, and protective efficacy against heterologous viral challenges compared to a single-prime immunization. These findings underscore the promise of intranasal vaccines employing tFliC-adjuvanted protein nanoparticles to induce robust immune responses in the respiratory system. The approach of slow delivery further enhances these responses, offering a compelling strategy for addressing influenza epidemics. This research advances the development of mucosal vaccines that could provide broader and more effective protection against influenza, contributing to global public health efforts.Methods
Ethics statements
This study was conducted in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. The animal facility at Georgia State University holds accreditation from the American Association for Accreditation of Laboratory Animal Care. All animal procedures were approved by the Georgia State University Institutional Animal Care and Use Committee under protocol No. A22029.Fabrication and characterization of double-layered SDAD protein nanoparticles
We fabricated and characterized the double-layered nanoparticles as described previously.34 Briefly, 200 μg of NP protein was mixed with four times the volume of ethanol (100%, 1 mL min−1) while stirring for 20 min. The nanoparticles were pelleted by centrifugation at 20000 × g for 20 min, sonicated (40%-amp, 3 s on and 3 s off), and re-suspended in DPBS (250 μL). The NP core nanoparticle solution was mixed with a tenfold molar of excess SDAD ((NHS-SS-diazirine) (succinimidyl 2-((4,4′-azipentanamido)ethyl)-1,3′-dithiopropionate)) for 30 min. Then, Tris–HCl (15 μL, 1 M, PH 8.0) buffer was added to quench the reaction for 5 min. The excessive SDAD was removed by washing with DPBS and centrifuging at 20000 × g for 20 min. HA3 (200 μg) or HA3 + tFliC (160 + 40 μg respectively) proteins were incubated with SDAD-conjugated NP core nanoparticles for 1 hour under UV light (350 nm). Then, the nanoparticles were purified by centrifugation at 20000 × g for 20 min, resuspension, and sonication in 200 μL DPBS. The individual HN and THN SDAD protein nanoparticle was treated with 2.5% 2-mercaptoethanol in SDS-PAGE loading buffer at 100 °C for 5 mins and then characterized by 10% SDS-PAGE followed by Coomassie blue staining. The nanoparticle sizes and morphology were determined by dynamic light scattering analysis (Malvern Zetasizer) and Tescan Vega's 3rd generation scanning electron microscopy (SEM). Fluorescent nanoparticle samples were prepared following previously established methods.48 Briefly, 10 mg of the SDAD protein nanoparticles were dissolved in 1 mL of 0.1 M sodium bicarbonate buffer. While stirring, 3 μL of the amine-reactive dye (Alexa Fluor 488 or Alexa Fluor 700 NHS Ester, Thermo Fisher) dissolved in DSMO at 10 mg mL−1 was slowly added to the protein solution, followed by a 1-hour incubation at room temperature. The fluorescently labeled particles were mixed with regular nanoparticles in a 1:3 ratio.
TLR5 signaling activity
TLR5 signaling activity was measured as previously described.49 Briefly, pUNO1-hTLR5 and reporter pGL4.32 plasmids were used to transfect the HEK 293 T cells using Lipofectamine 3000. Seventy-two hours later, the transfected cells were treated with serially diluted soluble tFliC and tFliC conjugated THN, ranging from 2 μg mL−1 to 31.25 ng mL−1 for stimulation. After 5 h incubation, 100 μL of the Steady-Glo luciferase assay reagents were added to each well and transferred into a white plate. The luciferase activity was measured by Promega GloMax Discover Microplate Reader.In vitro cellular uptake assays
JAWS II cells were cultured in the alpha minimum essential medium containing ribonucleosides, deoxyribonucleosides, 4 mM L-glutamine, 1 mM sodium pyruvate, 5 ng mL−1 recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF), and 20% fetal bovine serum (FBS). Then, cells were seeded at 1 × 105 cells per well in a 24-well cell culture plate. The cells were treated with fluorescently labeled protein nanoparticles at a concentration of 100 μg mL−1, using DPBS as the negative control. After a 24-hour incubation, the cells were washed with PBS, fixed, and permeabilized using the BD fixation/permeabilization buffer at 4 °C for 20 min. Subsequently, the cells were stained with DAPI at room temperature for 30 min. The results were analyzed using a Keyence BZ-X710 fluorescence microscope. Fluorescent microscopy with the BZ-X710 (Keyence) was used to examine the in vitro uptake of protein nanoparticles and the soluble HA3 protein by JAWS II dendritic cells, with DPBS as a negative control. This was complemented by precise quantitative measurements of mean fluorescence intensity per cell, using the BZ-X analysis software (Keyence).Biodistribution of protein nanoparticles
To evaluate the distribution of intranasally administered fluorescent nanoparticles in mouse organs, we employed the in vivo imaging system (IVIS). Protein nanoparticles labeled with Alexa Fluor 700 were administered intranasally, and the mice were euthanized at specific time points (24, 48, and 120 hours post-administration). Organs, including the hearts, lungs, kidneys, and spleen, were harvested and placed on transparent Petri dishes for ex vivo imaging. The images were collected using a PerkinElmer IVIS Spectrum system (Caliper Life Sciences). The fluorescent intensity of the nanoparticles in the tissues was quantified by analyzing the images with Living Image software (PerkinElmer) to determine the fluorescent radiant efficiency.Bone marrow dendritic cell maturation and cytokine secretion
BMDC maturation and cytokine secretion were measured as previously described.50 Briefly, bone marrow cells were isolated and seeded at 106 cells per mL in complete RPMI 1640 medium (cRPMI) supplemented with GM-CSF (20 ng mL−1) and cultured for two days. Two days after the initial culture, half of the culture medium was replaced with fresh GM-CSF-supplemented cRPMI media and continued cultured for one more day. The non-adherent and loosely attached cells were centrifuged at 250 × g for 5 min, resuspended, and cultured in fresh culture medium with GM-CSF for three additional days. Then, the cells were collected and seeded at 106 cells per well in a 24-well cell culture plate, followed by treatments with HN, sTHN, and THN particles for 18 hours. Untreated cells were used as negative controls. To assess BMDC maturation, cells were first incubated with Zombie Aqua dye (Zombie Aqua Fixable Viability Kit, BioLegend) to distinguish between live and dead cells. The cells were stained with anti-mouse CD11c-APC, CD40-PE, CD80-FITC, and CD86-APC/Cy7 surface antibodies. Following extensive washing, the cells were analyzed using the BD LSR Fortessa Cell Analyzer. The expression levels of TNF-α, IL-1β, and IL-6 in the BMDC-cultured supernatant were determined by coating ELISA plates with 4 μg mL−1 of cytokine-specific capture antibodies and incubating them overnight at 4 °C. Then, 50 μL of supernatant was added to each well and incubated for 2 hours at 37 °C. This was followed by incubation with biotin-conjugated detection antibodies and HRP-conjugated streptavidin.Immunization and influenza infection
Female BALB/c mice at 6–8 weeks of age received the priming and boosting immunizations on day 0 and day 28, respectively, by intranasal immunization of 35 μL of vaccine formulations. There were four groups for the initial part of the study: (1) HA3/NP nanoparticles (HN, 12 μg), (2) HA3/NP nanoparticles with soluble tFliC (sTHN, 12 μg of HN and 3 μg of tFliC), (3) HA3-tFliC/NP nanoparticles with tFliC adjuvanted on the protein surface (THN, 15 μg), (4) PBS. Twenty-one days after the boost immunization, immune sera from the different groups were collected. These mice were intranasally challenged 4 weeks post boosting immunization, with 5 × LD50 of A/Aic, H3N2 or 3 × LD50 of A/Phil, H3N2. Mouse body weights were monitored for 14 days after infection.The impact of sequential immunizations was assessed by intranasally immunizing mice with 35 μL of the vaccine formulations (THN, 15 μg) twice, with a 4-week interval between doses. For the priming immunization, the mice group received (1) PBS, (2) the traditional single-prime immunization with 15 μg of THN, and (3) an 8-day sequential-prime immunization with THN consisting of 5 doses. Mice received five doses of 3 μg each over 8 days to ensure extended exposure to the vaccine components. Immunized mice were intranasally challenged with 3 × LD50 of A/Phil, H3N2 4 weeks post-boosting immunization.
Antigen-specific antibodies (enzyme-linked immunosorbent assay, ELISA)
Antibody ELISA was performed as previously described51 to determine the protein (HA3, HA4, HA7, HA10, and HA3 stem) or virus (Aic, Phil, Wis, or rSH) -specific IgG or IgA antibody titers in sera, nasal washes or BALF samples post-immunization. Briefly, ELISA plates were coated with target antigens (4 μg mL−1) overnight at 4 °C in PBS. Then, the antigen-coated plates were blocked with 2% BSA for 1 hour at 37 °C. After blocking, sera, nasal wash, and BALF samples were serially diluted and added to the plates for 2 hours of incubation at 37 °C. After washing, HRP-conjugated anti-mouse IgG antibodies (IgG, IgG1, and IgG2a) or IgA antibodies were applied to the plates and incubated for 1 hour. Finally, the HRP conjugated antibodies were reacted with TMB subtracts, and 1 M H2SO4 stopped the reactions. The plates were measured by Bioreader-6000-E (Biosys) at the absorbance of 450 nm. The highest dilution exhibiting an OD450 value twice that of the naive group was considered as the endpoint titer.Enzyme-linked immunospot assay (ELISPOT)
The cellular immune responses were detected by ELISPOT assays, as previously described.52 Briefly, to determine the number of cytokines secreting T cells by ELISPOT assays, one million splenocytes were seeded onto the 96-well ELISPOT plates that were coated with anti-mouse IFN-γ and IL4-specific monoclonal antibodies with different stimulators. Three days after culturing, the cytokine-secreting cells were visualized by biotin-conjugated IFN-γ or IL4-detection antibodies and horseradish peroxidase (HRP)-conjugated streptavidin antibody using a Bioreader-6000-E. Also, to evaluate the antibody-secreting cells (ASCs), splenocytes and bone marrow cells were added to the plates pre-coated with HA proteins. Then, the colonies were visualized by HRP-conjugated anti-mouse IgG antibody.Flow cytometry
Immunized mice were euthanized four weeks post-boosting immunization for lung collection to determine the tissue-resident memory B cell populations in the lung. The samples were prepared as previously described.34 Lung tissues were treated with Collagenase type 4 at a concentration of 1 mg mL−1 and DNase I at 30 μg mL−1 in RPMI 1640 media, incubated at 37 °C for 30 minutes. Then, lung samples were homogenized through a 70 μm cell strainer and centrifuged at 1500 rpm for 5 minutes at 4 °C. After removing the supernatant, cells were washed with FACS buffer (DPBS with 2% FBS) and stained with anti-mouse CD19-APC, B220-AF700, IgD-FITC, IgM-PE/Cy7, CD69-PE, CD38-Pacific Blue, CD16/32 antibodies, and Zombie NIR dye. Mediastinal lymph nodes (mLNs) from different groups were collected and examined by flow cytometry 14 days after the primary immunization. The cells from mLNs were divided into two parts and stained with different combinations of antibodies as follows: (1) anti-mouse B220-AF700, GL7-PerCP/Cy5.5, CD95-FITC, CD38-Pacific Blue, CD16/32 antibodies, and Zombie NIR dye and (2) anti-mouse CD4-APC, CD62L-FITC, CD44-BV421, PD1-PE/Cy7, CXCR5-PE, CD16/32 antibodies, and Zombie NIR dye combinations. Cells were recorded using the BD LSR Fortessa Cell Analyzer.Data availability
The data supporting the results of this article, “Double-Layered Protein Nanoparticles Conjugated with Truncated Flagellin Induce Improved Mucosal and Systemic Immune Responses in Mice,” are available on Mendeley Data at https://data.mendeley.com/preview/vhmyybwrc4?a=abe54560-d1a7-46b3-9e10-9d7976bcc678. This includes all data related to the fabrication, characterization, and immunogenicity assessments of the double-layered protein nanoparticles. Additional supporting data and materials have been included in the ESI,† section of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the US NIH/National Institute of Allergy and Infectious Diseases under grants R01AI101047 and R01AI143833 to B.-Z.W. The content in this study is solely the author's responsibility and does not necessarily represent the official views of the funders. SEM images were acquired using the instruments and services in the Imaging Core Facility at Georgia State University. The graphical abstract was created with https://biorender.com/.References
- B. K. Jha, R. Pandit, R. Jha and K. D. Manandhar, Heliyon, 2020, 6, e03304 CrossRef PubMed.
- C. M. Trombetta, O. Kistner, E. Montomoli, S. Viviani and S. Marchi, Vaccines, 2022, 10(5), 714 CrossRef CAS.
- R. F. Nuwarda, A. A. Alharbi and V. Kayser, Vaccines, 2021, 9(9), 1032 CrossRef CAS.
- S.-S. Wong and R. J. Webby, Clin. Microbiol. Rev., 2013, 26, 476–492 CrossRef CAS.
- J. A. Lewnard and S. Cobey, Vaccines, 2018, 6(2), 28 CrossRef PubMed.
- A. C. Tricco, A. Chit, C. Soobiah, D. Hallett, G. Meier, M. H. Chen, M. Tashkandi, C. T. Bauch and M. Loeb, BMC Med., 2013, 11, 153 CrossRef PubMed.
- F. Krammer, A. García-Sastre and P. Palese, Cold Spring Harbor Perspect. Biol., 2018, 10(7), a028845 CrossRef PubMed.
- J. R. Baker, Jr., M. Farazuddin, P. T. Wong and J. J. O'Konek, J. Allergy Clin. Immunol., 2022, 150, 1–11 CrossRef PubMed.
- M. M. Wilk and K. H. G. Mills, Front. Immunol., 2018, 9, 1860 CrossRef PubMed.
- J. K. Kim, S. M. Kang, R. W. Compans and B. Z. Wang, Expert Opin. Drug Discovery, 2021, 16, 213–216 CrossRef.
- Z. Staneková and E. Varečková, Virol. J., 2010, 7, 351 CrossRef PubMed.
- E. Kirkpatrick, X. Qiu, P. C. Wilson, J. Bahl and F. Krammer, Sci. Rep., 2018, 8, 10432 CrossRef PubMed.
- C. Jiao, B. Wang, P. Chen, Y. Jiang and J. Liu, Front. Immunol., 2023, 14, 1086297 CrossRef CAS.
- A. B. Arunachalam, P. Post and D. Rudin, npj Vaccines, 2021, 6, 144 CrossRef CAS.
- M. W. LaMere, H. T. Lam, A. Moquin, L. Haynes, F. E. Lund, T. D. Randall and D. A. Kaminski, J. Immunol., 2011, 186, 4331–4339 CrossRef CAS PubMed.
- A. Rak, I. Isakova-Sivak and L. Rudenko, Vaccines, 2023, 11(12), 1747 CrossRef CAS.
- D. F. Hoft and R. B. Belshe, Mo Med., 2014, 111, 321–325 Search PubMed.
- S. L. Epstein, Am. J. Epidemiol., 2018, 187, 2603–2614 CrossRef PubMed.
- C. Dong and B.-Z. Wang, Adv. NanoBiomed Res., 2022, 2, 2100122 CrossRef CAS PubMed.
- S. Liu, M. Hu, X. Liu, X. Liu, T. Chen, Y. Zhu, T. Liang, S. Xiao, P. Li and X. Ma, Vaccines, 2023, 12(1), 30 CrossRef.
- F. Lamontagne, V. Khatri, P. St-Louis, S. Bourgault and D. Archambault, Vaccines, 2022, 10(11), 1920 CrossRef CAS.
- M. Zhu, R. Wang and G. Nie, Hum. Vaccines Immunother., 2014, 10, 2761–2774 CrossRef PubMed.
- X. Fang, H. Lan, K. Jin, D. Gong and J. Qian, Cancers, 2022, 14(16), 3842 CrossRef CAS PubMed.
- Y. Fan and J. J. Moon, Vaccines, 2015, 3, 662–685 CrossRef CAS PubMed.
- Y. Song, F. Mehl and S. L. Zeichner, Vaccines, 2024, 12(2), 191 CrossRef CAS.
- E. Kehagia, P. Papakyriakopoulou and G. Valsami, Vaccine, 2023, 41, 3589–3603 CrossRef CAS.
- E. Gianchecchi, A. Manenti, O. Kistner, C. Trombetta, I. Manini and E. Montomoli, Influenza Other Respir. Viruses, 2019, 13, 429–437 CrossRef CAS PubMed.
- K. Schulze, T. Ebensen, L. A. Babiuk, V. Gerdts and C. A. Guzman, Nanomedicine, 2017, 13, 2169–2178 CrossRef CAS.
- K. M. Cirelli, D. G. Carnathan, B. Nogal, J. T. Martin, O. L. Rodriguez, A. A. Upadhyay, C. A. Enemuo, E. H. Gebru, Y. Choe, F. Viviano, C. Nakao, M. G. Pauthner, S. Reiss, C. A. Cottrell, M. L. Smith, R. Bastidas, W. Gibson, A. N. Wolabaugh, M. B. Melo, B. Cossette, V. Kumar, N. B. Patel, T. Tokatlian, S. Menis, D. W. Kulp, D. R. Burton, B. Murrell, W. R. Schief, S. E. Bosinger, A. B. Ward, C. T. Watson, G. Silvestri, D. J. Irvine and S. Crotty, Cell, 2019, 177, 1153–1171.e1128 CrossRef CAS.
- B. S. Ou, O. M. Saouaf, J. Baillet and E. A. Appel, Adv. Drug Delivery Rev., 2022, 187, 114401 CrossRef CAS.
- B. Z. Wang, H. S. Gill, S. M. Kang, L. Wang, Y. C. Wang, E. V. Vassilieva and R. W. Compans, Clin. Vaccine Immunol., 2012, 19, 1119–1125 CrossRef CAS.
- B. Zhao, J. Yang, B. He, X. Li, H. Yan, S. Liu, Y. Yang, D. Zhou, B. Liu, X. Fan, M. Zhong, E. Zhang, F. Zhang, Y. Zhang, Y.-Q. Chen, S. Jiang and H. Yan, Cell Rep., 2021, 36, 109401 CrossRef CAS PubMed.
- A. H. López-Yglesias, C.-C. Lu, X. Zhao, T. Chou, T. VandenBos, R. K. Strong and K. D. Smith, ImmunoHorizons, 2019, 3, 422–432 CrossRef PubMed.
- W. Zhu, J. Park, T. Pho, L. Wei, C. Dong, J. Kim, Y. Ma, J. A. Champion and B.-Z. Wang, Small, 2023, 19, 2301801 CrossRef CAS PubMed.
- R. Mudgal, S. Nehul and S. Tomar, Hum. Vaccines Immunother., 2020, 16, 2921–2931 CrossRef CAS PubMed.
- E. C. Lavelle and R. W. Ward, Nat. Rev. Immunol., 2022, 22, 236–250 CrossRef CAS.
- Y. Wang, L. Deng, S. M. Kang and B. Z. Wang, Expert Rev. Vaccines, 2018, 17, 967–976 CrossRef CAS PubMed.
- A. Khalaj-Hedayati, C. L. L. Chua, P. Smooker and K. W. Lee, Influenza Other Respir. Viruses, 2020, 14, 92–101 CrossRef.
- J. H. Rhee, K. Khim, S. Puth, Y. Choi and S. E. Lee, Curr. Opin. Virol., 2023, 60, 101330 CrossRef CAS PubMed.
- I. A. Hajam, P. A. Dar, I. Shahnawaz, J. C. Jaume and J. H. Lee, Exp. Mol. Med., 2017, 49, e373 CrossRef CAS PubMed.
- L. Deng, T. Mohan, T. Z. Chang, G. X. Gonzalez, Y. Wang, Y.-M. Kwon, S.-M. Kang, R. W. Compans, J. A. Champion and B.-Z. Wang, Nat. Commun., 2018, 9, 359 CrossRef PubMed.
- G. A. Roth, V. C. T. M. Picece, B. S. Ou, W. Luo, B. Pulendran and E. A. Appel, Nat. Rev. Mater., 2022, 7, 174–195 CrossRef CAS PubMed.
- K. Elumalai, S. Srinivasan and A. Shanmugam, Biomed. Technol., 2024, 5, 109–122 CrossRef CAS.
- J. H. Lee, H. J. Sutton, C. A. Cottrell, I. Phung, G. Ozorowski, L. M. Sewall, R. Nedellec, C. Nakao, M. Silva, S. T. Richey, J. L. Torres, W.-H. Lee, E. Georgeson, M. Kubitz, S. Hodges, T.-M. Mullen, Y. Adachi, K. M. Cirelli, A. Kaur, C. Allers, M. Fahlberg, B. F. Grasperge, J. P. Dufour, F. Schiro, P. P. Aye, O. Kalyuzhniy, A. Liguori, D. G. Carnathan, G. Silvestri, X. Shen, D. C. Montefiori, R. S. Veazey, A. B. Ward, L. Hangartner, D. R. Burton, D. J. Irvine, W. R. Schief and S. Crotty, Nature, 2022, 609, 998–1004 CrossRef CAS.
- H. H. Tam, M. B. Melo, M. Kang, J. M. Pelet, V. M. Ruda, M. H. Foley, J. K. Hu, S. Kumari, J. Crampton, A. D. Baldeon, R. W. Sanders, J. P. Moore, S. Crotty, R. Langer, D. G. Anderson, A. K. Chakraborty and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6639–e6648 CrossRef CAS.
- M. Beukema, S. Gong, K. Al-Jaawni, J. J. de Vries-Idema, F. Krammer, F. Zhou, R. J. Cox and A. Huckriede, Front. Immunol., 2023, 14, 1249902 CrossRef CAS PubMed.
- X. Gao, K. Luo, D. Wang, Y. Wei, Y. Yao, J. Deng, Y. Yang, Q. Zeng, X. Dong, L. Xiong, D. Gong, L. Lin, K. Pohl, S. Liu, Y. Liu, L. Liu, T. H. O. Nguyen, L. F. Allen, K. Kedzierska, Y. Jin, M. R. Du, W. Chen, L. Lu, N. Shen, Z. Liu, I. A. Cockburn, W. Luo and D. Yu, eLife, 2023, 12, e82217 CrossRef CAS PubMed.
- L. Deng, T. Z. Chang, Y. Wang, S. Li, S. Wang, S. Matsuyama, G. Yu, R. W. Compans, J. D. Li, M. R. Prausnitz, J. A. Champion and B. Z. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7758–e7767 CAS.
- C. Wang, W. Zhu and B. Z. Wang, Int. J. Nanomed., 2017, 12, 4747–4762 CrossRef CAS.
- J. Helft, J. Böttcher, P. Chakravarty, S. Zelenay, J. Huotari, B. U. Schraml, D. Goubau and C. Reis e Sousa, Immunity, 2015, 42, 1197–1211 CrossRef CAS PubMed.
- W. Zhu, L. Wei, C. Dong, Y. Wang, J. Kim, Y. Ma, G. X. Gonzalez and B. Z. Wang, Mol. Ther. Nucleic Acids, 2022, 30, 421–437 CrossRef CAS PubMed.
- C. Dong, Y. Wang, W. Zhu, Y. Ma, J. Kim, L. Wei, G. X. Gonzalez and B. Z. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 6331–6342 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00287c |
| This journal is © The Royal Society of Chemistry 2024 |