
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced force microscopy as a novel method for the study of microbial nanostructures
Josh
Davies-Jones
 *a,
Philip R.
Davies
*a,
Arthur
Graf
a,
Dan
Hewes
a,
Katja E.
Hill
b and
Michael
Pascoe
ac
*a,
Philip R.
Davies
*a,
Arthur
Graf
a,
Dan
Hewes
a,
Katja E.
Hill
b and
Michael
Pascoe
ac
aCardiff Catalysis Institute, Cardiff School of Chemistry, Cardiff University, Cardiff, CF10 3A, UK. E-mail: Daviesja21@cardiff.ac.uk; Daviespr@cardiff.ac.uk; PascoeMJ@cardiff.ac.uk; GrafA2@cardiff.ac.uk; HewesDG@cardiff.ac.uk
bAdvanced Therapies Group, School of Dentistry, Cardiff University, Cardiff, CF14 4XY, UK. E-mail: Hillke1@cardiff.ac.uk
cSchool of Pharmacy and Pharmaceutical Sciences, Cardiff University, Cardiff, CF10 3BN, UK. E-mail: PascoeMJ@cardiff.ac.uk
First published on 28th November 2023
Abstract
A detailed comparison of the capabilities of electron microscopy and nano-infrared (IR) microscopy for imaging microbial nanostructures has been carried out for the first time. The surface sensitivity, chemical specificity, and non-destructive nature of spectroscopic mapping is shown to offer significant advantages over transmission electron microscopy (TEM) for the study of biological samples. As well as yielding important topographical information, the distribution of amides, lipids, and carbohydrates across cross-sections of bacterial (Escherichia coli, Staphylococcus aureus) and fungal (Candida albicans) cells was demonstrated using PiFM. The unique information derived from this new mode of spectroscopic mapping of the surface chemistry and biology of microbial cell walls and membranes, may provide new insights into fungal/bacterial cell function as well as having potential use in determining mechanisms of antimicrobial resistance, especially those targeting the cell wall.
Introduction
Antimicrobial resistance (AMR) and the spread of multi-drug resistant microorganisms remains one of the major global health threats facing us today, requiring an in-depth understanding of bacterial and fungal properties at the nanoscale level to try and develop strategies to combat them. Until recently, studying the chemistry of bacteria at this scale presented substantial challenges. A diverse array of analytical tools, each with its unique strengths and limitations, including electron microscopy, optical microscopy, vibrational spectroscopy, and force microscopy techniques can be used to achieve this.1–5 This paper introduces an emerging technique, photo-induced force microscopy (PiFM), which combines vibrational spectroscopy with nanoscale topography, as a cutting-edge approach to explore the nanoscale chemistry of individual microbial cells and their sub-cellular features.Vibrational spectroscopy offers the capability of distinguishing between specific biomolecular components which play important roles in a wide range of biological processes, through their characteristic absorption bands in the 900–1900 cm−1 “fingerprint” region of the IR spectrum.6 IR spectroscopy is often combined with optical microscopy to identify the chemistry of physical features e.g. the presence of the aromatic amino acid tyrosine in beta sheets,7,8 which has a characteristic absorption band at about 1655 cm−1 due to vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in its phenol group.9–11 Other macromolecules such as lipids, carbohydrates, and nucleic acids can also be identified by IR spectroscopy. For example, the absorption bands at about 2850 cm−1 and 2950 cm−1 arise from the C–H stretching vibrations of fatty acids and other lipids, while the absorption bands at about 1020 cm−1 and 1080 cm−1 are often attributed to the C–O–C and C–O stretching vibrations of carbohydrates.12–14 The value of this chemically-specific information has been recognised for many years.15–17 However, for optical techniques such as infrared microscopy and confocal Raman microscopy, the diffraction limit of light imposes a maximum resolution of about a μm, thereby preventing the nanoscale imaging necessary to fully characterise biological features.18,19 Furthermore, infrared spectroscopy typically has penetration depths in biological samples of up to 1 μm, and therefore can only provide information averaged over the “bulk” of a sample rather than surface specific information.20
O bond in its phenol group.9–11 Other macromolecules such as lipids, carbohydrates, and nucleic acids can also be identified by IR spectroscopy. For example, the absorption bands at about 2850 cm−1 and 2950 cm−1 arise from the C–H stretching vibrations of fatty acids and other lipids, while the absorption bands at about 1020 cm−1 and 1080 cm−1 are often attributed to the C–O–C and C–O stretching vibrations of carbohydrates.12–14 The value of this chemically-specific information has been recognised for many years.15–17 However, for optical techniques such as infrared microscopy and confocal Raman microscopy, the diffraction limit of light imposes a maximum resolution of about a μm, thereby preventing the nanoscale imaging necessary to fully characterise biological features.18,19 Furthermore, infrared spectroscopy typically has penetration depths in biological samples of up to 1 μm, and therefore can only provide information averaged over the “bulk” of a sample rather than surface specific information.20
One method to circumvent Abbe's diffraction limit is the use of “near-field” methods based on atomic force microscopy (AFM) which has proven useful in the imaging of the mechanical properties of cells and interactions between cells and their environment.21 AFM also has the advantage over other techniques such as transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS), of operating under ambient conditions, with minimal sample preparation needed. What AFM lacks is the chemical specificity needed to understand the distribution of specific molecular components on the sample surface.22,23
PiFM bridges the gap between high spatial resolution and chemical specificity by combining the strengths of AFM and vibrational spectroscopy24 and, as a result, holds immense potential for unveiling the chemistry of cellular surfaces at the nanometre scale. The technique also operates non-destructively, eliminating the need for high-energy electrons or other damaging radiation forms. Additionally, the system functions under ambient conditions rather than the ultra-high vacuums required for electron microscopies.25 With a lateral resolution of 5 nm and a penetration depth of less than 50 nm, PiFM is uniquely situated to study the nanoscale chemistry of biological samples.24
In AFM, one of the natural vibrational resonances (eigenmodes) of a cantilever with a sharp tip, is used to measure the topography of a sample surface, with the spatial resolution primarily dictated by the sharpness of the tip; usually around about 5 nm.26,27 Advanced techniques like bimodal AFM employ multiple eigenmodes to achieve increased resolution and measure additional properties such as deformation and elastic modulus.28
In contrast, in PiFM, the first eigenmode of the tip is used to measure the surface topography of the sample, while the second eigenmode is employed to detect the photoinduced force generated on the tip by oscillations of species on the surface of the sample. Almajhadi et al.29 demonstrated that the PiFM signal is a result of opto-mechanical damping of the amplitude of the cantilever motion because of the excitation of surface molecules at their vibrational optical resonance.30,31 Since the molecular excitation is a dipole–dipole based phenomenon, the result is a nanometre resolved vibrational spectroscopy that correlates exactly with dipole–dipole based far-field infrared spectroscopy. PiFM differs from other techniques such as photo-thermal infrared microscopy (PTIR or AFM-IR) and nano-FTIR in that it operates in a non-contact mode, measuring van der Waals forces and interactions without physical contact between the tip and the sample.30,31 In contrast, AFM-IR relies on mechanical detection of thermal expansion upon contact, while nano-FTIR uses near-field optics for high-resolution IR spectroscopy. Some of the key aspects of these techniques have been identified in Table 1.
| AFM-IR32 | PiFM24,29 | s-SNOM33 | |
|---|---|---|---|
| Operating mode | Operates in contact mode. The sample is illuminated from below with an IR laser via total internal reflection. When the wavelength of light matches a material absorbance band, the sample expands. This expansion is detected using the AFM cantilever. | Operates in non-contact mode. It relies on tip–sample force interactions. An excitation laser illuminates the sample surface creating the photo-induced force detected via a modulation of the AFM oscillating amplitude. | Based on an apertureless near-field optical microscope design. It utilizes a broadband (white-light) laser source instead of a fixed-wavelength laser. |
| Spatial resolution | ∼50–100 nm. This is because the spatial resolution is not determined by the tip radius alone, but also by the thickness of the sample via mechanical coupling to surrounding material. | ∼5–10 nm, Limited by the tip diameter because the tip-enhanced field profile is much smaller than the physical tip profile. | ∼1–20 nm. The spatial resolution is not determined by the tip radius alone, but also by the thickness of the sample via mechanical coupling to surrounding material. |
| Force detection | Relies on short-range repulsive forces through direct contact. | Background free measurement of attractive van der Waals forces. | Relies on short-range repulsive forces through direct contact. |
Scattering scanning near-field optical microscopy (s-SNOM) is a technique where light is elastically scattered from a sharp AFM tip and optically detected.34,35 SNOM has been used successfully in the study of several soft matter materials at the nanoscale, such as cells, bacteria, and viruses.36–38 While this technique is promising, unlike FM-IR, nano-FTIR and PiFM, it lacks the ability to directly correspond to IR spectroscopy. However, recent applications have seen it successfully paired with IR spectroscopy.39
The potential for AFM-IR in imaging individual cells and deciphering the chemistry of microbial samples has recently been demonstrated by Kochan et al.40,41 They utilised spectral bands from cell wall components to highlight how AFM-IR was able to distinguish between Gram-positive and Gram-negative bacteria, as well as identifying distinct differences in chemical composition between isogenic antibiotic susceptible and resistant Staphylococccus aureus strains, with vancomycin intermediate S. aureus (VISA) and daptomycin resistant cells shown to possess increased non-intracellular carbohydrates and phospholipid content respectively compared to their susceptible counterparts.40,41 Although individual bacterial cells were successfully imaged using AFM-IR, its limited resolution hampered its ability to directly investigate the nanoscale features of the cell membrane. Therefore, while these papers represent significant progress in the field of nano vibrational spectroscopy and imaging, there are still critical gaps in the current research.29 One notable limitation is the under-utilization of the nanoscale resolution of the technique. To date, this aspect (possessed by techniques like TEM) has remained largely unexplored due to the inability of AFM-IR to penetrate the microbial surface. So, an important advantage of PiFM over these applications is its high spectral resolution of 1 cm−1, which can assist in the deconvolution of fine spectral components, which are lost in the 8 cm−1 resolution found in AFM-IR in these applications.40,41
In the present study, we employed conventional TEM preparation methods to highlight the unique potential of PiFM in imaging biological specimens, while directly comparing it to TEM for the first time. We selected three opportunistic pathogens for the study: S. aureus (Gram-positive), Escherichia coli (Gram-negative), and Candida albicans (yeast), as representative cell types.
Materials and methods
Microbial media and culture conditions
Standard strains of S. aureus NCTC 10788, E. coli ATCC 10536 and C. albicans ATCC 10231 were cultured on tryptone soya agar (E&O Laboratories) for 24 hours at 37 °C (bacteria) or 48 hours at 32 °C (C. albicans). Following incubation, isolated colonies were resuspended in sterile tryptone sodium chloride (TSC; 8.5 g L−1 NaCl, 1 g L−1 tryptone) and dispersed by vortexing with 3 mm glass beads. Using a spectrophotometer, A630 was adjusted to 0.5, to yield microbial suspensions of approximately 108 colony forming units (CFU) per ml (bacteria) or 107 CFU per ml (C. albicans).Fixation and embedding of microbial samples
Samples were fixed by adding 1 part cell suspension to 9 parts fixing solution (2 wt% glutaraldehyde in 0.1 M sodium cacodylate, pH 7.2). After 24 hours, fixed suspensions were centrifuged at 100g for 5 minutes. After removal of the supernatant, pellets were resuspended 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with 4% (w/v) molten (50 °C) low melting point agarose (ThermoFisher Scientific) in polypropylene microcentrifuge tubes and allowed to cool. The agarose blocks were cut into 1 mm3 sections and post-fixed in 1% osmium tetroxide for 2 hours. The samples were then washed with distilled water and block stained with uranium acetate (2%) for 2 hours. After washing with distilled water, the blocks were dehydrated in a series of propan-2-ol (50, 70, 90 and 100%) for 15 minutes each. Dehydrated blocks were transferred to 50% TAAB embedding resin (TER; TAAB Laboratories Equipment Ltd) in propan-2-ol for 30 minutes and then transferred to 100% TER for 1 hour, three times. Samples were then embedded in 100% TER at 60 °C for 24 hours in truncated polypropylene moulds (TAAB Laboratories Equipment Ltd). Samples were cut into 100–300 nm thick sections using a diamond knife, collected onto 300 mesh copper grids. Some variations in the thickness of sections can be expected due to differences in physical properties between the resin and biological matter.42,43
:1 with 4% (w/v) molten (50 °C) low melting point agarose (ThermoFisher Scientific) in polypropylene microcentrifuge tubes and allowed to cool. The agarose blocks were cut into 1 mm3 sections and post-fixed in 1% osmium tetroxide for 2 hours. The samples were then washed with distilled water and block stained with uranium acetate (2%) for 2 hours. After washing with distilled water, the blocks were dehydrated in a series of propan-2-ol (50, 70, 90 and 100%) for 15 minutes each. Dehydrated blocks were transferred to 50% TAAB embedding resin (TER; TAAB Laboratories Equipment Ltd) in propan-2-ol for 30 minutes and then transferred to 100% TER for 1 hour, three times. Samples were then embedded in 100% TER at 60 °C for 24 hours in truncated polypropylene moulds (TAAB Laboratories Equipment Ltd). Samples were cut into 100–300 nm thick sections using a diamond knife, collected onto 300 mesh copper grids. Some variations in the thickness of sections can be expected due to differences in physical properties between the resin and biological matter.42,43
Transmission electron microscopy (TEM)
For TEM imaging, the sections were subsequently stained with lead citrate and examined in a JEOL 2100 TEM (Jeol ltd, Japan) at 200 kV. Post-acquisition, the images were processed using ImageJ, a public domain program developed by the National Institutes of Health (NIH) to improve image brightness and contrast.Photoinduced force microscopy (PiFM)
In the PiFM analysis, samples were prepared as unstained sections supported on 300 lines per inch mesh copper grids. Imaging and spectral data were acquired using a Vista One nano-IR microscope & spectrometer (Molecular Vista Inc, USA) equipped with a Bloc 780–1930 cm−1 quantum-cascade laser (QCL) and Vistascan Version 28 (Molecular Vista Inc, USA). Non-contact high-resolution PtIr-coated (NCHR) cantilevers were used in the measurements. These cantilevers were initially sourced from Molecular Vista 3.2 release 18 (Molecular Vista Inc, USA) and had a spring constant of 45 N m−1 and a resonance frequency of 335 kHz.All measurements were conducted on three microbial cells per species in sideband mode, providing a 20 nm penetration depth, with a spectral resolution of 1 cm−1. Cells were selected randomly from those whose cytoplasmic membrane and cytoplasm were clearly apparent at the focal plane. Subsequently, both spectra and images were subjected to analysis using Surfaceworks 3.0 Release 32 (Molecular Vista Inc, USA). Further spectral subtractions were carried out using OMNIC (Thermo Fisher Scientific), and data fitting was performed with CasaXPS version 2.3.2428.44 To gain insights into microbial species differentiation, Principal Component Analysis (PCA) was applied to nine spectra, each derived from an average of 20 repetitions. The spectra were organized into a matrix and subjected to analysis in MATLAB (The MathWorks Inc., 2022, R2022b). The PCA approach in this study was specifically tailored to differentiate microbial species based on their unique spectral features. This approach differs from the PCA technique employed in e.g. Otter et al. (2023), where the primary objective was to generate component maps.45 The distinct aim of our study was to underscore the ability of PiFM to discriminate between microbial species through their individual spectral characteristics, rather than mapping their spatial distribution.
Results and discussion
Comparison of FTIR and PiFM spectroscopy
Conventional antibiotic susceptibility testing using antibiogram tests requires 18–24 hours to yield results, due to limits on the rates of bacterial growth within laboratories. Infrared spectroscopy is currently being developed to try to improve these diagnostic times. IR spectroscopy has been successfully used on urine samples from urinary tract infections to detect susceptibility and resistance to extended spectrum β-lactamases (ESBLs), fluoroquinolones and aminoglycosides in a range of bacteria including E. coli, Klebsiella pneumoniae and Proteus mirabilis46–49 with accuracy levels as high as 89% reported.A comparison of typical IR spectra of the three different cell types with that recorded by PiFM at a specific point on the surface is shown in Fig. 1. The data shows that there is a broad agreement between the FTIR spectra of organic species at cell surfaces reported in the literature50–52 and the PiFM data, although some important differences between the two spectra were observed. For example, PiFM analysis of C. albicans revealed a broad signal at 1220 cm−1, which was absent in the reference spectra and corresponds to C–O stretches in free nucleotides. Since these samples were sectioned, it is possible that PiFM may directly assay intracellular contents, producing higher proportions of typical DNA or nucleotide stretches. This effect was seen across all three of the samples tested. Most of the differences observed between FTIR and PiFM were slight shifts in positions, or the presence of additional peaks in the PiFM spectra within areas which previously produced broad signals. One such example occurs in the 1000–1100 cm−1 region of both the S. aureus and C. albicans spectra, with the PiFM spectra showing 2 peaks where only 1 broad peak was observed within the reference FTIR spectrum, possibly consisting of a combination of the peaks that are separately resolved in the PiFM spectra. The fundamental frequency resolution of PiFM is ∼1 cm−1, similar to that of a standard FTIR spectrometer. The better resolution of the PiFM spectra is therefore likely to be due to the high lateral resolution of the PiFM. Depending on the laser frequency, a typical ATR-FTIR system samples an area between 2.5 to 25 μm in diameter, 250 to 2500 times larger than the area sampled by PiFM. Since many bacteria are typically 1 μm or less in length, FTIR spectra therefore reports an average over the entire cell, whereas PiFM provides information on specific points on the cell, and thus is capable of identifying subtle changes in the micro- and nanostructure of the surface.
 | ||
| Fig. 1 (A) Mean PiFM (red line) and FTIR spectra (black dotted line) for E. coli, S. aureus and C. albicans with reference spectra taken from the literature.50–52 (B) Table comparing PiFM spectra against general FTIR assignments for microbial samples. | ||
Comparison between PiFM and TEM imaging of bacterial and yeast cell sections
For a fair comparison between the two imaging methods, we used the same sample preparation method for both TEM and PiFM, embedding the cells in epoxy resin and cutting sections using a microtome. Since PiFM can only measure the spectral signal of compounds present within the first ∼200 nm of a sample, sectioning the bacteria was necessary to study their cellular contents, although the cellular surface may also be studied without such intrusive manipulation. The PiFM images in Fig. 2 and 3 were found to match well with previously reported ATR-FTIR fingerprints of the three species.50,53,54 With a theoretical resolution of 0.2 nm, TEM produced high resolution images of all three species of microbes examined within this study and, was clearly able to distinguish cellular structures such as the cell wall of S. aureus and the vacuole and nucleus of C. albicans (Fig. 2).55 In contrast, the PiFM images have an approximate pixel size of 5 nm, thus providing both spectral and morphological information. Of particular importance, is the ability of PiFM to image the outer membrane of the Gram-negative bacterium E. coli, which is mostly composed of extracellular polymers and carbohydrates, and appears as a bright blue halo around the cell (Fig. 2A) which is not distinguishable in the TEM image (Fig. 2B). In S. aureus, the cell wall structure is clearly visible in the PiFM images, with the peptidoglycan constituent, comprised of β-(1,4) linked N-acetylglucosamine and N-acetylmuramic acid, shown as lilac in colour (Fig. 2C) in contrast to the TEM image (Fig. 2D). In addition, when studying C. albicans (Fig. 2E), the cell wall is mostly formed of chitin and β (1,3)-glucan and appears as turquoise in colour in the combined image, with the organelles particularly highlighted in the TEM image (Fig. 2F). The background of the PiFM images (which varies between images from blue to lilac) is a consistent signal due to background absorption across the sample resulting from the epoxy resin. This was therefore discounted from our analysis when exploring the relative intensities of each component.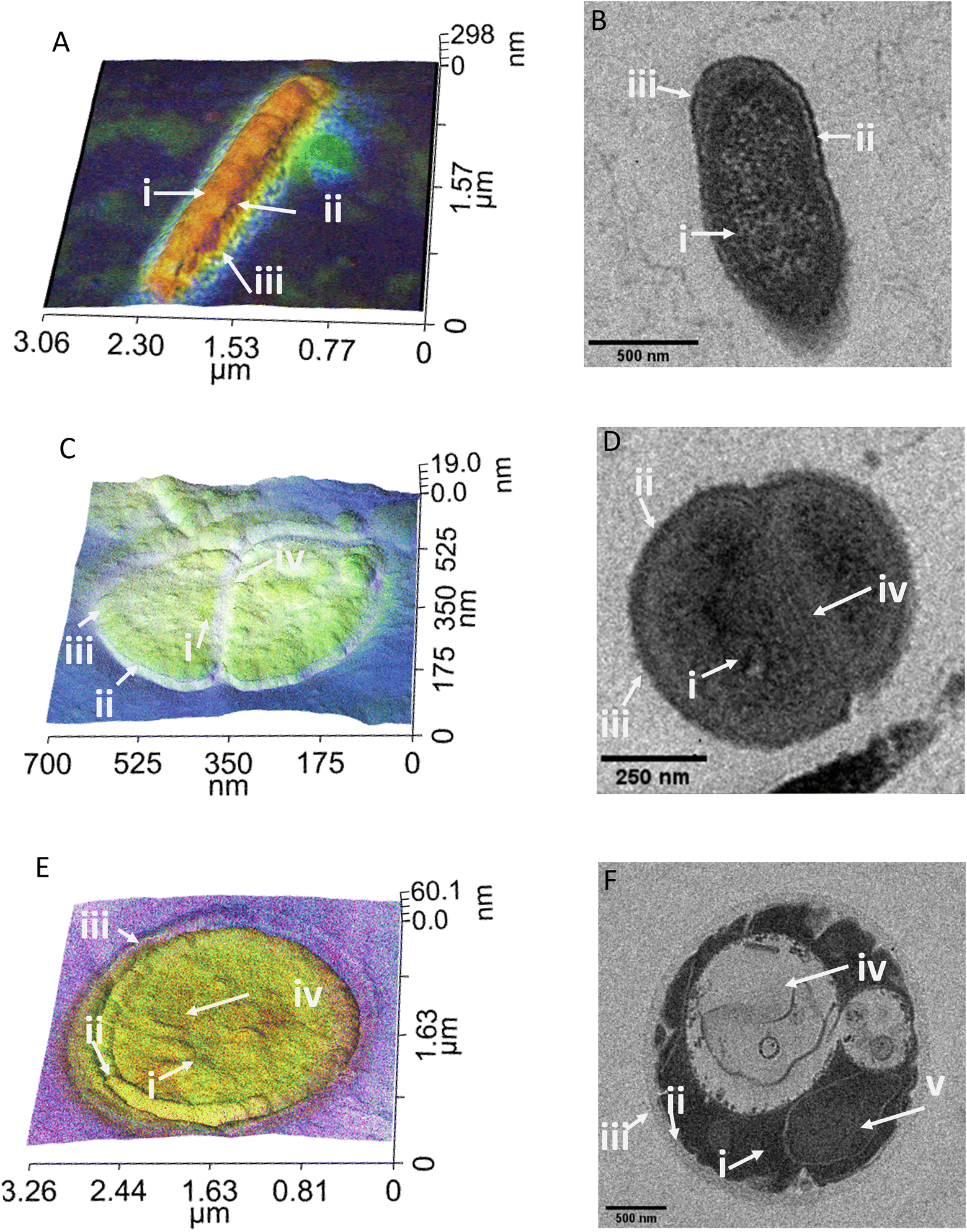 | ||
| Fig. 2 Comparison of PiFM spectral images with transmission electron micrographs (TEM) of cross-sectioned cell preparations of the three different microbes. In the PiFM images (A, C and E), colours correspond to the intensities of peaks at approximately 1665, 1555 and 1100–1075 cm−1, which are assigned to amide I (red), amide II (green) and carbohydrate/phospholipids (blue) respectively. (A and B) E. coli, with arrows identifying (i) nucleoid & cytoplasm, (ii) cytoplasmic membrane, (iii) cell wall, outer membrane & capsule. (C and D) S. aureus with arrows identifying (i) cytoplasm, (ii) plasma membrane, (iii) cell wall (iv) septum. (E and F) C. albicans with arrows identifying (i) cytoplasm, (ii) plasma membrane, (iii) cell wall, (iv) vacuole, (v) nucleus. | ||
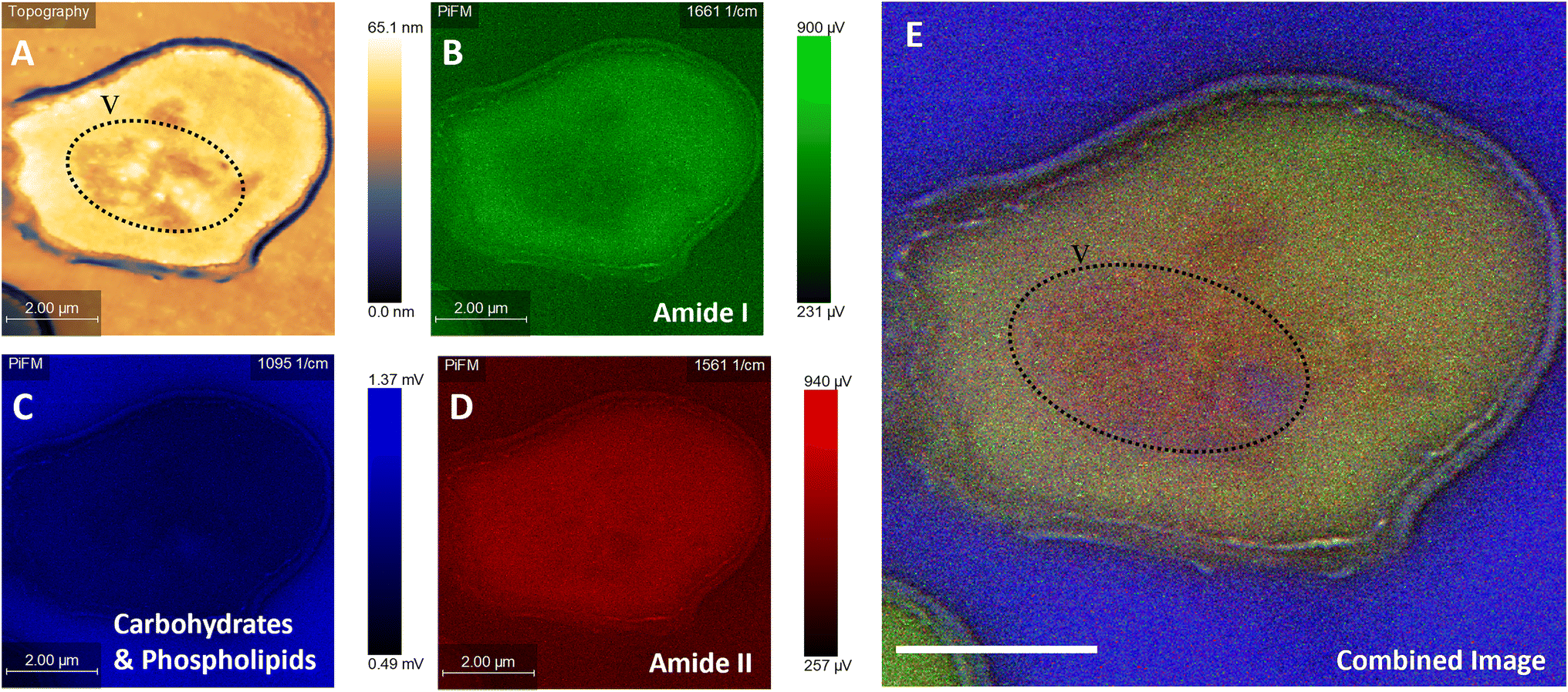 | ||
| Fig. 3 PiFM micrographs and spectral images of a damaged C. albicans cell, the source of the damage is unknown but could be caused through the sectioning procedure. (A) Topographical image, (B) hyperspectral image taken at 1661 cm−1, (C) spectral image taken 1095 cm−1, (D) spectral image taken at 1561 cm−1 and (E) combined image of B–D. The vacuole is marked with “V” and damaged areas of the cell wall are highlighted in yellow. | ||
A particular drawback of PiFM is its inability to penetrate through the entire sample, resulting in a less clear differentiation between the cytoplasm and the cellular machinery of e.g., C. albicans (Fig. 2). However, this is not always the case as illustrated in Fig. 3, where several organelles can be observed (outlined with a dotted line), perhaps because they are close to the cell surface. Bacteria are simple prokaryotic cells with no nucleus (the nucleoid being an irregularly shaped region within the cell containing the genetic material), whilst yeasts are eukaryotic, possessing a differentiated nucleus and a variety of other organelles. Vacuoles occupy up to 20% of the C. albicans cell and are thought to be involved in many cellular functions which enhance its ability to adapt to stress and changes in host environment during infection, so the ability to image and study these directly is important.56 The vacuoles are filled with hydrolase enzymes, amino acids, cations and are the main store of polyphosphates within a yeast cell.57,58
Fig. 3 illustrates the usefulness of PiFM for the direct study of organelles within the yeast cell, demonstrated by the large area of heterogenous chemistry in the cell centre occupying roughly 25% of the intracellular space, imaged as a drop in topography of ∼20 nm. It is hypothesised that this area of the cell (identified with a dotted line) is a vacuole, which was filled with high ratios of amide II and phosphate contributions (Fig. 3A and E). This matches well with the theoretical content of a vacuole; the blue areas corresponding to polyphosphate stores and the red corresponding to amines (Fig. 3C and D). Surprisingly, a lower amide I contribution was also noted in this area (Fig. 3B). Amino acids such as lysine, glycine and tyrosine are known to adsorb in the 1620 and 1550 cm−1 region for amide I and amide II respectively. It could therefore be postulated that these areas may correspond to dissociated states of free amino acids and proteins present within the acidic vacuole of the yeast cell resulting from subtle changes in their bonding, thus reducing the contribution of the amide I peak, or perhaps shifting it to a lower wavenumber than 1660 cm−1.
The cell in Fig. 3 is clearly damaged, with the plasma membrane noticeably pulled away from the cell wall, (seen as the light blue line running 200 nm parallel to the cell wall). The plasma membrane is a mosaic of phospholipid molecules and indirectly bound proteins.59,60 In areas where the wall and membrane were damaged, there appears to be a higher proportion of the 1095 cm−1 contribution, compared to areas of the cell with a more rounded and intact plasma membrane. In contrast, in areas of greater plasma membrane integrity, larger components of amides I and II were noted. The areas of damage represent a significant shift in phospholipid organisation, resulting in a higher contribution to the 1095 cm−1 signal from PO4. Hence, the ability of the PiFM to directly image and characterise the chemistry of different organelles and areas of the cell is a significant advantage over that of TEM, offering label-free imaging and avoiding the need for extraction of cellular contents (Fig. 3E).
Principal component analysis (PCA)
To assess the ability of PiFM to distinguish between different microbial species, we conducted both PCA and hierarchical cluster analyses on the vibrational spectra recorded from the different microbes (Fig. 4). Both analyses showed that the system was effective in differentiating between the three species studied. The dendrogram (Fig. 4A) provides a visual representation of the hierarchical cluster analysis performed on the vibrational spectra of the different microbial species. A dendrogram shows the relationship and similarities between objects or clusters. In this case, it demonstrates the dissimilarity or proximity between the microbial species studied. The “distance” on the x-axis of the dendrogram represents the measure of dissimilarity or proximity between the objects or clusters, successfully illustrating the similarity of vibrational spectra within each microbial species. The linkage function group objects are based on their closest distances, forming clusters, and assigning unique index values to the newly formed clusters. In this dendrogram, the clustering shows that PiFM could effectively distinguish between bacteria and fungi, as well as between Gram-positive and Gram-negative bacterial species. It is important to note that, whilst PCA is a useful technique for demonstrating differences in data sets, further exploration of different species and growth conditions is necessary to fully understand the ability of PiFM to differentiate between microbial species. Factors such as pH, temperature, and culture conditions can all influence the biochemical profile of microorganisms, and individual differences between microbes of the same genera and species should also be considered. Therefore, further analysis and method development is required to validate the effectiveness of the technique for microbial species differentiation.61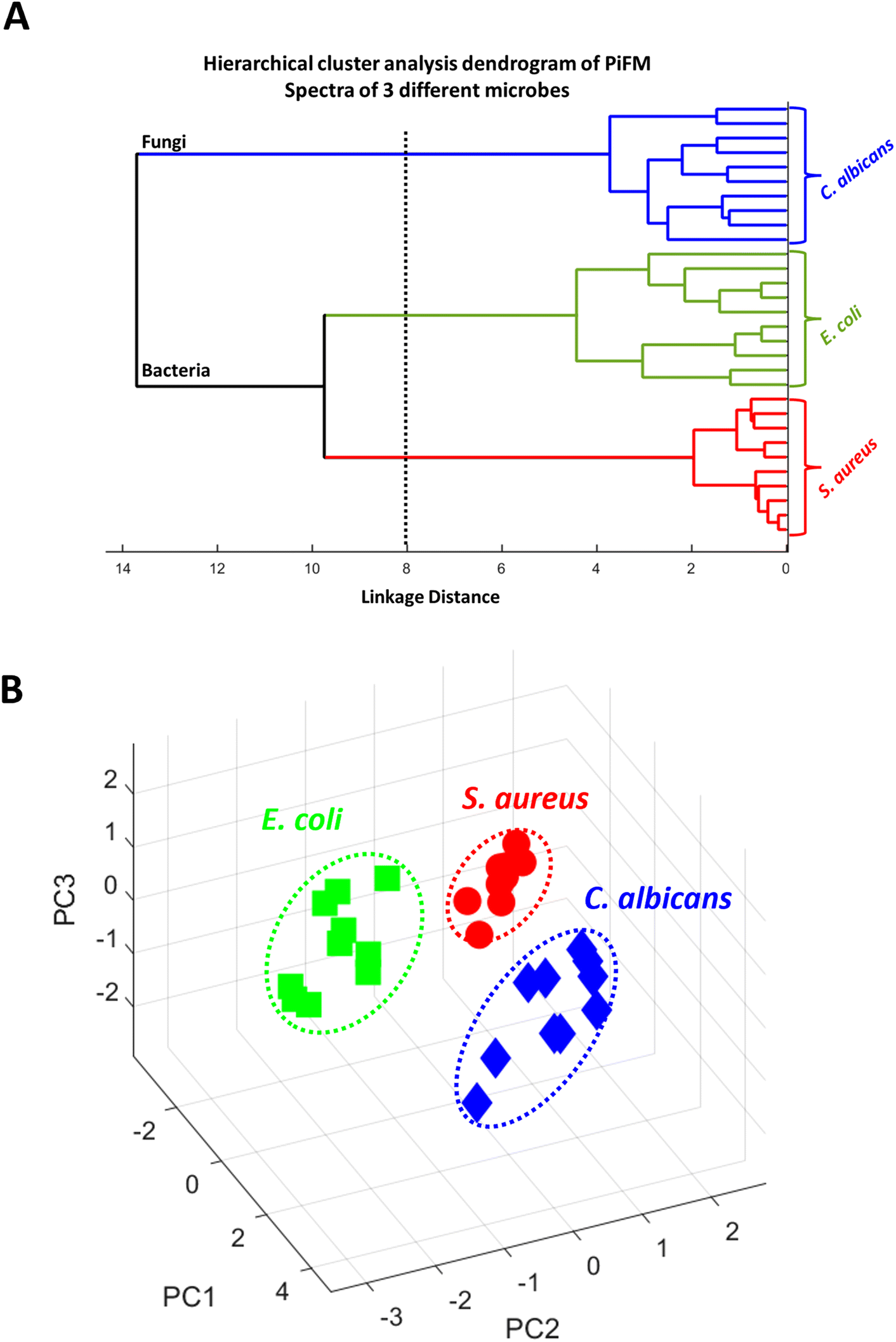 | ||
| Fig. 4 (A) Hierarchical cluster plot dendrogram analyses of the vibrational spectra recorded from the different microbes C. albicans, E. coli and S. aureus (n = 10). The “distance” on the x-axis of a dendrogram represents the measure of dissimilarity or proximity between objects or clusters. (B) Principal component (PC) analysis of the data in A for C. albicans, E. coli and S. aureus. PC1, PC2 and PC3 representing 53%, 21% and 12% of the score respectively. | ||
PCA illustrated a few key areas within the spectra that differed between the three microbial species (Fig. 4B). PC1 was responsible for 53% of the differentiation of the data with particularly interesting differentiation coming in the 1669, 1511, 1225 and 1190 cm−1 regions. These correspond to changes in the amide I, amide II, free nucleotides, and carbohydrates respectively, implying that the majority of PC1 arose from changes in the proteins and amino acids present in each species. PC2 (21%) was mostly characterised by changes in the 1730, 1595, 1425 and 1090 cm−1 regions, of which the majority can be attributed to changes in the lipid and phospholipid structures of the microbes. PC3 only corresponded to 12% of the differentiation between the species, but was mostly characterised by peaks in the 1120, 1295, 1400 and 1730 cm−1 regions which are only loosely connected. Here, regions 1120 cm−1 and 1400 cm−1 can be attributed to changes in the structure of polysaccharides such as chitin or glucans, and the peaks at 1295 cm−1 and 1730 cm−1 probably correspond to changes in the amide III and lipid esters respectively. A mix of contributions characterised by the carbohydrates, proteins and lipids is not surprising since they correspond to a high proportion of the composition of a microbial sample, and with large changes in side-chains, conformation and functionality, it was expected that these biomolecules would be very important for differentiation between the three cell types.62,63 Although these results are extremely encouraging, comparisons between microbes of the same species have yet to be performed to confirm the validity of the technique, demonstrating that further analysis and method development is still required.
Spectral imaging
The most significant advantage of PiFM for the analysis of microbes is demonstrated by the IR spectral profiles across the different samples tested at a spatial resolution of <20 nm (Fig. 5–7). The cell wall and cytoplasmic membrane of C. albicans has been widely studied in recent years, and its structure described as sequential layers of mannoproteins (outer layer) and glucans mixed with chitins (inner layer) followed by a cell membrane mostly constructed from phospholipids.60,64 PiFM spectra (Fig. 5A–D) were taken at four specific sites of the cell, namely: the outer cell wall, the inner cell wall, the cell membrane, and the cytoplasm (Fig. 5E). However, these spectra did not exhibit the significant differences previously observed in the spectral images. This may be due to the data processing performed to remove peaks corresponding to the epoxy resin, causing a reduction in the sensitivity of the PiFM spectra. Nevertheless, some notable key areas of difference still support the changes seen within the spectral images.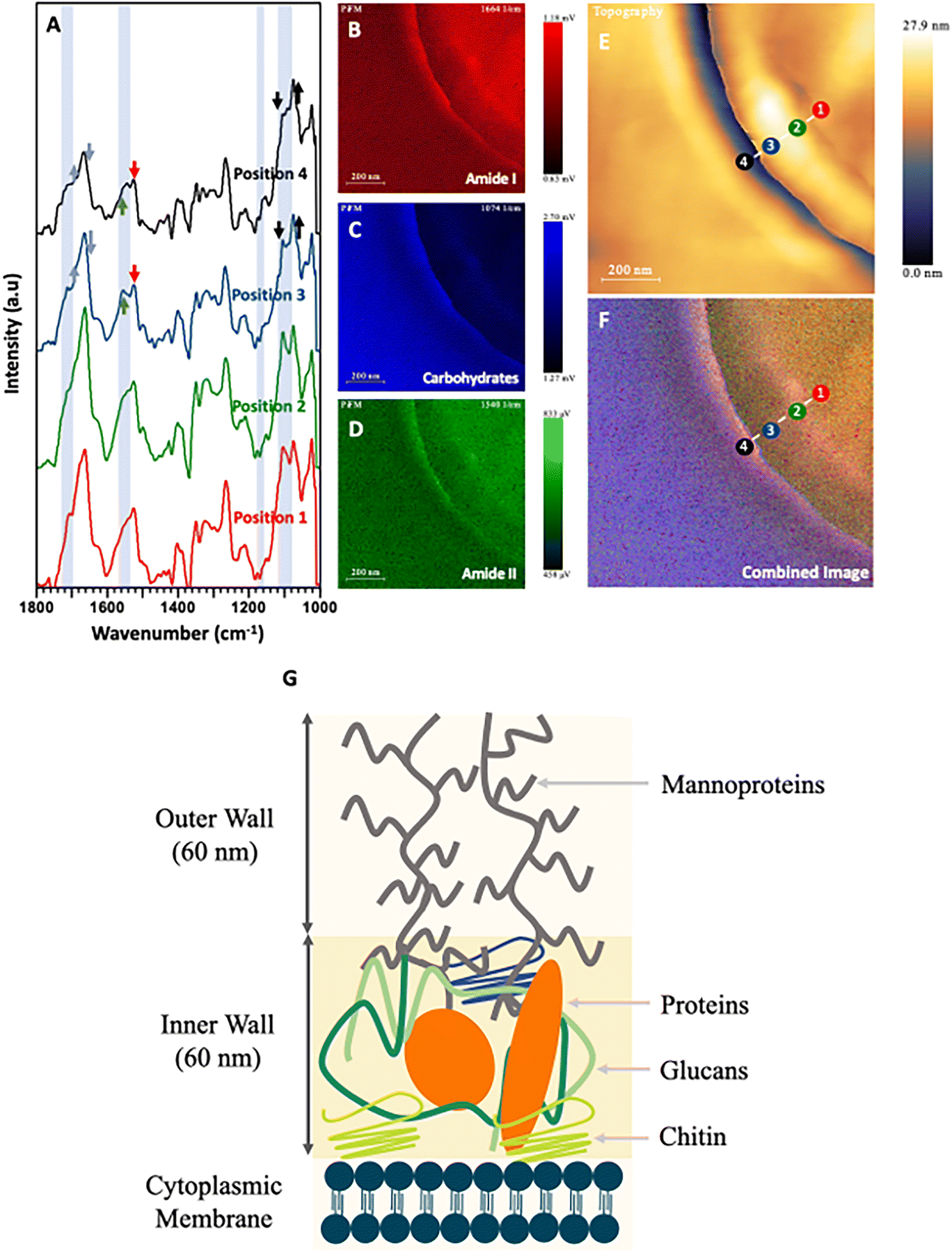 | ||
| Fig. 5 (A) PiFM spectra taken from four positions (1 to 4) across the cell wall of a sectioned C. albicans cell where each position and colour correspond to the indicated position and colour in (E) and (F). (B) Spectral map of 1664 cm−1 intensity. (C) Spectral map of 1074 cm−1 intensity. (D) Spectral map of 1540 cm−1 intensity. (E) Topographical map of C. albicans across the cell wall. (F) Combined image of B–D. (G) Diagram representing the cell wall of C. albicans based on Lenardon et al. (2020).65 | ||
 | ||
| Fig. 6 (A) PiFM spectra taken from four positions (1 to 4) across a single E. coli cell section, where each position and colour correspond to the indicated position and colour in (E) and (F). (B) Spectral map of 1664 cm−1 intensity. (C) Spectral map of 1074 cm−1 intensity. (D) Spectral map of 1540 cm−1 intensity. (E) Topographical map of E. coli across the cell wall. (F) Combined image of B–D. (G) Diagram representing the cell wall of Gram-negative bacteria Clifton et al. (2013) and Pajerski et al. (2019).66,67 | ||
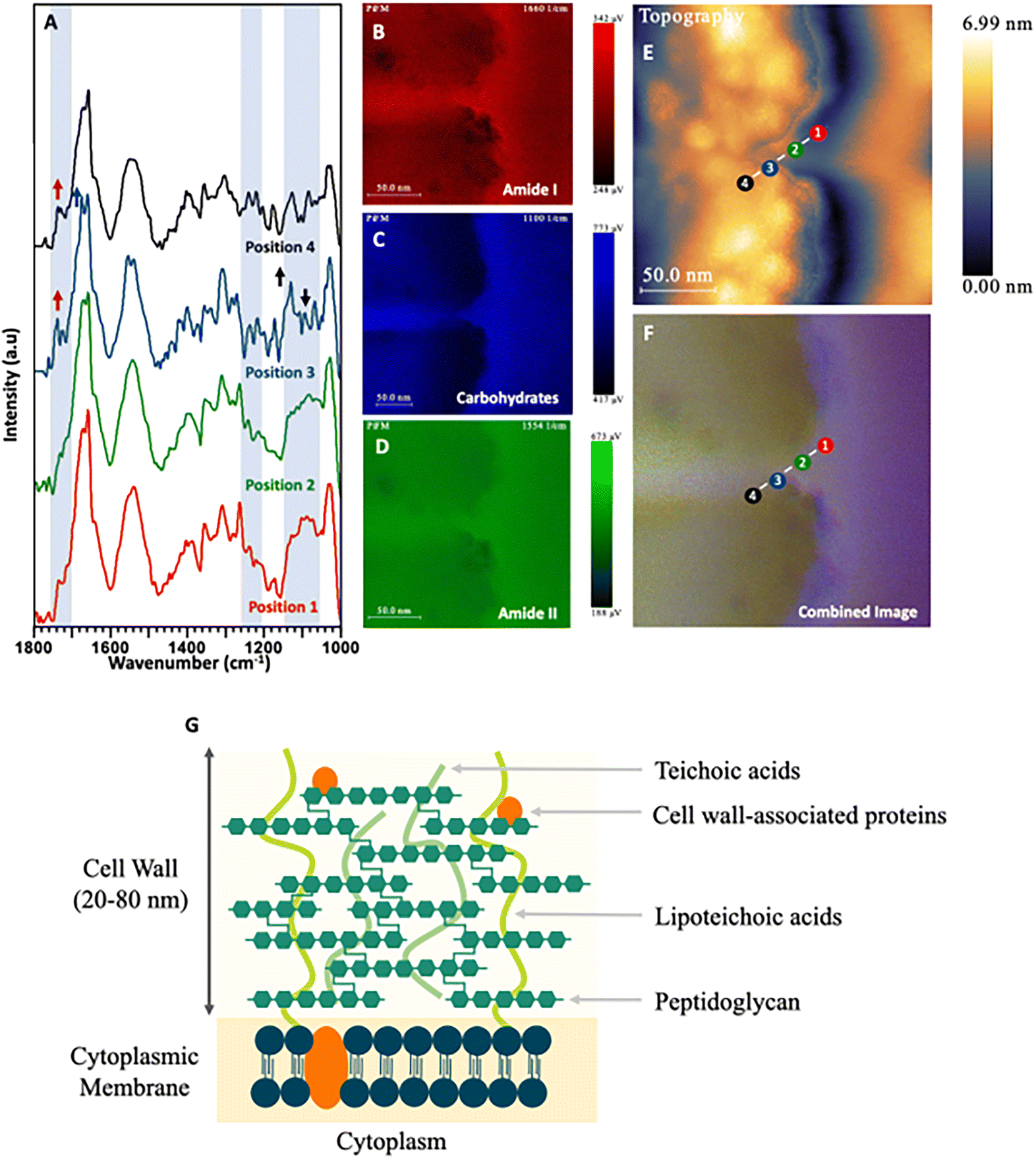 | ||
| Fig. 7 (A) PiFM spectra taken from four positions (1 to 4) across the cell wall and cytoplasmic membrane of a S. aureus cell section where each position and colour corresponds to the indicated position and colour in (E) and (F). (B) Spectral map of 1664 cm−1 intensity. (C) Spectral map of 1074 cm−1 intensity. (D) Spectral map of 1540 cm−1 intensity. (E) Topographical map of S. aureus across the cell wall. (F) Combined image of B–D. (G) Diagram representing the cell wall of Gram-positive bacteria based on Lovering et al. (2010) and Pajerski et al. (2019).67,68 | ||
Firstly, positions 3 and 4 at the outer and inner cell wall showed higher absorptions in the 1025, 1095 and 1110 cm−1 regions when compared to the amide I region. These correspond to glucan polysaccharides and phospholipids. Position 4 shows a large shoulder at 1735 cm−1 corresponding to esters present within the phospholipids. Furthermore, a peak at 1225 cm−1 shows increased levels of intensity towards the centre of the cell. This corresponds to free nucleotides present within the cytoplasm for DNA and RNA replication. The last, most notable difference in the PiFM spectra is demonstrated at position 4 (outer cell wall). Here, two components corresponding to the amide II region at 1530 and 1565 cm−1 were observed. Region 1530 cm−1 displayed a relative increase in intensity compared to region 1565 cm−1 when progressing from position 1 to position 4. While the cause of this change is not clear, it is likely due to changes in the conformation of the amides and proteins present within the inner and outer cell wall. These differences between different structures can clearly be seen in the combined image (Fig. 5F). The outermost layer of the cell wall is characterised as being rich in glycoproteins (mannoprotein), (predominantly coloured in red and green) corresponding to the amide I and amide II functionalities present in the mannoprotein coating. The next layer (inner cell wall) is mostly comprised of polysaccharides. This can be observed in the ratio between the amide peaks and those of the 1074 cm−1 polysaccharide peak, (which mostly corresponds to phosphate within the phospholipids) but will also have contributions from the beta glucans forming the structural component of the cell wall. The next (cell membrane) layer is a matrix of glucans, proteins, and chitin65,69 (which appears as a strong red colour in the combined image), corresponding to the additional amide contribution from the proteins in this region. Lastly, a thin blue line (between positions 1 & 2) can be observed corresponding to the phospholipids present in the cytoplasmic membrane, before moving into the cytoplasmic region of the fungal cell.
While the changes in the PiFM spectra of the yeast C. albicans were not particularly marked, this was not the case for the bacterium E. coli (Fig. 6A–D). Positions 1 & 4 were on the outer membrane of the bacterium, while positions 2 & 3 were towards the centre of the cell, close to the nucleoid and cytoplasm (Fig. 6E and F). The structure of the Gram-negative bacterial cell envelope is well understood and consists of an inner (cytoplasmic) and outer membrane comprised of lipid bilayers, which encapsulate an intermembrane space (periplasm) containing a thin loosely cross-linked peptidoglycan layer.70,71 The cell wall of Gram-negative bacteria is considerably thinner than that of Gram-positive species, with the peptidoglycan anchored to the outer membrane via murein lipoprotein, (LPP) which is the most abundant protein in E. coli cells.72
The outer membrane is composed of lipopolysaccharide (LPS) and phospholipids. The LPS layer is formed of a long polysaccharide chain (O antigen) which is embedded into the outer membrane via a phospholipid called lipid A.73,74 Consequently, the intense and broad peaks centred at approximately 1100 cm−1 and 1235 cm−1 may be ascribed to both the carbohydrate rich O antigen of LPS and P–O stretching in lipid A, as reported by Barkleit et al.75 these areas being visualised as a blue halo in the outermost layer of the cell (Fig. 6). The PiFM spectra taken at positions 1 & 4 show intense peaks at both 1095 cm−1 and 1235 cm−1, again in direct agreement with Barkleit et al.75 There is an intensity contribution from amide I and amide II in this region, which is to be expected (and was also seen in the IR signal) and probably corresponds to outer membrane proteins. When comparing the intensities of the LPS to the amide I and II, it was found to only be localised around the outermost layer. As the peptidoglycan layer is closely associated with the outer membrane and is comprised of complex carbohydrates41 which absorb in similar areas to that of the LPS, (at 1105 and 1060 cm−1), this signal probably contributes to the intensity observed in the 1095 cm−1 region (Fig. 6C). The inner part of the cell corresponds to the cytoplasm and nucleoid and was almost entirely devoid of any 1095 cm−1 intensity. Instead, this area was characterised by large contributions from amide I and amide II bands corresponding to the proteins and DNA present within the bacterium.
In contrast to Gram-negative bacteria, Gram-positive species such as S. aureus lack an outer membrane and do not produce LPS. Instead, the single (lipid-rich) cytoplasmic membrane is surrounded by a thick cell wall comprised of peptidoglycan and negatively charged teichoic acids (TA) to which it is anchored (Fig. 7A–D). Positions 1 & 2 of the PiFM micrograph (Fig. 7E and F) sit upon this region, with the chemistry at these positions found to be very similar, with a broad peak between 1100–1050 cm−1 corresponding to the membrane phospholipids. In contrast to E. coli, however, a change in the intensity and full width half maxima of the polysaccharide areas was observed, which may be ascribed to the TA component of the cell wall (visible as a purple halo around the cell and along the septum between the dividing daughter cells). This layer is as distinct in the spectral map as in the topography image (Fig. 2C). The major difference in the transition between positions 1 & 2 and 3 & 4 is the emergence of a sharp peak at 1120 cm−1 and a shoulder at 1735 cm−1; these being ascribable to glycogen and lipoproteins respectively. Although positions 3 & 4 sit inside the cytoplasm of the S. aureus cell, they remain on the inner edge of the cytoplasmic membrane, which may explain the only slight variation in the spectra. The spectral imaging shows clearly that the inside of the bacteria is again mostly formed of amide I and amide II from the proteins and constituents of the cell within the cytoplasm.
This study successfully analysed Gram-positive, Gram-negative and yeast cells using PiFM, yielding high-resolution 3-dimensional topographical images and direct chemical mapping of specific points across the cell surface. Although bacterial peptidoglycan isolated from Enterococcus faecium has previously been analysed using PiFM (Wang et al. 2022), whole cell preparations as shown here have not been studied before.76 Employing techniques that can visualise the structural biology of a cell can be used to aid our understanding of AMR, as well as having the potential to facilitate the development of new strategies to overcome it. Whilst this study investigated the structure of strains which were known to be susceptible to antibiotics, antimicrobials such as β-lactams, vancomycin, polymyxins and a number of antimicrobial peptides are known to target cell surface structures.77,78 Using PiFM, treatment effects on these structures may now be potentially visualised and studied. Additionally, AMR to these antimicrobials could also similarly be studied, providing important, previously unseen insights into the biochemical and structural changes to the cell surface which may have arisen as a consequence of resistance. Only by more fully understanding AMR can we hope to combat its catastrophic effects.
Conclusion
This study demonstrates the powerful ability of PiFM to study the nanostructure of microbial samples directly, with applications as wide as colony formation behaviour and mechanisms of antimicrobial resistance. While TEM can provide higher resolution imaging, the molecular information provided by PiFM is particularly informative on the chemistry of the system. We have demonstrated that PiFM is able to precisely image the fine and complex biochemistry of cells, directly corroborating the findings of the established literature without the need for either labels or staining. We believe that this approach will become an important tool for the direct study of cell membranes, organelles, and artificial cell systems; the underlying biochemistry of which play key roles in cellular homeostasis, host–pathogen interactions, and susceptibility to antimicrobial agents.Author contributions
JDJ – conceptualisation, methodology, validation, investigation, data curation, writing, funding acquisition. PRD – conceptualisation, supervision, writing – review and edit and funding acquisition. AG – writing review and methodology. DH – methodology and visualization. KH – supervision, writing – review and editing. MP – methodology, validation, and investigationConflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Christopher Von Ruhland for embedding samples. The PiFM spectrometer was acquired with the EPSRC grant EP/V05399X/1. The European Regional Development Fund (ERDF) and the Welsh European Funding Office (WEFO) part-funded the Cardiff Catalysis Institute Microscopy facility. This manuscript was produced with the assistance of a large language model (LLM).References
- K. Kochan, C. Nethercott, J. Taghavimoghaddam, Z. Richardson, E. Lai, S. A. Crawford, A. Y. Peleg, B. R. Wood and P. Heraud, Anal. Chem., 2020, 92, 8235–8243 CrossRef CAS PubMed.
- J. Zahller and P. S. Stewart, Antimicrob. Agents Chemother., 2002, 46, 2679 CrossRef CAS.
- A. Hisada, E. Matsumoto, R. Hirano, M. Konomi, J. Y. Bou Khalil, D. Raoult and Y. Ominami, Sci. Rep., 2023, 13, 11258 CrossRef CAS PubMed.
- T. Lithgow, C. J. Stubenrauch and M. P. H. Stumpf, Nat. Rev. Microbiol., 2023, 21, 502–518 CrossRef CAS PubMed.
- R. S. Brzozowski, M. L. White and P. J. Eswara, J. Visualized Exp., 2019, 153 Search PubMed.
- C. L. M. Morais, K. M. G. Lima, M. Singh and F. L. Martin, Nat. Protoc., 2020, 15, 2143–2162 CrossRef CAS PubMed.
- S. Koide and S. S. Sidhu, ACS Chem. Biol., 2009, 4, 325 CrossRef CAS.
- N. K. Tonks, Nat. Rev. Mol. Cell Biol., 2006, 7, 833–846 CrossRef CAS.
- A. Martínez, J. Haavik, T. Flatmark, J. L. R. Arrondo and A. Muga, J. Biol. Chem., 1996, 271, 19737–19742 CrossRef.
- Y. Wang, Y. Chang, L. Yin, Y. Xue, Z. Li and C. Xue, Food Bioprocess Technol., 2016, 9, 621–627 CrossRef CAS.
- S. A. Bhat and S. Ahmad, J. Mol. Struct., 2016, 1105, 169–177 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS.
- D. Chapman, J. Am. Oil Chem. Soc., 1965, 42, 353–371 CrossRef CAS.
- E. Wiercigroch, E. Szafraniec, K. Czamara, M. Z. Pacia, K. Majzner, K. Kochan, A. Kaczor, M. Baranska and K. Malek, Spectrochim. Acta, Part A, 2017, 185, 317–335 CrossRef CAS PubMed.
- D. Cozzolino, Food Anal. Methods, 2022, 15, 1390–1396 CrossRef.
- I. Pupeza, M. Huber, M. Trubetskov, W. Schweinberger, S. A. Hussain, C. Hofer, K. Fritsch, M. Poetzlberger, L. Vamos, E. Fill, T. Amotchkina, K. V. Kepesidis, A. Apolonski, N. Karpowicz, V. Pervak, O. Pronin, F. Fleischmann, A. Azzeer, M. Žigman and F. Krausz, Nature, 2020, 577, 52–59 CrossRef CAS PubMed.
- C. R. Baiz, B. Błasiak, J. Bredenbeck, M. Cho, J. H. Choi, S. A. Corcelli, A. G. Dijkstra, C. J. Feng, S. Garrett-Roe, N. H. Ge, M. W. D. Hanson-Heine, J. D. Hirst, T. L. C. Jansen, K. Kwac, K. J. Kubarych, C. H. Londergan, H. Maekawa, M. Reppert, S. Saito, S. Roy, J. L. Skinner, G. Stock, J. E. Straub, M. C. Thielges, K. Tominaga, A. Tokmakoff, H. Torii, L. Wang, L. J. Webb and M. T. Zanni, Chem. Rev., 2020, 120, 7152–7218 CrossRef CAS PubMed.
- Y. Phal, L. Pfister, P. S. Carney and R. Bhargava, J. Phys. Chem. C, 2022, 126, 9777–9783 CrossRef CAS.
- E. Abbe, Arch. Mikrosk. Anat., 1873, 9, 413–418 CrossRef.
- P. Lasch and D. Naumann, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 814–829 CrossRef CAS PubMed.
- M. Krieg, G. Fläschner, D. Alsteens, B. M. Gaub, W. H. Roos, G. J. L. Wuite, H. E. Gaub, C. Gerber, Y. F. Dufrêne and D. J. Müller, Nat. Rev. Phys., 2019, 1, 41–57 CrossRef.
- K. Umeda, S. J. Mcarthur and N. Kodera, Microscopy, 2023, 72, 151–161 CrossRef CAS PubMed.
- A. Miranda, A. I. Gómez-Varela, A. Stylianou, L. M. Hirvonen, H. Sánchez and P. A. A. De Beule, Nanoscale, 2021, 13, 2082–2099 RSC.
- J. A. Davies-Jones and P. R. Davies, Mater. Chem. Front., 2022, 6, 1552–1573 RSC.
- L. M. Otter, M. W. Förster, E. Belousova, P. O'Reilly, D. Nowak, S. Park, S. Clark, S. F. Foley and D. E. Jacob, Geostand. Geoanal. Res., 2021, 45, 5–27 CrossRef CAS.
- A. M. Joshua, G. Cheng and E. V. Lau, Appl. Surf. Sci. Adv., 2023, 17, 100448 CrossRef.
- G. M. McClelland, R. Erlandsson and S. Chiang, Rev. Prog. Quant. Nondestr. Eval., 1987, 1307–1314 Search PubMed.
- R. Garcia and R. Proksch, Eur. Polym. J., 2013, 49, 1897–1906 CrossRef CAS.
- M. A. Almajhadi, S. M. A. Uddin and H. K. Wickramasinghe, Nat. Commun., 2020, 11, 5691 CrossRef CAS PubMed.
- D. Nowak, W. Morrison, H. K. Wickramasinghe, J. Jahng, E. Potma, L. Wan, R. Ruiz, T. R. Albrecht, K. Schmidt, J. Frommer, D. P. Sanders and S. Park, Sci. Adv., 2016, 2, e1501571 CrossRef.
- A. A. Sifat, J. Jahng and E. O. Potma, Chem. Soc. Rev., 2022, 51, 4208–4222 RSC.
- J. Mathurin, A. Deniset-Besseau, D. Bazin, E. Dartois, M. Wagner and A. Dazzi, J. Appl. Phys., 2022, 131, 10901 CrossRef CAS.
- D. V. Kazantsev, E. V. Kuznetsov, S. V. Timofeev, A. V. Shelaev and E. A. Kazantseva, Phys.-Usp., 2017, 60, 259 CrossRef CAS.
- M. Moo-Young, Comprehensive Biotechnology, 2019, ch. 1.41, pp. 561–571 Search PubMed.
- A. Lewis, A. Radko, N. Ben Ami, D. Palanker and K. Lieberman, Trends Cell Biol., 1999, 9, 70–73 CrossRef CAS.
- A. Rygula, T. Oleszkiewicz, E. Grzebelus, M. Z. Pacia, M. Baranska and R. Baranski, Spectrochim. Acta, Part A, 2018, 197, 47–55 CrossRef CAS PubMed.
- J. Wang, L. Peng, D. Han, T. Zheng, T. Chang and H. L. Cui, Front. Microbiol., 2023, 14, 1195448 CrossRef PubMed.
- S. Gamage, M. Howard, H. Makita, B. Cross, G. Hastings, M. Luo and Y. Abate, PLoS One, 2018, 13, e0199112 CrossRef PubMed.
- Q. Zhu, R. Zhou, J. Liu, J. Sun and Q. Wang, Nanomaterials, 2021, 11, 1353 CrossRef CAS PubMed.
- K. Kochan, C. Nethercott, D. Perez-Guaita, J. H. Jiang, A. Y. Peleg, B. R. Wood and P. Heraud, Anal. Chem., 2019, 91, 15397–15403 CrossRef CAS PubMed.
- K. Kochan, D. Perez-Guaita, J. Pissang, J. H. Jiang, A. Y. Peleg, D. McNaughton, P. Heraud and B. R. Wood, J. R. Soc., Interface, 2018, 15, 20180115 CrossRef.
- A. P. Fellows, M. T. L. Casford and P. B. Davies, J. Microsc., 2021, 284, 189–202 CrossRef CAS PubMed.
- N. Matsko and M. Mueller, J. Struct. Biol., 2004, 146, 334–343 CrossRef CAS.
- N. Fairley, CasaXPS Manual: 2.3.15 Spectroscopy, 2009 Search PubMed.
- L. M. Otter, K. Eder, M. R. Kilburn, L. Yang, P. O’Reilly, D. B. Nowak, J. M. Cairney and D. E. Jacob, Nat. Commun., 2023, 14, 2254 CrossRef CAS PubMed.
- M. Suleiman, G. Abu-Aqil, U. Sharaha, K. Riesenberg, O. Sagi, I. Lapidot, M. Huleihel and A. Salman, Analyst, 2021, 146, 1421–1429 RSC.
- G. Abu-Aqil, M. Suleiman, U. Sharaha, L. Nesher, I. Lapidot, A. Salman and M. Huleihel, Spectrochim. Acta, Part A, 2023, 295, 122634 CrossRef CAS PubMed.
- U. Sharaha, M. Suleiman, G. Abu-Aqil, K. Riesenberg, I. Lapidot, A. Salman and M. Huleihel, Anal. Chem., 2021, 93, 13426–13433 CrossRef CAS PubMed.
- U. Sharaha, G. Abu-Aqil, M. Suleiman, K. Riesenberg, I. Lapidot, M. Huleihel and A. Salman, J. Biophotonics, 2023, 16, e202200198 CrossRef CAS PubMed.
- M. J. Fiołka, S. Mieszawska, P. Czaplewska, A. Szymańska, K. Stępnik, W. Sofińska-Chmiel, T. Buchwald and K. Lewtak, Sci. Rep., 2020, 10, 16352 CrossRef.
- A. Dazzi, R. Prazeres, F. Glotin and J. M. Ortega, Infrared Phys. Technol., 2006, 49, 113–121 CrossRef CAS.
- S. Wongthong, P. Tippayawat, M. Wongwattanakul, P. Poung-ngern, L. Wonglakorn, A. Chanawong, P. Heraud and A. Lulitanond, World J. Microbiol. Biotechnol., 2020, 36, 1–12 CrossRef.
- B. Feng, H. Shen, F. Yang, J. Yan, S. Yang, N. Gan, H. Shi, S. Yu and L. Wang, Spectrochim. Acta, Part A, 2022, 279, 121369 CrossRef CAS PubMed.
- H. Lamprell, G. Mazerolles, A. Kodjo, J. F. Chamba, Y. Noël and E. Beuvier, Int. J. Food Microbiol., 2006, 108, 125–129 CrossRef CAS PubMed.
- A. Muller and J. Grazul, J. Electron Microsc., 2001, 50, 219–226 CrossRef.
- Q. Lv, L. Yan and Y. Jiang, Front. Microbiol., 2021, 12, 3774 Search PubMed.
- T. Tsuji and T. Fujimoto, Biochem. Soc. Trans., 2018, 46, 1047–1054 CrossRef CAS.
- S. C. Li and P. M. Kane, Biochim. Biophys. Acta, Mol. Cell Res., 2009, 1793, 650–663 CrossRef CAS.
- J. Suchodolski, J. Muraszko, P. Bernat and A. Krasowska, Microorganisms, 2019, 7, 378 CrossRef CAS PubMed.
- V. Cabezón, A. Llama-Palacios, C. Nombela, L. Monteoliva and C. Gil, Proteomics, 2009, 9, 4770–4786 CrossRef.
- J. Lever, M. Krzywinski and N. Altman, Nat. Methods, 2017, 14, 641–642 CrossRef CAS.
- A. N. Spitsyn, D. V. Utkin, O. S. Kuznetsov, P. S. Erokhin, N. A. Osina and V. I. Kochubei, Opt. Spectrosc., 2021, 129, 135–148 CrossRef CAS.
- D. Alexandrakis, G. Downey and A. G. M. Scannell, J. Agric. Food Chem., 2008, 56, 3431–3437 CrossRef CAS.
- R. Garcia-Rubio, H. C. de Oliveira, J. Rivera and N. Trevijano-Contador, Front. Microbiol., 2020, 10, 2993 CrossRef.
- M. D. Lenardon, P. Sood, H. C. Dorfmueller, A. J. P. Brown and N. A. R. Gow, Cell Surf., 2020, 6, 100047 CrossRef CAS.
- L. A. Clifton, M. W. A. Skoda, E. L. Daulton, A. V. Hughes, A. P. Le Brun, J. H. Lakey and S. A. Holt, J. R. Soc., Interface, 2013, 10, 20130810 CrossRef PubMed.
- W. Pajerski, D. Ochonska, M. Brzychczy-Wloch, P. Indyka, M. Jarosz, M. Golda-Cepa, Z. Sojka and A. Kotarba, J. Nanopart. Res., 2019, 21, 1–12 CrossRef CAS.
- A. L. Lovering, L. Y. C. Lin, E. W. Sewell, T. Spreter, E. D. Brown and N. C. J. Strynadka, Nat. Struct. Mol. Biol., 2010, 17, 582–589 CrossRef CAS.
- A. Cassone, Nat. Rev. Microbiol., 2013, 11, 884–891 CrossRef CAS.
- M. Mathelié-Guinlet, A. T. Asmar, J. F. Collet and Y. F. Dufrêne, Nat. Commun., 2020, 11, 1789 CrossRef.
- T. J. Silhavy, N. Ruiz and D. Kahne, Nat. Rev. Microbiol., 2006, 4, 57–66 CrossRef CAS PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- A. Steimle, I. B. Autenrieth and J. S. Frick, Int. J. Med. Microbiol., 2016, 306, 290–301 CrossRef CAS.
- J. L. Ding and B. Ho, Subcell. Biochem., 2010, 53, 187–208 CAS.
- A. Barkleit, H. Foerstendorf, B. Li, A. Rossberg, H. Moll and G. Bernharda, Dalton Transactions, 2011, 40, 9868–9876 RSC.
- H. Wang, Q. Xie and X. G. Xu, Adv. Drug Delivery Rev., 2022, 180, 114080 CrossRef CAS.
- A. Farkas, G. Maróti, A. Kereszt and É. Kondorosi, Front. Microbiol., 2017, 8, 241211 Search PubMed.
- M. F. Chellat, L. Raguž and R. Riedl, Angew. Chem., Int. Ed., 2016, 55, 6600–6626 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |