
Ov-rich γ-MnO2 enhanced electrocatalytic three-electron oxygen reduction to hydroxyl radicals for sterilization in neutral media†
Yingnan
Qin
a,
Tongzhu
Han
b,
Ligang
Chen
*c,
Kexin
Yan
d,
Jing
Wang
e,
Ning
Wang
*a and
Baorong
Hou
a
aKey Laboratory of Advanced Marine Materials, Key Laboratory of Marine Environmental Corrosion and Bio-fouling, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, 266071, China. E-mail: wangning@qdio.ac.cn
bMarine Bioresource and Environment Research Center, Key Laboratory of Marine Eco-Environmental Science and Technology, First Institute of Oceanography, Ministry of Natural Resources, Qingdao, 266061, China
cState Power Investment Corporation Hydrogen Energy Company, Limited, Beijing 102600, China. E-mail: ligangchen0310@163.com
dCollege of Environmental and Safety Engineering, Qingdao University of Science and Technology, Qingdao 266042, PR China
eCollege of Chemical Engineering, Qingdao University of Science and Technology, Qingdao 266042, PR China
First published on 28th August 2024
Abstract
Marine biofouling severely limits the development of the marine economy, and reactive oxygen species (ROS) produced by electrocatalytic antifouling techniques could inactivate marine microorganisms and inhibit the formation of marine biofouling. Compared with an electro-Fenton reaction, a three-electron oxygen reduction reaction (3e− ORR) could generate a hydroxyl radical (˙OH) in situ without the limitation of pH and iron mud pollutants. Herein, Ov-rich γ-MnO2 is designed to enhance the 3e− ORR performance in neutral media and exhibits excellent sterilization performance for typical marine bacteria. DFT calculation reveals that Ov is beneficial to the “end-on” adsorption and activation of O2, and the Mn site could accept the electrons from *OOH and promote its further reduction to form ˙OH; Ov and Mn sites together guarantee the high 3e− ORR efficiency. In addition, liquid chromatography–tandem mass spectrometry (LC–MS/MS) proves the vast formation of ˙OH in the primary reaction stage, which is the key to sterilization. This work explores the reaction mechanism of the 3e− ORR in neutral media and provides the possibility for the application of electrocatalysis technology in the treatment of marine biofouling pollution.
New conceptsIn this study, we have developed a novel Ov-rich γ-MnO2 catalyst that exhibits remarkable three-electron oxygen reduction reaction (3e− ORR) performance in neutral media. Moreover, the as-prepared MnO2-800A catalyst effectively inactivates two typical marine bacteria, Pseudomonas aeruginosa and Staphylococcus aureus, within a short duration. Typically, the adherence of microorganisms to marine service equipment prompts the development of marine biofouling. Our proposed 3e− ORR method is advantageous as it generates H2O2 and various reactive oxygen species (ROS) on-site, offering a more convenient and efficient alternative to traditional anti-fouling techniques. Theoretical calculations reveal that the presence of Ov enhances the “end-on” adsorption of O2 and accelerates the activation process of oxygen. Additionally, the attractive electronic interaction between the Mn site and *OOH leads to the elongation of the O–O bond, favoring the formation of ˙OH radicals. This study sheds significant insight into the mechanism of the 3e− ORR and its antibacterial properties, laying a theoretical foundation for effective marine biofouling protection. |
1. Introduction
The attachment of marine biofouling on the surface of service equipment shortens the useful time and incurs severe economic losses.1 Compared with traditional physical and chemical antifouling technologies,2,3 electrocatalytic antifouling technology has been regarded as an alternative in marine biofouling prevention,4–6 depending on the reactive oxygen species (ROS) released during an electrochemical reaction.7,8 As the strongest oxidant (the oxidation potential is 2.80 V), hydroxyl radicals (˙OH) are usually generated by activation of H2O2 through an electro-Fenton (EF) reaction,9,10 but the secondary pollution of iron mud and the harsh pH conditions (pH 2–3) severely limit its broader applications.11,12 In contrast to the EF reaction, the three-electron oxygen reduction reaction (3e− ORR) would in situ generate ˙OH by regulating the number of electron transfer without the assistance of Fe2+.13–15 During the 3e− ORR process, H2O2 was first generated through the 2e− pathway (eqn (1)), and subsequently activate H2O2 to ˙OH via additional one-electron reduction (eqn (2)).16–18 Thus, it is important to construct 3e− ORR catalysts with multiple active sites to facilitate the selectivity and activation of H2O2 and ensure the efficient generation of ˙OH.19–21| O2 + 2H+ + 2e− → H2O2 | (1) |
| H2O2 + e− → ˙OH + OH− | (2) |
In addition, it is worth noting that the activation of H2O2 is critical for the 3e− ORR pathway.18,31 In general, transition metal-based materials (TMs) (such as Fe, Cu, and Mn based materials) are affiliative to O, and the metal centers have been regarded as active sites to activate H2O2 for the efficient production of OH radicals.32–35 Among them, MnO2 could accelerate the decomposition of H2O2 to release O2 and accompany the formation and the quenching of ˙OH simultaneously (eqn (3) and (4)),36 and the reaction between ˙OH and organics in wastewater is expected to compete with the formation of O2.37,38 Previous studies have shown that the weaker electronegativity of Mn could lead to charge redistribution in dual-site MnCu/C catalysts, and the electron bridge Mn–N/O–Cu boosts the efficient activation of H2O2 and the formation of ˙OH.39 However, the H2O2 activation mechanism of MnO2-based materials is still unclear.
 | (3) |
| 2˙OH → 1/2O2 + H2O | (4) |
2. Experimental
Materials
Manganese(II) chloride tetrahydrate (MnCl2·4H2O, 99.99%), LC/MS-grade formic acid and Nafion solution (5 wt%) were purchased from Shanghai Macklin Biochemical Co., Ltd. Potassium hexacyanocobaltate(III) (K3[Co(CN)6], 98%) and ceric sulfate (CeSO4, 97%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Ammonium hydroxide (NH3·H2O, AR), sulfuric acid (H2SO4, AR), sodium chloride (NaCl, AR), agar powder (BR), tryptone (BR), sodium phosphate dibasic dodecahydrate (NaH2PO4·12H2O, AR), sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O, AR), 30% hydrogen peroxide (H2O2, GR), and ethanol (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Carbon fiber paper (CFP) was purchased from Suzhou Shengernuo Technology Co., Ltd. Yeast extract powder (BR) was purchased from Beijing Aoboxing Biotechnology Co., Ltd. Salicylic acid (>99.0%) and HPLC-grade methanol were purchased from Merck (Darmstadt. Germany). All chemicals were used as received without further purification. Water (18 MΩ cm) used in all experiments was prepared by passing through an ultra-pure purification system.Experimental section
Synthesis of MnO2-xA. The as-prepared Mn3O4-x were soaked in 0.5 M H2SO4 at 60 °C for 2 h, and the precipitates were collected by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min. The final products were donated as MnO2-700A, MnO2-800A, and MnO2-900A.
000 rpm for 5 min. The final products were donated as MnO2-700A, MnO2-800A, and MnO2-900A.
Characteristics
The morphologies of as-prepared catalysts were obtained by scanning electron microscopy (SEM, NOVA NANOSEM450, FEl, USA) and transmission electron microscopy (TEM, FEI Ta-los F200C, Thermo Fisher Scientific, Czechia), and energy dispersive X-ray spectrometry (EDS, OXFORD X-MaxN50, Oxford Instruments Technology Ltd, China) was used to demonstrate the contribution of each element. Powder X-ray diffraction (PXRD, Bruker AXS D8 DISCOVER diffractometer) was used to detect the crystal structure of as-prepared catalysts. The chemical state and composition were analyzed by X-ray photoelectron spectroscopy (SSI SProbe XPS spectrometer). Raman spectra were measured by Renishaw using a Raman microscope with an argon ion laser beam. The absorbance data of the spectrophotometer were collected on an ultraviolet-visible (UV-vis) spectrophotometer (Hitachi U-3900H, Hitachi High-Technologies Co., Japan).Trace the generative process of ˙OH
i–t tests were conducted in 40 mL of 3.5% NaCl solution containing 10 μM salicylic acid (SA), after certain reaction times (0, 30, 60, 90, and 120 min). 1 mL of the electrolyte was taken out to work as the sample of LC–MS/MS. SA in electrolyte samples was analyzed using an optimized on-line solid phase extraction and liquid chromatography-tandem mass spectrometry (on-line SPE-LC–MS/MS) system to trace the generation of ˙OH within different reaction times. A schematic of the online SPE-LC–MS/MS is presented in Fig. S1 (ESI†). Each 100 μL of electrolyte wad loaded into an Agilent Zorbax Eclipse XDB-C8 (2.1 mm × 12.5 mm; 5 μm) online SPE column and eluted with 1 mL of 0.5% (v/v) formic acid solution with methanol at a flow rate of 0.5 mL min−1. SA from the SPE column was loaded onto a Zorbax Extend C18 (3.0 mm × 150 mm, 3.5 μm) reversed-phase HPLC column using 0.5% (v/v) formic acid solution and methanol as the mobile phase. At a flow rate of 0.3 mL min−1, the elution gradient was linearly increased from 20% to 50% methanol for 5 min, and then linearly increased to 100% methanol for 2 min. Details of the online SPE-LC–MS/MS conditions are listed in Tables S1 and S2 (ESI†), and the quantitative procedures are explained in the section Electrochemical properties.3. Results and discussion
Structure and morphology characterization
The as-prepared MnO2-xA (x refers to calcination temperature, x = 700, 800, 900) were synthesized by the self-template process. The precursor, Co, Mn-PBA, was first assembled via coprecipitation, which was a uniform polyhedron with an average size of 1 μm, confirmed using a scanning electron microscope (SEM) as shown in Fig. S2 (ESI†). After retreatment in ammonia, the uniform polyhedron broke into octahedral particles (Fig. 1a, Fig. S3a and b, ESI†). With further calcination, the octahedral particles aggregated into an irregular polyhedron with a larger size (Fig. S4, ESI†). Acid treatment could tear these polyhedrons into much smaller particles (Fig. 1b and c and Fig. S5–S7, ESI†), and with the increase in heat treatment temperature, a needle-like structure gradually emerged in these particles. The lattice spacings of as-prepared catalysts were recorded using a high-resolution transmission electron microscope (HRTEM). Lattice spacings of 0.308 and 0.502 nm were assigned to the (112) and (101) facets of Mn3O4-800 (Fig. 1d), and lattice spacings of 0.240 and 0.264 nm were assigned to the (131) and (031) facets of γ-MnO2 (Fig. 1e). The element distributions of MnO2-800A (Fig. 1f) were intuitively displayed by energy dispersion spectra (EDS), and Mn, O, and C atoms were equally distributed over the catalyst. | ||
| Fig. 1 (a) The SEM image of Mn3O4-800. (b) SEM and (c) TEM images of MnO2-800A. (d) and (e) HRTEM images of Mn3O4-800 and MnO2-800A. (f) SEM-EDS images of MnO2-800A. | ||
To investigate the crystal structure of as-prepared catalysts, the powder X-ray diffraction (XRD) pattern showed that Mn3O4-800 was transferred into MnO2 after pyrolysis and acid treatment (Fig. 2a). The characteristic peaks at 32.3° and 36.1° represented the (103) and (211) lattice planes of Mn3O4-800, and the peaks located at 22.1°, 37.1°, 42.3° and 55.9° corresponded to the (120), (131), (300), and (160) lattice planes of γ-MnO2.38,41 XRD and HRTEM together demonstrated the formation of γ-MnO2. It is worth noting that, with the increase of calcination temperature, the phase of as-prepared catalysts gradually transferred from Mn3O4-800 into MnO2 (Fig. S8, ESI†). The chemical compositions and bonding states of as-prepared catalysts were identified by X-ray photoelectron spectroscopy (XPS). As shown in survey-scan spectra (Fig. S9a, ESI†), Mn, O, and C coexisted in as-prepared catalysts. The oxidation state of Mn can be identified by the magnitude of the peak-to-peak splitting of the Mn 3s peak. In Fig. 2b and Table S3 (ESI†), the ΔEMn3s value of Mn3O4-800 is 5.51 eV, the average state of Mn is 2.67 which was calculated using a linear equation 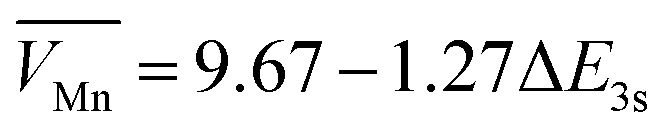 , and close to the theoretical valence, indicating the formation of Mn3O4-800.42 The ΔEMn3s values of as-prepared MnO2-xA are around 4.48 eV, illustrating the Mn valence of +4, further proving the generation of MnO2.41,43 In O 1s spectra (Fig. 2c and Fig. S9b, ESI†), the position at 529.7, 530.7, 531.7, and 533.4 eV is assigned to the characterization peak of Mn–O, Ov, C–O, and C
, and close to the theoretical valence, indicating the formation of Mn3O4-800.42 The ΔEMn3s values of as-prepared MnO2-xA are around 4.48 eV, illustrating the Mn valence of +4, further proving the generation of MnO2.41,43 In O 1s spectra (Fig. 2c and Fig. S9b, ESI†), the position at 529.7, 530.7, 531.7, and 533.4 eV is assigned to the characterization peak of Mn–O, Ov, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. The content of Ov in each catalyst was calculated by integrating the peak area, as shown in Table S4 (ESI†); calcination would reduce the Ov content, and the percentage of Ov decreased from 26.5% to 19.4%, 17.6%, and 15.5% with calcination temperatures of 700, 800, and 900 °C. More importantly, the positive shift of Mn 2p3/2 in Fig. 2d and Fig. S9c (ESI†) shows tighter bonding between Mn and O, which is consistent with the reduction of Ov in the O 1s spectrum.
O, respectively. The content of Ov in each catalyst was calculated by integrating the peak area, as shown in Table S4 (ESI†); calcination would reduce the Ov content, and the percentage of Ov decreased from 26.5% to 19.4%, 17.6%, and 15.5% with calcination temperatures of 700, 800, and 900 °C. More importantly, the positive shift of Mn 2p3/2 in Fig. 2d and Fig. S9c (ESI†) shows tighter bonding between Mn and O, which is consistent with the reduction of Ov in the O 1s spectrum.
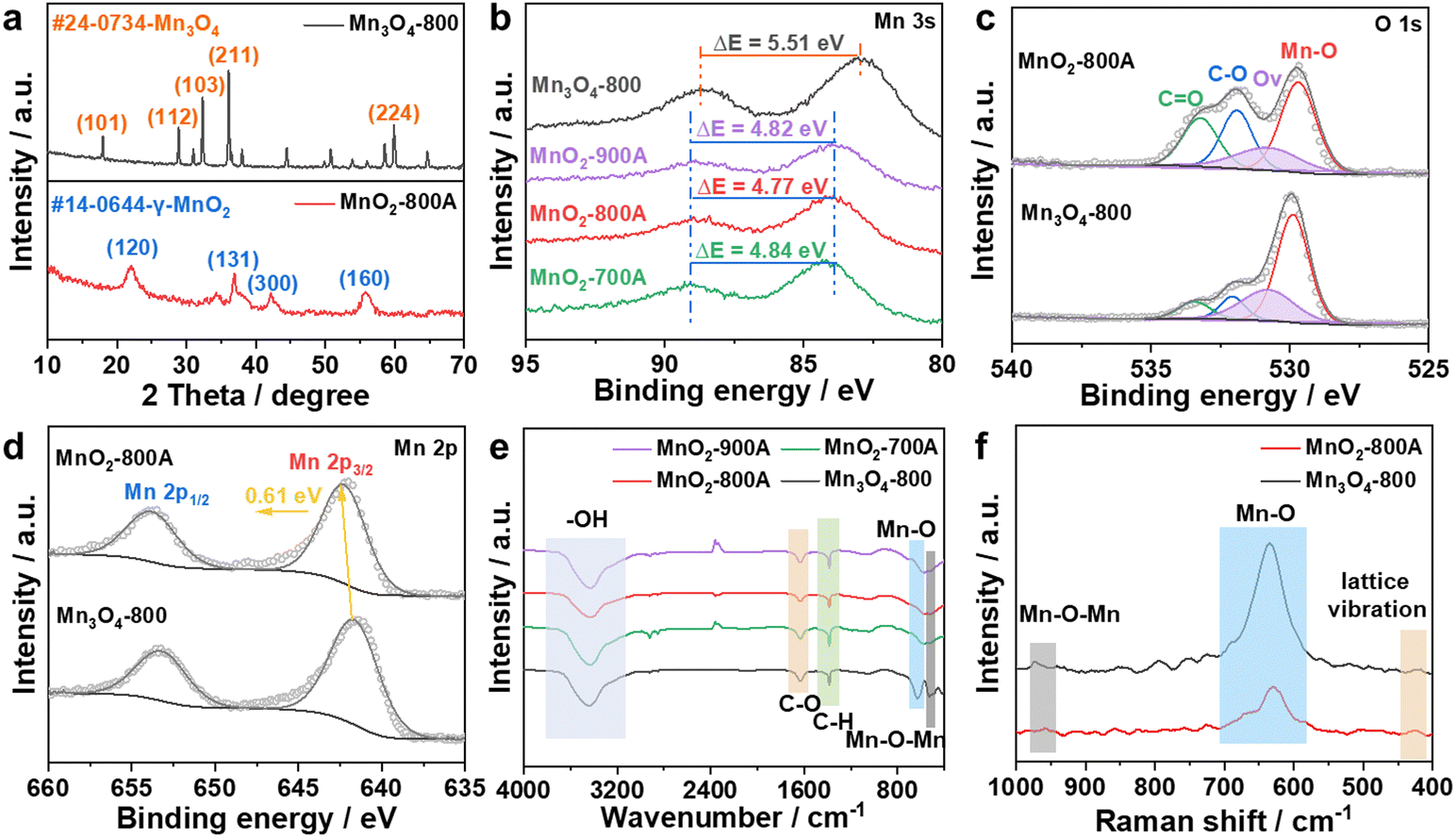 | ||
| Fig. 2 (a) XRD patterns of as-prepared catalysts. XPS spectra of as-prepared catalysts. (b) Mn 3s, (c) O 1s, and (d) Mn 2p. (e) FT-IR and (f) Raman spectra of as-prepared catalysts. | ||
Fourier transform infrared spectroscopy (FT-IR) (Fig. 2e) shows typical characteristic peaks of MnO2: the peak at 3425.01 cm−1 is assigned to the stretching vibration of hydroxyl or bound water, obvious stretching vibration of C–O, and bending vibration of C–H signals appeared at 1631.99 and 1384.25 cm−1, and in the fingerprint region, the specific peaks at around 600 and 400 cm−1 are related to vibration of Mn–O and Mn–O–Mn.44 Compared to Mn3O4-800, the peak intensity of MnO2 catalysts is significantly reduced, showing a change in the lattice structure and the formation of Ov. In the meantime, the shrinkage of the peak intensity in Raman spectroscopy (Fig. 2f) also proved the deficiency of O.45
Electrochemical properties
The intrinsic ORR performance of as-prepared catalysts was first evaluated using a traditional three-electrode system, with a catalyst ink uniformly coated onto the glassy carbon electrode of a rotating ring disk electrode (RRDE) as the working electrode, a saturated calomel electrode (SCE) as the reference electrode, and a carbon rod as the counter electrode, in O2-saturated 3.5% NaCl as the electrolyte. The double electrode layer capacitance of linear scanning curves (LSV) has been revised by N2-saturated tests; the applied potential of the Pt ring was set to 1.2 V (vs. RHE). As shown in Fig. 3a and Fig. S10 and S11a (ESI†), MnO2-800A has a more positive onset potential and a larger current than the other as-prepared catalysts, indicating its superior electrocatalytic properties. The corresponding H2O2 selectivity and the number of electron transfer (n) were calculated based on LSV curves. MnO2-800A exhibited the highest H2O2 selectivity of 42% and a stable n of 3.16, indicating a three-electron transfer ORR pathway (Fig. 3b and Fig. S11b and c, ESI†). The Tafel slope is a critical indicator to evaluate the kinetics of oxygen molecular activation (Fig. 3c and Fig. S11d, ESI†). Compared with Mn3O4-800 (105.23 mV dec−1), MnO2-700A (196.03 mV dec−1) and MnO2-900A (245.41 mV dec−1), the Tafel slope of MnO2-800A was the smallest (71.16 mV dec−1), which indicated the fastest oxygen molecular activation on the surface of MnO2-800A and ensured the 3e− ORR pathway going smoothly. The stability of as-prepared catalysts was measured in O2-saturated 3.5% solution at 0.1 V (vs. RHE) (Fig. S12, ESI†). For MnO2-800A, the current density decreased dramatically in the initial stage and tends to be stable after 5 h, and finally held at around 5 mA cm−2, indicating the excellent 3e− ORR stability of MnO2-800A. In contrast, the current density of Mn3O4-800 drops dramatically after 10 h test. Although MnO2-700A and MnO2-900A show more stable current densities, they are comparatively lower. Consequently, MnO2-800A emerges as the catalyst with the most impressive 3e− ORR stability among all the samples.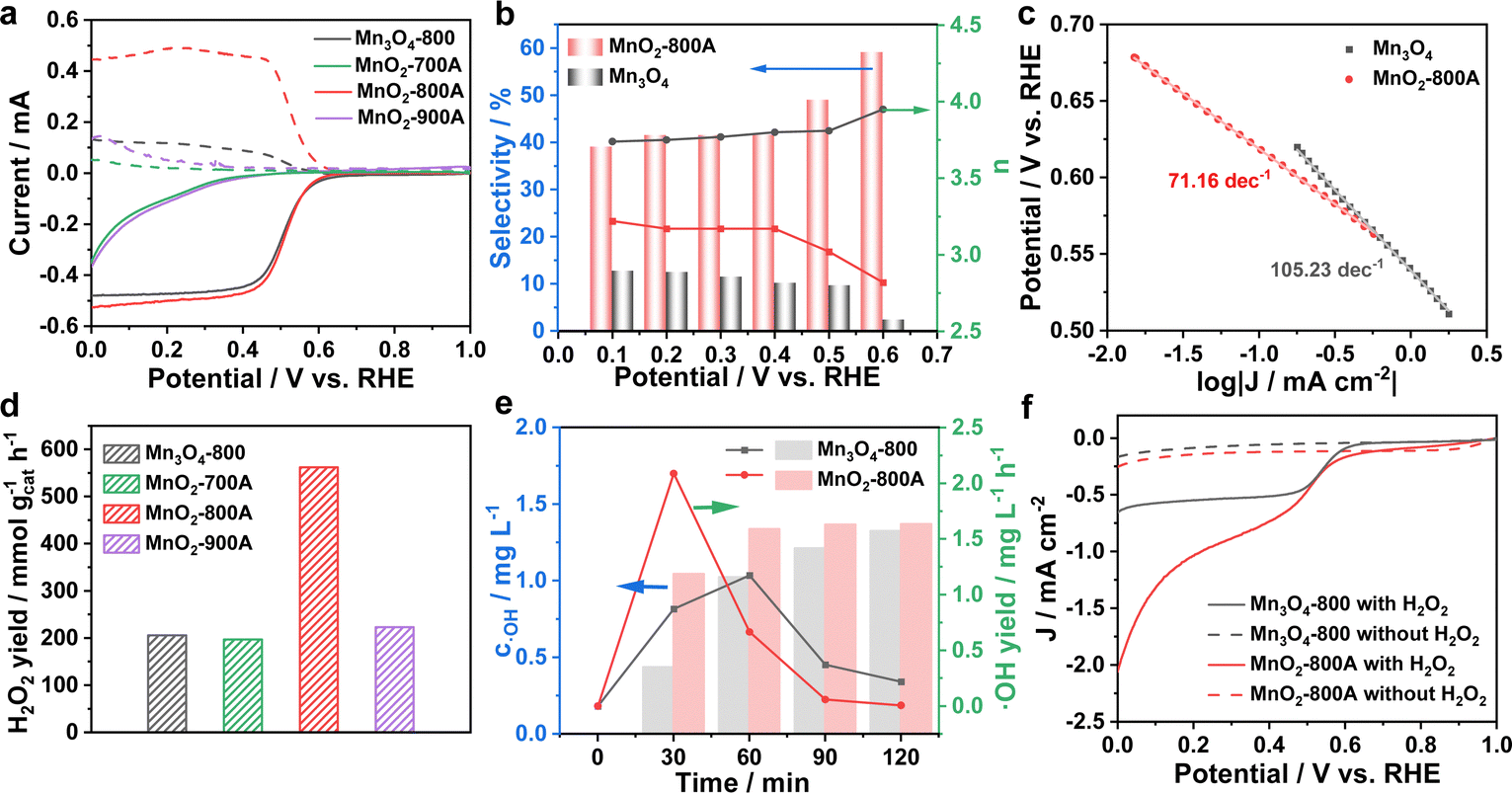 | ||
| Fig. 3 Electrochemical performance of Mn3O4-800 and MnO2-800A. (a) RRDE polarization curves. (b) Selectivity and the number of electron transfer. (c) Tafel curves. (d) H2O2 yield rates. (e) The concentration and yield rate of ˙OH within different reaction times. (f) LSV curves before and after the addition of H2O2. | ||
The formation of H2O2 is a crucial step in the 3e− ORR process, and the H2O2 yields were detected by UV-visible spectrometry with Ce(SO4)2 as the color indicator, based on the reaction between H2O2 and Ce4+. The gathering of H2O2 was conducted by the i–t test in O2-saturated 3.5% NaCl solution, with a fixed applied potential of 0.1 V (vs. RHE) for 2 h. The electrolyte was mixed after the reaction with Ce(SO4)2 standard solution (0.5 mM) and allowed to react for another 2 h; the H2O2 yield would be calculated using the standard curve (Fig. S13, ESI†) and eqn (S3) and (S4) (ESI†). As shown in Fig. 3d and Table S5 (ESI†), the H2O2 yields of Mn3O4-800, MnO2-700A, MnO2-800A, and MnO2-900A were assigned to 205.6, 196.9, 562.2 and 233.1 mmol gcat−1 h−1, respectively, indicating the best 2e− ORR performance of MnO2-800A among the as-prepared catalysts. Besides the excellent 2e− ORR performance, the smooth process of the 3e− ORR relays on the efficient activation of H2O2. An optimized on-line solid phase extraction and liquid chromatography–tandem mass spectrometry (on-line SPE-LC–MS/MS) system was used to trace ˙OH within different reaction times, based on the reaction between ˙OH and salicylic acid (SA) (Fig. S14, ESI†). As shown in Fig. S15a (ESI†), the peak at 9.854 min was identified as SA, and a good linear relationship with the peak area and the content was found in the mass range of 0.025–25 ng, which was used to calculate a calibration curve with y = 7682 + 18071x (R2 = 0.99655) (Fig. S15b, ESI†). It is worth noting that there were a few points outside the calibration curve (Table S6, ESI†); thus, points A and B located at (25, 449895) and (125, 1332858) were used as the standard point to calculate the concentration of SA by single-point quantification. The concentration of SA and the related concentration of ˙OH produced within certain time are shown in Fig. S15c (ESI†) and Fig. 3e. MnO2-800A exhibited a faster yield rate (2.09 mg L−1 h−1) and a higher ˙OH concentration (1.04 mg L−1) than Mn3O4-800 (yield rate: 0.87 mg L−1 h−1; ˙OH concentration: 0.44 mg L−1) in the initial 30 min, showing the efficient ˙OH production of MnO2 in a short time period by the 3e− pathway ORR. To further investigate the H2O2 activation ability of MnO2-800A, LSV curves were measured in N2-saturated 3.5% NaCl solution with or without 2.4 mM H2O2. As shown in Fig. 3f, the curves can reach a much higher current density when the electrolyte contains H2O2, indicating the activation process of H2O2. More importantly, MnO2-800A has a more positive onset potential and higher current density than Mn3O4-800, implying that MnO2-800A is more efficient at activating H2O2.
Reaction mechanism
To gain better insight into the reaction mechanism of the 3e− ORR on the as-prepared catalysts, density functional theory (DFT) calculations46,47 were carried out on Ov-Mn3O4 (101) and Ov-γ-MnO2 (300) models (Fig. S16, ESI†). The adsorption and activation of O2, a prerequisite of the ORR, thus, the adsorption energies of O2 (ΔE*O2) and the length of O–O bond (LO–O) at the active site were first calculated to expose the influence of Ov in adsorbing O2. In Fig. 4a and b, the ΔE*O2 significantly decreased (−9.98 to −2.03 eV for Mn3O4 and −12.24 to −1.43 eV for MnO2) and the LO–O increased (1.25 to 1.30 Å for Mn3O4 and 1.27 to 1.41 Å for MnO2), and the reason for this phenomenon could be explained as the Ov favored the “end-on” adsorption of O2 (Fig. 4c). In the “end-on” configuration, only one O atom was combined with the active site, which was responsible for the decrease of ΔE*O2. Besides, the elongation of the O–O bond indicated that O2 was preferably activated over Ov-γ-MnO2 (300).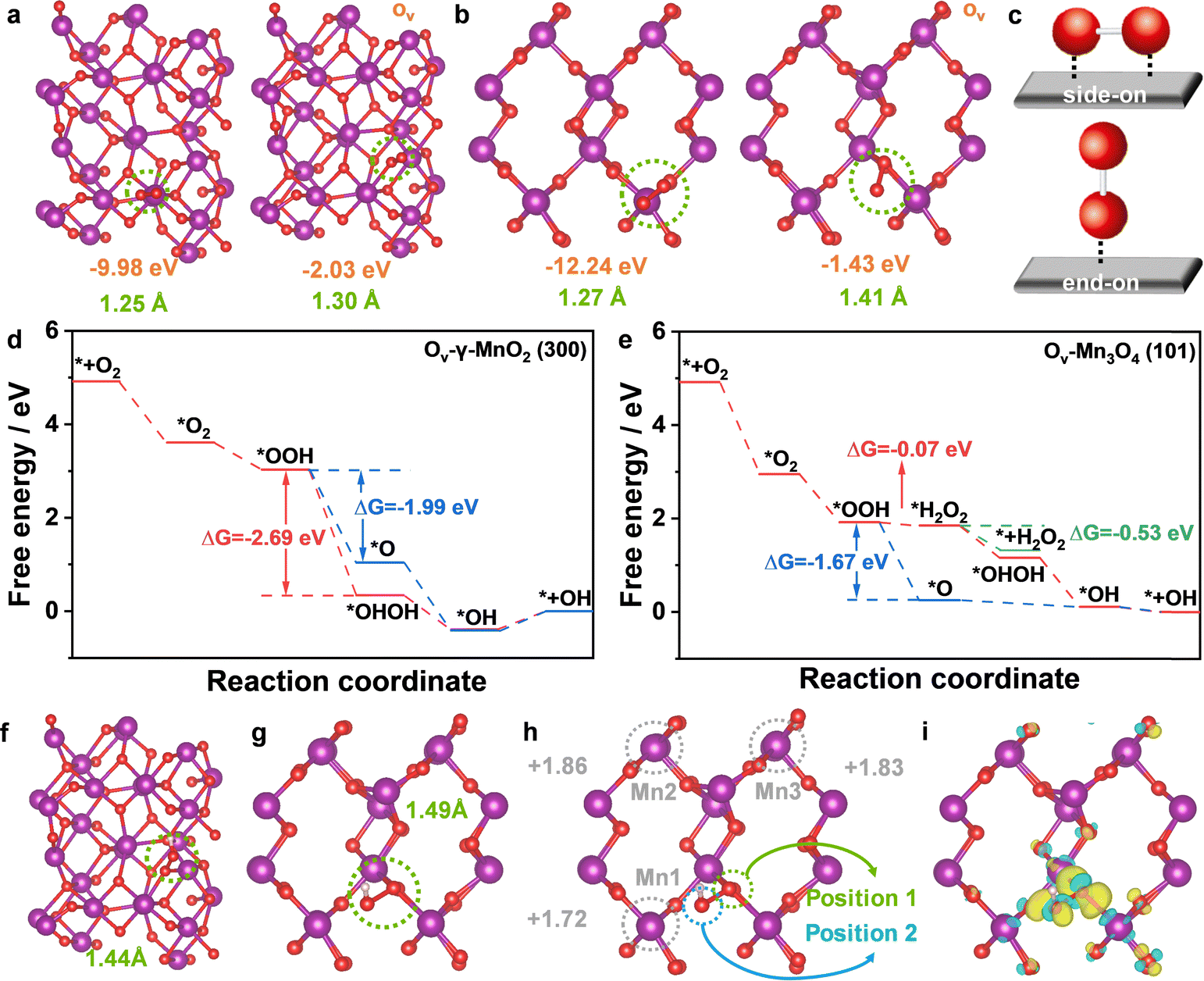 | ||
| Fig. 4 (a) and (b) The O2 adsorption energy (orange) and corresponding length of the O–O bond (green) in Mn3O4, Ov-Mn3O4, γ-MnO2, and Ov-γ-MnO2 models. (c) Illustration of “side-on” and “end-on” adsorption of O2 on catalysts. The Gibbs free energy diagram for the 3e− ORR on Ov-γ-MnO2 (300) (d) and Ov-Mn3O4 (101) models (e). Atomic configurations of *OOH and the corresponding O–O bond lengths on Ov-Mn3O4 (f) and Ov-γ-MnO2 (g). Bader charge analysis (h) and differential charge analysis (i) of *OOH adsorbed Ov-γ-MnO2 (the yellow region: the electron-generating region; the light-blue region: the electro-loss region). | ||
And then, the adsorbed O2 (*O2) protonated to *OOH, as the most important intermediate in the ORR process, the dissociation and adsorption of *OOH would determine the reaction pathway of the ORR (Fig. S17, ESI†). When the H+ dissociated from water attacks the O at position 1, it is more likely to generate *OHOH (two *OH adsorbed on Ov and Mn sites respectively) and benefit to the 3e− ORR pathway; in contrast, *OOH will break into *O and H2O, and it is more inclined to the 4e− ORR pathway. The free energy diagrams were further conducted to intuitively reveal the selection process of the ORR pathway. For Ov-γ-MnO2 (Fig. 4d and Fig. S18, ESI†), the change in Gibbs free energy (ΔG) values to generate *O and *OHOH were −1.99 and −2.69 eV, respectively. Simultaneously, the formation of *OH was an exothermic progress, but there was an extra energy barrier to overcome for desorption, indicating that it was difficult for *OH to reduce to H2O, and it was more favourable for Ov-γ-MnO2 to produce ˙OH via the 3e− ORR pathway. However, the ΔG*O and ΔG*H2O2 values for Ov-Mn3O4 were −1.67 and −0.07 eV, respectively, which indicate that the formation of H2O2 on Ov-Mn3O4 was more difficult than the formation of H2O, showing the preferable 4e− ORR pathway for Ov-Mn3O4 (Fig. 4e and Fig. S19, ESI†).
The origin of the excellent 3e− ORR performance of the Ov-γ-MnO2 (300) site was ulteriorly investigated by the projected density of state (PDOS) analysis (Fig. S20, ESI†). The d-band center of Ov-γ-MnO2 was at 2.59 eV, which is more negative than that of Ov-Mn3O4 (2.97 eV), indicating the weaker adsorption of intermediate oxygen-containing species on Ov-γ-MnO2, which is consistent with the results shown in the reaction step diagram. In Fig. 4f and g, the O–O bond of adsorbed *OOH on the Ov-γ-MnO2 (300) site was extended to 1.49 Å, much longer than that on the Ov-Mn3O4 (101) site (1.44 Å), which would accelerate the breakage of the O–O bond. Bader charge analysis was used to analyse the charge distribution and transfer between the catalysts and the adsorbed intermediates.30 As shown in Fig. 4h, the valent state for the Mn1 site (the Mn site close to *O on the position of *OOH) was +1.72, far below the similar Mn sites (+1.86 for the Mn2 site and +1.83 for the Mn3 site) in Ov-γ-MnO2. Combined with the differential charge analysis (Fig. 4i), the yellow region near the O atom on position 2 of *OOH was shifted toward the Mn1 site, showing the corresponding transformation of electrons. Thus, the interaction between Mn1 and the O atom on position 2 promoted the *OH adsorption on Mn1, rather than releasing H2O2 directly, improving the 3e− ORR selectivity for Ov-γ-MnO2.
Antibacterial performances and mechanisms
Pseudomonas aeruginosa (P. aeruginosa) and Staphylococcus aureus (S. aureus) served as the typical strains to evaluate the antibacterial efficacy of as-prepared catalysts. After incubating at 37 °C in the LB medium for 24 h, the bacteria solution was diluted with 0.1 M phosphate buffer solution (PBS) to 107 cfu mL−1. The electrochemical test was conducted in 35 mL of O2-saturated bacteria solution, employing MnO2-800A coated carbon paper with a loading mass of 0.2 mg cm−2 as the working electrode. The applied potential was fixed at 0.1 V (vs. RHE), and aliquots of 100 μL were sampled at intervals of 10, 30, 60, 120, and 180 minutes during the i–t test. These samples were then diluted progressively and spread onto nutrient agar plates, which were incubated at 37 °C for an additional 24 hours. In Fig. S21a and S22 (ESI†), the colony number of P. aeruginosa varied from 92 to 52, 18, 2, 0, and 0 with the passage of time, achieving a disinfection rate of 97.8% after 60 minutes. And for S. aureus, the colony number decreased from 56 to 17, 2, 0, 0, and 0 at the subsequent time points, and exhibited 96.4% of the disinfection rate in 30 min (Fig. S21b and S22, ESI†). To assess the significance of ˙OH in the disinfection process, isopropanol (IPA, 1% in 0.1 M PBS) was used as the quencher to eliminate ˙OH, and the colony numbers remained largely unaffected (Fig. S23, ESI†). As shown in Fig. S22 (ESI†), the sterilization efficiencies significantly compromised upon the addition of IPA, with less than 20% bacteria being inactivated after the 180 min i–t test.The generation of ROS has been identified as a pivotal factor in the efficient sterilization of H2O2. To delve deeper into the 3e− ORR sterilization mechanism of MnO2-800A, the generation of ˙OH in this system was investigated using a combination of the DFT calculation and LC–MS/MS. The DFT calculation illuminated that, in contrast to the formation of *O leading to H2O, the interaction between the Mn site and *OOH facilitates the cleavage of the O–O bond, thereby promoting the formation of ˙OH. The subsequent quantitative analysis of ˙OH via LC–MS/MS confirmed a substantial production of ˙OH during the initial reaction stage, ensuring the superior antimicrobial capabilities of MnO2-800A.
Based on the aforementioned findings, the proposed 3e− ORR sterilization mechanism of MnO2-800A was inferred as fellows: Initially, O2 was adsorbed on the Ov site of γ-MnO2, undergoing reduction to form the *HOOH intermediate. Subsequently, *HOOH decomposed into *OH and produced plenty of ˙OH through further reduction. Ultimately, ˙OH attacked and inactivated the bacterial, thereby achieving sterilization.
4. Conclusions
In summary, Ov-rich γ-MnO2 catalysts synthesized by calcination and acid oxidation of Co, Mn-PBA exhibit excellent 3e− ORR performance in neutral media. Among them, MnO2-800A revealed the highest 2e− ORR selectivity of 42%, an electron transfer number of 3.16, a H2O2 yield rate of 562.2 mmol gcat−1 h−1, and a higher ˙OH yield rate in the initial reaction stage. Meanwhile, MnO2-800A had high disinfection rates of 97.8% and 96.4% for typical marine bacteria (P. aeruginosa and S. aureus) in 60 and 30 min, respectively. DFT calculations proved that the activation of O2 was enhanced by Ov, and the electron transfer from *OOH to Mn could break the O–O bond and accelerate the formation of ˙OH. Inferred by the consequences of the DFT calculation and LC–MS/MS, the plentiful ˙OH formed in the initial 3e− ORR stage is the key to sterilization performance. This work dives into how Ov-rich γ-MnO2 worked in the 3e− ORR in neutral media and provides a theoretical foundation for marine biofouling prevention.Author contributions
Yingnan Qin: conceptualization/validation, methodology, writing – original draft preparation, and funding acquisition; Tongzhu Han: methodology and writing – review and editing; Ligang Chen: theoretical calculations; Kexin Yan: methodology; Jing Wang: funding acquisition; Ning Wang: funding acquisition; Baorong Hou: conceptualization.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 42006046, 42106051, and U2106206), the Hebei Province Key Research and Development Plan (No. 22373501D), and the China Postdoctoral Science Foundation (No. 2023M733526).Notes and references
- P.-Y. Qian, A. Cheng, R. Wang and R. Zhang, Nat. Rev. Microbiol., 2022, 20, 671–684 CrossRef PubMed.
- A. Kumar, A. AL-Jumaili, O. Bazaka, E. P. Ivanova, I. Levchenko, K. Bazakaaef and M. V. Jacob, Mater. Horiz., 2021, 8, 3201–3238 RSC.
- H. Jin, L. Tian, W. Bing, J. Zhao and L. Ren, Prog. Mater. Sci., 2022, 124, 100889 CrossRef.
- K. Yan, Y. Qin, J. Xue, J. Wang, G. Wang, N. Wang and Y. Chu, Chem. Eng. J., 2024, 484, 149363 CrossRef.
- N. Wang, S. Ma, R. Zhang, L. Wang, Y. Wang, L. Yang, J. Li, F. Guan, J. Duan and B. Hou, Adv. Sci., 2023, 10, e2302446 CrossRef.
- Y. Zeng and G. Wu, Chin. J. Catal., 2021, 42, 2149–2163 CrossRef.
- Y. Tu, W. Tang, L. Yu, Z. Liu, Y. Liu, H. Xia, H. Zhang, S. Chen, J. Wu, X. Cui, J. Zhang, F. Wang, Y. Hub and D. Deng, Sci. Bull., 2021, 66, 720–726 CrossRef PubMed.
- T. Richards, J. H. Harrhy, R. J. Lewis, A. G. R. Howe, G. M. Suldecki, A. Folli, D. J. Morgan, T. E. Davies, E. J. Loveridge, D. A. Crole, J. K. Edwards, P. Gaskin, C. J. Kiely, Q. He, D. M. Murphy, J.-Y. Maillard, S. J. Freakley and G. J. Hutchings, Nat. Catal., 2021, 4, 575–585 CrossRef.
- X. Wen, X. Zhang, M. Wang, C. Yuan, J. Lang, X. Li, H. Wei, D. Mandler and M. Long, Appl. Catal., B, 2024, 342, 123437 CrossRef.
- C. Liu, D. He, H. Yang, K. Zhang, X. Zhou, T. Zhang and J. Qu, Chem. Eng. J., 2023, 477, 146835 CrossRef.
- X. Shen, F. Xiao, H. Zhao, Y. Chen, C. Fang, R. Xiao, W. Chu and G. Zhao, Environ. Sci. Technol., 2020, 54, 4564–4572 CrossRef PubMed.
- T. Liu, S. Xiao, N. Li, J. Chen, X. Zhou, Y. Qian, C. H. Huang and Y. Zhang, Nat. Commun., 2023, 14, 2881 CrossRef PubMed.
- H. Kim, J. Lim, S. Lee, H. H. Kim, C. Lee, J. Lee and W. Choi, Environ. Sci. Technol., 2019, 53, 2918–2925 CrossRef PubMed.
- J. Zhang, S. Qiu and F. Deng, J. Hazard. Mater., 2024, 465, 133261 CrossRef.
- F. Miao, M. Gao, X. Yu, P. Xiao, M. Wang, Y. Wang, S. Wang and X. Wang, Electrochem. Commun., 2020, 113, 106687 CrossRef CAS.
- M. Li, L. Bai, S. Jiang, M. Sillanpaa, Y. Huang and Y. Liu, J. Hazard. Mater., 2023, 452, 131352 CrossRef PubMed.
- R. Ren, X. Shang, Z. Song, C. Li, Z. Wang, F. Qi, A. Ikhlaq, J. Kumirska, E. Maria Siedlecka and O. Ismailova, Chem. Eng. J., 2023, 474, 145545 CrossRef.
- Y. Sun, P. Cai, D. Yang and X. Yao, Chem Catal., 2022, 2, 679–692 CrossRef.
- L. Xie, P. Wang, W. Zheng, S. Zhan, Y. Xia, Y. Liu, W. Yang and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2307989120 CrossRef.
- F. Xiao, Z. Wang, J. Fan, T. Majima, H. Zhao and G. Zhao, Angew. Chem., Int. Ed., 2021, 60, 10375–10383 CrossRef CAS PubMed.
- G. Zhao, T. Chen, A. Tang and H. Yang, J. Mater. Chem. A, 2024, 12, 7227–7236 RSC.
- G. Huo, X.-W. Wang, Z.-B. Zhang, Z. Song, X.-M. Kang, M.-X. Chen, Q. Wang, X.-Z. Fu and J.-L. Luo, J. Energy Chem., 2020, 51, 81–89 CrossRef.
- H. Huang, A. Huang, D. Liu, W. Han, C. H. Kuo, H. Y. Chen, L. Li, H. Pan and S. Peng, Adv. Mater., 2023, 35, e2303109 CrossRef PubMed.
- Y. Aoki, K. Takase, H. Kiuchi, D. Kowalski, Y. Sato, H. Toriumi, S. Kitano and H. Habazaki, J. Am. Chem. Soc., 2021, 143, 6505–6515 CrossRef PubMed.
- C. Bao, H. Wang, C. Wang, X. Zhang, X. Zhao, C.-L. Dong, Y.-C. Huang, S. Chen, P. Guo, X. She, Y. Sun and D. Yang, J. Hazard. Mater., 2023, 441, 129922 CrossRef.
- M. Xu, J. Wei, X. Cui, J. Li, G. Pan, Y. Li, Z. Jiang, X. Niu, N. Cui and J. Li, Chem. Eng. J., 2023, 455, 140922 CrossRef.
- R. Gao, L. Pan, Z. Li, C. Shi, Y. Yao, X. Zhang and J. J. Zou, Adv. Funct. Mater., 2020, 30, 1910539 CrossRef.
- S. Ding, Y. Zhang, F. Lou, M. Li, Q. Huang, K. Yang, B. Xia, C. Tang, J. Duan, M. Antonietti and S. Chen, Mater. Today Energy, 2023, 38, 101430 CrossRef CAS.
- Z. Deng, L. Li, Y. Ren, C. Ma, J. Liang, K. Dong, Q. Liu, Y. Luo, T. Li, B. Tang, Y. Liu, S. Gao, A. M. Asiri, S. Yan and X. Sun, Nano Res., 2022, 15, 3880–3885 CrossRef CAS.
- C. Wang, Y. Lei, Q. Lv, P. Wang, W. Kong, F. Wan and W. Chen, Appl. Catal., B, 2022, 315, 121547 CrossRef CAS.
- C. Ling, X. Liu, H. Li, X. Wang, H. Gu, K. Wei, M. Li, Y. Shi, H. Ben, G. Zhan, C. Liang, W. Shen, Y. Li, J. Zhao and L. Zhang, Angew. Chem. Int. Ed., 2022, 61, e202200670 CrossRef CAS PubMed.
- Z. Chen, G. Liu, S. Yu, L. Yang, L. Zheng, Z. Wei and S. Luo, Chem. Eng. J., 2023, 474, 145581 CrossRef CAS.
- C. Wang, W. Zhang, J. Wang, P. Xia, X. Duan, Q. He, I. Sirés and Z. Ye, Appl. Catal., B, 2024, 342, 123457 CrossRef CAS.
- C. Sun, J. Wang, C. Gu, C. Wang, S. Sun and P. Song, Chem. Eng. J., 2023, 452, 139592 CrossRef.
- P. Huang, Q. Chang, G. Jiang, K. Xiao and X. Wang, Sep. Purif. Technol., 2023, 306, 122582 CrossRef.
- D. Li, T. Sun, L. Wang and N. Wang, Electrochim. Acta, 2018, 282, 416–426 CrossRef.
- C. Qu, Y. G. Li, S. J. Meng, X. H. Li, S. J. Zhang and D. W. Liang, J. Hazard. Mater., 2022, 434, 128923 CrossRef PubMed.
- S. Zhang, J. Lv, R. Han and S. Zhang, Colloids Surf., A, 2022, 651, 129807 CrossRef CAS.
- D. Shang, S. Wang, J. Li, S. Zhan, W. Hu and Y. Li, Small, 2024, e2311984, DOI:10.1002/smll.202311984,.
- C. Wang, Y. Zeng, X. Xiao, S. Wu, G. Zhong, K. Xu, Z. Wei, W. Su and X. Lu, J. Energy Chem., 2020, 43, 182–187 CrossRef.
- Y. Dai, Y. Men, J. Wang, S. Liu, S. Li, Y. Li, K. Wang and Z. Li, Colloids Surf., A, 2021, 627, 127216 CrossRef CAS.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- N. A. Fathy, S. E. El-Shafey, O. I. El-Shafey and W. S. Mohamed, J. Environ. Chem. Eng., 2013, 1, 858–864 CrossRef CAS.
- Z. Deng, C. Ma, S. Yan, K. Dong, Q. Liu, Y. Luo, Y. Liu, J. Du, X. Sun and B. Zheng, J. Mater. Chem. A, 2021, 9, 20345–20349 RSC.
- Z. Deng, C. Ma, S. Yan, K. Dong, Q. Liu, Y. Luo, Y. Liu, J. Du, X. Sun and B. Zheng, J. Mater. Chem. A, 2021, 9, 20345–20349 RSC.
- W. Xu, N. Apodaca, H. Wang, L. Yan, G. Chen, M. Zhou, D. Ding, P. Choudhury and H. Luo, ACS Catal., 2019, 9, 5074–5083 CrossRef.
- X. Qi, T. Yang, P. Li and Z. Wei, Nano Mater. Sci., 2023, 5, 287–292 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. Fig. S1–S23. See DOI: https://doi.org/10.1039/d4nh00289j |
| This journal is © The Royal Society of Chemistry 2024 |