
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic-bioinformatic natural product-inspired peptides†
Samantha
Nelson
a and
Elizabeth I.
Parkinson
 *ab
*ab
aBorch Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, Indiana 47906, USA. E-mail: eparkins@purdue.edu
bJames Tarpo Jr. and Margaret Tarpo Department of Chemistry, Purdue University, West Lafayette, Indiana 47906, USA
First published on 30th October 2024
Abstract
Covering: 2016 to 2024
Natural products, particularly cyclic peptides, are a promising source of bioactive compounds. Nonribosomal peptide synthetases (NRPSs) play a key role in biosynthesizing these compounds, which include antibiotic and anticancer agents, immunosuppressants, and others. Traditional methods of discovering natural products have limitations including cryptic biosynthetic gene clusters (BGCs), low titers, and currently unculturable organisms. This has prompted the exploration of alternative approaches. Synthetic-bioinformatic natural products (syn-BNPs) are one such alternative that utilizes bioinformatics techniques to predict nonribosomal peptides (NRPs) followed by chemical synthesis of the predicted peptides. This approach has shown promise, resulting in the discovery of a variety of bioactive compounds including peptides with antibacterial, antifungal, anticancer, and proteasome-stimulating activities. Despite the success of this approach, challenges remain especially in the accurate prediction of fatty acid incorporation, tailoring enzyme modifications, and peptide release mechanisms. Further work in these areas will enable the discovery of many bioactive peptides that are currently inaccessible.
 Samantha Nelson | Samantha Nelson received their bachelor's degree in chemistry and a minor in mathematics from McPherson College in 2019. While at McPherson, they performed undergraduate research with Profs. Manjula Koralegedara and Allan Ayella on antibacterial and insect repellant properties of essential oils from plant materials. They then went on to perform doctoral research with Prof. Elizabeth Parkinson at Purdue University, where they attained their PhD in Medicinal Chemistry and Molecular Pharmacology in 2024. During their time in the Parkinson laboratory, Sam's research interests focused on bioinformatics predictions of cryptic nonribosomal peptide biosynthetic gene clusters, as well as chemical synthesis and biological evaluation of the predicted molecules. |
 Elizabeth I. Parkinson | Elizabeth (Betsy) Parkinson attended Rhodes College, where she obtained her BS in chemistry in 2010. She conducted graduate research with Prof. Paul Hergenrother at the University of Illinois at Urbana-Champaign (UIUC) on the synthesis of bioactive natural products and their mechanisms of action. After obtaining her PhD in 2015, she joined the laboratory of Prof. William Metcalf in Microbiology at UIUC for her postdoctoral studies, where she studied the biosynthesis of natural products from Streptomyces. Betsy started her independent career as an assistant professor in the Departments of Chemistry and Medicinal Chemistry and Molecular Pharmacology at Purdue University in the Fall of 2018. In her lab, research focuses on the discovery of natural products from cryptic biosynthetic gene clusters and elucidating the function of natural product biosynthetic enzymes. |
1. Introduction
Natural products are a bountiful source of bioactive compounds, with many being utilized as therapeutics. As of 2019, approximately two-thirds of FDA-approved small molecules are derived from natural products,1 highlighting the significance of natural products in the drug discovery pipeline. Bacteria, particularly Actinomycetota, are a prolific source of bioactive compounds,2 with over 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bacterial natural products discovered to date.3,4
000 bacterial natural products discovered to date.3,4
Among the bioactive natural products, cyclic peptides are a promising class of therapeutics, with over 40 FDA-approved cyclic peptides, many derived from natural products, targeting a range of bioactivities.5–9 Since 2000, approximately one cyclic peptide has been approved each year,6–8 highlighting their growing importance in the pharmaceutical industry. Compared to their linear counterparts, cyclic peptides typically exhibit heightened bioactivity due to enhanced cell permeability, binding affinity, and proteolytic stability.10–14
In biological systems, peptide natural products are primarily synthesized via two main pathways: ribosomally synthesized and post-translationally modified peptides (RiPPs)15 and nonribosomal peptide synthetases (NRPSs).16 RiPPs have been recently reviewed elsewhere.17–19 This review focuses on NRPSs, which produce nonribosomal peptides (NRPs) with potential for a broad range of therapeutic applications, including antifungals such as caspofungin, antibiotics such as daptomycin and colistin, chemotherapeutics such as actinomycin D, and immunosuppressants such as cyclosporine (Fig. 1). Despite the considerable number of NRPs discovered thus far, bioinformatics analyses of sequenced bacterial genomes suggest that there remains immense potential for unearthing novel bioactive molecules from Actinomycetota.20–22
 | ||
| Fig. 1 A selection of FDA approved NRPs with their approval date, bioactivity, and species of origin. | ||
Traditional methods of natural product discovery, such as phenotypic screening,23 co-cultivation,24 and one strain many compounds (OSMAC),25 have yielded valuable insights, but suffer from limitations such as low yields, high rediscovery rates, and time intensive isolations.26,27 To address these challenges, alternative approaches such as heterologous expression of biosynthetic gene clusters (BGCs) and promoter engineering have been employed.28 However, challenges persist, including choice of chassis and promoters, as well as frequent low yields.
Recently, a new technique was developed, which focuses on bioinformatics predictions of NRP structures followed by chemical synthesis and biological testing to generate bioactive natural product-inspired metabolites (Fig. 2A). This method was initially developed by Brady and coworkers and was named the synthetic-bioinformatic natural product (syn-BNP) approach.29 The goal of the syn-BNP method is not to generate the exact natural product. Instead, it aims to quickly and more easily access the core scaffold of the natural product that is hopefully “close enough” in that it retains the biological activity of the natural product. This goal is well supported by the many natural product derivatives that are used as medicines (e.g. eribulin, doxycycline, rezafungin, as well as many others). While there are natural products that are directly used as medicines, most must be modified to improve properties such as pharmacokinetics or decrease toxicity. This is well evidenced by the number of natural products (71) versus natural product derivatives (356) approved by the FDA as drugs from 1981 to 2019.1 The use of direct chemical synthesis allows access to many (oftentimes between 10 and a few hundred) peptides on reasonable scales for screening (usually a few milligrams). This is in contrast to isolation where one or a few molecules are isolated, oftentimes at submilligram quantities resulting in limited numbers of biological assays being performed. Additionally, the synthetic approach allows for easy modification for rapid structure activity relationship analyses and is generally easier to scale-up than isolation for further analyses.
 | ||
| Fig. 2 (A) General workflow of the syn-BNP approach. (B) General NRPS machinery. Rectangles denote modifications installed by tailoring enzymes. Abbreviations: adenylation (A), thiolation (T), condensation (C), thioesterase (TE), biosynthetic gene cluster (BGC). | ||
Overall, the syn-BNP approach complements more historical approaches including bioactivity-guided fractionation of organisms (either collected or cultured) as well as heterologous expression. In particular, it overcomes many of the limitations of these more traditional approaches, including supply limitations, costly rediscovery of known natural products, the high percentage of currently unculturable organisms, and the challenges of finding appropriate hosts for heterologous expression. However, syn-BNP is currently limited by our ability to predict natural product structures, with highly modified bioactive molecules likely being missed by this approach. It also limits the source of the natural products explored. While it is possible to apply the syn-BNP method to fungal or plant genomes, it is more challenging to predict natural product structures when biosynthetic genes are not clustered, thus limiting our ability to apply the syn-BNP method to these organisms. As natural product structure predictions improve, we expect that syn-BNP will be more widely applied to natural products from different domains and kingdoms. Syn-BNP, along with other BGC targeted approaches such as heterologous expression, also suffers from the fact that we are often unable to predict whether a natural product will be bioactive based solely on its BGC. For this reason, there is a need to screen molecules from a wide variety of BGCs in a large number of assays to identify bioactive natural products. While this is very challenging using heterologous expression, this is an area where the syn-BNP method excels. Specifically, the synthetic approach of syn-BNP allows for rapid generation of medium sized libraries of natural product-like molecules for biological testing. Additionally, the fact that syn-BNPs are pure molecules is another advantage compared to traditional screening of extracts. In particular, it avoids the challenges of crude extracts or fractions, such as molecules that directly interfere with screens (e.g. pan-assay interference compounds, PAINS) or compounds that synergize and lose activity upon isolation. Since the initial inception of the syn-BNP approach in 2016, the technique has resulted in the identification of many bioactive molecules such as antibacterial, antifungal, and anticancer compounds (ESI Table 1†), showcasing its potential in expanding the repertoire of bioactive molecules derived from natural sources. In this review, we cover the syn-BNP method and papers published on the topic from 2016 to August 2024.
2. Bioinformatics prediction of NRPs
NRPSs are multimodular enzyme complexes that possess distinct domains responsible for loading and coupling amino acids. The essential domains include adenylation (A), thiolation (T), and condensation (C) domains (Fig. 2B). The A domain selects and activates the amino acid via adenylation. These activated amino acids are then loaded onto the phosphopantetheinyl arm of the T (also known as the peptidyl carrier protein, PCP) domain, which is responsible for carrying the growing peptide chain. Finally, the C domain is responsible for peptide bond formation between the amino acids on the upstream and downstream T domains. In addition to the essential domains, several other domains are often present on NRPSs including epimerization (E, epimerizes L amino acids to D amino acids), methylation (M, often N-methylates an amino acid), heterocyclization (Cy, cyclodehydrates between a Cys, Ser, or Thr and backbone carbonyl), reduction (R, offloads the peptide as an aldehyde) and thioesterase (TE, offloads peptide as either a carboxylic acid, lactone, or lactam) domains.16,30,31 These additional domains and in trans tailoring enzymes facilitate additional modifications to give the final NRP.2.1. Strategies for peptide structure prediction
Accurate predictions of amino acid sequences of NRPs are essential for the syn-BNP method to be successful. Many methods for adenylation domain specificity predictions have been developed over the years. Initially, Stachelhaus developed a predictive method based on the A domain active site residues.32 It was discovered that certain residues (positions 235, 236, 239, 278, 299, 301, 322, 330, 331, and 517) are essential for substrate selectivity. The amino acids at these locations within the substrate binding pocket generally correlate with the class of amino acids that are loaded. This study helped identify rules that can be utilized to predict the amino acids that will be loaded by a specific adenylation domain. More recently, tools such as AntiSMASH,33,34 PRISM,35,36 SANDPUMA,37 Norine,38 and NRPSPredictor2 (ref. 39) (Table 1) leverage machine learning techniques, including profile hidden Markov models, to predict the amino acid associated with the adenylation domain. These models are robust and widely employed for predicting the amino acid affiliated with a specific A domain.58| Bioinformatics tools | Reference paper |
|---|---|
| AntiSMASH v.6.1 | 40 |
| AntiSMASH v.5.1.2 | 41–45 |
| AntiSMASH v.5.0 | 46–49 |
| AntiSMASH v.3.0 | 50 |
| AntiSMASH v.2.0 | 29 and 51–53 |
| PRISM 4 | 47–50 |
| Minowa | 54 and 55 |
| NRPSPredictor2 | 54 and 55 |
| Stachelhaus | 55 and 56 |
| SANDPUMA | 45 and 56 |
| Norine | 45 |
| MIBiG v.2.0 | 45 |
| In-house method | 41, 42, 45 and 57 |
In addition to predicting the amino acid sequence, it is necessary to predict other modifications to the NRP, such as those performed by the non-essential domains and in trans tailoring enzymes. Both antiSMASH and PRISM provide valuable insights into these modifications. For instance, they both predict E, M, and Cy domains within the NRPS, enabling epimerization, methylation, and heterocycle formation to be predicted. They can also predict starter C (Cs) domains, which are present at the beginning of the initiation module. Cs domains are typically indicative of a lipopeptide, a distinct class of NRPs with an N-terminal fatty acid tail. However, accurate prediction of the identity of the chain remains challenging. These programs can also predict the presence of likely peptide tailoring enzymes, such as hydroxylases, glycosyltransferases, and halogenases but are typically not able to accurately predict how these tailoring enzymes will modify the NRP. For this reason, BGCs with multiple tailoring enzymes are usually avoided in the syn-BNP method.
Once the linear NRP is predicted, determining the method of release becomes crucial. Two primary enzymes have been utilized to date in the syn-BNP approach: traditional TEs and penicillin-binding protein (PBP)-like TEs (PBP-TEs). Traditional TE domains often act in cis and catalyze the release of peptides from the final T domain of the NRPS, either through hydrolysis or cyclization. Several types of cyclization are possible such as: head-to-tail, head-to-sidechain, or head-to-fatty acid. While a single TE will usually only perform one of these offloading methods, predicting this process based solely on TE sequence remains a challenge.59 This results in many predicted NRPs for each NRPS BGC. For the syn-BNP approach with traditional TE containing NRPSs, either the linear peptide is exclusively synthesized29,53–55 or multiple cyclized products per predicted linear NRP (i.e., head-to-tail, head-to-sidechain, and/or head-to-fatty acid) are produced.51,52 This can make the synthesis process laborious as multiple derivatives must be synthesized to identify the correct molecule of interest. While the majority of NRPS BGCs found to date end in a traditional TE domain, other offloading methods exist. One such method is via PBP-TEs, which act in trans to the NRPS. Multiple PBP-TEs have been studied (e.g., SurE,60,61 Ulm16,62 PenA,63 WolJ,64 and DsaJ65) with several studies having utilized derivatives of their native substrates to determine the scope of the PBP-TEs. To date, these studies have only found PBP-TEs to catalyze head-to-tail cyclization.60,62,63,66 Therefore, when utilizing a PBP-TE for bioinformatics predictions, only one peptide needs to be synthesized per linear prediction.47–50
2.2. Sources of biosynthetic gene clusters
To generate bioinformatics predictions, genome libraries are essential. For the syn-BNP approach, three major sources of genomes have been investigated: genomes from isolated soil bacteria, soil metagenomes, and the human microbiome. For the studies that used genomes from isolated bacteria, most have relied on either the GenBank67 or the Joint Genome Institute (JGI)68 database (Table 2). Others have focused on particular bacterial species such as Streptomyces sp. H-KF8 (ref. 50) and Paenibacillus mucilaginosus K02.54 The soil metagenome and the microbiome projects are particularly interesting sources because they do not require the isolation of an organism. Given that approximately 1% of bacterial species are thought to be currently culturable, the natural products in the other 99% of bacterial species are currently inaccessible.69 The culture-independent nature of the syn-BNP method facilitates access to these otherwise very challenging to attain molecules.| Repository | Mechanism | Reference paper |
|---|---|---|
| GenBank | Amoebicidal, antibiotic, antifungal, chemotherapeutic, proteasome stimulator | 41–45, 47–49, 51, 53 and 57 |
| Joint genome institute | Antibiotic | 57 |
| Human microbiome project | Antibiotic, chemotherapeutic | 29, 52 and 55 |
| Human oral microbiome database | Antibiotic, chemotherapeutic | 29, 52 and 55 |
| Soil metagenome | Antibiotic, chemotherapeutic | 40, 46 and 56 |
The work with soil metagenomes has exclusively been performed by Brady and co-workers using their extensive soil metagenome libraries.29,52,59 For the microbiome work, genomes have primarily been accessed from resources like the Human Microbiome Project70 and the Human Oral Microbiome Database.71 These diverse sources of microorganisms have led to the identification of unique and novel syn-BNPs. Given the vast amount of genome sequences that exist and the continuing growth of these libraries, we hypothesize that there are still many bioactive syn-BNPs yet to be discovered.
3. Synthesis of NRPs
The second step of the syn-BNP method is the chemical synthesis of the predicted peptides. Given that syn-BNP has only been performed on NRPS substrates, except for the NRPS-PKS hybrid albicidin, the major steps of the synthesis are the generation of the linear peptide substrate and the subsequent cyclization strategy, if applicable. The following sections delve into the synthetic techniques employed in these areas. Another important area, which will not be discussed in depth here, is the synthesis of non-canonical amino acids. The synthesis of syn-BNPs can be greatly complicated by predictions of non-commercially available non-canonical amino acids. Continued development of efficient methods to access non-canonical amino acids will help to expedite these syntheses.72,733.1. Linear peptide synthesis
The synthesis of linear peptides is often the first step of syn-BNP synthesis. Solid-phase peptide synthesis (SPPS)74 is nearly always employed (Fig. 3A). The exceptions to this are the highly modified lapcin and PABA-containing syn-BNPs, which were synthesized using batch methods.40,43,46 Many fantastic reviews of SPPS exist75–77 and thus we will only outline the methods used to generate syn-BNPs. Chlorotrityl or Wang resin is most often employed because either can be used to generate C-terminal carboxylic acids, yielding completed linear peptides that can be further activated for cyclization reactions if desired.77 Specific reagents for SPPS have included common coupling reagents HATU, HBTU, Oxyma, or PyBOP for amide bond formation with piperidine for Fmoc-deprotection. Once the full linear peptide is synthesized, it can be cleaved from the resin using HFIP or TFA, depending on whether a side chain protected or fully deprotected peptide is desired. N-terminal modifications such as fatty acid installation occur during SPPS (e.g. cilagicin,41 lapcin,43 macolacin44). While these methods work well for generation of linear peptides, future studies should consider greener alternatives to SPPS, which requires the use of large amounts of toxic organic solvents as well as explosive coupling reagents and allergenic additives.78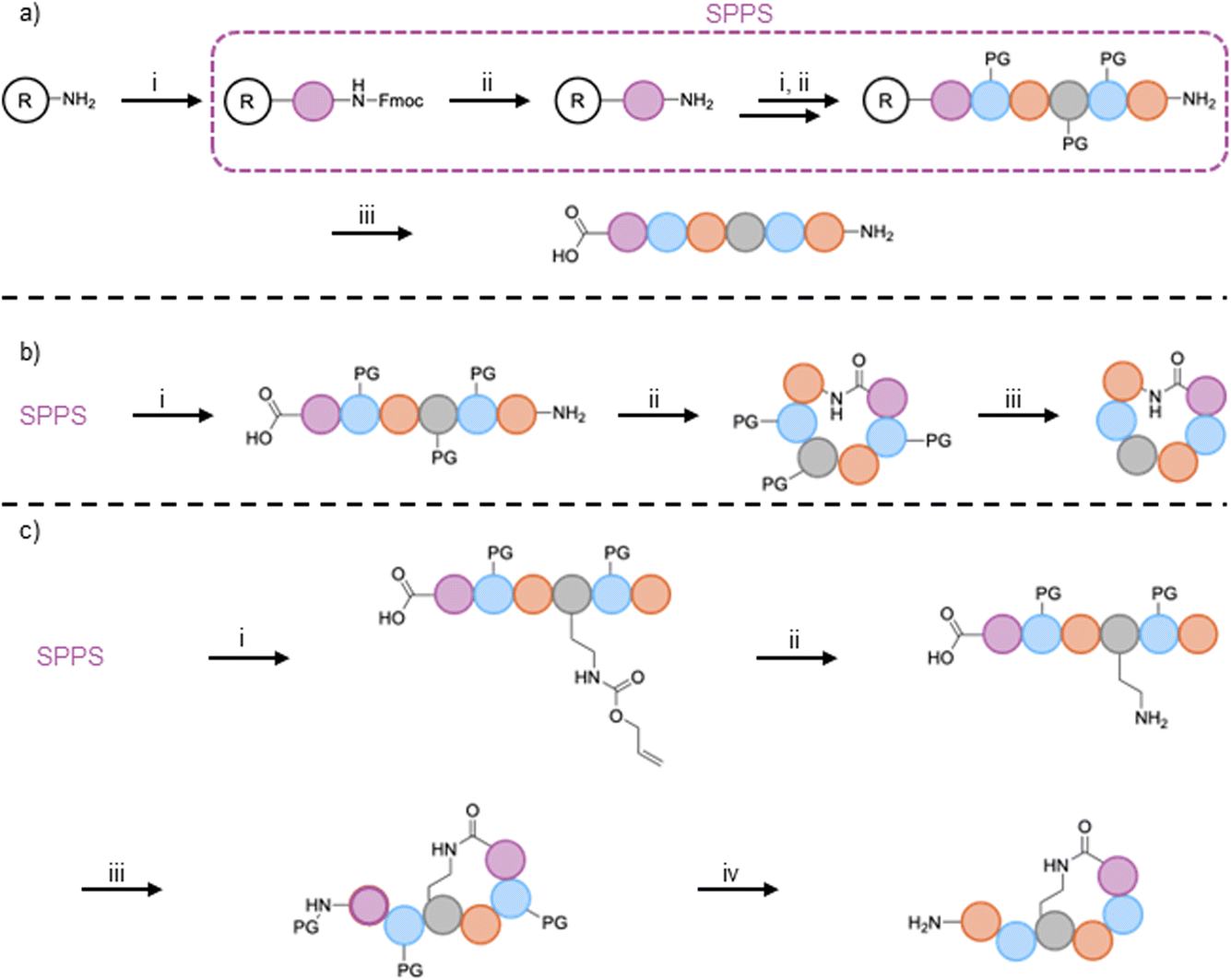 | ||
| Fig. 3 (A) General scheme for synthesizing linear peptides. Starting with a resin pre-loaded with the C-terminal amino acid, the steps for SPPS include: (i) activation of the amino acid with an amide coupling reagent and (ii) deprotection of the Fmoc-protecting group with piperidine. Steps i and ii are repeated until all amino acids are coupled. (iii) Removal of the protecting groups and cleavage of the peptide from the resin. (B) General scheme for synthesizing head-to-tail cyclized peptides. After completion of SPPS: (i) cleavage of the peptide from resin using a method that retains sidechain protecting groups, (ii) cyclization of the peptide using a coupling agent, and (iii) global deprotection of the peptide. (C) General scheme for synthesizing head-to-sidechain cyclized peptides. After completion of SPPS with the desired coupling sidechain having an orthogonal protecting group: (i) deprotection of the allyloxycarbonyl group, (ii) cyclization with a coupling agent, and (iv) cleavage from resin and global deprotection to obtain the final product. Acronyms: solid-phase peptide synthesis (SPPS), resin (R), protecting group (PG). | ||
3.2. Cyclization strategies
Many of the syn-BNPs generated to date are cyclic peptides, including head-to-tail, head-to-sidechain, and head-to-fatty acid cyclic peptides. Head-to-tail cyclized peptides are generally made using chlorotrityl resin followed by cleavage from the resin with HFIP or TFA and DIEA, to retain the sidechain protecting groups and thus prevent unwanted sidechain cyclization (Fig. 3B). The specific conditions for the TFA deprotection are important to retain the protecting groups on the designated sidechains, typically using short exposure to 1% TFA in DCM with neutralization by DIEA.41,44,51,52 Head-to-tail cyclization is then catalyzed using PyBOP or HATU in solution, usually at low concentrations to avoid oligomerization. After successful cyclization, the sidechain protecting groups are removed with TFA resulting in the final cyclic peptide.47–49Head-to-sidechain and head-to-fatty acid syntheses both rely on orthogonal protecting strategies (Figs. 3C, e.g., an allyloxycarbonyl (Alloc) protecting group for an amine that can be removed using palladium, leaving the other Fmoc-protected amines protected). This ensures a selective cyclization of the previously Alloc-protected amine with the activated carboxyl group on the peptide. While utilization of the orthogonal protecting group strategy for peptide cyclization can be employed off-resin, performing the cyclization on-resin is often advantageous due to the pseudodilution effect.79 Specifically, peptides are much less likely to oligomerize, thus giving higher yields of correctly cyclized products.41,42,44,45,51,52,56,57 One example of on-resin cyclization for syn-BNPs added a monomethoxytrityl protected lysine to the C-terminus of the linear peptide during SPPS. Glutaric anhydride was then coupled to the N-terminus using DMAP. Cleavage of the monomethoxytrityl group on the lysine was performed using TFA, and on-resin cyclization was performed with PyBOP or HATU resulting in the final product.50
4. Examples of syn-BNP libraries and bioactivities
4.1. Unbiased syn-BNP predictions and bioactivities
After predicting and synthesizing the syn-BNP libraries, they are screened for bioactivities. This screening process has yielded peptides exhibiting a wide spectrum of bioactivities (Table 2 and ESI Table 1†), underscoring the versatility and potential of this technique. | ||
| Fig. 4 Structures of a subset of the bioactive syn-BNPs identified from unbiased libraries. Under each structure is the name, species of origin and bioactivity of the molecule. Blue indicates residues with differing predictions. Red is the cyclization site. | ||
In another early syn-BNP study, a library of 288 linear syn-BNPs from the NCBI database yielded 5 antibiotic agents with MIC values less than 8 μg mL−1 against ESKAPE pathogens.53 The most potent compound, syn-BNP 1 (Fig. 4), was discovered from the Paenibacillus mucilaginosus K02 genome. Syn-BNP 1 demonstrated activity against Gram-positive bacteria, as well as the Gram-negative Acinetobacter baumanii. This compound mimics tridecaptins, with 5 of the 13 amino acids being shared. Interestingly, no resistance to syn-BNP 1 could be developed via direct plating or serial passaging methods. Because this is often indicative of a membrane based mechanism, syn-BNP 1 was tested for its ability to depolarize cells using a fluorescence DiBAC4 assay. Similar to tridecaptin, no depolarization of the membrane was observed. Tridecaptin has previously been shown to bind lipid II, and it is hypothesized that syn-BNP 1 is acting similarly. However, additional experimentation is needed to verify this hypothesis. Derivatives of syn-BNP 1 were explored in a follow up paper where differences in amino acid predictions as well as alternate fatty acid chains were investigated.54 Interestingly, the choice of fatty acid chain greatly affected the activity profile. For example, replacement of the decanoyl of syn-BNP 1 (aka compound 1F) with an acetyl group (compound 4F, Fig. 4) resulted in decreased potency against most of the Gram-positive strains and greatly increased potency against the Gram-negative strains. 4F showed good selectivity for bacterial cells over mammalian cells and strong activity in a rat cutaneous model of multidrug resistant A. baumannii. To explore the mechanism of 4F, resistant mutants were developed using A. baumannii ATCC 17978. These mutants generally grew slower and had a different morphology compared to the parent strains. Sequencing revealed that mutations occurred in the cell membrane phospholipid metabolism network (e.g. phospholipase A and D and phosphatidyltransferase), suggesting that 4F targets the membrane. Interestingly, several mutations in the adeS gene, which encodes for a histidine kinase in a two-component regulatory system, and in the lipopolysaccharide biosynthesis pathway further supported the membrane as a target of 4F. Additionally, while only moderate membrane depolarization was observed upon treatment with 4F, extensive pore formation was observed.
Another unbiased syn-BNP library was developed from bacterial genomes found in GenBank.51 After excluding known NRPS biosynthetic gene clusters as well as clusters containing a large number of tailoring enzymes or fewer that 3 adenylation domains, 96 linear peptide predictions were explored. This work differed from previous publications in that it focused on cyclic peptides. Specifically, for each linear peptide predicted, a head-to-tail (or head-to-fatty acid for predicted N-acyl containing peptides) and head-to-side chain cyclic peptides were explored. This resulted in 157 cyclic peptides, which were screened against ESKAPEE pathogens and Mycobacterium tuberculosis. Impressively, this relatively small library resulted in 9 bioactive syn-BNP cyclic peptides (4 Gram-positive specific and 5 with both Gram-positive and Gram-negative activity), with 7 showing minimal to no cytotoxicity to mammalian cells. These syn-BNP cyclic peptide antibiotics (syCPAs) were found to have various mechanisms of action including cell lysis (syCPA 12, 102, and 123), membrane depolarization (syCPA 63), inhibition of cell wall biosynthesis (syCPA 4), and ClpP protease dysregulation (syCPA 116). Three active molecules (syCPA 2, 144, and 153) currently have undetermined mechanisms of action (Fig. 4).
Overall, these findings suggest that syn-BNPs are a great source of antibiotics with interesting mechanisms that have the potential to target multidrug-resistant bacteria. Future screening efforts should continue to explore and expand on this activity. Additionally, in the future, standardized conditions for testing peptides for antibiotic activity should be developed. Many are screened using conditions originally optimized for small molecules. These conditions can sometimes result in degradation of the peptides and thus the community may be missing some promising compounds.
4.2. Targeted derivative predictions
While many of the papers utilizing the syn-BNP method employ an unbiased approach (i.e. synthesizing any NRP with between 4 or 5 and 13 amino acids),47–54 some have employed a targeted strategy to identify more potent analogs of either a certain peptide class or molecule. Nearly all of these studies, with the exception of the head-to-tail cyclized peptides, have focused on antibiotic activity. Below we have described some of the targeted approaches, specifically focusing on those that went after specific NRP classes or NRPs containing certain non-canonical amino acids.000 bacterial genomes in Genbank using the Cs domain as the query. From this tree, they identified a clade with no characterized lipopeptides that fell into a larger group with Cs domains from known antibiotic BGCs. As it is difficult to predict the release of an NRP by a TE, they synthesized two linear and six cyclic peptides identified in this clade. The fatty acid utilized for each peptide family was determined based on the lipid most commonly observed in the most closely related Cs domain. For example, when peptides were similar to the paenibactins, myristic acid was utilized. This led to the discovery of cilagicin (Fig. 5), a dodecalipodepsipeptide antibiotic with potent activity against Gram-positive, antibiotic-resistant pathogens.42 Cilagicin was found to bind both undecaprenyl phosphate (C55:P) and undecaprenyl pyrophosphate (Cff:PP), essential carrier lipids required for the translocation of cell wall intermediates, resulting in disrupted cell wall synthesis. To date, no resistance has been developed to cilagicin, likely due to its ability to bind to multiple non-peptidic targets.42 Furthermore, cilagicin did not demonstrate cytotoxic activity against HEK-293 cells at up to 64 μg mL−1.57 Due to this very promising activity, cilagicin was evaluated for in vivo efficacy in neutropenic mouse thigh models with S. aureus and S. pyrogens, where it demonstrated very promising reductions in bacterial burden. Unfortunately, high serum binding likely reduced efficacy in vivo.42
 | ||
| Fig. 5 Structures of bioactive syn-BNPs identified by the lipopeptide and n-cinnamoyl targeted approaches. Under each structure is the name, species of origin and bioactivity of the molecule. | ||
To discover peptides with similar or enhanced activity compared to cilagicin, a screen was conducted using both the Joint Genomic Institute (JGI) and GenBank databases to identify compounds with highly homologous BGCs to the cilagicin BGC.57 Of the 25 they screened, only five highly similar (>50% shared amino acids) NRPs were identified, with two being identical to cilagicin. The three predicted NRPs (paenilagicin, bacilagicin, and virgilagicin) were found to have similar, if slightly less potent, Gram-positive antibiotic activity (0.12 to 8 μg mL−1) to cilagicin. Interestingly, cilagicin, paenilagicin, and virgilagicin did not lead to resistance after prolonged antibiotic exposure.57 More recently, Brady and coworkers have put forth efforts to optimize cilagicin for clinical development. The myristic acid at the N-termini was replaced with a biphenyl moiety, which in turn reduced the serum binding and improved in vivo efficacy. A robust structure activity relationship campaign identified a derivative called dodecacilagicin which improved potency, reduced serum binding, and exhibited low MICs.82
000 strains screened, over 15000 BGCs contained Cs domains, and 395 were predicted to have ≥5 amino acids with 2 or more amino acids being positively charged. Analysis of uncharacterized BGCs that were in the same Cs clade as known cationic lipopeptides led to the discovery of derivatives of laterocidine (syn-CNRLP1, -2, and -3), tridecaptin (syn-CNRPL4, -5, and -6), and paenibacterin (syn-CNRLP7, Fig. 6). The cationic lipopeptides syn-CNRLP1, -5, and -7 displayed antibiotic activity against ESKAPEE pathogens ranging from 0.25 to 64 μg mL−1. The laterocidine derivative, syn-CNRLP1, was active against Gram-negative bacteria, while the tridecaptin derivative syn-CNRLP5 and the paenibacterin derivative syn-CNRLP7 exhibited activity against both Gram-negative and Gram-positive bacteria. Interestingly, tridecaptin A was only active against Gram-negative bacteria, demonstrating that the syn-BNP method was able to identify syn-CNRLP5 as a more broad-spectrum tridecaptin. Similarly, while syn-CNRLP7 has weaker activity against Gram-negative bacteria compared to paenibacterin, it has increased activity against WT S. aureus, three strains of multidrug-resistant S. aureus, and three strains of vancomycin-resistant E. faecium. The mechanism of action for these molecules is currently unknown,41 but given that they are cationic lipopeptides it is reasonable to assume that their activity is likely at least in part due to membrane interactions.
 | ||
| Fig. 6 Structures of syn-BNPs identified by the cationic lipopeptide targeted approach. Under each structure is the name, species of origin and bioactivity of the molecule. | ||
Another cationic lipopeptide syn-BNP with antibiotic activity is macolacin (Fig. 6), which has potent activity against ESKAPEE pathogens, including those resistant to colistin.44 Colistin is a cationic lipopeptide that works by binding to lipid A in lipopolysaccharides, resulting in membrane disruption and cell death. Recently, resistance to colistin has been rising, in large part due to the plasmid-borne mobilized colistin resistance gene mcr-1, which encodes phosphoethanolamine transferase.88 This transferase modifies lipid A with the positively charged phosphoethanolamine, resulting in the loss of colistin binding. Brady and co-workers specifically searched for colistin-like BGCs. 35 syn-BNPs from colistin-like BGCs were synthesized and tested for activity against pairs of colistin-sensitive and colistin-resistant K. pneumoniae and A. baumannii. This resulted in the discovery that macolacin has similar potency as colistin against colistin-sensitive strains and much improved activity against colistin-resistant strains (∼16-fold, single-digit μg mL−1 activities). Additionally, macolacin has improved activity against strains with a 4-amino-4-deoxy-L-arabinose modification compared to colistin and other polymyxins. Due to these very promising results, derivatives of macolacin were synthesized, including one that replaced the linear fatty acid tail with a biphenyl. Biphenyl-macolacin proved to be more potent than macolacin in vitro and was shown to be effective in vivo in a neutropenic thigh mouse model of infection using colistin-resistant A. baumannii strains.44
 | ||
| Fig. 7 Structures of bioactive NRPs identified by the head-to-tail and p-aminobenzoic acid (PABA) containing targeted approach. | ||
When natural products are screened for activities, they are most often tested in phenotypic assays for their antimicrobial and anticancer activities. However, many other important targets and interesting assays exist. To date, the SNaPP library has been explored in a handful of other activity assays including proteasome stimulation and antiamoebic activity.
The accumulation of disordered proteins such as Tau and α-synuclein has been implicated in neurodegenerative diseases such as Alzheimer's94,95 and Parkinson's Disease.96–98 Stimulation of the 20S core particle (CP) of the proteasome has shown promise in enhancing the clearance of highly disordered and misfolded proteins through ubiquitin-independent proteolysis.99 Screening of the SNaPP library in an in vitro proteasome stimulation assay resulted in the discovery of molecules that lead to selective degradation of misfolded and highly disordered protein α-synuclein at 10 μM.49 These molecules have been renamed the Cyclic Peptide Proteaseome Stimulators (CyPPSs). Several CyPPSs (CyPPS-1, -13, -14, and -23, Fig. 7) are cell-permeable and stimulate cytosolic 20S CP at 10 μM in cell culture. Additionally, they exhibit no cytotoxic effects at up to the highest concentration tested (15 μM).49
Balamuthia mandrillaris is a pathogenic free-living amoeba that has a fatality rate of approximately 92%.100 A primary screen of the SNaPP library targeting the free-living amoebas Balamuthia mandrillaris, Acanthamoeba castellanii, and Naegleria fowleri was conducted.47 Eight hits were identified for Balamuthia mandrillaris, sixteen for Acanthamoeba castellanii, and one against Naegleria fowleri at 16 μg mL−1. One hit, pNP-43 (Fig. 7), displayed significant potency against Balamuthia mandrillaris with an IC50 of 4.6 μM. Furthermore, this molecule exhibited no cytotoxicity, showing no activity against A549 cells nor causing hemolysis at 100 μM.47 Overall, these two examples demonstrate that syn-BNPs have activities outside of the traditionally tested antimicrobial and anticancer activities. This should inspire further testing of these molecules in additional assays in the future.
In addition to derivatives of albicidin and cystobactamid, Brady and co-workers also investigated novel NRPS BGCs with PABA A domains found in their metagenomic library.43 They identified an NRPS BGC encoding a decapeptide with a Cs domain, suggesting it is a lipopeptide. The NRPS BGC also contained two Cy domains and an FMN-dependent dehydrogenase, suggesting the presence of two thiazole rings. Overall, this prediction was for a unique NRP, which they named lapcin (Fig. 7). Lapcin was successfully synthesized, but heterologous expression of the BGC was unsuccessful, preventing confirmation of the actual natural product.43 Interestingly, lapcin was found to be a potent anticancer agent (nM to pM IC50) with generally good selectivity for cancerous over non-cancerous cell lines. It was particularly potent against cell lines with mutant p53. Unlike the other PABA-containing natural products albicidin and cystobactamid, lapcin had little-to-no DNA gyrase inhibition nor antibiotic activity. Instead, it was found to be a potent and selective human topoisomerase inhibitor. Another decalipopeptide similar to lapcin (tapcin) was also identified from a soil metagenome library that has a BGC with high similarity to the lapcin BGC.43 The two major differences of the new BGC are the lack of a serine at the two site and an additional Cy domain. The molecule resulting from this BGC was named tapcin and contains three thiazole rings. Similar to lapcin, tapcin is a potent chemotherapeutic through inhibition of human topoisomerase.40 Tapcin has similar efficacy to the clinically used topoisomerase irinotecan in both hollow fiber models and xenograft murine models of colorectal adenocarcinoma.
 | ||
| Fig. 8 Structures of bioactive NRPs identified by the cationic menaquinone-binding antibiotics targeted approach. | ||
5. Future direction
Synthetic-bioinformatic natural product-inspired peptides have proven to be a valuable tool in identifying novel peptides with unique mechanisms of action and bioactivity.29,40–53,55–57 This approach has led to the discovery of compounds with antibiotic,29,41,42,44–46,48,50,51,53–57 anticancer,40,43,52,56 amoebicidal,47 and proteasome stimulation activity.49 Many of these compounds are potent, and some have demonstrated activity in cellulo and/or in vivo. However, several challenges persist.One such challenge arises from the presence of a Cs domain in the NRPS, indicating the presence of a fatty acid at the N terminus.110 To the best of our knowledge, current NRPS predictions cannot accurately determine which fatty acid will be added to the NRP, complicating the design and synthesis of the predicted peptide. When only a few peptides are being targeted, the synthesis of several peptides, each with a different fatty acid chain, is manageable. However, simplification become more necessary in cases where a large library is being synthesized, often resulting in the same fatty acid being added to all the peptides predicted to possess a starter Cs domain.51 This approach likely limits the number of identified bioactive peptides, since the identity of the fatty acid often has a large effect on the bioactivity of the molecule.111,112 Improvements in bioinformatics predictions of Cs fatty acid specificity will enable access to syn-BNPs that more closely resemble the true natural products, and are thus more likely to have interesting bioactivities.
Another challenge for accurate syn-BNP predictions is tailoring enzymes. Tailoring enzymes frequently are located within the same genomic neighborhood as the NRPS and are responsible for the synthesis of non-canonical amino acids or the modification of the NRP during or after its assembly. Common tailoring enzymes include halogenases (halogenation), glycosyltransferases (addition of a sugar moiety), hydroxylases (addition of hydroxyl), and P450s (various oxidation reactions).113 Unfortunately, precise predictions of the modifications that these tailoring enzymes will introduce is challenging. Currently, many researchers refrain from incorporating modifications due to the inability to predict if or how the tailoring enzymes will act. Since many of these modifications are essential to bioactivity (e.g. the antibiotic activity of vancomycin and proteasome inhibition by salinosporamide A both are dependent on the incorporation of halogen atoms by halogenases),114 their exclusion likely limits the discovery of bioactive molecules. As predictions for these tailoring enzymes improve, their incorporation into syn-BNP predictions will likely aid in the discovery of bioactive compounds. However, the requirement for modified non-canonical amino acids is likely to complicate the synthesis of these molecules.
Finally, the release of an NRP from an NRPS by a TE domain presents another significant challenge for predictions. While there are a handful of programs that attempt to predict the cyclization method of an NRP based on the peptide structure,115 the methods developed to date are based on small libraries and are thus biased and lack accuracy.59 This necessitates the synthesis of multiple peptides per predicted linear substrate when a traditional TE is present. Improvements of these predictions, perhaps through machine learning analyses of known TEs, will help to overcome this challenge, further streamlining the syn-BNP approach.
Overall, to address all of these challenges, more data about the biosynthesis of known molecules combined with predictions, likely through machine learning models, will enhance the efficiency and accuracy of peptide design and synthesis in the future.
6. Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.7. Author contributions
S. N. and E. I. P. conceptualized and wrote the article.8. Conflicts of interest
The authors have no conflicts of interest to report.9. Acknowledgments
We would like to acknowledge E. Brajkovich, Z. Budimir, P. Jeannette, and N. Moriarty for their perceptive editing and insightful comments. This work was funded by a grant from the National Institutes of Health (1R35GM138002-01 to E. I. P) and the Frederick N. Andrews Fellowship from the Department of Medicinal Chemistry and Molecular Pharmacology at Purdue University (to S. N.).10. References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- E. Patridge, P. Gareiss, M. S. Kinch and D. Hoyer, Drug Discovery Today, 2016, 21, 204–207 CrossRef CAS PubMed.
- A. S. Abdel-Razek, M. E. El-Naggar, A. Allam, O. M. Morsy and S. I. Othman, Processes, 2020, 8, 470 CrossRef CAS.
- J. A. van Santen, E. F. Poynton, D. Iskakova, E. McMann, T. A. Alsup, T. N. Clark, C. H. Fergusson, D. P. Fewer, A. H. Hughes, C. A. McCadden, J. Parra, S. Soldatou, J. D. Rudolf, E. M-L Janssen, K. R. Duncan and R. G. Linington, Nucleic Acids Res., 2021, 50, D1317–D1323 CrossRef PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- H. Zhang and S. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC.
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. G. de la Torre, Pharmaceuticals, 2024, 17, 243 CrossRef CAS.
- O. Al Musaimi, D. Al Shaer, F. Albericio and B. G. de la Torre, Pharmaceuticals, 2023, 16, 336 CrossRef.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS.
- S. Jackson, W. Degrado, A. Dwivedi, A. Parthasarathy, A. Higley, J. Krywko, A. Rockwell, J. Markwalder, G. Wells, R. Wexler, S. Mousa and R. Harlow, J. Am. Chem. Soc., 1994, 116, 3220–3230 CrossRef CAS.
- J. J. Devlin, L. C. Panganiban and P. E. Devlin, Science, 1990, 249, 404–406 CrossRef CAS PubMed.
- K. Deyle, X.-D. Kong and C. Heinis, Acc. Chem. Res., 2017, 50, 1866–1874 CrossRef CAS PubMed.
- X. Ji, A. L. Nielsen and C. Heinis, Angew. Chem., Int. Ed., 2024, 136, e202308251 CrossRef.
- D. A. Price, H. Eng, K. A. Farley, G. H. Goetz, Y. Huang, Z. Jiao, A. S. Kalgutkar, N. M. Kablaoui, B. Khunte, S. Liras, C. Limberakis, A. M. Mathiowetz, R. B. Ruggeri, J. M. Quan and Z. Yang, Org. Biomol. Chem., 2017, 15, 2501–2506 RSC.
- M. Montalbán-Lopez, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. José Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, J. A. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef.
- I. P. M. Pfeiffer, M. P. Schröder and S. Mordhorst, Nat. Prod. Rep., 2024, 41, 990–1019 RSC.
- H. Li, W. Ding and Q. Zhang, RSC Chem. Biol., 2024, 5, 90–108 RSC.
- C. Ongpipattanakul, E. K. Desormeaux, A. Dicaprio, W. A. Van Der Donk, D. A. Mitchell and S. K. Nair, Chem. Rev., 2022, 122, 14722–14814 CrossRef CAS PubMed.
- A. Gavriilidou, S. A. Kautsar, N. Zaburannyi, D. Krug, R. Müller, M. H. Medema and N. Ziemert, Nat. Microbiol., 2022, 7, 726–735 CrossRef CAS PubMed.
- E. Kalkreuter, S. A. Kautsar, D. Yang, C. D. Bader, C. N. Teijaro, L. L. Fluegel, C. M. Davis, J. R. Simpson, L. Lauterbach, A. D. Steele, C. Gui, S. Meng, G. Li, K. Viehrig, F. Ye, P. Su, A. F. Kiefer, A. Nichols, A. J. Cepeda, W. Yan, B. Fan, Y. Jiang, A. Adhikari, C.-J. Zheng, L. Schuster, T. M. Cowan, M. J. Smanski, M. G. Chevrette, L. P. S. De Carvalho and B. Shen, bioRxiv, preprint, 2023, DOI:10.1101/2023.12.14.571759.
- B. S. Jian, S. L. Chiou, C. C. Hsu, J. Ho, Y. W. Wu and J. Chu, ACS Chem. Biol., 2023, 18, 476–483 CrossRef CAS PubMed.
- J. G. Moffat, F. Vincent, J. A. Lee, J. Eder and M. Prunotto, Nat. Rev. Drug Discovery, 2017, 16, 531–543 CrossRef CAS.
- Z. Q. Tan, H. Y. Leow, D. C. W. Lee, K. Karisnan, A. A. L. Song, C. W. Mai, W. S. Yap, S. H. E. Lim and K. S. Lai, Open Biotechnol. J., 2019, 13, 18–26 Search PubMed.
- H. B. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 Search PubMed.
- A. Y. Alwali, D. Santos, C. Aguilar, A. Birch, L. Rodriguez-Ordunea, C. B. Roberts, R. Modi, C. Licona-Cassani and E. I. Parkinson, J. Ind. Microbiol. Biotechnol., 2024, 51, kuae014 CAS.
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 Search PubMed.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509–523 CAS.
- J. Chu, X. Vila-Farres, D. Inoyama, M. Ternei, L. J. Cohen, E. A. Gordon, B. V. B. Reddy, Z. Charlop-Powers, H. A. Zebroski, R. Gallardo-Macias, M. Jaskowski, S. Satish, S. Park, D. S. Perlin, J. S. Freundlich and S. F. Brady, Nat. Chem. Biol., 2016, 12, 1004–1006 CAS.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS.
- K. Blin, S. Shaw, A. M. Kloosterman, Z. Charlop-Powers, G. P. Van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2021, 49, W29–W35 CrossRef CAS.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS.
- M. A. Skinnider, C. A. Dejong, P. N. Rees, C. W. Johnston, H. Li, A. L. H. Webster, M. A. Wyatt and N. A. Magarvey, Nucleic Acids Res., 2015, 43, 9645–9662 CAS.
- M. A. Skinnider, N. J. Merwin, C. W. Johnston and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef CAS.
- M. G. Chevrette, F. Aicheler, O. Kohlbacher, C. R. Currie and M. H. Medema, Bioinformatics, 2017, 33, 3202–3210 CrossRef CAS PubMed.
- A. Flissi, E. Ricart, C. Campart, M. Chevalier, Y. Dufresne, J. Michalik, P. Jacques, C. Flahaut, F. Lisacek, V. Leclère and M. Pupin, Nucleic Acids Res., 2020, 48, D465–D469 CAS.
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef.
- Z. Wang, A. Kasper, M. Takahashi, A. Morales Amador, A. Bhattacharjee, J. Kan, Y. Hernandez, M. Ternei and S. F. Brady, Angew. Chem., Int. Ed., 2024, 63, e202317187 CrossRef CAS PubMed.
- Z. Wang, B. Koirala, Y. Hernandez and S. F. Brady, Org. Lett., 2022, 24, 4943–4948 CrossRef CAS PubMed.
- Z. Wang, B. Koirala, Y. Hernandez, M. Zimmerman and S. F. Brady, Science, 2022, 376, 991–996 CrossRef CAS.
- Z. Wang, N. Forelli, Y. Hernandez, M. Ternei and S. F. Brady, Nat. Commun., 2022, 13, 842 CrossRef CAS.
- Z. Wang, B. Koirala, Y. Hernandez, M. Zimmerman, S. Park, D. S. Perlin and S. F. Brady, Nature, 2022, 601, 606–611 CrossRef CAS PubMed.
- L. Li, B. Koirala, Y. Hernandez, L. W. MacIntyre, M. A. Ternei, R. Russo and S. F. Brady, Nat. Microbiol., 2022, 7, 120–131 CrossRef CAS.
- Z. Wang, A. Kasper, R. Mehmood, M. Ternei, S. Li, J. S. Freundlich, S. F. Brady, Z. Wang, A. Kasper, R. Mehmood, M. Ternei, S. F. Brady, S. Li and J. S. Freundlich, Angew. Chem., Int. Ed., 2021, 60, 22172–22177 CrossRef CAS.
- C. Lu, S. Nelson, G. Coy, C. Neumann, E. I. Parkinson and C. A. Rice, bioRxiv, preprint, 2024, DOI:10.1101/2024.05.03.592372.
- M. A. Hostetler, C. Smith, S. Nelson, Z. Budimir, R. Modi, I. Woolsey, A. Frerk, B. Baker, J. Gantt and E. I. Parkinson, ACS Chem. Biol., 2021, 16, 2604–2611 CrossRef CAS PubMed.
- S. Nelson, T. J. Harris, C. S. Muli, M. E. Maresh, B. Baker, C. Smith, C. Neumann, D. J. Trader and E. I. Parkinson, ChemBioChem, 2023, 25, e202300671 CrossRef PubMed.
- L. I. Beyer, A.-B. Schäfer, A. Undabarrena, I. Mattsby-Baltzer, D. Tietze, E. Svensson, A. Stubelius, M. Wenzel, B. Cámara and A. A. Tietze, ACS Infect. Dis., 2023, 10, 79–92 CrossRef PubMed.
- J. Chu, B. Koirala, N. Forelli, X. Vila-Farres, M. A. Ternei, T. Ali, D. A. Colosimo and S. F. Brady, J. Am. Chem. Soc., 2020, 142, 14158–14168 CrossRef CAS.
- J. Chu, X. Vila-Farres and S. F. Brady, J. Am. Chem. Soc., 2019, 141, 15737–15741 CrossRef CAS PubMed.
- X. Vila-Farres, J. Chu, D. Inoyama, M. A. Ternei, C. Lemetre, L. J. Cohen, W. Cho, B. B. Vijay Reddy, H. A. Zebroski, J. S. Freundlich, D. S. Perlin and S. F. Brady, J. Am. Chem. Soc., 2017, 139, 1404–1407 CrossRef CAS.
- X. Vila-Farres, J. Chu, M. A. Ternei, C. Lemetre, S. Park, D. S. Perlin and S. F. Brady, mSphere, 2018, 3, e00528 CrossRef CAS PubMed.
- J. Chu, X. Vila-Farres, D. Inoyama, R. Gallardo-Macias, M. Jaskowski, S. Satish, J. S. Freundlich and S. F. Brady, ACS Infect. Dis., 2018, 4, 33–38 CrossRef CAS PubMed.
- L. W. MacIntyre, B. Koirala, A. Rosenzweig, A. Morales-Amador and S. F. Brady, Org. Lett., 2024, 26, 4433–4437 CrossRef CAS.
- A. F. Rosenzweig, Z. Wang, A. Morales-Amador, K. Spotton and S. F. Brady, ACS Infect. Dis., 2023, 9, 2394–2400 CrossRef CAS PubMed.
- K. Blin, H. U. Kim, M. H. Medema and T. Weber, Brief. Bioinform, 2019, 20, 1103–1113 CrossRef CAS.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- Y. Zhou, X. Lin, C. Xu, L. Li, H. Deng and H.-W. Lin Correspondence, Cell Chem. Biol., 2019, 26, 737–744 CrossRef CAS.
- T. Kuranaga, K. Matsuda, A. Sano, M. Asakazu Kobayashi, A. Ninomiya, K. Takada, S. Matsunaga and T. Wakimoto, Angew. Chem., Int. Ed., 2018, 130, 9591–9595 CrossRef.
- Z. L. Budimir, R. S. Patel, A. Eggly, C. N. Evans, H. M. Rondon-Cordero, J. J. Adams, C. Das and E. I. Parkinson, Nat. Chem. Biol., 2023, 20, 120–128 CrossRef PubMed.
- K. Matsuda, K. Fujita and T. Wakimoto, J. Ind. Microbiol. Biotechnol., 2021, 48, kuab023 CrossRef CAS.
- M. Kobayashi, K. Fujita, K. Matsuda and T. Wakimoto, J. Am. Chem. Soc., 2023, 145, 3270–3275 CrossRef CAS.
- Q. Li, Y. Song, X. Qin, X. Zhang, A. Sun and J. Ju, J. Nat. Prod., 2015, 78, 944–948 CrossRef CAS.
- S. Son, Y.-S. Hong, M. Jang, K. T. Heo, B. Lee, J.-P. Jang, J.-W. Kim, I.-J. Ryoo, W.-G. Kim, S.-K. Ko, B. Y. Kim, J.-H. Jang and J. S. Ahn, J. Nat. Prod., 2017, 80, 3025–3031 CrossRef CAS.
- D. A. Benson, M. Cavanaugh, K. Clark, I. Karsch-Mizrachi, J. Ostell, K. D. Pruitt and E. W. Sayers, Nucleic Acids Res., 2018, 46, D41–D47 CrossRef CAS PubMed.
- I. V. Grigoriev, H. Nordberg, I. Shabalov, A. Aerts, M. Cantor, D. Goodstein, A. Kuo, S. Minovitsky, R. Nikitin, R. A. Ohm, R. Otillar, A. Poliakov, I. Ratnere, R. Riley, T. Smirnova, D. Rokhsar and I. Dubchak, Nucleic Acids Res., 2012, 40, D26–D32 CrossRef CAS.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS.
- J. Peterson, S. Garges, M. Giovanni, P. McInnes, L. Wang, J. A. Schloss, V. Bonazzi, J. E. McEwen, K. A. Wetterstrand, C. Deal, C. C. Baker, V. Di Francesco, T. K. Howcroft, R. W. Karp, R. D. Lunsford, C. R. Wellington, T. Belachew, M. Wright, C. Giblin, H. David, M. Mills, R. Salomon, C. Mullins, B. Akolkar, L. Begg, C. Davis, L. Grandison, M. Humble, J. Khalsa, A. Roger Little, H. Peavy, C. Pontzer, M. Portnoy, M. H. Sayre, P. Starke-Reed, S. Zakhari, J. Read, B. Watson and M. Guyer, Genome Res., 2009, 19, 2317–2323 CrossRef.
- T. Chen, W. H. Yu, J. Izard, O. V. Baranova, A. Lakshmanan and F. E. Dewhirst, Database, 2010, 2010, baq013 CrossRef PubMed.
- S. I. Toh, C. L. Lo and C. Y. Chang, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2023, 79, 193–199 CrossRef CAS PubMed.
- D. J. Atkinson, B. J. Naysmith, D. P. Furkert and M. A. Brimble, Beilstein J. Org. Chem., 2016, 12, 2325 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- J. A. Moss, Curr. Protoc. Protein Sci., 2005, 40 DOI:10.1002/0471140864.ps1807s40.
- K. G. Varnava and V. Sarojini, Chem.–Asian J., 2019, 14, 1088–1097 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- M. S. Donia, B. Wang, D. C. Dunbar, P. V Desai, A. Patny, M. Avery and M. T. Hamann, Int. J. Mol. Sci., 2021, 22, 3973 CrossRef.
- I. Nouioui, A. Zimmermann, O. Hennrich, S. Xia, O. Rössler, R. Makitrynskyy, J. Pablo Gomez-Escribano, G. Pötter, M. Jando, M. Döppner, J. Wolf, M. Neumann-Schaal, C. Hughes and Y. Mast, Front. Bioeng. Biotechnol., 2024, 12, 1255151 CrossRef.
- A. Rosenzweig, K. Spotton, A. Bhattacharjee, A. Morales-Amador and S. F. Brady, ACS Infect. Dis., 2024, 10, 1536–1544 CrossRef CAS.
- C. Zhang and M. R. Seyedsayamdost, Angew. Chem., Int. Ed., 2020, 59, 23005–23009 CrossRef CAS.
- J. Shi, C. L. Liu, B. Zhang, W. J. Guo, J. Zhu, C.-Y. Chang, E. J. Zhao, R. H. Jiao, R. X. Tan and H. M. Ge, Chem. Sci., 2019, 10, 4839–4846 RSC.
- Q. Liu, Z. Liu, C. Sun, M. Shao, J. Ma, X. Wei, T. Zhang, W. Li and J. Ju, Org. Lett., 2019, 21, 2634–2638 CrossRef CAS PubMed.
- S. Biswas, J. M. Brunel, J. C. Dubus, M. Reynaud-Gaubert and J. M. Rolain, Expert Rev. Anti-Infect. Ther., 2012, 10, 917–934 CrossRef CAS PubMed.
- S. A. Cochrane and J. C. Vederas, Med. Res. Rev., 2016, 36, 4–31 CrossRef CAS PubMed.
- Y.-Y. Liu, C. E. Chandler, L. M. Leung, C. L. Mcelheny, R. T. Mettus, R. M. Q. Shanks, J.-H. Liu, D. R. Goodlett, R. K. Ernst, Y. Doi, C. Liu, C. Ce, L. Lm, M. Cl, M. Rt, S. Rmq and J.-H. Liu, Antimicrob. Agents Chemother., 2017, 61, e00580 Search PubMed.
- N. A. Magarvey, B. Haltli, M. He, M. Greenstein and J. A. Hucul, Antimicrob. Agents Chemother., 2006, 50, 2167–2177 CrossRef CAS.
- H. He, R. T. Williamson, B. Shen, E. I. Graziani, H. Y. Yang, S. M. Sakya, P. J. Petersen and G. T. Carter, J. Am. Chem. Soc., 2002, 124, 9729–9736 CrossRef CAS PubMed.
- Y. Song, Q. Li, X. Liu, Y. Chen, Y. Zhang, A. Sun, W. Zhang, J. Zhang and J. Ju, J. Nat. Prod., 2014, 77, 1937–1941 CrossRef CAS PubMed.
- F. Xu, B. Nazari, K. Moon, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2017, 139, 9203–9212 CrossRef CAS.
- K. Takada, A. Ninomiya, M. Naruse, Y. Sun, M. Miyazaki, Y. Nogi, S. Okada and S. Matsunaga, J. Org. Chem., 2013, 78, 6746–6750 CrossRef CAS PubMed.
- K. Gadhave, N. Bolshette, A. Ahire, R. Pardeshi, K. Thakur, C. Trandafir, A. Istrate, S. Ahmed, M. Lahkar, D. F. Muresanu and M. Balea, J. Cell. Mol. Med., 2016, 20, 1392–1407 CrossRef CAS PubMed.
- G. Palmieri, E. Cocca, M. Gogliettino, R. Valentino, M. Ruvo, G. Cristofano, A. Angiolillo, M. Balestrieri, M. Rossi and A. Di Costanzo, J. Alzheimer's Dis., 2017, 60, 1097–1106 CAS.
- L. Stefanis, Cold Spring Harbor Perspect. Med., 2012, 2, a009399 Search PubMed.
- K. St. P. McNaught and P. Jenner, Neurosci. Lett., 2001, 297, 191–194 CrossRef CAS.
- K. St. P. McNaught, R. Belizaire, O. Isacson, P. Jenner and W. C. Olanow, Exp. Neurol., 2003, 179, 38–46 CrossRef CAS PubMed.
- R. A. Coleman, R. Mohallem, U. K. Aryal and D. J. Trader, RSC Chem. Biol., 2021, 2, 636–644 RSC.
- J. R. Cope, J. Landa, H. Nethercut, S. A. Collier, C. Glaser, M. Moser, R. Puttagunta, J. S. Yoder, I. K. Ali and S. L. Roy, Clin. Infect. Dis., 2019, 68, 1815–1822 CrossRef PubMed.
- S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noëll, I. Pieretti, M. Hügelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Süssmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef CAS PubMed.
- S. Baumann, J. Herrmann, R. Raju, H. Steinmetz, K. I. Mohr, S. Hüttel, K. Harmrolfs, M. Stadler and R. Müller, Angew. Chem., Int. Ed., 2014, 53, 14605–14609 CrossRef CAS PubMed.
- J. M. Johnston and E. M. Bulloch, Curr. Opin. Struct. Biol., 2020, 65, 33–41 CrossRef CAS PubMed.
- H. Hamamoto, M. Urai, K. Ishii, J. Yasukawa, A. Paudel, M. Murai, T. Kaji, T. Kuranaga, K. Hamase, T. Katsu, J. Su, T. Adachi, R. Uchida, H. Tomoda, M. Yamada, M. Souma, H. Kurihara, M. Inoue and K. Sekimizu, Nat. Chem. Biol., 2014, 11, 127–133 CrossRef PubMed.
- H. Itoh, K. Tokumoto, T. Kaji, A. Paudel, S. Panthee, H. Hamamoto, K. Sekimizu and M. Inoue, J. Org. Chem., 2018, 83, 6924–6935 CrossRef CAS PubMed.
- M. Sang, H. Wang, Y. Shen, N. Rodrigues De Almeida, M. Conda-Sheridan, S. Li, Y. Li and L. Du, Org. Lett., 2019, 21, 6432–6436 CrossRef CAS PubMed.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. H. Huson, BMC Evol. Biol., 2007, 7, 1–15 CrossRef.
- N. Sakura, T. Itoh, Y. Uchida, K. Ohki, K. Okimura, K. Chiba, Y. Sato and H. Sawanishi, Bull. Chem. Soc. Jpn., 2004, 77, 1915–1924 CrossRef CAS.
- H. Tsubery, I. Ofek, S. Cohen and M. Fridkin, Peptides, 2001, 22, 1675–1681 CrossRef CAS.
- C. T. Walsh, Nat. Prod. Rep., 2023, 40, 326–386 RSC.
- C. S. Neumann, D. G. Fujimori and C. T. Walsh, Chem. Biol., 2008, 15, 99–109 CrossRef CAS PubMed.
- P. Agrawal and D. Mohanty, Bioinformatics, 2021, 37, 603–611 CrossRef CAS.
- C. T. Walsh, Nat. Prod. Rep., 2023, 40(2), 326–386 RSC.
- C. S. Neumann, D. G. Fujimori and C. T. Walsh, Chem. Biol., 2008, 15(2), 99–109 CrossRef CAS.
- P. Agrawal and D. Mohanty, Bioinformatics, 2021, 27(5), 603–611 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4np00043a |
| This journal is © The Royal Society of Chemistry 2025 |