
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopically labelled hydroxylamino-triazine (BHT) siderophores toward the quantification of iron(III), vanadium(V) and uranium(VI) hard metal ions†
Angelos
Amoiridis
 a,
Michael
Papanikolaou
a,
Chryssoula
Drouza
b,
Themistoklis A.
Kabanos
*c and
Anastasios D.
Keramidas
*a
a,
Michael
Papanikolaou
a,
Chryssoula
Drouza
b,
Themistoklis A.
Kabanos
*c and
Anastasios D.
Keramidas
*a
aUniversity of Cyprus, Department of Chemistry, 2109, Nicosia, Cyprus. E-mail: akeramid@ucy.ac.cy
bDepartment of Agricultural Sciences, Biotechnology and Food Science, Cyprus University of Technology, Limassol 3036, Cyprus. E-mail: chryssoula.drouza@cut.ac.cy
cDepartment of Chemistry, Section of Inorganic and Analytical Chemistry, University of Ioannina, 45110 Ioannina, Greece. E-mail: tkampano@uoi.gr
First published on 31st July 2024
Abstract
Based on the strong binding properties of hydroxylamino-1,3,5-triazine (BHT) for hard metal ions, a novel spectroscopically labelled triazine-hydroxylaminato ligand, N,N′-(6-(9H-carbazol-9-yl)-1,3,5-triazine-2,4-diyl)bis(N-methylhydroxylamine) (H2cbht), was synthesized. Reaction of H2cbht with FeIII, VV and UVI resulted in the synthesis of [UVIO2(cbht)(DMF)(MeOH)], 1, (PPh4)2([UVIO2(Hcbht)2]·2[UVIO2(Hcbht)(cbht)]), 2, (PPh4)2[(UVIO2)3(μ-cbht)2(cbht)2], 3, Na[VVO2(cbht)], 4, and (PPh4)[FeIII(cbht)2], 5. The complexes were characterized by X-ray crystallography showing strong chelation of the metal ions with the BHT chelating moiety H2cbht. The DMSO/DMF solutions of the complexes were characterized by NMR, UV-vis and electrochemistry, confirming the high affinity of H2cbht for FeIII, VV or UVI. The luminescence properties of the carbazole group were retained in the BHT adduct of the H2cbht ligand. Interaction of the metal ions with H2cbht resulted in quenching of the luminescence of H2cbht. Stern–Volmer plots of luminescence vs. concentration of the metal ions are linear and suitable for the determination of the concentration of metal ions in nM range concentrations. Benesi–Hildebrand plots show that the metal-to-ligand ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 while the formation of the metal complexes exhibits high association constants.
:1 while the formation of the metal complexes exhibits high association constants.
Introduction
The removal of hard metal ions (referring to the hard–soft acid base theory, HSAB) from the environment, human body, and radioactive wastes produced by nuclear industries and metal mining from seawater has motivated scientists toward the synthesis of new, strong and selective chelators, including siderophores.1–27 A group of non-toxic siderophores, based on an N,N′-disubstituted bis(hydroxyamino)-1,3,5-triazine (BHT) moiety (Scheme 1), shows explicit preference to binding hard metal ions, such as FeIII, TiIV, VV, UVI and MoVI.3,10–14,28–31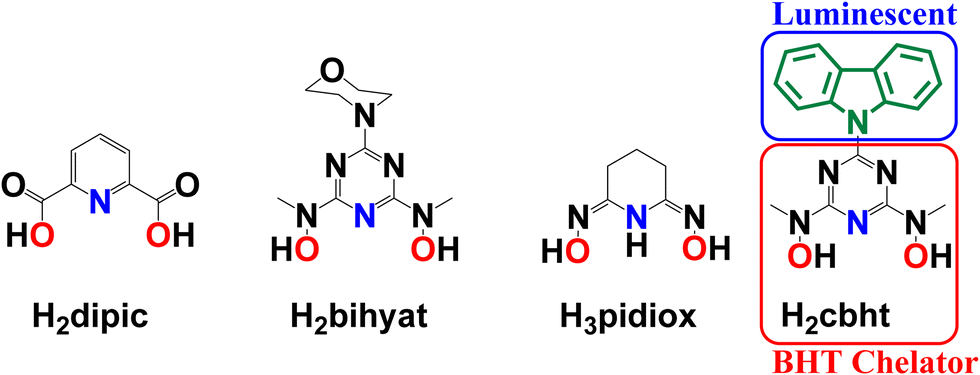 | ||
| Scheme 1 Molecular drawings of H2dipic, H2bihyat, H3pidiox and H2cbht. | ||
Highly sensitive and specific probes for various metal ions have been synthesised and intensively investigated by scientists due to their potential applications in environmental sciences and biological systems for understanding their roles in different chemical and biological processes.32–53 In particular, the concentration of metal ions can be increased in humans through industrial pollution and war pollution; for example, using depleted uranium in ammunition,18,54–57 release of unregulated metal ions in organisms connected to certain diseases58,59 and the use of biologically active metal complexes as drugs. For example, vanadium complexes have shown such activity, mainly as antidiabetic and anticancer drugs.60–78
An advantage of BHT-type ligands is their easy modification by replacing the R group (Scheme 1) while keeping the same chelating moiety. The type of R group can directly influence the electron-donating ability of the chelator, thus modifying the electronic properties31 of the coordinated metal ions. Subsequently, R groups with optical and/or electrochemical properties will provide BHT-type ligands with sensing capabilities.
The chelating group controls selectivity and affinity towards the metal ions. The selectivity and thermodynamic stability of H2bihyat (Scheme 1) for [VVO2]+ and [UVIO2]2+ were found to be superior to those of other hard donor ligands, such as pyridine-2,6-dicarboxylic acid (H2dipic; Scheme 1) and amidoxime (H3pidiox, Scheme 1), indicating that BHT-type ligands are the best candidates for the development of sensors for these metal ions.
Herein, we describe the synthesis of N,N′-(6-(9H-carbazol-9-yl)-1,3,5-triazine-2,4-diyl)bis(N-methylhydroxylamine) (H2cbht, Scheme 1), a carbazole covalent adduct with a BHT chelating group. The organic molecule H2cbht retains the luminescence and redox properties of carbazole and is the first luminescent BHT ligand to be reported. Reactions of H2cbht with UVIO22+, VVO43− and FeIII in organic solvents result in the synthesis of [UVIO2(cbht)(DMF)(MeOH)], 1, (PPh4)2([UVIO2(Hcbht)2]·2[UVIO2(Hcbht)(cbht)]), 2, (PPh4)2[(UVIO2)3(μ-cbht)2(cbht)2], 3, Na[VVO2(cbht)], 4, and (PPh4)[FeIII(cbht)2], 5. The complexes were characterized in the solid state by X-ray crystallography. The speciation in solution and thermodynamic stability of the metal complexes were determined by 1D and 2D 1H NMR spectroscopy and electrochemistry. The coordination of the metal ions to H2cbht results in the quenching of luminescence. The Stern–Volmer graphs of the quenching of the luminescence are linear, indicating that the plots can be used for UVIO22+, VVO43− and FeIII quantification in nM level concentration. The large association constants calculated from the luminescence spectra confirm the high thermodynamic stability of the complexes.
Experimental section
All the chemicals were of reagent grade purity and were provided by Sigma-Aldrich unless otherwise stated. THF was dried over a sodium wire and distilled just prior to use.n-BuLi in hexane is flammable and ignited in the presence of air.
Synthesis of 9-(4,6-dichloro-1,3,5-triazin-2-yl)-9H-carbazole (dctc)
To a solution of carbazole (2.84, 17 mmol) in dry THF (40 mL), a solution (2.5 M) of n-BuLi in hexane (6.8 mL, 17 mmol) was added over 10 minutes at −40 °C, under an argon atmosphere. The resulting mixture was stirred at room temperature for 30 minutes under argon flow and added to a stirred solution of cyanuric chloride (3.12 g, 17 mmol) in THF (40 mL) using a dropping funnel over 30 minutes at 0 °C. The red mixture was allowed to reach room temperature and then refluxed overnight. After that, the solvent was removed under reduced pressure and acetone (20 mL) was added to the red-white residue. The mixture was stirred for 30 minutes in an ice bath, and a white solid (4.82 g) was collected by filtration and used without further purification. Yield: 90% (based on carbazole). 1H NMR (CDCl3, 500 MHz, 25 °C) δ (ppm): 8.56 (2H, d, J = 8.43 Hz, carbazole), 7.99 (2H, d, J = 7.60 Hz, carbazole), 7.52 (2H, td, J = 7.22, 1.33 Hz, carbazole), 7.45 (2H, td, J = 7.40, 0.87 Hz, carbazole). 13C NMR (CDCl3, 125.7 MHz, 25 °C) δ (ppm): 171.42, 163.93, 138.63, 128.19, 127.93, 125.56, 120.25, and 119.36. IR(ATR): v(BHT ring) 723, 758 1475, 1500, 1535 cm−1. Elemental analysis of C15H8Cl2N4 (Mr = 315.2): found C, 56.99; H, 2.52; N, 17.83; calculated: C, 57.17; H, 2.56; N, 17.78.Synthesis of N,N′-(6-(9H-carbazol-9-yl)-1,3,5-triazine-2,4-diyl)bis(N-methylhydroxylamine) (H2cbht)
To a stirred aqueous solution (2 mL) of N-methylhydroxylamine hydrochloride (1.33 g, 15.9 mmol), an aqueous solution (2 mL) of NaOH (0.630 g, 15.9 mmol) was added dropwise at 0 °C, and the resulting solution was slowly added to a solution of dctc (1.25 g, 3.97 mmol) in THF (20 mL) at 0 °C. Then, the solution was allowed to reach room temperature (25 °C) and refluxed overnight, after which a white solid was formed, which was filtered, washed with THF (5 mL) and distilled water (2 × 5 mL), and dried under vacuum to give 1.08 g of the ligand H2cbht. Yield: 81% (based on dctc). 1H NMR (DMSO-d6, 500 MHz, 25 °C) δ (ppm): 9.90 (2H, s, N–OH), 9.12 (2H, d, J = 8.35 Hz, carbazole), 8.17 (2H, d, J = 7.69 Hz, carbazole), 7.49 (2H, td, J = 7.17, 1.31 Hz, carbazole), 7.37 (2H, td, J = 7.15, 0.87 Hz, carbazole), 3.45 (6H, s, N–CH3). 13C NMR (DMSO-d6, 125.7 MHz, 25 °C) δ (ppm): 166.94, 163.91, 138.69, 126.71, 125.22, 122.47, 119.80, 118.09, and 38.51. IR(ATR): v(BHT ring) 723, 758 1392, 1438, 1641 cm−1. Elemental analysis of C17H6N6O2 (Mr = 336.4): found: C, 59.89; H, 4.85; N, 24.24; calculated: C, 60.71; H, 4.79; N, 24.99.Synthesis of [UVIO2(cbht)(DMF)(MeOH)], 1
H2cbht (0.0760 g, 0.226 mmol) and triethylamine (63.0 μL, 0.452 mmol) were sequentially added to a stirred DMF (5 mL) solution of [UVIO2(NO3)2(H2O)2]·4H2O (0.113 g, 0.226 mmol), which yielded a dark brown solution. XRD quality single crystals of 1 (0.116 g) were obtained by layering methanol (6 mL) to an undisturbed brown solution. Yield: 74% (based on H2cbht). 1H NMR (DMSO-d6, 500 MHz, 25 °C) δ (ppm): 8.95 (2H, d, J = 8.40 Hz, carbazole), 8.21 (2H, d, J = 7.70 Hz, carbazole), 7.55 (2H, td, J = 7.30, 1.50 Hz, carbazole), 7.38 (2H, td, J = 7.80, 0.80 Hz, carbazole), 3.79 (s, 6H, N–CH3). 13C NMR (DMSO-d6, 125.7 MHz, 25 °C) δ (ppm): 162.34, 160.67, 159.46, 138.82, 126.71, 124.94, 122.12, 119.78, 117.34, and 38.20. IR(ATR): v(BHT ring) 719, 752, 771, 1442, 1535, 1639 cm−1, v(U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 905 cm−1. Elemental analysis of C20H23N7O6U (Mr = 695.2): found: C, 34.39; H, 3.48; N, 13.99; calculated: C, 34.54; H, 3.33; N, 14.10.
O), 905 cm−1. Elemental analysis of C20H23N7O6U (Mr = 695.2): found: C, 34.39; H, 3.48; N, 13.99; calculated: C, 34.54; H, 3.33; N, 14.10.
Synthesis of (PPh4)2([UVIO2(Hcbht)2]·2[UVIO2(Hcbht)(cbht)])·2MeOH·2Et2O·2H2O, 2,·2MeOH·2Et2O·2H2O and (PPh4)2[(UVIO2)3(μ-cbht)2(cbht)2]·DMF, 3·DMF
H2cbht (0.0760 g, 0.226 mmol), [UVIO2(NO3)2(H2O)2]·4H2O (0.0570 g, 0.113 mmol) and triethylamine (378 μL, 2.71 mmol) were dissolved in 3 mL of DMF. To the stirred brown solution solid, PPh4Cl (0.169 g, 0.452 mmol) was added in one portion. The vapour diffusion of diethylether into the undisturbed brown solution resulted in two types of single crystals suitable for X-ray structure analysis. The needle-type crystals correspond to (PPh4)2([UVIO2(Hcbht)2]·2[UVIO2(Hcbht)(cbht)])·2MeOH·2Et2O·2H2O (complex 2·MeOH·Et2O·2H2O) and the block-type crystals correspond to (PPh4)2[(UVIO2)3(μ-cbht)2(cbht)2]·DMF (complex 3·DMF). The two complexes crystallize together, as shown in Fig. S1 (ESI†). The crystals were manually separated under a microscope. Yield: 0.0252 g, 17% (2·MeOH·Et2O·2H2O) and 0.0065 g, 6% (3·DMF). IR(ATR) of 2: v(BHT ring) 727, 754, 777, 1390, 1516 cm−1, v(UO), 901 cm−1. Elemental analysis of C160H160N36O24P2U3 (2·2MeOH·2Et2O·2H2O, Mr = 3729.29): found: C, 51.68; H, 4.51; N, 13.27; calculated C, 51.28; H, 4.30; N, 13.46. Elemental analysis of C119H103N25O15P2U3 (3·DMF, Mr = 2897.91): found: C, 49.87; H, 3.71; N, 11.92; calculated: C, 49.30; H, 3.58; N, 12.08.
Synthesis of Na[VVO2(cbht)]·3H2O, 4·3H2O
To a stirred solution of H2cbht (0.0760 g, 0.226 mmol) and triethylamine (63.0 μL, 0.452 mmol) in DMF (5 mL), an aqueous solution (2 mL) of NaVVO3 (0.0300 g, 0.226 mmol) was added, and the colour of the solution changed to yellow. The yellow solution was allowed to stand undisturbed at room temperature, and after 4 hours, XRD-quality orange crystals of complex 4·3H2O (0.056 g) were obtained. Yield: 50% (based on H2cbht). 1H NMR (DMSO-d6, 500 MHz, 25 °C) δ (ppm): 8.88 (2H, d, J = 8.50 Hz, carbazole), 8.19 (2H, d, J = 7.39 Hz, carbazole), 7.53 (2H, td, J = 7.32, 1.22 Hz, carbazole), 7.38 (2H, td, J = 7.08, 0.92 Hz, carbazole), 3.46 (s, 6H, N–CH3). 13C NMR (DMSO-d6, 125.7 MHz, 25 °C) δ (ppm): 162.45, 157.34, 138.73, 126.85, 125.18, 122.48, 119.74, 117.69, and 35.82. IR(ATR): v(BHT ring) 721, 758, 721, 1442, 1525, 1590, 1650 cm−1, v(VO), 916 cm−1. Elemental analysis of C17H20N6NaO7V (Mr = 494.07): found: C, 41.38; H, 3.99; N; calculated: C, 16.87; 41.31; H, 4.08; N, 17.00.
Synthesis of (PPh4)[FeIII(cbht)2]·9H2O, 5·9H2O
H2cbht (0.076 g, 0.226 mmol) and Et3N (378 μL (2.71 mmol) were dissolved in 3 mL of DMF. [FeIII(NO3)3]·9H2O 0.0460 g (0.113 mmol) was added to the DMF solution, and the colour of the solution turned dark purple. The addition of 1 mL of water to the stirred solution resulted in the formation of a small amount of a purple solid, which was re-dissolved by warming the solution to 100 °C. Then, the warm solution was slowly cooled to room temperature (20 °C) and left undisturbed for 3 days, after which small dark purple crystals (0.244 g) of 5·9H2O suitable for X-ray structure analysis were precipitated. Yield: 75% (based on H2cbht). Elemental analysis of C58H66FeN12O13P (Mr = 1225.40): found: C, 57.42; H, 5.30; N, 13.88; calculated: C, 56.82; H, 5.43; N, 13.71.Results and discussion
Synthesis of the ligand H2cbht and complexes 1–5·9H2O
A slightly modified method reported in the literature79 was used for the synthesis of dctc. The synthesis of dctc, shown in Scheme 2, is based on the nucleophilic substitution of cyanuric chloride and takes place in two steps. The first step involves the deprotonation of the cyclic amine nitrogen by a very strong base (n-BuLi), and the second step involves the substitution of one chlorine atom of cyanuric chloride with a nitrogen atom of the deprotonated carbazole. All glassware used for the synthesis of dctc was flame-dried, and all processes were performed under an inert atmosphere using high-purity argon flow. The synthesis of the ligand H2cbht, depicted in Scheme 2, is based on the nucleophilic substitution of the two remaining chlorine atoms of dctc with N-methylhydroxylamine hydrochloride in a THF/H2O solution previously neutralized with NaOH. The ligand H2cbht is insoluble in water in the pH range 1–14.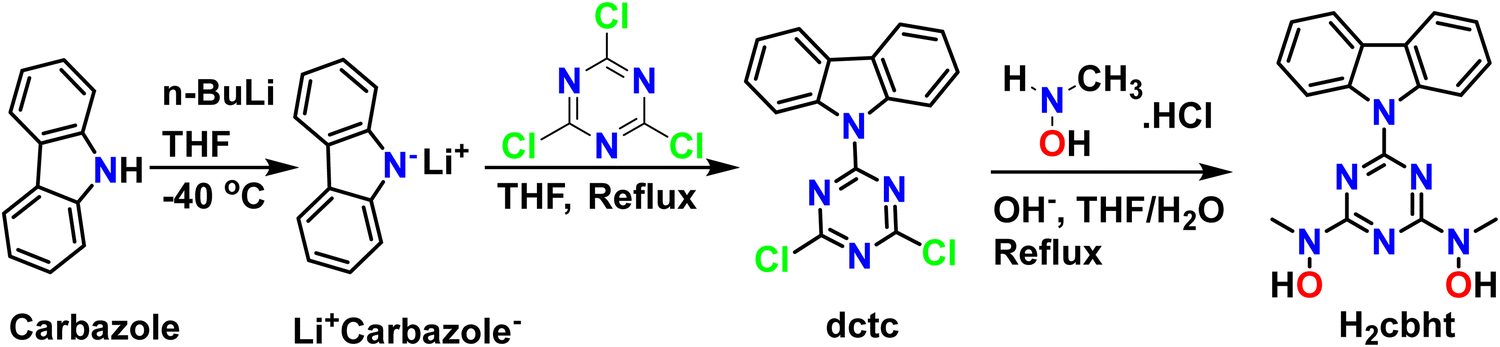 | ||
| Scheme 2 Synthesis of organic molecules dctc and H2cbht. | ||
The synthesis of uranium(VI) (1, 2, and 3) and vanadium(V) (4)/iron(III) (5) compounds is a one-pot synthesis, as shown in Schemes S1 and S2 (ESI†), respectively. Reaction of equivalent quantities of UVIO22+ and H2cbht with two equivalents of Et3N leads to the formation of complex 1, while the reaction of UVIO22+ and H2cbht in a molar ratio 1:2 in the presence of excess Et3N (12 equivalents) and Ph4PCl results in the formation of compounds 2 and 3. 1H NMR spectroscopy revealed that complexes 1, 2 and 3 are in equilibrium in solutions containing UVIO22+ and H2cbht. Addition of Et3N and/or H2cbht shifts the equilibrium towards 2 and 3. Attempts to optimize the synthetic procedure in order to obtain 2 and 3 separately were unsuccessful. Reactions of VVO43− or FeIII with H2cbht give only 4 (VVO2+–cbht2− 1:1) or 5 (FeIII–cbht2− 1:2), respectively. Complexes 1–5 are soluble in organic solvents, including DMF and DMSO.
Characterization of the complexes in the solid state
The synthesized compounds have been characterized in the solid state by Fourier transform infrared spectroscopy and X-ray crystallography. The infrared spectra and assignments of the characteristic peaks are shown in Fig. S2 (ESI†). A summary of the crystallographic data and the final refinement details for complexes 1–5·9H2O are provided in Tables S1 and S2 (ESI†). The interatomic distances and bond angles relevant to UVI, VV and FeIII coordination spheres are listed in Tables S3 and S4 (ESI†). Ortep plots of the crystal structures of the uranium(VI) complexes 1–3·DMF and 4·3H2O–5·9H2O are shown in Fig. 1 and 2, respectively.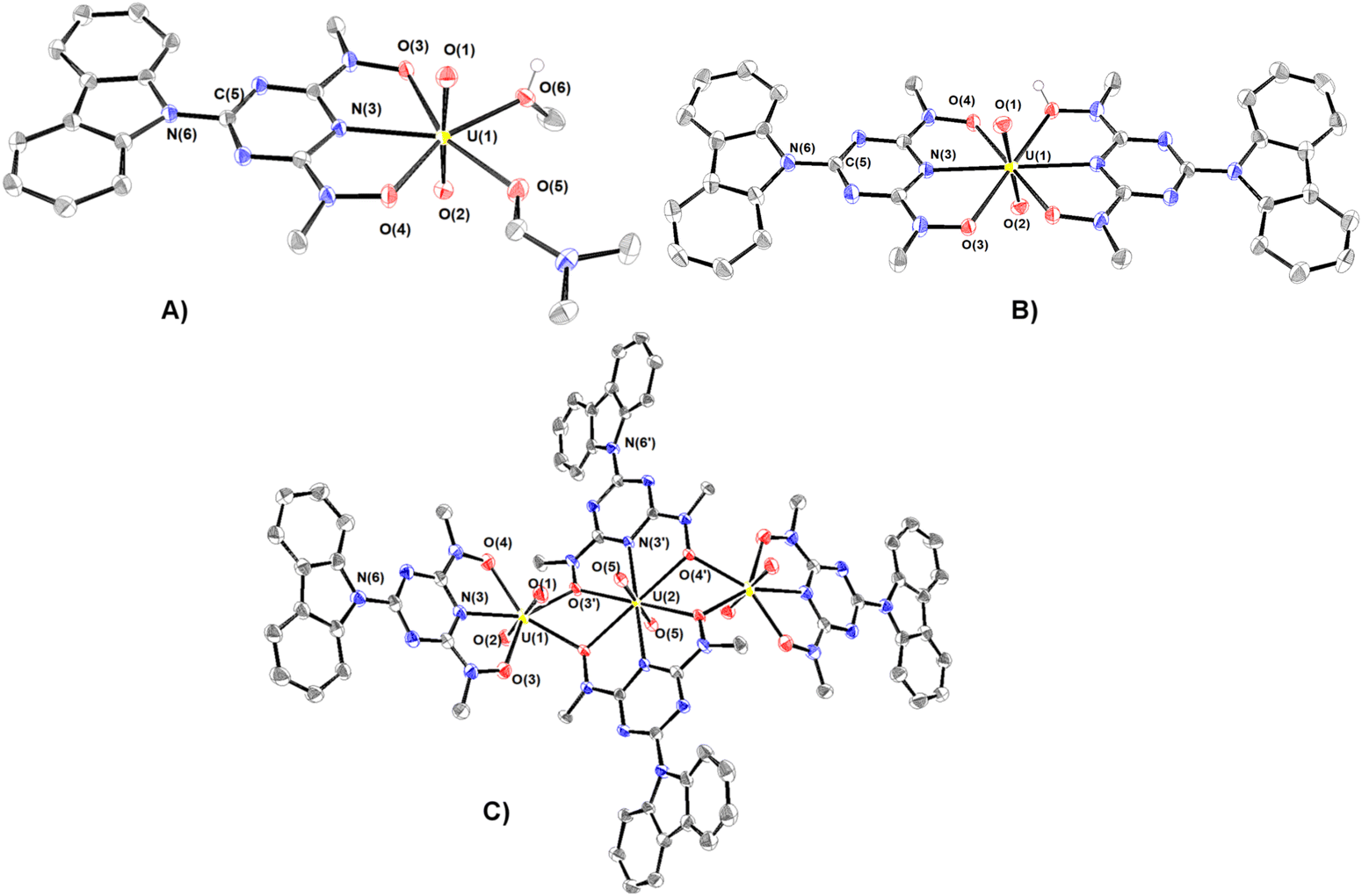 | ||
| Fig. 1 ORTEP plots of 1 (A) and anions of 2 (B) (only one of the three uranyl molecules of 2 is shown for clarity) and 3 (C) with 50% thermal ellipsoids. Hydrogen atoms were omitted for clarity. Bond lengths (Å): (A) U–O(1) = 1.780(2), U–O(2) = 1.776(2), U–O(3) = 2.415(2), U–O(4) = 2.299(2), U–N(3) = 2.433(2), U–O(5) = 2.400(2), and U–O(6) = 2.382(2). (B) U–O(1) = 1.781(3), U–O(2) = 1.781(3), U–O(3) = 2.478(3), U–O(4) = 2.479(3), U–N(3) = 2.530(4). (C) U(1)–O(1) = 1.777(3), U(1)–O(2) = 1.793(3), U(1)–O(3) = 2.309(3), U(1)–O(4) = 2.319(3), U(1)–N(3) = 2.430(4), U(1)–O(3′) = 2.438(3), U(1)–O(4′) = 2.454(3), U(2)–O(5) = 1.781(3), U(2)–O(3′) = 2.466(3), U(2)–O(4′) = 2.503(3), and U(2)–N(3′) = 2.530(3). | ||
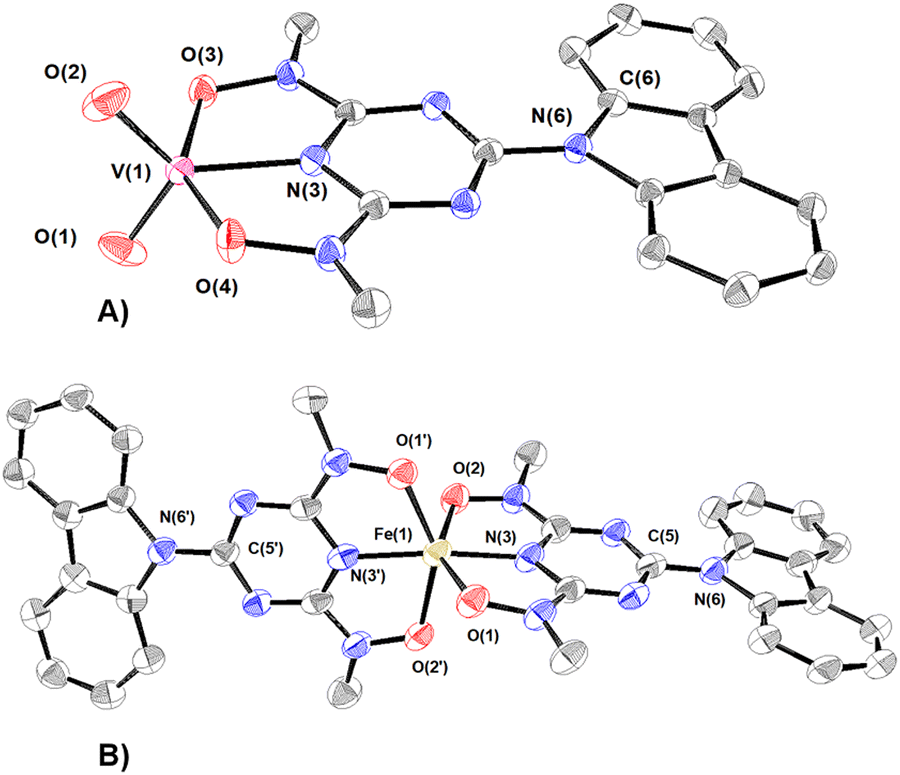 | ||
| Fig. 2 ORTEP plots of the anions of 4 (A) and 5 (B) with 50% thermal ellipsoids. Hydrogen atoms were omitted for clarity. Bond lengths (Å): (A) V(1)–O(1) = 1.632(1), V(1)–O(2) = 1.631(1), V(1)–O(3) = 2.014(1), V(1)–O(4) = 1.971(1), and V(1)–N(3) = 1.984(1). (B) Fe(1)–O(1) = 2.029(3), Fe(1)–O(1′) = 2.043(3), Fe(1)–O(2) = 2.119(3), Fe(1)–O(2′) = 2.114(3), Fe(1)–N(3) = 1.980(3), and Fe(1)–N(3′) = 1.997(4). | ||
The uranium(VI) atom in complex 1 adopts a pentagonal bipyramidal structure, with two terminal oxido groups, O(1) and O(2) [dmean(UO) ∼ 1.778 Å], occupying the two axial positions, whereas the triazine nitrogen atom N(3), [d(U–Ntr) = 2.433 Å], two deprotonated hydroxylamine hydroxyls O(3) and O(4), and two oxygen atoms O(6) and O(7) of methanol and DMF molecules, lie in the equatorial plane. dU–O(3) (2.415 Å) is significantly longer than dU–O(4) (2.299 Å), reflecting the stronger trans effect of the DMF carbonyl than the methanol oxygen donor atoms.
The coordination environment of the three UVI in the three hydrogen-bonded UVI–Hcbht− molecules of complex 2 are hexagonal bipyramids with two oxido groups, O(1) and O(2) [d(UO) = 1.781(3) Å] occupying the axial positions (Fig. 1(B) and Fig. S3, ESI†). The equatorial plane of the two uranium atoms is defined by the triazine nitrogen atom N(3) and the two hydroxylamine oxygen atoms O(3) and O(4) of cbht2− and Hcbht− ligands, whereas the third central UVI is defined by the donor atoms of two Hcbht− ligands. Although some of the hydroxylamine oxygen atoms are deprotonated and others are not, the four UVI–Oh bond lengths (∼2.479 Å) are indistinguishable. The protons of the hydroxylamine oxygen atom form a strong hydrogen bond [(H)O–O(4) = 2.549 (3) Å] with the deprotonated hydroxylamine oxygen atom [O(4)] of a neighbouring molecule, and vice versa, bringing the three uranyl molecules of 2 in close proximity, almost perpendicular to each other [O(1)U⋯UO(1) torsion angle ∼115°]. The two equatorial triazine-hydroxylaminate parts of the ligands exert strong trans effect31 to each other, resulting in significant lengthening of the four UVI–Oh bonds in comparison with the corresponding UVI–Oh bonds in 1.
The structure of the anion of 3 (Fig. 1(C)) consists of two distorted pentagonal and a hexagonal bipyramidal uranyl unit bridged by two cbht2− ligands in a centrosymmetric trimer [U(2) is the center of symmetry]. Each of the uranyl group, U(1)O22+ and its symmetry-related U(1A)O22+, is bonded to a tridentate cbht2− ligand through two deprotonated hydroxylamine hydroxyls and the central triazine nitrogen atom while both are bridged to each other through the U(2)O22+ group and with the hydroxylamine hydroxyls of the two cbht2− ligated to U(2). The three uranium(VI) atoms are arranged in a linear fashion [U(1A)–U(2)–U(1) = 180°]. The trinuclear complex (Et3NH)2[(UVIO2)3(μ-bihyat)2(bihyat)2] has a linear arrangement similar to that of the three UVI metal atoms.13
This mode of bridging action of cbht2− type ligands is the first example to be reported. The N atoms of the hydroxylamines in the cbht2− ligands of 3 have a flat trigonal geometry; therefore, they are sp2-hybridized. This is in agreement with the ligands of 3, which exhibits resonance structure B (Scheme 3). In marked contrast, in the complex (Et3NH)2[(UVIO2)3(μ-bihyat)2(bihyat)2], the N atoms of the bridged hydroxylamines have a trigonal pyramidal geometry; thus, they are sp3-hybridized.13
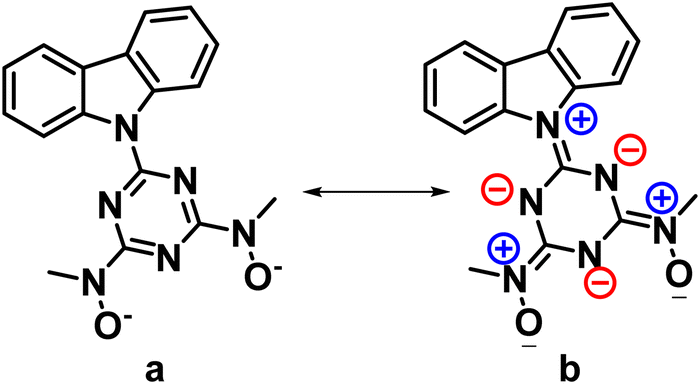 | ||
| Scheme 3 Resonance structures of cbht2−. | ||
The resonance structure B for complexes 1–3 is also supported by the short C(5)–N(6) bond distances [dmean C(5)–N(6) ∼ 1.39 Å], revealing a double bond character. In addition, the carbazole moiety is coplanar with the triazine ring. The only exception is the plane defined by the carbazole of the bridging cbht2− in 3, which forms an angle of 22.6° with the plane defined by triazine, accompanied by a 0.01 Å lengthening of the C(5)–N(6) bond.
The vanadium(V) centre in 4 adopts a distorted trigonal bipyramidal configuration (τ = 0.67; τ = {[O(3)–V(1)–O(4)] − [O(1)–V(1)–O(2)]}/60)80 and is bonded to the cbht2− ligand through the triazine nitrogen N(3) [d(V–Ntr) = 1.997(2) Å], the two deprotonated hydroxylamine hydroxyl groups O(3) and O(4) [dmean(V–Oh) = 1.993(2) Å] and two cis oxido groups O(1) and O(2) [dmean(VO) = 1.631(2) Å]. The vanadium(V) atom is displaced above the equatorial plane defined by two oxido groups and the triazine nitrogen atoms by 0.0216(3) Å. Similar to the crystal structure of 1, the bond length of V–Ntr is one of the shortest reported in the literature [N(3)–V = 1.984 Å]. Most of the five-coordinated vanadium(V) complexes have a square pyramidal geometry.81–83 Strong σ-donor atoms in an anti-position to VO cause distortion from square pyramidal to trigonal bipyramidal geometry by tilting the oxido groups; thus, the σ-donor and the oxido atoms avoid sharing the same orbital. The trigonal bipyramidal configuration of 4 confirms the strong-donating properties of the triazine heterocyclic N atom.
The coordination environment of FeIII in 5 is a distorted octahedron with two cbht2− ligands bonded to the metal ion. Two hydroxylamine hydroxyls, O(1) and O(2), occupy the axial positions. The equatorial plane is defined by Ntr, two Oh donor atoms of one cbht2− ligand and the Ntr donor atom of the other cbht2− ligand. The FeIII ion is 0.0781 Å above the equatorial plane. The mean Fe–Oh bond distance is 1.993 Å.
The M–N bond lengths of UVI, VV and FeIII, similar to those of H2cbht hard tridentate ligands, are shown in Table 1. It is apparent that the metal ion–N bond distances increase significantly, going from 1:1 to 1:2 (M:L) complexes.
| Ligand/bond length (Å) | UVI–N (1:1) |
UVI–N (1:2) |
VV–N (1:1) |
FeIII–N (1:1) |
FeIII–N (1:2) |
|---|---|---|---|---|---|
| cbht2− | 2.433(2) | 2.530 | 1.977(2) | — | 2.076(3) |
| bihyat2− | 2.436(4)13 | 2.518(5)13 | 1.993(3)14 | — | 1.976(2)29 |
| qtn4− | 2.435(4)31 | — | 1.997(2)31 | — | — |
| pdl4− | 2.441(6)31 | — | 2.005(2)31 | — | — |
| enl4− | — | — | 1.992(3)31 | — | — |
| dipic2− | 2.520(6)84 | 2.641(2)84 | 2.086(2)85 | 2.062(1)86 | |
| Hpidiox2− | — | 2.56387 | 1.988(5)88 | 2.005(8)89 | 2.030(1)89 |
In all crystal structures, the flat carbazole moiety forms strong π–π bonds with the triazine moiety of a neighboring molecule. The distances between the planes defined by the interacting carbazole and triazine range from 3.250 to 3.360 Å. In the crystal structure of 4, π–π bonding arranges the molecules in a linear chain with π-interacting ligands inside and VVO2+ moieties on the outside of the chain. The VVO2+oxido moieties interact with the vanadium atoms of a neighboring chain, arranging the chains on infinite planes. In contrast to 4, the crystal structures of 1, 2, 3 and 5 show π-interactions to be only between the two organic ligands of two different molecules, avoiding polymeric structures.
NMR spectroscopy
The organic molecules and complexes 1–4·3H2O were characterized using 1H, 13C and 51V NMR (complex 4) spectroscopy in DMSO-d6 solutions. The 1H and 13C NMR spectra of the organic molecules are shown in Fig. S4–S7 (ESI†). The 1H and 13C chemical shifts of complexes 1–4·3H2O are listed in Table 2. For the DMSO-d6 solutions of complex 1, it was expected that the unidentate DMF and H2O ligands in the coordination sphere of 1 would be replaced by DMSO-d6. Thus, in the DMSO-d6 solution, complex 1 is converted to [UVIO2(cbht)(DMSO-d6)2]. The 1H NMR spectrum of the free ligand H2cbht in DMSO-d6 solution exhibited peaks at 3.45 and 7.40, 7.49, 8.17 and 9.14 ppm, which were assigned to the methyl hydroxylamino and aromatic carbazole protons, respectively (Fig. 3). Upon complexation with the metal ions, the peaks are shifted to a lower field, except for the peak at 9.14 ppm [H(8)], which is shielded. The uranium(VI) ion causes a significantly larger shifting than vanadium(V), which is in line with the NMR spectra of complexes of the two metals with BHT-type ligands (Table 2).31 The 51V NMR spectrum of 4·3H2O in DMSO-d6 exhibited broad peaks at −500 ppm, typical for this type of compound.14,31| Compound/1H ppm (13C ppm) | H(1) | H(5) | H(6) | H(7) | H(8) |
|---|---|---|---|---|---|
| H2cbht | 3.45(39.18) | 8.17(119.60) | 7.37(122.47) | 7.49(125.22) | 9.14(118.10) |
| 1 | 3.80(38.20) | 8.21(119.78) | 7.40(122.12) | 7.56(126.71) | 8.96(117.34) |
| 2 | 3.83(38.54) | 8.21(119.78) | 7.40(121.79) | 7.56(126.42) | 8.96(117.01) |
| 3A + 3B | 3.83(38.54), 3.89(38.51), 4.60(41.35) | 8.21(119.78), 8.20(119.68), 8.28(119.68) | 7.40(121.79), 7.55(121.79), 7.38(121.79) | 7.56(126.42), 7.66(126.36), 7.44(126.49) | 8.96(117.01), 8.96(117.01), 9.01(117.01) |
| 4 | 3.46(35.82) | 8.21(119.74) | 7.38(122.48) | 7.53(126.85) | 8.90(117.69) |
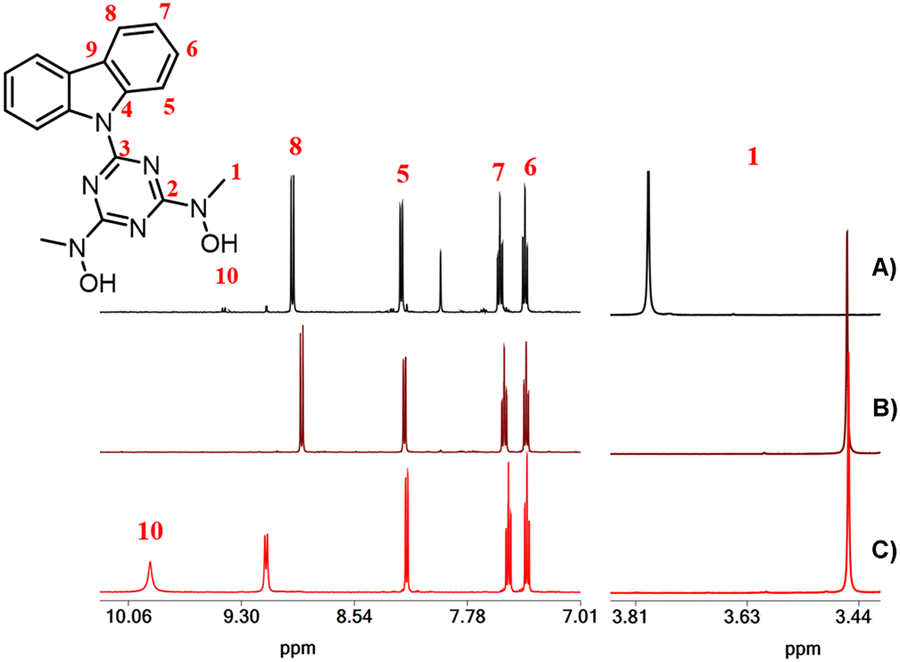 | ||
| Fig. 3 1H NMR spectra of DMSO-d6 solutions of (A) 1, (B) 4·3H2O and (C) H2cbht. | ||
The DMSO-d6 solutions of a mixture of crystals of 2 and 3 exhibited peaks from the free ligand, 1, 2 and 3. This suggests that all compounds are in equilibrium in the solution. To further examine the reaction and correctly assign the 1H NMR peaks, we reproduced the species in DMSO-d6 solutions by reacting 1 with H2cbht and Et3N. The 1H NMR spectrum of the solution confirmed the presence of the free ligand, 1, 2 and 3 in equilibrium (eqn (1) and (2)). The addition of either Et3N or H2cbht drives the reaction to the right, increasing the concentrations of 2 and 3, thus permitting the correct assignment of the peaks (Table 2).
| [UVIO2(cbht)(DMSO-d6)2] (1) + H2cbht + Et3N ⇌ [UVIO2(Hcbht)(cbht)]− (2) + Et3NH+ | (1) |
| 3[UVIO2(cbht)(DMSO-d6)2] (1) + H2cbht + 2Et3N ⇌ [UVI3O6(μ-cbht)2(cbht)2]2− (3) + 2Et3NH+ | (2) |
Compound 2 exhibited a peak at 3.83 ppm, which was assigned to the methyl protons of methylhydroxylamine. The chemical shifts of the carbazole protons of 2 were the same as those of 1. In the aromatic region, Complex 3 exhibited three peaks for the methylhydroxylamine protons at 3.83, 3.89 and 4.60 ppm. However, the highly symmetric crystal structure of 3 shows only two types of –CH3, Ha and Hb (Scheme 4(A), molecule 3A). The ratio of the integral of the peak at 3.89 ppm vs. the peak at 4.60 ppm is constant at 2.2, independent of the experimental conditions. Complex 3, which is similar to the trinuclear complex [UVI3O6(μ-bihyat)2(bihyat)2]2−, crystallizes with two N-hydroxylamine atoms in sp3 instead of sp2 hybridization. The –CH3 attached to the sp3 N is more deshielded than those on the sp2 N atom. The shift in the 1H NMR peaks of the –CH3 groups supports equilibrium between species 3A and 3B, as shown in Scheme 4. The aliphatic region of the EXSY spectrum exhibited off-diagonal peaks between (i) the –CH3 peak of 2 + H(b) of 3 and the –CH3 peak of the free ligand and (ii) the peaks at 3.89 and 4.60 ppm. The former exchange process occurred between the ligands of 3 and the free ligand. The exchange between the cbht2− of 2 and the free ligand is not supported based on the fact that the exchange between 2 and 1 was not observed. The off-diagonal cross-peaks between the peaks at 3.89 and 4.60 ppm are much more intense than the exchange peaks of 3 with the free ligand, indicating a much faster intramolecular exchange process. Intramolecular changes in the structure of the hydroxylamine nitrogen atom from flat trigonal sp2 to tetrahedral sp3 are the only processes that do not require intermolecular exchange with the free ligand. Furthermore, the carbazole protons of the bridging cbht2− in 3B should be asymmetric. The 2D {1H} COSY and EXSY (Fig. 4 and Fig. S8, ESI†) of the aromatic region show that the carbazole proton peaks split due to the asymmetry of the ligand and fast exchange. Apparently, the NMR results confirm that 3 in DMSO-d6 solutions exists in the form of two fast-exchanging tautomeric forms, 3A and 3B.
 | ||
| Scheme 4 (A) Equilibrium between tautomers 3A and 3B, and (B) sp2 and sp3 N structures. | ||
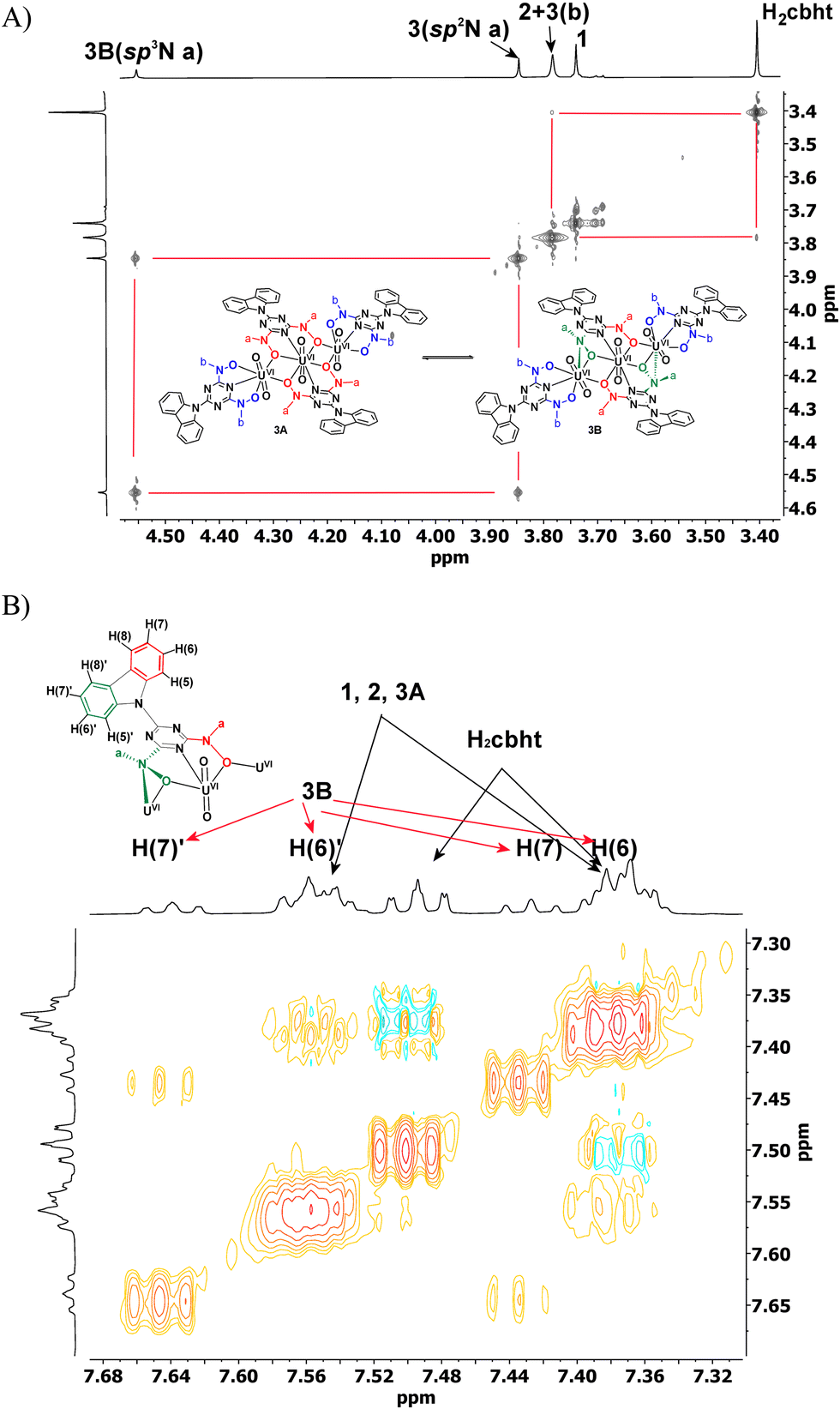 | ||
| Fig. 4 (A) 2D {1H} grEXSY spectrum of the aliphatic region and (B) 2D {1H} grEXSY spectrum of part of the aromatic region of a DMSO-d6 solution of 1 (6.00 μmol), H2cbht (6.0 μmol) and triethylamine (72 μmol). | ||
Luminescence and UV-vis
The luminescence and UV-vis data of the DMSO solution of the ligand and complexes 1, 4·3H2O and 5·9H2O are listed in Table 3. Solutions of the free ligand H2cbht in DMSO emit blue light after excitation at 338 nm (Fig. S9, ESI†). The shape of the emission spectrum (two peaks at 348 and 364 nm) is the same as that of carbazole (two peaks at 338 and 351 nm), slightly shifted at lower energies. The DMSO solutions of the complexes did not emit light.| Compound/peak | Intra-ligand transitions | CT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ε | nm | ε | nm | ε | nm | ε | nm | ε | nm | |
| H2cbht | 6.0 × 104 | 276 | 4.2 × 104 | 291 | 2.3 × 104 | 312 | 2.9 × 104 | 323 | — | — |
| 1 | 6.7 × 104 | 272 | 5.5 × 104 | 288 | 2.9 × 104 | 315 | 3.1 × 104 | 323 | 3.1 × 104 | 326 |
| 4·3H2O | 6.3 × 104 | 276 | 4.8 × 104 | 291 | 3.8 × 104 | 313 | 4.8 × 104 | 325 | — | — |
| 5·9H2O | 6.7 × 104 | 277 | 5.5 × 104 | 291 | 3.4 × 104 | 314 | 3.9 × 104 | 325 | 3.6 × 103 | 540 |
The UV-vis spectrum of the DMSO solution of H2cbht showed strong peaks at 276, 291 (sh), 312 and 323 nm. The shoulder at 290 nm originates from triazine intra-ligand electronic transitions, which are commonly observed in triazines without the chromophore.31 The peak at 323 nm is assigned to carbazole intra-ligand transitions. In addition to the peak at 323 nm, the UV-vis spectrum of the non-substituted carbazole shows a peak at 336 nm. In H2cbht, this peak collapsed into a broad shoulder (∼335 nm).
The solution of 1 exhibited a LMCT peak at 326 nm, in addition to the ligand peaks. The intensity of the UV-vis peaks of the bound ligand of 4·3H2O in the solution increased by 50% compared to the spectrum of the solution of the free ligand and shifted slightly to higher wavelengths. A broad signal at 320–380 nm was assigned to LMCT transitions. The CT transition of 5·9H2O appeared as a broad peak at 540 nm and is responsible for the deep purple colour of the complex.
Titration of 7.42 × 10−5 M H2cbht with the metal ions shows a linear increase of CT transitions with increasing concentration of the metal ion. The absorption of UVIO22+ and VVO43− with H2cbht is maximum when the ratio between the metal ions and ligand is 1:1 (Fig. S10 and S11, ESI†). This agrees with the 1H NMR experiments, showing the 1:1 species to be the most stable at these conditions. In contrast, regarding the FeIII complex, the 1:2 metal to ligand species is stable in solution (Fig. S12, ESI†), suggesting that the crystallographically characterized complex 5·9H2O is the main species.
Electrochemistry
The cyclic voltammographs (CVs) of 1, 4·3H2O and 5·9H2O are shown in Fig. 5. All complexes show ligand-centred oxidation waves at 886, 968 and 653 mV vs. NHE for 1, 4·3H2O and 5·9H2O, respectively. The oxidation of the ligand on the electrode surface results in the emergence of daughter peaks at −474 and 136 mV vs. NHE for 1 and 4·3H2O, respectively, assigned to the reduction of the oxidized ligand. Interestingly, the potential for the oxidation of the ligand is linear and analogous to the energy of the LMCT transitions of the complexes (Fig. S13, ESI†).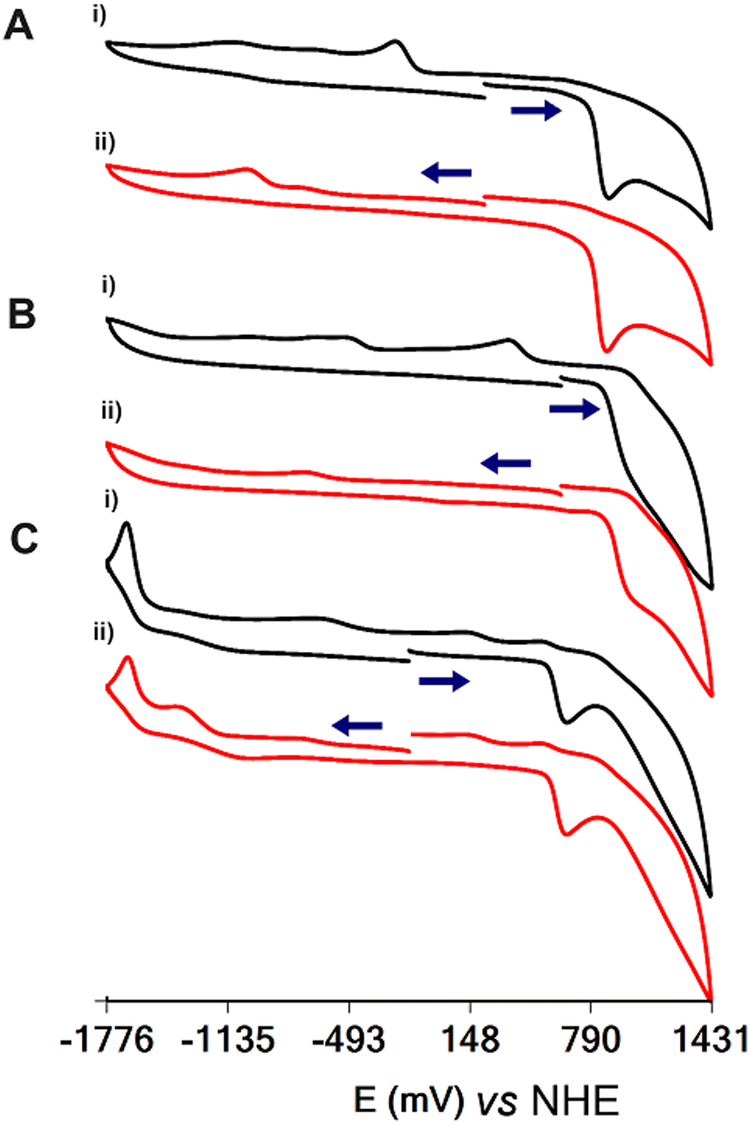 | ||
| Fig. 5 Cyclic voltammograms of DMSO:H2O (9:1) solutions of (A) 1 (1.00 mM), (B) 4·3H2O (1.00 mM) and (C) 5·9H2O (1.00 mM). (i) The scan starts towards a higher voltage and (ii) the scan starts towards a lower voltage. The supporting electrolyte is 0.1 M But4NClO4. Glassy carbon was used as the working electrode, platinum wire as the auxiliary electrode and Ag/AgCl (0.20 V vs. NHE) as the reference electrode. | ||
In addition to the oxidation of the ligand, the CVs show an irreversible wave assigned to the metal-centred reduction of the complexes at −1015, −729 and −1666 mV vs. NHE for 1, 4·3H2O and 5·9H2O, respectively.
Affinity of H2cbht for VVO43− and UVIO22+ based on 1H NMR spectroscopy and square wave voltammetry (SWV)
The 1H NMR spectra of the titration of a DMSO-d6 solution of 4·3H2O with UVIO22+ are shown in Fig. 6. The addition of UVIO22+ to DMSO-d6 solutions of 4·3H2O results in the disappearance of the peak of the hydroxylamine methyl protons of 4·3H2O and the appearance of a peak at 3.76 ppm assigned to 1 and a broad peak at 3.495 ppm. The 51V NMR spectra of the solutions of 4·3H2O show that its peak at −500 ppm is replaced by a broad peak at −542 ppm (assigned to inorganic vanadate, Fig. S14, ESI†) after addition of UVIO22+.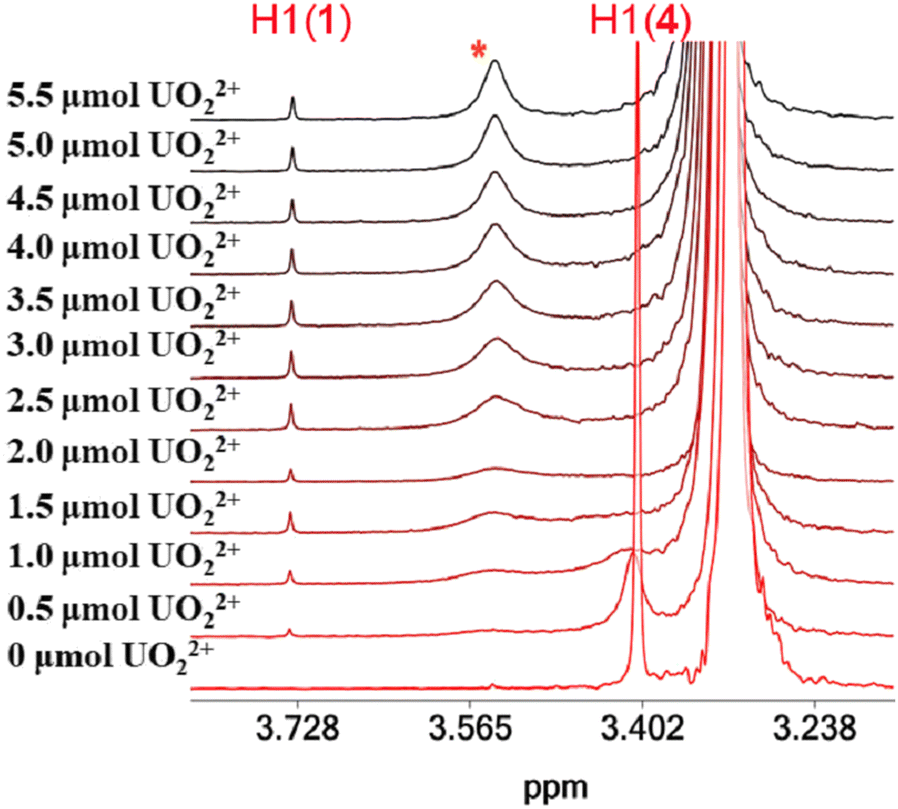 | ||
| Fig. 6 The aliphatic region of the 1H NMR spectra of solutions (DMSO-d6) containing 4·3H2O (2.27 μmol) and UVIO22+ (0–5.5 μmol). | ||
The titration of DMF solutions of 4·3H2O with UVIO22+ was monitored using SWV (Fig. 7). Inorganic UVIO22+ in DMF solutions gives two reduction waves at −310 mV and −750 mV vs. NHE, which can be separated easily from the reduction peak of 1 at −972 mV vs. NHE. The addition of UVO22+ to the solution of 4·3H2O resulted in the appearance of a peak at −972 mV, suggesting the formation of complex 1 in line with the 1H NMR results. However, the SWV also reveals the presence of inorganic UVIO22+ (Fig. 7). The presence of inorganic uranyl and vanadate in the solution indicates that the peak at 3.50 ppm originated from an inorganic UVO22+–VVO43− salt, which interacts weakly with H2cbht.13
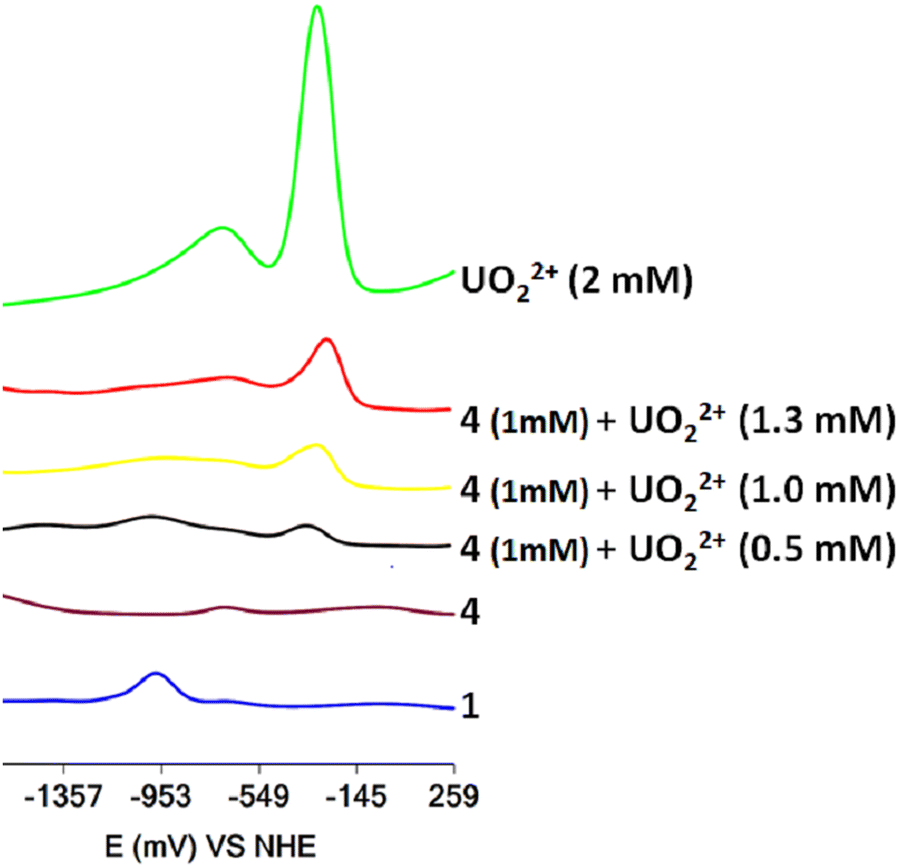 | ||
| Fig. 7 Square wave voltammograms of DMF solutions of complex 4·3H2O (1 mM) and portions of UVIO22+ (0–1.3 mM). The supporting electrolyte is 0.1 M But4NClO4. Glassy carbon was used as the working electrode, platinum wire as the auxiliary electrode and Ag/AgCl (0.20 V vs. NHE) as the reference electrode. | ||
The experiments reveal that H2cbht, under acidic conditions, is selective toward ligation to UVO22+ and confirms that H2cbht prefers to ligate UVIO22+ over VVO43− similar to H2bihyat.13 In addition, the UVO22+–VVO43− salt formation in H2cbht–UVO22+–VVO43− organic solutions is similar to the salt formed in H2bihyat–UVO22+–VVO43− aqueous solutions.13
The 1H NMR spectra of a DMSO-d6 solution of 4·3H2O (2.5 μmol), 2.5 μmol UVIO22+ and 5.0 μmol Et3N show only two methyl peaks assigned to 1 and 4·3H2O. The integrals of the two peaks are equal, suggesting that the ligand H2cbht, under alkaline conditions, binds UVIO22+ and VVO2+ with the same strength. Moreover, yellow solid precipitates and the 51V NMR spectrum of the solution show only the presence of complex 4·3H2O. The absence of a signal due to the released vanadate indicates that it forms a mixed UVIO22+–VVO43− yellow salt that precipitates out.31
Quantification of UVIO22+, VVO43− and FeIII based on luminescence experiments
The quenching of the luminescence of the ligand by the addition of the hard metal ions UVIO22+, VVO43− and FeIII was measured, and the data were fitted using the Stern–Volmer equation F0/F = 1 + [Mn+] × KSV, where F0 is the luminescence of the ligand, F is the measured luminescence after the addition of the metal ion, [Mn+] is the concentration of the metal ion, and KSV is the quenching constant (Fig. 8). Both UVIO22+ and VVO43− exhibit the same KSV of 10 × 103 mol−1 L. The KSV for FeIII is doubled compared with that of UVIO22+ and VVO43−, presumably due to the paramagnetism of FeIII.50 However, the luminescence data at an analyte concentration above 4 × 10−5 M show an upward curvature, which is attributed to the formation of the 1:2 FeIII–cbht2− complex in solution.90 The increase of e KSV with the number of the ligands ligating a single metal ion indicates that the quenching of H2cbht luminescence is initiated by the complexation with metal ions; therefore, the quenching follows a static than a collisional mechanism. The mechanism is also confirmed by the significant UV-vis changes in the absorption peaks during the titration of H2cbht with metal ions.91
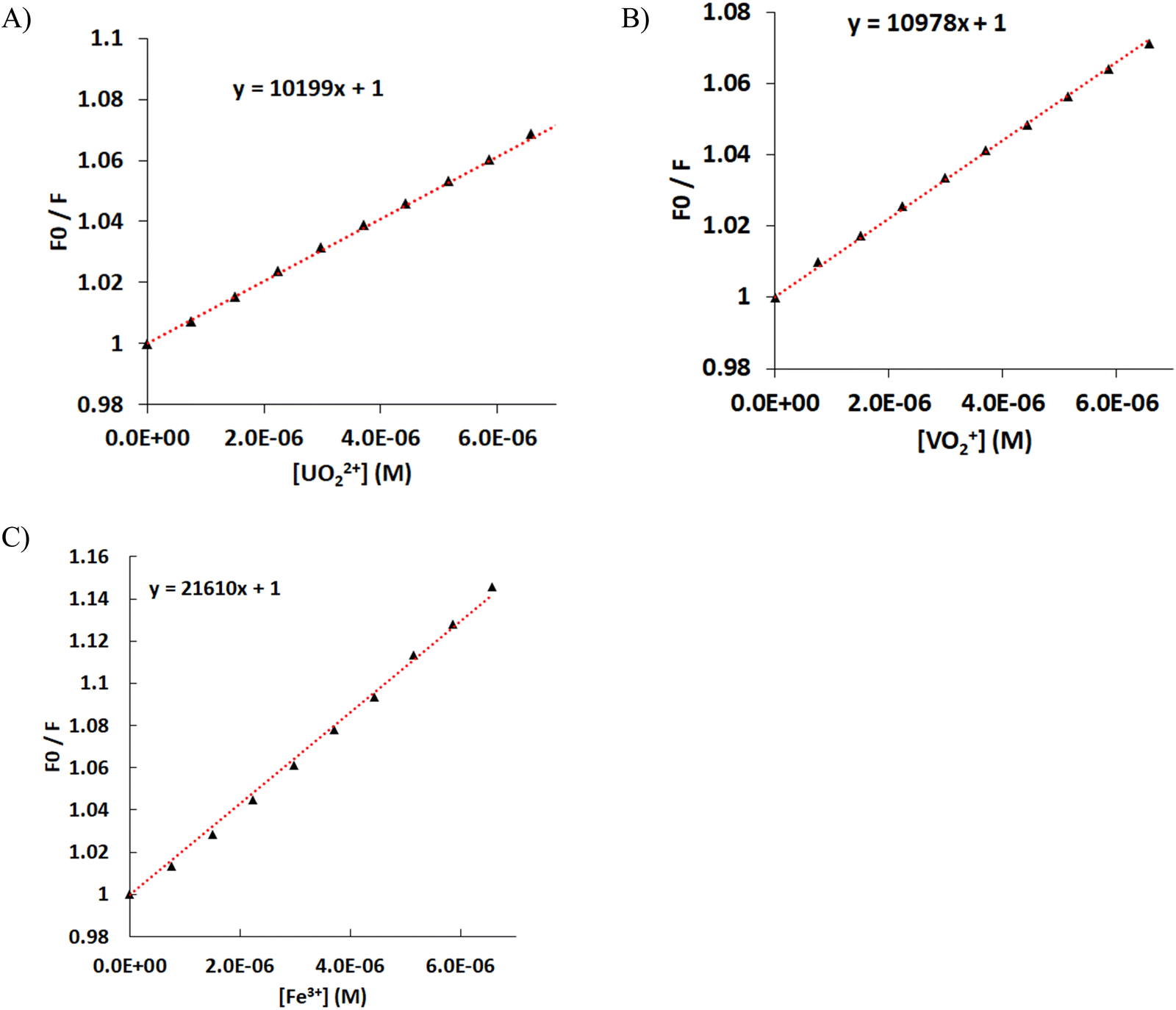 | ||
| Fig. 8 Stern–Volmer plots of H2cbht (3.74 × 10−5 M) and Et3N (14.8 × 10−5 M) DMSO 9:1 H2O solution after the addition of (A) UVIO22+, (B) VVO43−, and (C) FeIII. The data were fitted using the equation F0/F = 1 + [Mn+] × KSV. | ||
The association constant of the complexes was determined by the Benesi–Hildebrand method using fluorescence quenching data.35,92 The Benesi–Hildebrand equation of a 1:1 complex is 1/(F0 − F) = 1/{(F0 − F∞)K[Mn+]} + 1/(F0 − F∞), where F0 is the luminescence of the ligand, F∞ is the luminescence of the complex, F is the measured luminescence after the addition of the metal ion, [Mn+] is the concentration of the metal ion, and K is the association constant. The Benesi–Hildebrand plots of 1/(F0 − F) vs. 1/[Mn+] are straight lines (Fig. S15, ESI†), confirming that all metal complexes at concentrations of analyte below 4 × 10−5 are 1:1 in solution.
The log10(K) values for UVIO22+, VVO43− and FeIII 1:1 metal to ligand complexes were found to be 4.61, 4.72 and 4.59, respectively.
The quantification (LOQ) limits were calculated from the Stern–Volmer graphs and were found to be 8.9 × 10−7, 6.3 × 10−7 and 5.5 × 10−7 M for UVIO22+, VVO43− and FeIII, respectively. These values are in good agreement with the LOQ of other spectroscopic reagents, such as arsenazo, a typical spectroscopic reagent used for the quantification of UVIO22+.
Conclusions
In this work, we have demonstrated the design and synthesis of a spectroscopically labelled BHT-type ligand (H2cbht). The UVIO22+ reaction with H2cbht results in the synthesis of the mononuclear 1:1, 1:2, and trinuclear 3:4 metal-to-ligand complexes. The structural motifs of 2 and 3 were first observed for complexes of UVIO22+ with BHT-type ligands. VVO2+ complexes with H2cbht form trigonal bipyramidal structures instead of the expected square pyramidal structure, revealing the strong electron-donating ability of the triazine nitrogen donor atom. The 1:1 metal to ligand structures of UVIO22+ and VVO2+ and the crystallographically characterized 1:2 structure of FeIII were found to be the most stable in solution. 1H and 51V NMR spectroscopies and electrochemistry show that the ligand exhibits selectivity towards the coordination of uranium(VI) ions compared with vanadate in acidic organic solutions. In alkaline solutions, the binding strengths of H2cbht for both VV and UVI are the same. In this study, based on the quenching of the luminescence of BHT-type ligands upon ligation with metal ions, we have demonstrated for the first time that the quantification of hard metal ions has good repeatability, with low a LOQ (at nM concentration level). The high strength of BHT-type ligands in binding hard metal ions makes them ideal probes for the detection of UVIO22+, VVO2+ and FeIII. It is expected that the quantification limits of the hard metal ions using BHT chelators can reach values <1 nM, indicating that the binucleated BHT ligands31 form far more stable complexes than the mononucleated ones. Further work is underway to synthesize water-soluble binucleating BHT ligands to improve the LOQs and selectivity of the ligands for hard metal ions.
Author contributions
Angelos Amoiridis: formal analysis, investigation, validation, and writing – original draft. Michael Papanikolaou: formal analysis, investigation, and validation. Chryssoula Drouza: conceptualization, data curation, funding acquisition, methodology, and writing – review & editing. Themistoklis A. Kabanos: conceptualization and writing – review & editing. Anastasios D. Keramidas: conceptualization, data curation, funding acquisition, methodology, project administration, supervision, visualization, writing – original draft, and writing – review & editing.Data availability
The rest of the data are presented in the manuscript and ESI,† and hard copies are available from the authors on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the European Regional Development Fund and the Republic of Cyprus through the Research and Innovation Foundation (project: EXCELLENCE/0421/0520).Notes and references
- L. Février, F. Coppin, S. Pierrisnard, M. Bourdillon, L. V. Nguyen, N. Zaiter, S. Brandès, V. Sladkov, J. C. Chambron and M. Meyer, J. Environ. Radioact., 2021, 235–236, 106645 CrossRef PubMed.
- M. E. Kirby, J. L. Sonnenberg, A. Simperler and D. J. Weiss, J. Phys. Chem. A, 2020, 124, 2460–2472 CrossRef CAS PubMed.
- A. S. Ivanov, B. F. Parker, Z. Zhang, B. Aguila, Q. Sun, S. Ma, S. Jansone-Popova, J. Arnold, R. T. Mayes, S. Dai, V. S. Bryantsev, L. Rao and I. Popovs, Nat. Commun., 2019, 10, 819 CrossRef CAS PubMed.
- G. J. P. Deblonde, A. Ricano and R. J. Abergel, Nat. Commun., 2019, 10, 2438 CrossRef PubMed.
- A. Sornosa-Ten, P. Jewula, T. Fodor, S. Brandès, V. Sladkov, Y. Rousselin, C. Stern, J. C. Chambron and M. Meyer, New J. Chem., 2018, 42, 7765–7779 RSC.
- M. K. Lawson, M. Valko, M. T. D. Cronin and K. Jomová, Curr. Pharmacol. Rep., 2016, 2, 271–280 CrossRef CAS.
- L. Mullen, C. Gong and K. Czerwinski, J. Radioanal. Nucl. Chem., 2007, 273, 683–688 CrossRef CAS.
- P. W. Durbin, B. Kullgren, J. Xu and K. N. Raymond, Health Phys., 1997, 72, 865–879 CrossRef CAS PubMed.
- C. X. Bullock, C. S. Jamieson, P. Moënne-Loccoz, B. Taylor, J. A. M. Gonzalez, E. A. Draves and L. Y. Kuo, Inorg. Chem., 2021, 60, 7762–7772 CrossRef CAS PubMed.
- M. Stylianou, V. A. Nikolakis, G. I. Chilas, T. Jakusch, T. Vaimakis, T. Kiss, M. P. Sigalas, A. D. Keramidas and T. A. Kabanos, Inorg. Chem., 2012, 51, 13138–13147 CrossRef CAS PubMed.
- E. Y. Tshuva and D. Peri, Coord. Chem. Rev., 2009, 253, 2098–2115 CrossRef CAS.
- T. Hermon and E. Y. Tshuva, J. Org. Chem., 2008, 73, 5953–5958 CrossRef CAS PubMed.
- S. Hadjithoma, M. G. Papanikolaou, E. Leontidis, T. A. Kabanos and A. D. Keramidas, Inorg. Chem., 2018, 57, 7631–7643 CrossRef CAS PubMed.
- V. A. Nikolakis, J. T. Tsalavoutis, M. Stylianou, E. Evgeniou, T. Jakusch, A. Melman, M. P. Sigalas, T. Kiss, A. D. Keramidas and T. A. Kabanos, Inorg. Chem., 2008, 47, 11698–11710 CrossRef CAS PubMed.
- D. Fan and Q. Fang, Int. J. Pharm., 2021, 597, 120306 CrossRef CAS PubMed.
- L. Götzke, G. Schaper, J. März, P. Kaden, N. Huittinen, T. Stumpf, K. K. K. Kammerlander, E. Brunner, P. Hahn, A. Mehnert, B. Kersting, T. Henle, L. F. Lindoy, G. Zanoni and J. J. Weigand, Coord. Chem. Rev., 2019, 386, 267–309 CrossRef.
- L. O. De Serrano, Biomol. Concepts, 2017, 8, 169–178 CAS.
- C. Drouza, V. Gramlich, M. P. Sigalas, I. Pashalidis and A. D. Keramidas, Inorg. Chem., 2004, 43, 8336–8345 CrossRef CAS PubMed.
- S. W. Smith, J. Med. Toxicol., 2013, 9, 355–369 CrossRef CAS PubMed.
- P. W. Durbin, Health Phys., 2008, 95, 465–492 CrossRef CAS PubMed.
- R. J. Abergel, P. W. Durbin, B. Kullgren, S. N. Ebbe, J. Xu, P. Y. Chang, D. I. Bunin, E. A. Blakely, K. A. Bjornstad, C. J. Rosen, D. K. Shuh and K. N. Raymond, Health Phys., 2010, 99, 401–407 CrossRef CAS PubMed.
- G. Szigethy and K. N. Raymond, Inorg. Chem., 2010, 49, 6755–6765 CrossRef CAS PubMed.
- G. Szigethy and K. N. Raymond, Inorg. Chem., 2009, 48, 11489–11491 CrossRef CAS PubMed.
- C. J. Sunderland, M. Botta, S. Aime and K. N. Raymond, Inorg. Chem., 2001, 40, 6746–6756 CrossRef CAS PubMed.
- S. T. Tsantis, D. I. Tzimopoulos, M. Holyńska and S. P. Perlepes, Int. J. Mol. Sci., 2020, 21, 555 CrossRef CAS PubMed.
- G. Szigethy and K. N. Raymond, Chem. – Eur. J., 2011, 17, 1818–1827 CrossRef CAS PubMed.
- O. A. Osin, S. Lin, B. S. Gelfand, S. L. J. Lee, S. Lin and G. K. H. Shimizu, Nat. Commun., 2024, 15, 2614 CrossRef CAS PubMed.
- I. Ekeltchik, J. Gun, O. Lev, R. Shelkov and A. Melman, Dalton Trans., 2006, 1285–1293 RSC.
- J. Gun, I. Ekeltchik, O. Lev, R. Shelko and A. Melman, Chem. Commun., 2005, 5319–5321, 10.1039/b508138f.
- D. Sun, G. Melman, N. J. LeTourneau, A. M. Hays and A. Melman, Bioorg. Med. Chem. Lett., 2010, 20, 458–460 CrossRef CAS PubMed.
- A. Amoiridis, M. Papanikolaou, M. Vlasiou, N. A. G. Bandeira, H. N. Miras, T. Kabanos and A. Keramidas, Inorg. Chem., 2023, 62, 19971–19985 CrossRef CAS PubMed.
- X. Wu, Q. Huang, Y. Mao, X. Wang, Y. Wang, Q. Hu, H. Wang and X. Wang, TrAC, Trends Anal. Chem., 2019, 118, 89–111 CrossRef CAS.
- E. V. Gogol, E. S. Denisov, I. V. Lunev, O. S. Egorova, L. Sharipova and Y. A. Gusev, IOP Conf. Ser. Mater. Sci. Eng., 2017, 225, 012251 CrossRef.
- S. Badakhshan, S. Ahmadzadeh, A. Mohseni-Bandpei, M. Aghasi and A. Basiri, BMC Chem., 2019, 13, 131 CrossRef PubMed.
- V. S. Jisha, A. J. Thomas and D. Ramaiah, J. Org. Chem., 2009, 74, 6667–6673 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419–4476 CrossRef PubMed.
- A. B. Descalzo, R. Martínez-Máñez, R. Radeglia, K. Rurack and J. Soto, J. Am. Chem. Soc., 2003, 125, 3418–3419 CrossRef CAS PubMed.
- B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- R. R. Avirah, K. Jyothish and D. Ramaiah, Org. Lett., 2007, 9, 121–124 CrossRef CAS PubMed.
- R. R. Avirah, K. Jyothish and D. Ramaiah, J. Org. Chem., 2008, 73, 274–279 CrossRef CAS PubMed.
- Y. Jia, D. Li and M. Hu, Mater. Today Chem., 2023, 30, 101518 CrossRef CAS.
- A. Hazra and P. Roy, Anal. Chim. Acta, 2022, 1193, 339378 CrossRef CAS PubMed.
- T. Kajinehbaf and N. Alizadeh, New J. Chem., 2022, 46, 1763–1769 RSC.
- V. G. Kanellis and C. G. dos Remedios, Biophys. Rev., 2018, 10, 1401–1414 CrossRef CAS PubMed.
- C. Varadaraju, G. Tamilselvan, I. V. M. V. Enoch, V. Srinivasadesikan, S.-L. Lee and P. M. Selvakumar, New J. Chem., 2018, 42, 3833–3839 RSC.
- X.-A. Zhang and W.-D. Woggon, J. Am. Chem. Soc., 2005, 127, 14138–14139 CrossRef CAS PubMed.
- M. Wang, Z. Liu, X. Zhou, H. Xiao, Y. You and W. Huang, Inorg. Chem., 2020, 59, 18027–18034 CrossRef CAS PubMed.
- W. Liu, X. Dai, Z. Bai, Y. Wang, Z. Yang, L. Zhang, L. Xu, L. Chen, Y. Li, D. Gui, J. Diwu, J. Wang, R. Zhou, Z. Chai and S. Wang, Environ. Sci. Technol., 2017, 51, 3911–3921 CrossRef CAS PubMed.
- S. Pal, N. Chatterjee and P. K. Bharadwaj, RSC Adv., 2014, 4, 26585–26620 RSC.
- D. Rehder, Inorg. Chim. Acta, 2020, 504, 119445 CrossRef CAS.
- D. Rehder, Inorganics, 2023, 11, 256 CrossRef CAS.
- D. Rehder, Inorg. Chim. Acta, 2023, 549, 121387 CrossRef CAS.
- A. Rump, C. Hermann, A. Lamkowski, T. Popp and M. Port, Arch. Toxicol., 2023, 97, 1577–1598 CrossRef CAS PubMed.
- G. Xiao and J. Button, J. Radioanal. Nucl. Chem., 2023, 332, 185–191 CrossRef CAS PubMed.
- S. Liu, S. Wang, Y. Zhao, J. Li, C. Shu, Y. Li, J. Li, B. Lu, Z. Xu, Y. Ran and Y. Hao, Chem. – Biol. Interact., 2023, 372, 110356 CrossRef CAS PubMed.
- A. D. Keramidas, M. P. Rikkou, C. Drouza, C. P. Raptopoulou, A. Terzis and I. Pashalidis, Radiochim. Acta, 2002, 90, 549–554 CrossRef CAS.
- J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2008, 130, 6898–6899 CrossRef PubMed.
- C. Deraeve, C. Boldron, A. Maraval, H. Mazarguil, H. Gornitzka, L. Vendier, M. Pitié and B. Meunier, Chem. – Eur. J., 2008, 14, 682–696 CrossRef CAS PubMed.
- D. C. Crans, A. D. Keramidas, H. Hoover-Litty, O. P. Anderson, M. M. Miller, L. M. Lemoine, S. Pleasic-Williams, M. Vandenberg, A. J. Rossomando and L. J. Sweet, J. Am. Chem. Soc., 1997, 119, 5447–5448 CrossRef CAS.
- K. Elvingson, A. D. Keramidas, D. C. Crans and L. Pettersson, Inorg. Chem., 1998, 37, 6153–6160 CrossRef CAS.
- S. B. Etcheverry, D. C. Crans, A. D. Keramidas and A. M. Cortizo, Arch. Biochem. Biophys., 1997, 338, 7–14 CrossRef CAS PubMed.
- I. Hadjiadamou, M. Vlasiou, S. Spanou, Y. Simos, G. Papanastasiou, E. Kontargiris, I. Dhima, V. Ragos, S. Karkabounas, C. Drouza and A. D. Keramidas, J. Inorg. Biochem., 2020, 208, 111074 CrossRef CAS PubMed.
- K. Ioannou, C. Eleftheriou, C. Drouza, K. S. Pafiti, T. Panayi, A. D. Keramidas, L. C. Zacharia and M. C. Vlasiou, J. Mol. Struct., 2022, 1257, 132582 CrossRef CAS.
- M. Loizou, P. Papaphilippou, M. Vlasiou, M. Spilia, D. Peschos, Y. V. Simos, A. D. Keramidas and C. Drouza, J. Inorg. Biochem., 2022, 235, 111911 CrossRef CAS PubMed.
- M. Aureliano, N. I. Gumerova, G. Sciortino, E. Garribba, C. C. McLauchlan, A. Rompel and D. C. Crans, Coord. Chem. Rev., 2022, 454, 214344 CrossRef CAS.
- D. C. Crans, N. E. Barkley, L. Montezinho and M. M. Castro, RSC Metallobiol., 2019, 2019, 169–195 Search PubMed.
- D. C. Crans, L. Henry, G. Cardiff and B. I. Posner, Met. Ions Life Sci., 2019, 19, 203–230 CAS.
- A. L. De Sousa-Coelho, M. Aureliano, G. Fraqueza, G. Serrão, J. Gonçalves, I. Sánchez-Lombardo, W. Link and B. I. Ferreira, J. Inorg. Biochem., 2022, 235, 111915 CrossRef CAS PubMed.
- G. Ferraro, M. Paolillo, G. Sciortino, F. Pisanu, E. Garribba and A. Merlino, Inorg. Chem., 2023, 62, 8407–8417 CrossRef CAS PubMed.
- S. V. Gayakwad, D. S. Wankhede, V. D. Ragole, S. G. Wanale, S. A. Dake and S. B. Maulage, Anti-Infective Agents, 2023, 21, 41–56 CrossRef.
- C. Ghosh, D. Patra, N. Bala, I. Majumder, N. Sepay, P. Mukhopadhyay, S. Das, R. Kundu, M. G. B. Drew, A. R. León, T. Ghosh and M. Pradhan, Biometals, 2022, 35, 499–517 CrossRef CAS PubMed.
- D. Rehder, Comprehensive Inorganic Chemistry II: From Elements to Applications, 2nd edn, 2013, vol. 3, pp. 819–834 Search PubMed.
- G. Sciortino, V. Ugone, D. Sanna, G. Lubinu, S. Ruggiu, J. D. Maréchal and E. Garribba, Front. Chem., 2020, 8, 345 CrossRef CAS PubMed.
- V. Ugone, D. Sanna, G. Sciortino, D. C. Crans and E. Garribba, Inorg. Chem., 2020, 59, 9739–9755 CrossRef CAS PubMed.
- K. Kostenkova, A. Levina, D. A. Walters, H. A. Murakami, P. A. Lay and D. C. Crans, Chem. – Eur. J., 2023, 29, e202302271 CrossRef CAS PubMed.
- A. Levina, A. Pires Vieira, A. Wijetunga, R. Kaur, J. T. Koehn, D. C. Crans and P. A. Lay, Angew. Chem., Int. Ed., 2020, 59, 15834–15838 CrossRef CAS PubMed.
- A. Levina, C. Uslan, H. Murakami, D. C. Crans and P. A. Lay, Inorg. Chem., 2023, 62, 17804–17817 CrossRef CAS PubMed.
- E. A. Ignatenko, A. A. Gorbunov, E. V. Shklyaeva and G. G. Abashev, Chem. Heterocycl. Compd., 2014, 50, 691–698 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356, 10.1039/DT9840001349.
- D. Rehder, J. Inorg. Biochem., 2008, 102, 1152–1158 CrossRef CAS PubMed.
- D. C. Crans, M. L. Tarlton and C. C. McLauchlan, Eur. J. Inorg. Chem., 2014, 4450–4468 CrossRef CAS.
- C. C. McLauchlan, B. J. Peters, G. R. Willsky and D. C. Crans, Coord. Chem. Rev., 2015, 301–302, 163–199 CrossRef CAS.
- J. M. Harrowfield, N. Lugan, G. H. Shahverdizadeh, A. A. Soudi and P. Thuéry, Eur. J. Inorg. Chem., 2006, 389–396, DOI:10.1002/ejic.200500671.
- B. S. Parajón-Costa, O. E. Piro, R. Pis-Diez, E. E. Castellano and A. C. González-Baró, Polyhedron, 2006, 25, 2920–2928 CrossRef.
- A. Pushpaveni, S. Packiaraj, S. Govindarajan, G. T. McCandless, C. F. Fronczek and F. R. Fronczek, Inorg. Chim. Acta, 2018, 471, 537–549 CrossRef CAS.
- G. Tian, S. J. Teat, Z. Zhang and L. Rao, Dalton Trans., 2012, 41, 11579–11586 RSC.
- C. J. Leggett, B. F. Parker, S. J. Teat, Z. Zhang, P. D. Dau, W. W. Lukens, S. M. Peterson, A. J. P. Cardenas, M. G. Warner, J. K. Gibson, J. Arnold and L. Rao, Chem. Sci., 2016, 7, 2775–2786 RSC.
- X. Sun, C. Xu, G. Tian and L. Rao, Dalton Trans., 2013, 42, 14621–14627 RSC.
- M. H. Gehlen, J. Photochem. Photobiol., C, 2020, 42, 100338 CrossRef CAS.
- S. Bano, A. Mohd, A. A. P. Khan and K. S. Siddiqi, J. Chem. Eng. Data, 2010, 55, 5759–5765 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2360878 (1), 2360879 (3·DMF), 2360880 (5·9H2O), 2360881 (2·MeOH·Et2O·2H2O), and 2360882 (4·3H2O). For ESI and crystallographic data in CIF or other electronic format, see DOI: https://doi.org/10.1039/d4nj02645d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |