
DON encapsulated carbon dot–vesicle conjugate in therapeutic intervention of lung adenocarcinoma by dual targeting of CD44 and SLC1A5†
Afreen
Zaman
,
Aparajita
Ghosh
,
Anup Kumar
Ghosh
and
Prasanta Kumar
Das
 *
*
School of Biological Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata – 700032, India. E-mail: bcpkd@iacs.res.in
First published on 28th October 2024
Abstract
Lung adenocarcinoma, recognized as one of the most formidable malignancies with a dismal prognosis and low survival rates, poses a significant challenge in its treatment. This article delineates the design and development of a carbon dot–vesicle conjugate (HACD–TMAV) for efficient cytotoxicity towards lung cancer cells by target selective delivery of the glutamine inhibitor 6-diazo-5-oxo-L-norleucine (DON) within CD44-enriched A549 cancer cells. HACD–TMAV is composed of hyaluronic acid-based carbon dots (HACDs) and trimesic acid-based vesicles (TMAV), which are bound via electrostatic interactions. TMAVs are formed by positively charged trimesic acid-based amphiphiles through H-type aggregation in water. HACDs were synthesized through a one-step hydrothermal route. The blue-emitting HACD–TMAV conjugate demonstrated selective bioimaging in CD44-overexpressed A549 lung cancer cells due to specific ligand–receptor interactions between HA and CD44. HACD–TMAV exhibited notably improved DON loading efficiency compared to individual nano-vehicles. HACD–TMAV-DON exhibited remarkable (∼6.0-fold higher) cytotoxicity against CD44-overexpressing A549 cells compared to CD44− HepG2 cells and HEK 293 normal cells. Also, DON-loaded HACD–TMAV showed ∼2.0-fold higher cytotoxicity against A549 cells compared to individual carriers and ∼4.5-fold higher cytotoxicity than by DON. Furthermore, HACD–TMAV-DON induced a ∼3.5-fold reduction in the size of 3D tumor spheroids of A549 cells. The enhanced anticancer effectiveness was attributed to starvation of the A549 cells of glutamine by dual targeting of glutamine metabolism and solute linked carrier family 1 member A5 (SLC1A5) through HA-linked CD44-mediated targeted delivery of DON. This led to over-production of reactive oxygen species (ROS) that induced apoptosis of cancer cells through downregulation of the PI3K/AKT/mTOR signaling cascade.
Introduction
Cancer remains a significant global health concern owing to the growing cases and deaths.1–3 Among cancer subtypes, lung cancer stands out as one of the deadliest with a survival rate of less than 15% over the past five years.4,5 Non-small cell lung cancer (NSCLC), particularly lung adenocarcinoma, is a leading cause of global lung cancer-related mortality.5–7 Current therapeutic options, including chemotherapy and radiotherapy, pose challenges such as adverse side effects, drug resistance, and noticeable damage to normal cells, especially in patients with compromised pulmonary systems.1,8 Therefore, innovative therapeutic interventions are indeed in demand to overcome these limitations and to render improved treatment for lung adenocarcinoma.In recent days, predictive tumor biomarkers are being utilized to improvise the diagnostic paradigm of lung cancer.9 Among these biomarkers, CD44 serves as a prognostic marker in aggressive cancer populations.9,10 CD44's pivotal role in cell-to-cell interaction, proliferation, migration, metastasis, and immune response underscores its suitability for target specific drug delivery. CD44 also acts as a receptor for hyaluronic acid (HA), which influences cancer progression and metastasis.11,12 Alternative lung cancer treatment involves targeting cancer-specific biochemical phenotypes, particularly those determined metabolically.13,14 In this regard, L-glutamine (Gln), identified as an essential nutrient, plays a crucial role in the growth and proliferation of NSCLC cells, regulating signal transduction and redox homeostasis.13,14 Therefore, concurrent targeting of both CD44- and glutamine-dependent metabolism may emerge as a potentially effective treatment strategy against lung adenocarcinoma.
Nanotechnology has emerged as a promising avenue in the battle against cancer.1 Among various nanocarriers, supramolecular self-assembled vesicles (VESs) have gained prominence because of their unique structural features with an inner domain and an outer aqueous domain separated by a hydrophobic membrane.15,16 Such characteristics allow efficient encapsulation of a diverse range (polarities) of therapeutic agents within vesicles.15,16 Alongside, zero-dimensional carbon dots (CDs) have been recognized as promising cellular transporters and diagnostic agents owing to their intrinsic fluorescence, water solubility and cytocompatibility.17–19 However, one of the major challenges CDs face is their cargo loading ability.19–21 On the other hand, vesicles are quickly eliminated from circulation due to uptake by the reticuloendothelial system.22
To overcome these challenges, amalgamation of CDs and self-assembled vesicles as multifunctional nanocomposites (CD–VES) may emerge as a novel approach by complementing the advantages of the individuals. In this context, nanoconjugates comprising vesicles and carbon nanotubes (CNTs) have been reported to exhibit potential as delivery vehicles.22,23 However, CNTs were found to be detrimental to normal cells too. In this regard, the CD–VES conjugates can serve as improved cellular transporters through their efficient drug loading ability and hold promise as diagnostic tools. Studies on CD–VES conjugates are comparatively limited and the specific advantages of CD–VES conjugates over individual carriers (CDs or VESs) remain to be emphasized. Integrating the strengths of both vesicles and CDs could offer a promising solution in cancer treatment.
Herein, we report the synthesis of a novel carbon dot–vesicle conjugate, denoted as HACD–TMAV, designed for targeted drug delivery against lung adenocarcinoma. The formation of HACD–TMAV involves the electrostatic interaction between a negatively charged hyaluronic acid-based carbon dot (HACDs) and a positively charged trimesic acid-based vesicle (TMAV). Physicochemical characterization of the blue-emitting HACD–TMAV conjugate was carried out by spectroscopy and microscopy. Leveraging hyaluronic acid as a natural ligand for the CD44 receptor, HACD–TMAV was utilized as a target specific diagnostic probe for CD44-overexpressing cancer cells.9,24,25 The glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON), known for its robust anticancer effects but limited by non-selective biodistribution and toxicities, was successfully encapsulated within the HACD–TMAV conjugate.26–28 This nano-formulation exhibited significantly enhanced drug encapsulating efficiency compared to individual nanocarriers. DON-loaded HACD–TMAV exhibited ∼2.0-fold higher cytotoxicity against A549 cells than individual carriers and ∼4.5-fold higher cytotoxicity than free DON. Moreover, it exhibited ∼6.0-fold higher cytotoxicity of A549 cells than normal cells (HEK 293) due to the specific delivery to CD44-overexpressing cells. The enhanced anticancer efficacy was attributed to glutamine deprivation, inhibiting solute linked carrier family 1 member A5 (SLC1A5) and downregulating the PI3/AKT/mTOR signaling pathway inducing apoptosis.29–31
Experimental section
Synthesis of triskelion amphiphile (TMAV)
TMAV was synthesized by following a reported method.15 Briefly, trimesoyl chloride (1 equiv. in DCM) was added dropwise to an ice cold solution of methyl ester of L-phenylalanine (3.5 equiv. in DCM) in the presence of triethyl amine (Et3N) and stirred for 18 h. DCM was evaporated and the product was purified by column chromatography using a 100–200 mesh silica gel and chloroform/methanol as an eluent. The corresponding methyl ester was hydrolyzed with NaOH to prepare the free acid, which was coupled with mono-Boc-protected 2,2′-(ethylenedioxy)bis(ethylamine) by treatment with DCC (3.3 equiv.), DMAP (3.3 equiv.), and HOBt (3.0 equiv.) in dry DCM. The product formed was worked up with 1 N HCl and then purified by column chromatography using a 100–200 mesh silica gel and chloroform/methanol as an eluent. The pure Boc-protected compound was treated with trifluoroacetic acid (10 equiv.) in dry DCM under magnetic stirring for 4 h to obtain the corresponding amine. The reaction mixture was concentrated in a rotary evaporator. The obtained yellow gummy material was thoroughly washed with hot ether to obtain purified triammonium trifluoroacetate salt. This salt was subjected to ion exchange on an Amberlite IRA-400 chloride resin column to obtain pure chloride (Scheme S1, ESI†). The synthesized trimesic acid-based amphiphile (TMAV) was characterized by 1H-NMR and mass spectroscopy (Fig. S1 and S2, ESI†).Preparation of vesicles
The synthesized amphiphile forms vesicles in pure Milli-Q water. The vesicular solution was prepared by dissolving 4 mg of trimesic acid-based amphiphile (TMAV) in pure Milli-Q water. All the microscopic and spectroscopic experiments were performed using this vesicular solution.Preparation of carbon dots (HACDs)
Hyaluronic acid-based carbon dots (CDs) were synthesized via a one-step hydrothermal method by dissolving 150 mg of sodium hyaluronate in 15 mL of Milli-Q water.24 The solution was heated in a 50 mL Teflon-equipped stainless-steel autoclave at 180 °C for 6 h in a hot air oven. The product obtained was syringe filtered through a 0.22 μm polytetrafluoroethylene (PTFE) membrane. A brown solution of carbon dots was obtained after filtration. The obtained solution was then dialyzed with a 1000 Da cellulose dialysis membrane for 6 h against water to yield purified carbon dots (HACDs). Finally, carbon dots were lyophilized and characterized by spectroscopic studies (Scheme S2, ESI†).Preparation of the HACD–TMAV conjugate
The triskelion amphiphile (1 mg) was dissolved in 500 μL of Milli-Q water to form the TMAV. HACDs (0.1 to 1 mg) were separately dissolved in 500 μL of Milli-Q water. The solution of HACDs was added dropwise to different TMAV vesicular solutions to form the HACD–TMAV conjugate via electrostatic interactions between the HACDs and the TMAV. Different weight ratios (w/w) of the TMAV and HACDs [(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1), (1:0.2), (1:0.3), (1:0.5), and (1:1)] were chosen to characterize the best effective weight ratio for conjugate formation. Subsequently, it was syringe filtered through a 0.22 μm polytetrafluoroethylene (PTFE) membrane. The so obtained solution was then lyophilized and stored at −20 °C for further use.
:0.1), (1:0.2), (1:0.3), (1:0.5), and (1:1)] were chosen to characterize the best effective weight ratio for conjugate formation. Subsequently, it was syringe filtered through a 0.22 μm polytetrafluoroethylene (PTFE) membrane. The so obtained solution was then lyophilized and stored at −20 °C for further use.
Atomic force microscopy (AFM)
The atomic force microscopy (AFM) analysis of the TMAV, HACDs and HACD–TMAV was performed by casting a drop (10 μL) of each of the sample on a fresh mica surface and respective images were obtained using an Asylum Research MFP-3D microscope in non-contact mode.CD44-mediated intracellular targeting of HACD–TMAV
A549 (CD44+), HepG2 (CD44−) and HEK 293 (CD44−) cells (1 × 105 cells per mL of each cell line) were incubated with HACD–TMAV (200 μg mL−1) for 6 h under humidified conditions (5% CO2 and 37 °C). Next, the cells were washed with PBS and subjected to fluorescence microscopy (20× magnification) for bioimaging. In another experiment, A549 cells were treated with an excess amount of sodium hyaluronate for 1 h prior to incubation with HACD–TMAV (200 μg mL−1) for 6 h under a humidified atmosphere. Next, the cells were washed with PBS and imaged under an Olympus IX83 inverted fluorescence microscope (20× magnification). In order to generalize the approach, intracellular uptake of HACD–TMAV to the Hep3B cancer cell line was studied. Furthermore, time-dependent bioimaging studies of A549 cells upon treatment with HACD–TMAV for 6 h, 12 h, 24 h and 48 h, were performed to validate the fluorescence stability of the HACD–TMAV conjugate under cellular imaging conditions over time.Loading of 6-diazo-5-oxo-L-norleucine (DON) in the HACD–TMAV conjugate
A glutamine antagonist, 6-diazo-5-oxo-L-norleucine (DON), was encapsulated in the TMAV, HACDs and HACD–TMAV, separately. In a classical experiment, the TMAV (3 mg) and DON (1 mg) were taken in a glass vial, followed by the addition of Milli-Q water (1 mL). The mixture was kept overnight at 4 °C under stirring conditions. Next, a Sephadex G-50 column (10 cm height and 1.2 cm diameter), pre-equilibrated with water, was used to separate the free DON from the DON-encapsulated TMAV using size exclusion column chromatography. The DON-encapsulated TMAV was eluted right after the void volume of the eluent. The filtration was carried out until the un-entrapped DON was gel-filtrated and removed completely. We collected the eluent in 2 mL fraction each, followed by absorbance measurement for all the fractions at 274 nm to confirm the presence of DON. The eluent was collected until no detectable absorbance of DON was obtained. Finally, the DON loading efficiency of TMAV was calculated using a standard calibration curve. DON was encapsulated within HACD–TMAV in a similar way to that mentioned above and its DON loading efficiency was determined using the same calibration curve of DON. For the encapsulation of DON within the HACDs, the HACDs (3 mg) and (1 mg) DON were taken in glass vials. Milli-Q water (1 mL) was added, followed by overnight stirring at 4 °C. The resulting solution was dialyzed for 6 h, followed by the measurement of absorbance at 274 nm to confirm the presence of DON. The loading capacity of the HACDs was calculated using the standard calibration curve of DON. The loading of DON in HACD–TMAV, the TMAV and the HACDs was further investigated by UV-vis and FTIR study.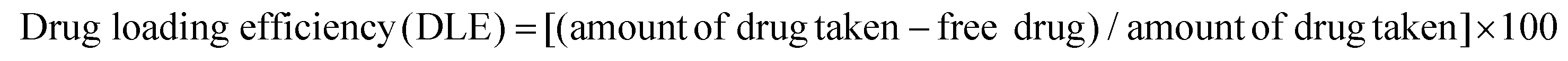 | (1) |
Cytotoxicity assay
The MTT assay was employed to study the cytotoxicity of HACD–TMAV-DON, HACD-DON, TMAV-DON and native DON. A549, HepG2 and HEK 293 cells (2 × 104 cells per well) were cultured in 96-well plates under a humidified atmosphere (5% CO2 and 37 °C). Upon 70% confluency, the cells were supplemented with fresh culture medium, followed by treatment with various concentrations (2–60 μg mL−1 for HepG2 and A549 and 20–640 μg mL−1 for HEK 293) of each nano-vehicle and free DON for 24–48 h. Next, the cells were washed with PBS and incubated with fresh medium containing 10 μL of MTT (5 mg mL−1 in PBS) for 4 h. The formazan crystal, so obtained from the dye incubation, was dissolved in 100 μL of DMSO, followed by the absorbance measurement at 570 nm using a SpectraMax iD5. Cell viability was calculated using eqn (2). Similarly, the cytotoxic potential of HACD–TMAV-DON was tested in the Hep3B cancer cell line that over expresses CD44 receptors to prove the general efficacy of the approach. | (2) |
Immunoblotting analysis
To explore the changes in protein translation through immunoblotting, A549 cells were cultured in 6 well plates at a cell density of 2 × 106 cells per well. The cells were separately incubated with HACD–TMAV, HACD–TMAV-DON, and native DON for 24 h. Following incubation, the wells were washed with PBS and the cells were trypsinized and centrifuged. The corresponding cell pellet so obtained was lysed at 4 °C with RIPA buffer (ThermoFisher Scientific). Bradford reagent (Sigma) was used to determine the protein concentration in the sample. Next, an equivalent amount of protein samples was prepared and subjected to SDS-PAGE protein gel electrophoresis and then transferred onto a polyvinylidene fluoride (PVDF) membrane. Following the successful transfer, blocking of the PVDF membrane was performed at room temperature for an hour. Next, the PVDF membrane was incubated with primary antibody overnight at 4 °C. Subsequently, the membrane was thoroughly washed with tris buffer saline containing 0.1% tween 20 (TBST) and subjected to secondary antibody incubation for 1 h at room temperature. Finally, the membrane was washed again with TBST and subsequently the protein bands were identified by electrochemiluminescence (ECL) assay.Apoptosis by flow cytometry
Flow cytometric assay was performed using annexin V-FITC/PI staining. In brief, A549 cells were treated with native DON, HACD-DON, TMAV-DON, HACD–TMAV and HACD–TMAV-DON for 24 h each at a concentration of 60 μg mL−1. Next, the cells were harvested and pelleted by centrifugation at 2000g for 5 min at room temperature and the cell pellet was washed with cold PBS. The cell pellet was re-suspended in binding buffer at a concentration of 1 × 106 cells per mL, followed by incubation with annexin V/FITC (5 μL) and PI (5 μL) in the dark for 15 min at room temperature. Following the incubation, binding buffer (400 μL) was added to each of the tubes and the samples were analyzed for HACD–TMAV-DON-induced cancer cell apoptosis using a BD FACS AriaTM III flow cytometer with an λex = 488 nm and λem = 533 ± 30 nm (FL-1) band-pass filter for annexin V-FITC and a 585 ± 40 nm (FL-2) band-pass filter for PI. The four quadrants of the scatter plot obtained from flow cytometry have their individual cell characteristics. Population of the cells in the Q3 quadrant displayed negative staining for both the annexin V-FITC and PI that corresponds to healthy viable cells, while annexin V-FITC positive – PI-negative populations of the Q4 quadrant represent the cells in early apoptosis and annexin V positive – PI-positive staining in the Q2 quadrant signifies that the cells are in late apoptosis. Necrosis cell populations were indicated in the Q1 quadrant that stain negative for annexin V-FITC and positive for PI.Nuclear condensation study by Hoechst 33342 staining
Nuclear condensation study was performed in A549 cells by staining the nuclei with Hoechst 33342 dye (ThermoFisher Scientific). In this context, A549 cells were incubated with HACD–TMAV-DON, HACD–TMAV, HACD-DON, TMAV-DON and DON alone for 24 h. Next, the cells were washed with PBS, followed by incubation with Hoechst 33342 dye for 10 min at room temperature in the dark. Following the incubation, the cells were thoroughly washed with PBS and imaged using an Olympus IX83 inverted fluorescence microscope (20× magnification) to observe the nuclear morphology.3D tumor spheroid formation
The cytotoxicity of HACD–TMAV-DON against A549 cells was further investigated by screening the anti-tumorigenic activity of the nano-formulation against the 3D tumor spheroids developed by the hanging drop method. In brief, fully confluent A549 cells were trypsinized with 0.05% trypsin–EDTA and centrifuged to obtain cell pellets that were re-suspended in DMEM. 20 μL (500 cells per μL) of cell suspension were cultured in DMEM (high glucose) containing 10% FBS and antibiotics (100 mg L−1 streptomycin and 100 IU mL−1 penicillin) in the lid of a 35 mm Petri dish under humidified conditions (5% CO2 and 37 °C). The cell suspension drops were then subjected to constant monitoring everyday under a microscope for viewing spheroid colony formation.Effect of HACD–TMAV-DON on the growth and proliferation of 3D-tumor spheroids
The dose-response effect of HACD–TMAV-DON was checked on the growth and progression of the 3D tumor spheroids. The cultured tumor spheroids were subjected to treatment with HACD–TMAV-DON at a dosage of 60 μg mL−1 under humidified conditions (5% CO2 and 37 °C). After the completion of drug incubation, the anti-tumorigenic potential of the nano-formulation was checked against morphological alterations in tumor spheroid dimensions at different time periods under a microscope. The alterations in the size of the tumor spheroids were calculated using Olympus CellSens Dimension software. The cytotoxicity and apoptotic potential of HACD–TMAV-DON against the 3D tumor spheroids were investigated by the MTT assay and the flow cytometric assessment of annexin V-FITC/PI staining. Moreover, flow cytometric assessment of the intra-tumoral ROS generation was studied in the 3D tumor spheroids following treatment with HACD–TMAV-DON.Assessment of the intracellular glutamine content
To investigate the amount of intracellular glutamine, an enzymatic reaction was performed according to the manufacturer's protocol. In this context, A549 cells were cultured at a density of 2 × 105 cells per well in six-well plates. After attaining 70% confluency, the cells were treated with HACD–TMAV, native DON and HACD–TMAV-DON at a concentration of 60 μg mL−1 for 24 h. The glutamine content in the cell lysates was then determined by measuring the absorbance for each group of the cells at 565 nm.Measurement of intracellular reactive oxygen species (ROS)
Dichlorofluorescein diacetate (DCFH-DA, Sigma) was used to detect reactive oxygen species (ROS) generation in A549 cells following exposure with HACD–TMAV, HACD–TMAV-DON and native DON for 8 h and 18 h, respectively. Following treatment with HACD–TMAV-DON and native DON, the cancer cells were washed with PBS, followed by incubation with DCFH-DA in the dark for 20 min. Next, the cells were washed with PBS thrice and subjected to fluorescence microscopy (10× magnification) for intracellular ROS generation which was indicated by the intensity of green fluorescence of 2′,7′-dichlorofluorescein (DCF) produced within the living cells.Results and discussion
In the contemporary landscape of novel strategies for combating lung adenocarcinoma, a promising avenue involves leveraging biomarkers to facilitate cancer cell-specific drug delivery.32,33 This approach holds the potential to mitigate the adverse effects on normal cells, thereby enhancing the overall clinical efficacy of the treatment. In pursuit to our objective, we aim to fabricate a nano-composite comprising self-assembled vesicle and fluorescent carbon dots to serve as a targeted cellular transporter with specific focus on delivering the encapsulated drug to cancer cells. In this context, we have synthesized a positively charged supramolecular self-assembled monolayer vesicle (VESs) using trimesic acid as a key component following the reported method (Fig. 1a and Scheme S1, ESI†).15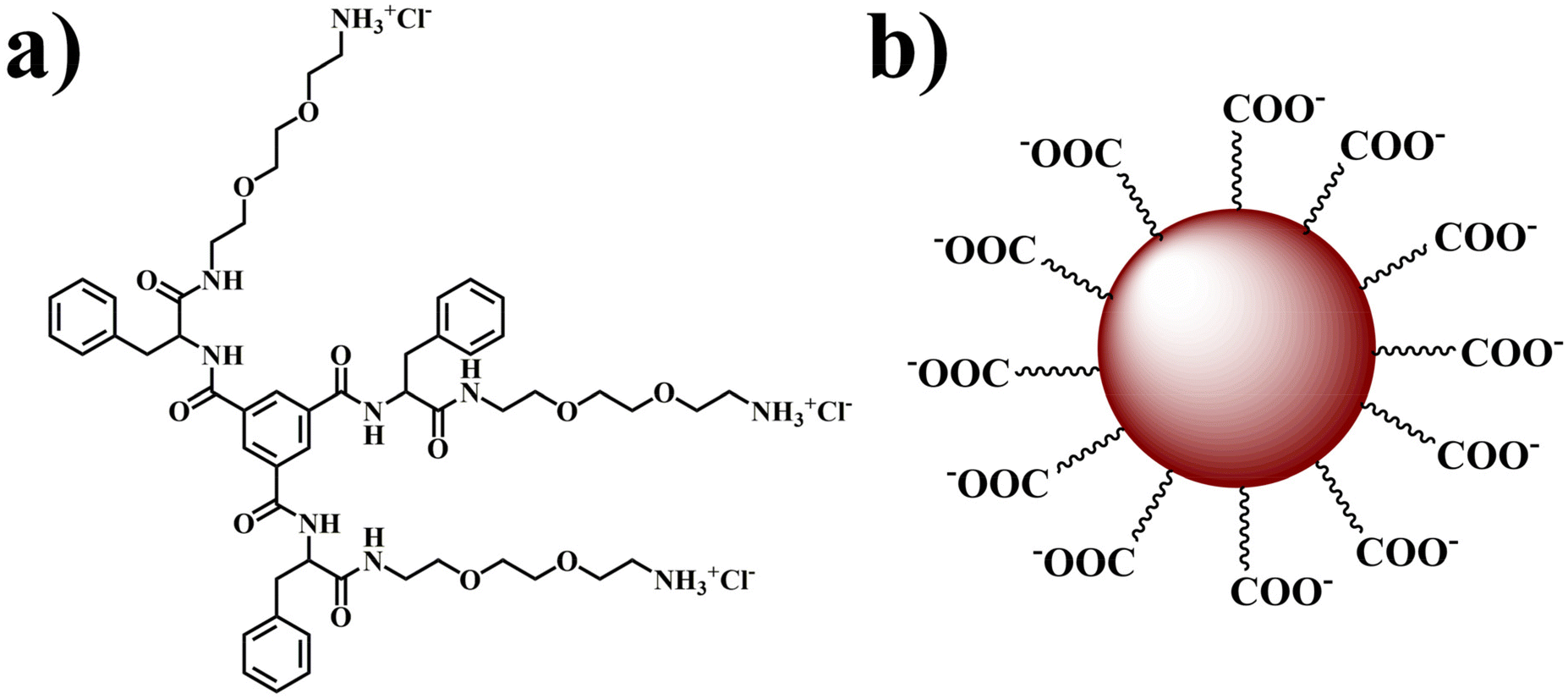 | ||
| Fig. 1 Molecular structures of (a) the TMAV and (b) HACDs. | ||
The choice of trimesic acid as the hydrophobic moiety stems from its tri-functionalized nature and the presence of an aromatic segment at its core, facilitating ordered stacking of phenyl rings during self-assembly.34,35 The remaining portion of the amphiphilic structure maintains its hydrophilicity, enabling spontaneous self-assembly in water.34,35 Concurrently, carbon dots (CDs) were derived from negatively charged hyaluronic acid (Fig. 1b). Hyaluronic acid (HA) was chosen as the precursor for carbon dot synthesis due to its specific binding affinity to the CD44 receptor, which is overexpressed in various solid tumors, including those in the breast, pancreas, brain, ovary, and lungs.36 HA also possess excellent biocompatibility, non-immunogenicity, biodegradability, and high hydrophilicity, rendering it an ideal candidate for synthesizing CDs.37–40 Our approach culminates in the development of a carbon dot–vesicle (CD–VES) conjugate (HACD–TMAV) through electrostatic interactions, where the surface of the trimesic acid-based vesicle (TMAV) would be enveloped by the hyaluronic acid-based carbon dots (HACD) (Scheme 1). The resulting conjugate with the HACDs at the outer component is expected to exhibit specific permeability towards cancer cells overexpressing the CD44 receptor.
 | ||
| Scheme 1 Schematic representation of the fabrication of the DON encapsulated carbon dot–vesicle conjugate (HACD–TMAV-DON) and its application as a cancer theranostic agent by dual targeting of CD44 and SLC1A5 via downregulation of the PI3K/AKT/mTOR signalling cascade. DON: 6-diazo-5-oxo-L-norleucine; SLC1A5: solute linked carrier family 1 member A5; GLS-1: glutaminase-1; GDH: glutamate dehydrogenase; ROS: reactive oxygen species. | ||
Characterization of the TMAV
The synthesized amphiphilic molecule spontaneously assembles into a vesicular structure (TMAV) in an aqueous environment. The critical aggregation concentration (CAC) of the TMAV amphiphile in aqueous medium was determined using the surface tension method by varying the concentration of TMAV from 0 to 3 mg mL−1 (Fig. 2a). It is well established that the surface tension of a liquid gradually decreases with an increasing concentration of surface-active agents. At the critical aggregation concentration (CAC), any further addition of the amphiphile does not alter the surface tension, signifying the initiation of self-organization at this point. The observed break point at 370 μg mL−1 (analogous to a previous report) indicated the commencement of self-aggregation of TMAV molecules at this specific concentration. | ||
| Fig. 2 (a) Plot of surface tension versus the concentration of the TMAV, (b) negatively stained TEM image, (c) AFM image, (d) FESEM image, (e) DLS plot of particle size distribution of the TMAV in water, (f) UV-vis spectra of the ANS-doped TMAV in different solvents, (g) FTIR spectra of the TMAV in the solid state (non-self-aggregated state) and D2O (self-aggregated state). | ||
The morphological characteristics of the TMAV were comprehensively investigated using transmission electron microscopy (TEM), field emission scanning electron microscopy (FESEM), and atomic force microscopy (AFM). The negatively stained TEM images of the TMAV revealed the formation of spherical self-assembled vesicles with a thin wall and a hollow core and size ranged from 100 to 150 nm (Fig. 2b). AFM images validated this spherical vesicular morphology with a diameter of ∼150 nm (Fig. 2c). The FESEM images provided additional confirmation of the spherical morphology of the self-assembled vesicles with an average size of ∼150 nm (Fig. 2d). Dynamic light scattering (DLS) experiments were conducted to determine the mean hydrodynamic diameter (Dh) of the vesicles, revealing a size distribution ranging from 120 to 150 nm (Fig. 2e and Fig. S3 ESI†), aligning well with the microscopic data. The zeta potential (ζ) of the vesicular solution was determined to be approximately +25 mV, indicating a positively charged surface. This positive charge contributes to the substantial stability of vesicular aggregates.
Next, we carried out spectroscopic studies to elucidate the mechanistic pathway guiding the self-assembly process, leading to vesicle formation. A hydrophobic probe ANS (1 × 10−5 M) was added into the aqueous solution of the TMAV (0.5 mg mL−1) to obtain solvent-dependent UV-vis spectra.22 The absorption maximum of the ANS–TMAV exhibited a discernible blue shift, transitioning from 370 nm to 348 nm (Fig. 2f) as the solvent system shifted from a non-self-assembled state (DMSO) to a self-assembled state (H2O). This observed blue shift in the absorption maximum signifies the self-organization of amphiphilic molecules through the face-to-face stacking of π moieties, initiating a sandwich-type arrangement referred to as H-type aggregation.41
The involvement of different non-covalent interactions including intermolecular hydrogen bonding (H-bonding), hydrophobic interactions, and π–π stacking during self-assembly was investigated by Fourier-transform infrared (FT-IR) spectroscopy. The FT-IR spectrum of the amphiphile displayed a characteristics sharp peak at 3440 cm−1 with a shoulder peak at 3252 cm−1 that corresponds to the stretching frequency the O–H/N–H group (Fig. 2g). The additional peaks at 2920 cm−1, 1651 cm−1, 1520 cm−1 and 1087 cm−1 are due to the stretching frequencies of C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –NHCO–, and C–O–C groups, respectively (Fig. 2g). However, the FT-IR spectrum of the TMAV in D2O showed a shift in the corresponding transmittance peak of the O–H/N–H group to a broad FT-IR peak centered at 3406 cm−1 (2920 cm−1–3715 cm−1) (Fig. 2g). The broadening of the O–H/N–H FT-IR peak upon dissolving the TMAV in D2O clearly signifies the participation of intermolecular hydrogen bonding between the carbonyl and amide groups present in the amphiphilic structure during the self-assembly-induced vesicle formation.
O, –NHCO–, and C–O–C groups, respectively (Fig. 2g). However, the FT-IR spectrum of the TMAV in D2O showed a shift in the corresponding transmittance peak of the O–H/N–H group to a broad FT-IR peak centered at 3406 cm−1 (2920 cm−1–3715 cm−1) (Fig. 2g). The broadening of the O–H/N–H FT-IR peak upon dissolving the TMAV in D2O clearly signifies the participation of intermolecular hydrogen bonding between the carbonyl and amide groups present in the amphiphilic structure during the self-assembly-induced vesicle formation.
Synthesis and characterization of HACDs
Hyaluronic acid-derived carbon dots (HACDs) were synthesized through a one-step hydrothermal method at 180 °C for 6 h, utilizing sodium hyaluronate as the precursor molecule dissolved in Milli-Q water.24 The resulting HACDs exhibited high solubility in water and its ζ-potential was determined to be −35 mV. This substantial negative ζ-potential indicates the remarkable stability of the HACDs in aqueous medium. The TEM and images of the HACDs showed a uniform size distribution of 3–5 nm (Fig. 3a and b). Fluorescence microscopic images of the HACDs exhibited blue-emitting dots, providing visual confirmation of the blue emissive properties inherent to the HACDs (Fig. 3c). The UV/Vis spectra of the HACDs showed an absorption maximum at 260 nm (Fig. 3d), attributed to the π–π* transitions arising from the presence of CC bonds in the HACDs. The emission behaviour of the HACDs (10 μg mL−1) was explored using fluorescence spectroscopy. The emission spectra exhibited an initial emission at 450 nm upon excitation at 260 nm. When the excitation wavelength increased from 260 nm to 360 nm (Fig. 3e), the emission spectra gradually experienced a blue shift in wavelength accompanied by an increased fluorescence intensity, indicative of excitation-dependent blue emission. The maximum fluorescence intensity of the HACDs was observed at 360 nm excitation with an emission peak at 437 nm (Fig. 3e). An aqueous solution of the HACDs showed blue fluorescence upon irradiation with UV light of 295 nm wavelength, which is in accordance with the emissive behavior of the HACDs (Fig. 3e inset). Furthermore, the FT-IR spectra of the HACDs showed a distinctive broad absorption peak centered at 3414 cm−1, attributed to the stretching vibration of the O–H/N–H group. The additional peaks at 2934 cm−1, 1619 cm−1, 1405 cm−1, and 1097 cm−1 corresponded to the stretching vibrations of C–H, CO, –NHCO–, and C–O–C groups, respectively (Fig. 3f). These FT-IR spectral features confirmed the presence of –NHCO–, C–O–C, hydroxyl, carbonyl, and carboxyl groups on the surface of the HACDs, exhibiting a coherent correlation with the FTIR spectra of hyaluronic acid (HA) (Fig. 3f). This observation suggests that the synthesized HACDs retained the structural properties of HA, which could be essential for their interaction with the CD44 receptor overexpressed in cancers. The relative quantum yield of the HACDs was determined to be 38% in reference to quinine hydrogen sulfate.
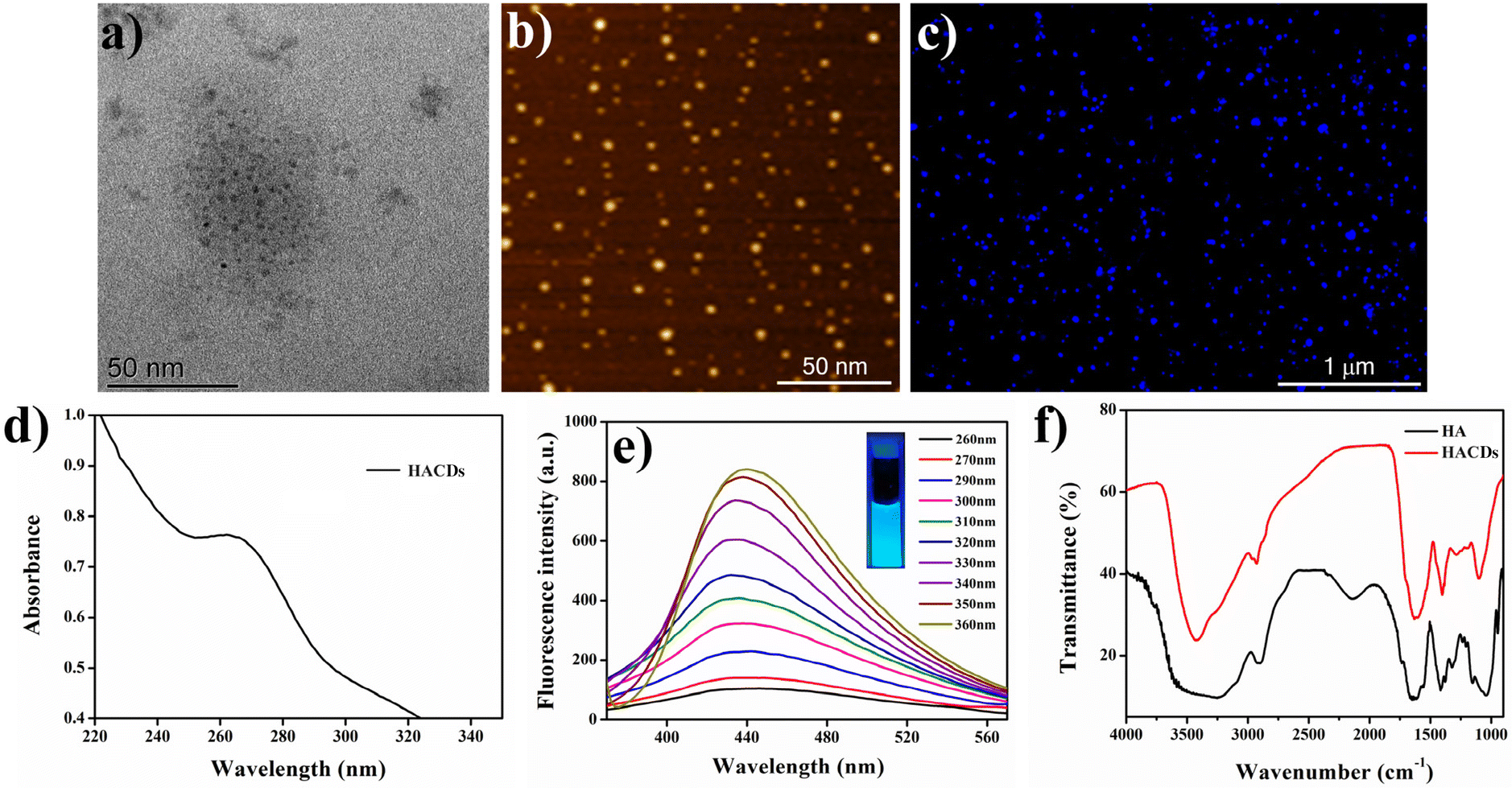 | ||
| Fig. 3 (a) TEM image, (b) AFM image, (c) fluorescence microscopic image, (d) UV-vis spectra, (e) excitation-dependent fluorescence spectra, and (f) FTIR spectra of the HACDs. | ||
Fabrication of the HACD–TMAV conjugate and its characterization
Next, we prepared a carbon dot–vesicle (CD–VES) conjugate (HACD–TMAV) by dissolving varying weight ratios of the TMAV and HACDs [(1:0.1), (1:0.2), (1:0.3), (1:0.5), and (1:1)]. The weight ratio at which the TMAV surface became completely saturated by the HACDs was determined through DLS and ζ-potential studies. The concentration of the HACDs varied from 100 μg mL−1 to 1 mg mL−1 by maintaining a constant concentration of the TMAV (1 mg mL−1). DLS analysis of the HACD–TMAV conjugate indicated a continuous increase in the size distribution of the nano-composite from ∼140 nm to 230 nm as the concentration of the TMAV/HACDs weight ratio increased from 1:0.1 to 1:0.5 (Fig. 4a, and Fig. S4, ESI†). However, there was a minimal change in the size distribution when the weight ratio was increased from 1:0.5 to 1:1 (Fig. 4a). This result suggested that the saturation of the TMAV surface by the HACDs reached at a 1:0.5 w/w ratio of the TMAV/HACDs or a 1:2 w/w ratio of the HACDs/TMAV. Furthermore, the ζ-potential values of the HACD–TMAV conjugate, maintaining various weight ratios [(1:0.1), (1:0.2), (1:0.3), (1:0.5), and (1:1)], were +5.83 mV, +2.59 mV, +2.11 mV, +0.21 mV, and −1.54 mV, respectively. The ζ-potential of the conjugate with a 1:0.5 w/w ratio of the TMAV and HACDs approached neutrality, indicating optimal formation of the conjugate at this weight ratio. The consistency between these experimental findings supports that a 1:0.5 w/w ratio of the TMAV/HACDs or a 1:2 w/w ratio of the HACDs/TMAV was ideal for further experiments involving the HACD–TMAV conjugate.
 | ||
| Fig. 4 (a) Plot of different weight ratios to form the HACD–TMAV conjugate vs. size distribution; (b) UV-vis spectra, (c) fluorescence spectra, and (d) FTIR spectra of the HACD–TMAV conjugate. | ||
After observing the initial results of CD–VES conjugate formation, we investigated its formation by UV-vis, fluorescence and Fourier transform infrared (FT-IR) spectroscopy. In the UV-vis spectroscopy, the absorption maximum of the HACDs (λmax = 260 nm) disappeared at a 1:2 w/w ratio of the HACDs and TMAV, suggesting the potential formation of the conjugate (Fig. 4b). The fluorescence spectroscopic study of HACD–TMAV displayed a reduction in the fluorescence intensity (λem = 437 nm) of the native HACDs (λex = 360 nm) in the presence of the TMAV at a 1:2 w/w ratio of the HACDs and TMAV (Fig. 4c). This quenching of fluorescence intensity served as a validation for the formation of a conjugate between the HACDs and the TMAV. Furthermore, we recorded the time-dependent fluorescence spectra of the HACD–TMAV conjugate, in Milli Q water and media used during cellular internalization, to study the fluorescence stability of HACD–TMAV over time (up to 8 days). These findings indicated that the relative fluorescence intensities (I/I0 at λem = 360 nm) of HACD–TMAV (Fig. S5a and b, ESI†) persisted almost unaltered and marginally diminished by ∼8–10% in both Milli-Q water and media after 8 days, suggesting superior stability of HACD–TMAV over time. The calculated relative quantum yield of the HACD–TMAV conjugate was determined to be 29%, referenced to quinine hydrogen sulfate. In the FTIR study of HACD–TMAV, a broad characteristic peak centered at 3444 cm−1 was observed, corresponding to the stretching frequency of –OH/–NH groups (Fig. 4d). Additional FTIR peaks at 1642 cm−1, 1410 cm−1, and 1097 cm−1 aligned with the stretching vibrations of CO, –NHCO–, and C–O–C groups, respectively. The FTIR spectra of the HACD–TMAV conjugate show all the characteristic peaks with respect to the native HACDs (Fig. 4d). Moreover, the broadening of peak centered at 3444 cm−1 in HACD–TMAV in comparison with the native HACDs, substantiated the conjugation of the HACDs with the TMAV via non-covalent electrostatic interactions. These comprehensive spectroscopic analyses provide a robust foundation for understanding the molecular interactions and structural characteristics of the HACD–TMAV conjugate.
Next, we investigated the conjugate formation by different microscopic techniques. The TEM images, devoid of staining, distinctly revealed the creation of spherical-shaped HACD–TMAV conjugates with an average diameter of 200 nm (Fig. 5a). The attachment of the HACDs onto the TMAV surface was clearly discernible in these TEM images. Real-time reverse phase contrast micrographs were employed to provide a more vivid visualization of the HACDs on the TMAV surface (Fig. 5b). In addition, FESEM and AFM experiments were performed for the HACD–TMAV conjugate. The FESEM images showcased the development of a spherical morphology with an average diameter of ∼200 nm (Fig. 5c) for the HACD–TMAV conjugate. The increase in size compared to that of the TMAV (∼150 nm) clearly indicated the conjugation of the HACDs on the TMAV surface. Consistent with the FESEM data, the AFM image confirmed the formation of the HACD–TMAV conjugate with an enlarged size of ∼200 nm (Fig. 5d). Furthermore, a fluorescence microscopic study was carried out to explore the blue fluorescence of the HACD–TMAV conjugate. The images vividly illustrated the creation of spherical-shaped HACD–TMAV conjugates, where the blue fluorescence of the HACDs was prominently visible on the surface (peripheral fluorescence) of the non-fluorescent TMAV (Fig. 5e and f). Hence, the blue fluorescent HACD–TMAV conjugate can serve as a fluorometric probe for bioimaging purposes. These images once again affirmed the presence of HACDs as the exterior component of the HACD–TMAV conjugate, ensuring the conjugate's ability to penetrate cancerous cells overexpressing the CD44 receptor.
 | ||
| Fig. 5 (a) TEM image (without staining), (b) real-time reverse phase contrast TEM image, (c) FESEM image, (d) AFM image, (e) fluorescence microscopic image, and (f) zoomed fluorescence microscopic image of the HACD–TMAV conjugate. | ||
Loading of 6-diazo-5-oxo-L-norleucine (DON) in the HACD–TMAV conjugate
After ensuring the successful formation of the HACD–TMAV conjugate, we loaded 6-diazo-5-oxo-L-norleucine (DON) separately into the HACD–TMAV conjugate, the HACDs, and the TMAV. DON was encapsulated in each of the nano-carriers (TMAV, HACDs, and HACD–TMAV conjugate) at a 3:1 (w/w) ratio using 3 mg of the nano-vehicle and 1 mg of DON. Separation of free DON from the loaded DON in the TMAV and the HACD–TMAV conjugate was achieved through size-exclusion column chromatography, employing a Sephadex G-50 column (details are provided in the Experimental section). Conversely, for the separation of free DON from DON-encapsulated HACDs, a dialysis process was employed. The calculated DON loading efficiency of the HACD–TMAV conjugate reached 91%, while for the individual TMAV and HACDs, it was observed to be 60% and 70%, respectively (measured with respect to a standard calibration curve of DON (Fig. S6a, ESI†)). The distinctive structure of the TMAV facilitated the loading of DON within the aqueous core, while in the case of the HACDs, the loading took place on the surface of the carbon dots through adsorption. The heightened DON loading capacity of the HACD–TMAV conjugate, compared to the individual nano-carriers, can be attributed to the synergistic accommodation of DON both in the inner aqueous core of the TMAV and on the surface of the HACDs. This dual-loading mechanism exhibited the cooperative and complementary nature of the HACD–TMAV conjugate, contributing to its enhanced efficiency in encapsulating DON.
The encapsulation of DON within the nano-vehicles (HACD–TMAV conjugate, TMAV, and HACDs) was investigated using UV-vis and FT-IR spectroscopy. DON exhibits a characteristic UV-vis absorption maximum at λmax = 274 nm. As depicted, the presence of the UV-vis absorbance peak at λmax = 274 nm (Fig. S6b, ESI†) in each nano-vehicle strongly indicated the successful loading of DON within the respective carriers. The FT-IR spectrum of DON showcased characteristic stretching peaks centered at νO–H/N–H = 3439 cm−1, νNN = 2542 cm−1, νCO = 1630 cm−1, νCN = 1378 cm−1, and νC–H = 1090 cm−1 (Fig. S6c, ESI†). In comparison, the FT-IR data of the HACD–TMAV-DON conjugate exhibited similar peaks at νO–H/N–H = 3435 cm−1, νNN = 2527 cm−1, νCO = 1625 cm−1, νCN = 1378 cm−1, and νC–H = 1090 cm−1 (Fig. S6c, ESI†). For TMAV-DON, the FT-IR peaks were observed at νO–H/N–H = 3431 cm−1, νNN = 2526 cm−1, νCO = 1643 cm−1, νCN = 1372 cm−1, and νC–H = 1096 cm−1 (Fig. S6d, ESI†). In the case of HACD-DON, the FT-IR peaks were observed at νO–H/N–H = 3442 cm−1, νNN = 2528 cm−1, νCO = 1632 cm−1, νCN = 1378 cm−1, and νC–H = 1091 cm−1 (Fig. S6d, ESI†). By comparing the FT-IR data of individual DON-loaded nano-vehicles with those of DON alone, it was evident that the presence of stretching peaks at νNN = 2542 cm−1 and νCN = 1378 cm−1 in the FT-IR spectrum of each DON-loaded nano-vehicle confirmed the successful loading of DON within the nano-carriers.
Cytocompatibility of the HACD–TMAV conjugate
Prior to the utilization of the synthesized HACD–TMAV conjugate as a biocompatible drug delivery vehicle, we have studied the time-dependent cytocompatibility of the HACD–TMAV conjugate against cancer cells A549 and HepG2 as well as for normal cells, HEK293. We have chosen A549 cells (NSCLC) as a positive control for CD44-overexpressed (CD44+) cancer cells, whereas HepG2 (CD44− cancer cells) and HEK 293 (normal cells) were chosen as negative controls. Each cell line was treated separately with varying concentrations of HACD–TMAV formulation (2–60 μg mL−1 for HepG2 and A549 and 20–640 μg mL−1 for HEK 293) for 24 and 48 h, under a humidified atmosphere (5% CO2 and 37 °C), respectively. The results obtained from the experiment suggested that the exposure of the HACD–TMAV conjugate to all three cell lines demonstrated negligible toxicity (Fig. S7, ESI†). Even at higher concentrations of the HACD–TMAV conjugate, over 90% of the cells remained viable for 24–48 h. Similarly, the biocompatibility of HACD–TMAV was checked in another cancer cell line over expressing the CD44 receptor (Hep3B) and the result so obtained suggested high biocompatibility of HACD–TMAV, as more than 90% cells were found to be viable (Fig. S8, ESI†). These findings demonstrate that the prepared HACD–TMAV conjugate is highly biocompatible and can be safely utilized for bio-medicinal applications.CD44-mediated intracellular uptake of the HACD–TMAV conjugate
To investigate the cellular internalization of the HACD–TMAV conjugate within the cancer cells, CD44+ A549 lung cancer cells were incubated with HACD–TMAV (200 μg mL−1) for 6 h, followed by observation under a fluorescence microscope. The fluorescence microscopic images of A549 cells displayed intense blue fluorescence, indicating the successful internalization of the HACD–TMAV conjugate within A549 cells without any loss of cellular integrity (Fig. 6a). Furthermore, the HACDs, being the exterior component of the HACD–TMAV conjugate, specifically bind with the CD44 receptor on the surface of A549 cells, leading to CD44-mediated internalization of the conjugate. To further confirm the CD44-mediated target-specific intracellular uptake, A549 cells were pre-incubated with an excess of HA for 2 h before incubating with the HACD–TMAV conjugate (200 μg mL−1) for 6 h and subsequently imaged under a fluorescence microscope. Images from this experiment displayed almost negligible fluorescence, indicating the failure of the HACD–TMAV conjugate to internalize in A549 cells under these conditions (Fig. 6b). This observation suggested the saturation of CD44 receptors on the surface of A549 cells by free HA, leaving almost no CD44 receptors exposed for binding with the HACD–TMAV conjugate. Furthermore, HepG2 cells (CD44− cancer cell line) and HEK 293 cells (CD44− non-cancerous cell line) were incubated with the HACD–TMAV conjugate (200 μg mL−1) for 6 h. The corresponding fluorescence microscopic images displayed almost no notable fluorescence, indicating the unsuccessful internalization of the HACD–TMAV conjugate inside cancerous HepG2 and non-cancerous HEK 293 cells that are deprived of CD44 receptors on its surface (Fig. 6c and d). These findings collectively underscored that the cellular internalization of the HACD–TMAV conjugate occurs specifically in CD44+ cancer cells through the interaction between the transmembrane CD44 receptor and HA available at the surface of the conjugate.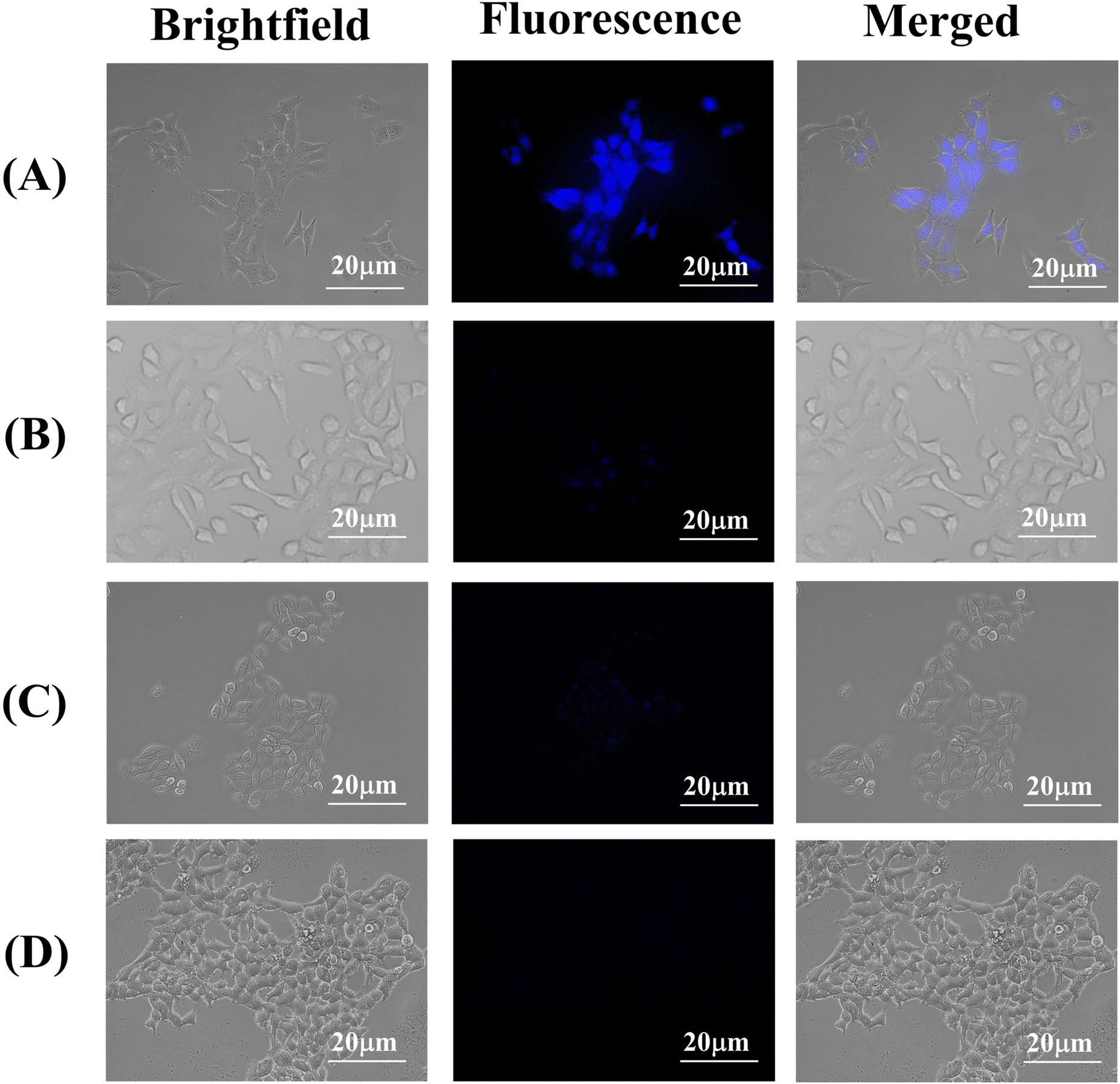 | ||
| Fig. 6 Bright-field, fluorescence and corresponding overlay images of (a) A549 cells, (b) A549 cells pre incubated with HA, (c) HepG2 cells and (d) HEK 293 cells incubated for 6 h with the HACD–TMAV conjugate (200 μg mL−1), respectively. Scale bars correspond to 20 μm. | ||
To further validate the cellular uptake of HACD–TMAV in cancer cells overexpressing CD44 receptors, a similar experiment was carried out in Hep3B cells, which is characterized by the high expression of the CD44 receptor. Here too, the successful internalization of HACD–TMAV in Hep3B cells was confirmed from the intense blue fluorescence inside the cells (Fig. S9, ESI†). This study reinforces the potential of HACD–TMAV for targeted bioimaging and therapeutic applications in CD44+ cancer cells. Moreover, we performed a time-dependent bio imaging study of A549 cell treated with HACD–TMAV to investigate the fluorescence stability of the HACD–TMAV conjugate under cellular imaging conditions over time. The fluorescence microscopic images of A549 cells obtained at different time points (6 h–48 h) validated the fluorescence stability of HACD–TMAV, owing to its intense blue fluorescence and comparable mean fluorescence intensities measured using Flow cytometry (Fig. S10, ESI†). These findings were found to be in full agreement with the data obtained from fluorescence microscopy.
Dose-dependent cytotoxic efficacy of the HACD–TMAV-DON conjugate against cancer cells
After selectively diagnosing CD44+ cancer cells by the HACD–TMAV conjugate, we explored the dose-dependent cytotoxic potential of the HACD–TMAV-DON formulation against cancer cells (A549 and HepG2) and non-cancerous cells (HEK 293) using the MTT assay. We also examined the dose-dependent cytotoxicities of native DON and DON-loaded individual nano-carriers (HACD-DON and TMAV-DON) against the cancer and the non-cancer cells. DON is known to exhibit a robust anticancer effect by inhibiting glutamine, an abundant amino acid in human blood. This inhibition suppresses the formation of tricarboxylic acid (TCA) cycle metabolites, crucial for the survival and proliferation of cancer cells. However, DON failed to receive approval as an anticancer drug during phase I and II clinical trials due to dose-limiting side effects such as nausea, vomiting, and toxicity to healthy tissues, especially the gastrointestinal (GI) tissue.42,43 In an effort to circumvent these limitations of DON, the targeted delivery of DON-encapsulated HACD–TMAV (HACD–TMAV-DON) to cancer cells could be a promising theranostic strategy minimizing its toxicity to healthy tissues.To this end, each cell line was treated separately with varying concentrations (2–60 μg mL−1 for HepG2 and A549 and 20–640 μg mL−1 for HEK 293) of HACD-DON, TMAV-DON, and HACD–TMAV-DON formulation for 24 and 48 h under a humidified atmosphere (5% CO2 and 37 °C), respectively. The quantity of loaded DON in these nano-formulations (3:1 w/w ratio) was determined based on the previously stated loading efficiency of each individual component (HACDs, TMAV, and HACD–TMAV conjugate). In the case of HEK 293 cells, the concentration of native DON was used up to 640 μg mL−1 to find its cytotoxicity. Notably, native DON showed meager cytotoxicity against all the cell lines, in particular, towards non-cancerous HEK 293 cells, even at its enhanced concentrations (320–640 μg mL−1) owing to its 70–75% cell viability. CD44− HepG2 cells exhibited around 78% cell viability following treatment with native DON at a concentration of 60 μg mL−1 for 48 h (Fig. 7a and b). Conversely, when CD44− HepG2 cells and non-cancerous HEK 293 cells were subjected to the HACD–TMAV-DON formulation, approximately 88% of CD44− HepG2 cells and more than 90% of HEK 293 cells were found to be alive across the diverse concentrations of the formulation (2–60 μg mL−1) for 48 h (Fig. 7a and b). Individual DON-loaded nano-carriers (HACD-DON and TMAV-DON) also exhibited no significant toxicity towards CD44− HepG2 cells and non-cancerous HEK 293 cells (>90% cell viability) for 48 h (Fig. 7a and b). The negligible toxicity of HACD–TMAV-DON towards CD44− HepG2 cells and non-cancerous HEK 293 cells is attributed to the unsuccessful intracellular uptake of the conjugate by both the cell lines lacking CD44 receptors on the cell surface.
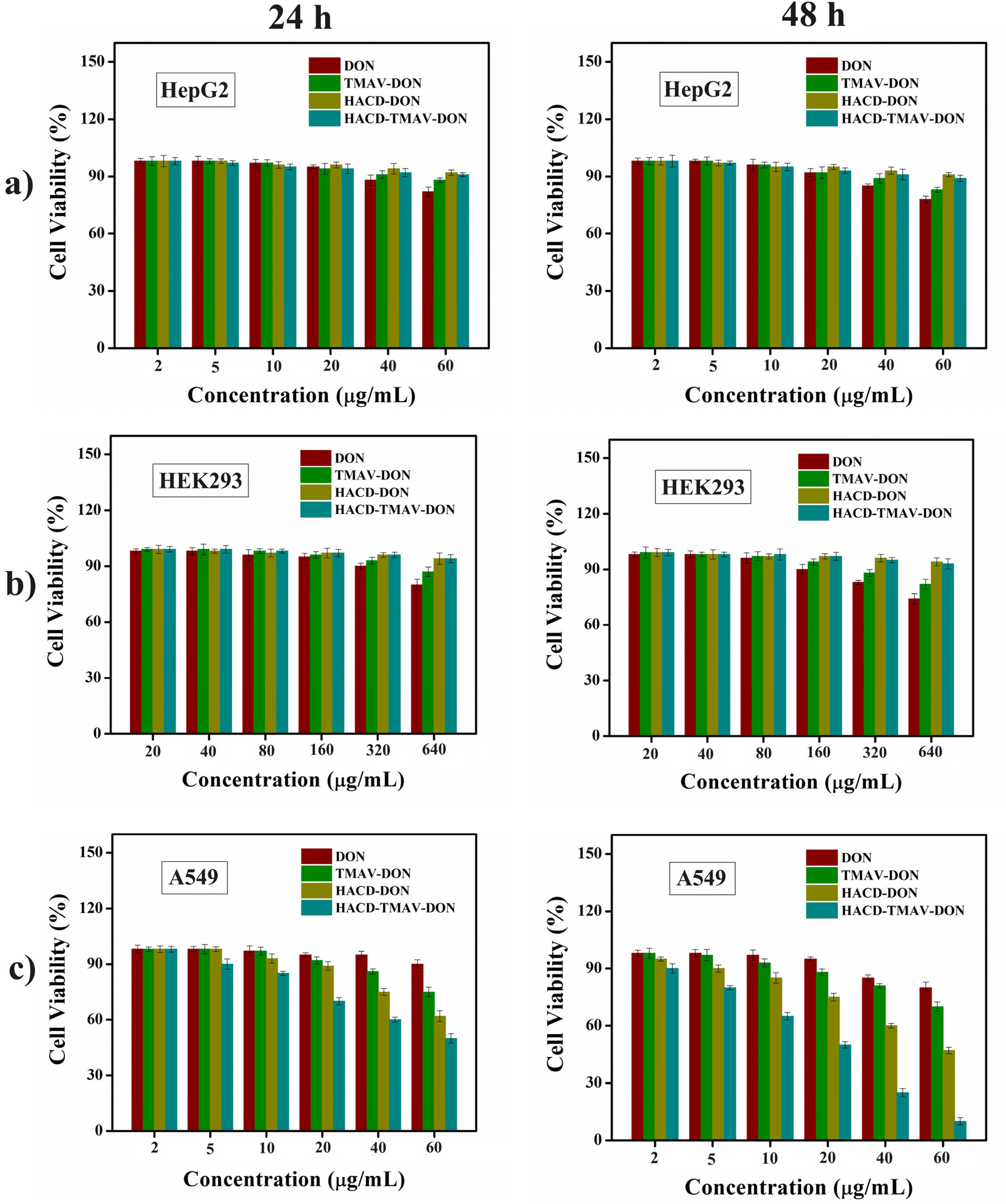 | ||
| Fig. 7 Cytotoxic potential (determined by the MTT assay) of native DON, TMAV-DON, HACD-DON, and HACD–TMAV-DON with increasing concentrations towards (a) HepG2 cells, (b) HEK 293 cells and (c) A549 cells for 24 h–48 h. Percentage experimental errors are within approximately ±3% in triplicate experiments. | ||
On the other hand, treatment of CD44+ A549 lung cancer cells with the HACD–TMAV-DON formulation (2.0–60 μg mL−1) led to a substantial reduction in cell viability (50% and 12%) when exposed for 24 and 48 h, respectively, at 60 μg mL−1 (Fig. 7c). The improved cytotoxic potential of the HACD–TMAV-DON formulation against CD44+ A549 cells, compared to those of CD44− HepG2 cells and HEK 293 cells (approximately ∼6.0-fold higher), was ascribed to the target-specific delivery of DON by the HACD–TMAV conjugate. In contrast, DON-loaded individual nano-carriers showed 62% and 47% cancer cell viability for 24 h and 48 h, respectively, for HACD-DON, and around 75% and 70% for TMAV-DON within the same timeframe (Fig. 7c). DON-encapsulated HACD–TMAV exhibited ∼2.0-fold higher cytotoxicity against A549 cells compared to individual carriers with an IC50 value of 54.7 μg mL−1 and 20 μg mL−1 for HACD-DON and HACD–TMAV-DON, respectively (Table S1 and Fig. S11, ESI†). The enhanced cytotoxicity of the HACD–TMAV-DON formulation compared to that of HACD-DON was ascribed to the comparably higher loading of DON by HACD–TMAV over the HACDs alone. The much-improved cytotoxicity of the HACD–TMAV-DON formulation towards CD44+ A549 cells compared to that of TMAV-DON was observed possibly due to better loading of DON in the HACD–TMAV conjugate and also owing to the presence of a target-specific moiety in the conjugate that was lacking in only the TMAV. Notably, exposure of CD44+ A549 cells to a comparable concentration of native DON (60 μg mL−1) exhibited meager cytotoxicity as ∼75–85% cancer cell was observed to be viable (Fig. 7c) for 24–48 h, due to its poor bioavailability in the absence of any cellular transporter. The HACD–TMAV-DON formulation exhibited ∼4.5-fold higher cytotoxic potential in CD44+ A549 cells compared to free DON. To demonstrate the generalized efficacy of this approach, we tested the cytotoxicity of the HACD–TMAV-DON formulation in Hep3B cancer cells, which also overexpresses CD44 receptors. The results indicated that the DON-loaded nanoconjugate exhibited a cytotoxic effect in Hep3B cells similar to that observed in A549 cells, with only a slight change in the percentage of cell viability (Fig. S12, ESI†). The observed cytotoxicity of CD44+ cancer cells demonstrated that the anti-cancer drug DON was efficiently cytotoxic towards cancer cells by encapsulating it within the HACD–TMAV conjugate at a much lower dose of 60 μg mL−1. The utilization of the HACD–TMAV-DON formulation at such a low dosage for being significantly cytotoxic towards CD44+ cancerous cells was attributed to the specific delivery of DON by the target-specific ligand-tethered nano-conjugate (HACD–TMAV), thereby significantly minimizing the drawbacks of DON (Scheme 1).
Mechanism of HACD–TMAV-DON-mediated cytotoxicity
The anticancer efficiency of the HACD–TMAV-DON formulation was explored by investigating the programmed cell death mechanisms in A549 cells upon exposure to the nano-formulation (60 μg mL−1). A549 lung adenocarcinoma cells were exposed to HACD–TMAV, HACD-DON, TMAV-DON, the HACD–TMAV-DON formulation, and native DON for 24 h. Following the completion of the drug treatments, the analysis was conducted through a flow cytometric assay utilizing annexin V-FITC and PI dye. Annexin V-FITC, a calcium-dependent phospholipid protein with high affinity for phosphatidylserine (PS), was employed for the detection of apoptotic cells with exposed PS. Propidium iodide (PI) served as a standard flow cytometric probe to identify necrotic cells. Four quadrants on the scatterplot were utilized to distinguish between viable and non-viable cells based on staining for both annexin V-FITC and PI (explained in the Experimental section). Interestingly, the Q3 quadrant exhibited negative staining for both annexin V-FITC and PI, corresponding to healthy cells, when CD44+ A549 cells were treated with only the HACD–TMAV conjugate (Fig. S13, ESI†). This result confirmed the high biocompatibility of the HACD–TMAV conjugate. Treatment of TMAV-DON with CD44+ A549 cells resulted in populations in Q3 and Q4 quadrants, suggesting that A549 cells underwent apoptosis through an early apoptotic pathway (Fig. S13, ESI†). Moreover, treatment of HACD-DON with CD44+ A549 cell results in apoptosis of A549 cells through early apoptotic pathways, as indicated by the population of Q3 and Q4 quadrants. The increased percentage of apoptotic cells in HACD-DON compared to TMAV-DON was attributed to the target-specific delivery of DON by the HACDs. In contrast, exposure to the HACD–TMAV-DON formulation for 24 h resulted in significant populations in Q4 and to a certain extent in Q2 quadrants, indicating that HACD–TMAV-DON induced apoptosis through an early to late apoptotic pathway (Fig. S13, ESI†). Conversely, exposure to native DON (60 μg mL−1) for 24 h resulted in the most population of Q3 and very few in the Q4 quadrant, demonstrating the limited bioavailability of DON to cancer cells in the absence of any cellular transporter. Therefore, this greater apoptotic potential of the HACD–TMAV-DON formulation was attributed to the presence of the targeting moiety hyaluronic acid and the significantly improved drug loading efficiency of the HACD–TMAV conjugate.Nuclear assessment of cancer cells following exposure to the HACD–TMAV-DON conjugate
After ensuring the apoptotic potential of HACD–TMAV-DON, we delved deeper into the apoptotic changes in the nuclei of CD44+ A549 cells following exposure to the nano-formulation.44 Specifically, we examined the impact of exposing A549 cells to the HACD–TMAV-DON formulation (60 μg mL−1) and the corresponding control groups mentioned earlier for 36 h. Cancer cell nuclei were stained with Hoechst 33342 dye, and subsequent fluorescence microscopic images were captured. Upon incubation with the HACD–TMAV-DON formulation, the nuclei of A549 cells exhibited signs of nuclear shrinkage, nuclear fragmentation, chromatin condensation, and the formation of apoptotic bodies (Fig. S14, ESI†). These observations suggested a loss of nuclear morphology, indicative of the progression of cancer cell apoptosis. In contrast, treatment with the native drug and the HACD–TMAV conjugate under similar experimental conditions displayed spherical-shaped healthy nuclei in A549 cells (Fig. S14, ESI†). These observations affirmed the high biocompatibility of the synthesized hybrid nanoconjugate, and highlighting the poor bioavailability of native DON. Furthermore, exposure to DON-loaded individual nano-vehicles (HACD-DON, TMAV-DON) induced negligible changes in the nuclear morphology of A549 cancer cells (Fig. S14, ESI†). This analysis further strengthened the notion that exposure to the HACD–TMAV-DON formulation resulted in robust apoptotic features within A549 cells, leading to a significant reduction in CD44+ A549 cell viability.Inhibition of SLC1A5 and GLS-1 expression in A549 cancer cells by HACD–TMAV-DON
The much-improved apoptotic potential of the nano-formulation HACD–TMAV-DON against A549 cells prompted us to further extrapolate the signaling mechanism that played a pivotal role in the CD44-mediated target-specific cytotoxic towards A549 cells. Cancer cells heavily rely on glutamine for its participation in the synthesis of TCA cycle metabolites essential for their survival and proliferation.45–47 Starving the cancer cells of glutamine, a crucial nutrient for energy production, leads to metabolic stress within these cells.48 Glutamine deprivation results in reduced ATP production, leading to the generation of ROS within cancer cells beyond threshold levels.48 Glutamine deficiency in cancer cells, achieved through a combination of inhibiting glutamine uptake via downregulation of the glutamine transporter (SLC1A5) and inhibiting glutamine metabolism, may synergistically enhance the ROS generation, triggering apoptosis in cancer cells.29,30,48,49 Glutaminolysis involves a series of enzymatic reactions, with glutaminase-1 (GLS-1) gaining particular significance as it participates in a key rate-determining step of converting glutamine to glutamate which ultimately converted to α-keto glutarate in the presence of glutamate dehydrogenase (GDH) enzyme (Scheme 1).45–47 Targeting glutaminolysis through the inhibition of glutamine holds considerable therapeutic promise in impeding the growth-promoting processes associated with tumor progression.45–47 DON, a reported glutamine inhibitor, hinders the activity of GLS.26 In this study, we investigated the expressions of SLC1A5 and GLS-1 in A549 cells following exposure to HACD–TMAV, native DON, and HACD–TMAV-DON for 24 h through immunoblotting analysis. The results indicated no alteration in the expression of both proteins (SLC1A5 and GLS1) when comparing the effects from the exposure of HACD–TMAV and native DON with the control alone (Fig. 8a). However, exposure of A549 cells to HACD–TMAV-DON for 24 h resulted in a significant downregulation in the expression of both SLC1A5 and GLS1 (Fig. 8a). Targeting SLC1A5 and GLS1 with HACD–TMAV-DON facilitated glutamine starvation in lung adenocarcinoma. Furthermore, the effect of HACD–TMAV-DON on the intracellular glutamine concentration of A549 cells was assessed using a glutamine assay kit (Sigma). The results indicated a significant reduction in the intracellular glutamine concentration content (53 μM) of A549 cells lysates upon exposure to HACD–TMAV-DON compared to the control cells (301 μM), cells treated with HACD–TMAV (298 μM), and native DON only (240 μM) (Fig. 8b and Fig. S15a, ESI†). Thus, glutamine starvation of A549 cells following exposure to HACD–TMAV-DON was achieved through the synergistic inhibition of glutamine metabolism and the glutamine transporter SLC1A5. DON's inhibitory action on GLS1 and SLC1A5 induces glutamine deprivation that possibly led to enhanced intracellular ROS-triggered cancer cell apoptosis (Scheme 1). | ||
| Fig. 8 (a) Immunoblotting of GLS1 and SLC1A5 in A549 cells following exposure to HACD–TMAV, native DON and HACD–TMAV-DON (β-actin levels are shown as loading control), (b) intracellular glutamine concentration in A549 cells when exposed to HACD–TMAV, native DON and HACD–TMAV-DON, (c) fluorescence microscopic images depicting intracellular ROS generation as indicated by the green fluorescence of DCF in A549 cells following incubation with HACD–TMAV, native DON, TMAV-DON, HACD-DON and HACD–TMAV-DON at 60 μg mL−1 of each formulation for 8 h and 18 h, and (d) immunoblotting analysis of CD44, pAMPK, PI3K, pAKT, and pmTOR levels in A549 cells in the presence of HACD–TMAV, native DON and HACD–TMAV-DON for 24 h. β-actin levels are shown as loading control. | ||
HACD–TMAV-DON targets CD44 and triggers ROS generation in A549 cells by activating PI3/AKT/mTOR signaling
Cancer cells are vulnerable to oxidative stress, which intensifies upon exceeding the threshold level, leading to ROS-mediated apoptosis.50 Glutaminolysis directly contributes to ROS homeostasis by exporting glutamate for glutathione (GSH) production.51 Glutamine deprivation in cancer cells disrupts redox homeostasis that caused enhanced intracellular ROS production and triggering apoptosis.52 To explore intracellular ROS production, A549 cells were treated with HACD–TMAV-DON, HACD–TMAV, TMAV-DON, HACD-DON, and native DON for 8–18 h, followed by staining with DCFH-DA dye. Upon internalization to A549 cells, DCFH-DA gets deacetylated to a non-fluorescent compound by cellular esterase, which is further oxidized to a green fluorescent compound, 2′,7′-dichlorofluorescein (DCF) in the presence of intracellular ROS. Notably, HACD–TMAV-DON (60 μg mL−1) exhibited enhanced ROS generation over time (8–18 h), as evidenced by the increased DCF fluorescence (Fig. 8c). HACD-DON and TMAV-DON showed significantly lower fluorescence, while native DON exhibited unnoticeable fluorescence, indicating negligible intracellular ROS generation at an individual dosage of 60 μg mL−1 (Fig. 8c and Fig. S15b, ESI†). Untreated control and HACD–TMAV-treated cells displayed feeble fluorescence, confirming minimal intracellular ROS generation (Fig. 8c and Fig. S15b, ESI†). Therefore, improved ROS levels in A549 cells following HACD–TMAV-DON exposure were attributed to glutamine starvation due to downregulated SLC1A5 and GLS1. Notably, selective glutamine deprivation took place only in A549 cells due to CD44-mediated target-specific delivery of DON by the HACD–TMAV-DON nano-formulation (Scheme 1).The amalgamation of HA and CD44 assists in the binding of adaptor molecules to the cytoplasmic region in CD44, and triggers multiple signaling pathways involved in cell adhesion, migration and proliferation, including phosphoinositide 3-kinase. Herein, we have explored the effect of HACD–TMAV-DON on the expression of CD44 for 24 h in A549 cell lysate via immunoblotting (Fig. 8d). It was observed that treatment with native DON imparted no alteration in the expression of CD44 in the cancer cells, indicating overexpressed CD44 because of the nonspecific delivery of the native drug to the tumor site (Fig. 8d). However, treatment with HACD–TMAV-DON for 24 h resulted in a significant downregulation in the expression of CD44 in A549 cells, indicative of a very low signaling of CD44 antibody (Fig. 8d). This signifies that the inhibition of CD44 expression by HACD–TMAV-DON in A549 cells can be ascribed to the CD44-mediated target specific delivery of DON to the tumor site by HACD–TMAV. Moreover, we studied the time-dependent expression of CD44 upon treating A549 cells with HACD–TMAV-DON for 6 h, 12 h and 24 h. The result so obtained suggested a time-dependent decrease in the expression of CD44, specifically at the 6-hour mark, CD44 levels remain relatively unchanged, allowing for efficient internalization of the conjugate. The downregulation becomes more pronounced at 12 and 24 hours, consistent with the drug's mechanism of action post-internalization. These findings motivated us for further investigations into the signaling mechanisms that play a critical role in the anticancer effect of the HACD–TMAV-DON formulation.
Multiple signaling pathways have been reported to date that are activated in the presence of an elevated intracellular ROS level and showed numerous impacts on the growth and progression of cancer cells.53 The PI3K/AKT/mTOR signaling pathway plays a crucial role in regulating cellular processes as well as most hallmarks of cancer, including cell proliferation, autophagy, apoptosis, angiogenesis, epithelial-to-mesenchymal transition (EMT), and chemoresistance.31,54–56 An extracellular ligand (for example, insulin or an insulin-like growth factor) binds to the cell-membrane receptor and thereby activates PI3K (phosphatidylinositol (3,4,5)-trisphosphate kinase), which in turn activates the downstream effector, AKT. The activation of AKT occurs through the phosphorylation process, which ultimately leads to the activation of mTOR (mammalian target of rapamycin), a key downstream effector of PI3K/AKT signaling. mTOR integrates many proteins and promotes survival, growth and progression of cancer cells. Herein, we used respective PI3K, AKT and mTOR antibodies along with their phosphorylated forms to study the alterations, if any, in the expression levels of these signaling proteins in A549 cells. The immunoblots so obtained from A549 cell lysates following exposure to HACD–TMAV-DON revealed downregulation of the PI3K/AKT/mTOR signaling cascade through inhibition of AKT phosphorylation that subsequently downregulated the phosphorylation of mTOR (Fig. 8d). However, exposure to HACD–TMAV exhibited no alteration in the expression of the PI3K/AKT/mTOR signaling cascade with respect to the control group, suggesting normal upregulated AKT as well as mTOR phosphorylation (Fig. 8d). On the other hand, the native DON has comparatively little influence on the expressions of PI3K and pAKT and consequently on p-m-TOR with respect to control cells. AMP-activated protein kinase (AMPK) senses cellular energy and activates pathways that are necessary in sustaining cellular energy response to this stress, thereby promoting metabolic reprogramming.57 Studies have shown that an enhanced intracellular ROS level activated AMPK through subsequent inhibition of AKT phosphorylation.57 In the present study, AMPK antibody and its phosphorylated form have been used in order to account for the changes in protein translation in A549 cells, exposed to HACD–TMAV-DON. Immunoblotting showed that the exposure of HACD–TMAV-DON resulted in activated pAMPK expression in A549 cells, indicating an elevated intra-cellular ROS level. This event triggered the anticancer efficacy of HACD–TMAV-DON to induce ROS-mediated cancer cell apoptosis (Fig. 8d). However, in the presence of only HACD–TMAV and the native DON alone, there is negligible activation of pAMPK, suggesting the inhibition of AMPK phosphorylation due to insignificant ROS generation (Fig. 8d). It is noteworthy that the apoptotic activity of HACD–TMAV-DON can be ascribed to the increased ROS generation in A549 cells through DON-induced inhibition of glutaminolysis via downregulation of the PI3K/AKT/mTOR signaling cascade (Scheme 1).
3D tumor spheroid formation and proliferation
The anti-tumoral impact of HACD–TMAV-DON was further investigated using a 3D tumor spheroid model by replicating the biochemical and biomechanical signals in the tumor microenvironment (TME) associated with tumor cell growth and proliferation.58,59 3D tumor spheroid models have attracted substantial attention for their ability to serve as a crucial bridge between in vitro experiments and in vivo studies.60,61 We established a 3D tumor spheroid model by cultivating A549 cells within a 3D culture module using the Hanging Drop method.62 Bright-field images captured after 72 h of culture displayed an initial tumor spheroid size of 294 μm, which remarkably expanded to approximately 1070 μm within the same timeframe (72 h). These spheroids exhibited a uniform spherical shape and robust cell–cell connectivity, as depicted in Fig. S16 (ESI†). The structural integrity of the cultured tumor spheroid was of significant importance, serving as a critical component in assessing the anti-tumor efficacy of HACD–TMAV-DON against 3D tumors characterized by an overexpression of CD44 receptors. The CD44-mediated intratumoral uptake of HACD–TMAV was evaluated by incubating the 3D tumor spheroids with HACD–TMAV for 6 h. The images obtained from fluorescence microscopy exhibited intense blue fluorescence within the tumor spheroids, indicating the successful internalization of HACD–TMAV (Fig. S17a–c, ESI†). To utilize HACD–TMAV in 3D spheroid models as a cellular transporter, we have investigated the time-dependent effect of HACD–TMAV (60 μg mL−1) on the 3D tumor spheroids. The result so obtained indicated negligible changes in the shape and size of the 3D tumor spheroids (Fig. S17d–g, ESI†) with an increase in time, suggesting the excellent biocompatibility of the HACD–TMAV conjugate.Effects of HACD–TMAV-DON on 3D tumor spheroids
We employed this 3D tumor model to investigate the anti-tumorigenic potential of HACD–TMAV-DON concerning the growth and proliferation of tumor spheroids at various time intervals. We incubated tumor spheroids with the most effective concentration of HACD–TMAV-DON (60 μg mL−1, as determined from the MTT assay in 2D cell culture) to examine its anti-proliferative properties in a time-dependent manner. As the incubation period increased, the size (1070 μm) and morphology of the tumor spheroids exhibited a gradual disintegration (Fig. 9a–e). Notably, exposure to HACD–TMAV-DON for 72 h resulted in a significant reduction in spheroid dimension (315 μm), indicating a remarkable loss of cells at a dosage of 60 μg mL−1. The disintegration of the tumor following treatment with HACD–TMAV-DON led to approximately 3.5-fold reduction in tumor size, reflecting the decreased cell viability and highlighting the potent anti-tumoral efficacy of the newly synthesized nano formulation, HACD–TMAV-DON.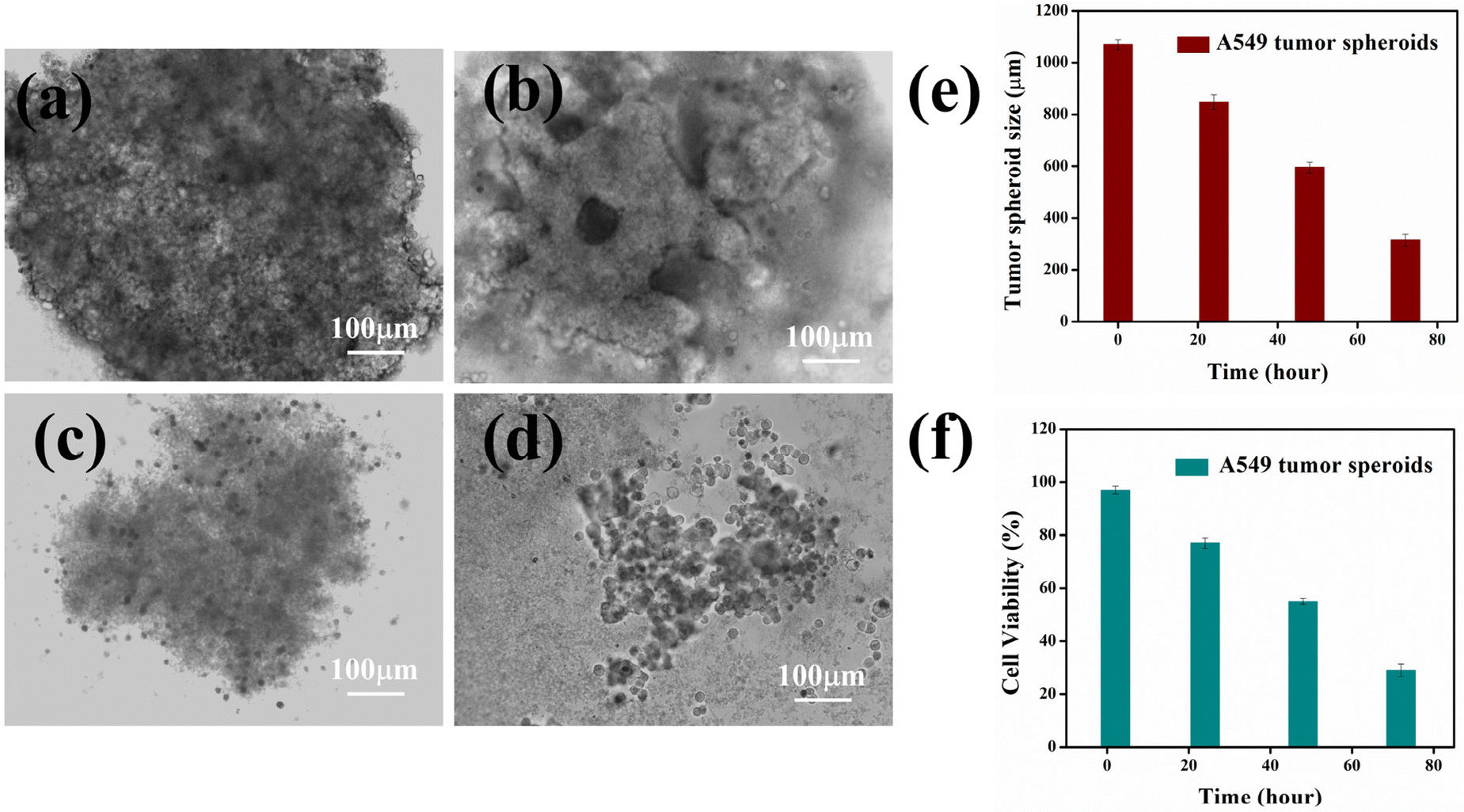 | ||
| Fig. 9 Time-dependent changes in the tumor spheroid morphology and diameter following exposure to HACD–TMAV-DON for (a) 0 h, (b) 24 h, (c) 48 h, and (d) 72 h; (e) time-course monitoring of the A549 tumor spheroid diameter; and (f) time-dependent anti tumoral effect of HACD–TMAV-DON (60 μg mL−1) on the tumor spheroid of A549 cells. The experimental errors were in the range of 1–3% in triplicate experiments. | ||
To validate the anti-proliferative efficacy of HACD–TMAV-DON on tumor spheroids, we conducted MTT assays. The results revealed a noteworthy reduction in the viability of A549 cells within the tumor spheroids when exposed to HACD–TMAV-DON (60 μg mL−1) at varying time intervals (Fig. 9f). Notably, at a dosage of 60 μg mL−1 of HACD–TMAV-DON, more than 70% of cancer cells were observed to be eliminated after 72 h (Fig. 9f). The findings obtained from the MTT assay further reinforced the anti-tumorigenic potential of the newly developed HACD–TMAV-DON against the 3D tumor spheroids.
HACD–TMAV-DON-induced ROS-triggered apoptosis in 3D tumour spheroids
After conducting antiproliferative investigations on the 3D tumor spheroids, we proceeded to assess the apoptotic potential of HACD–TMAV-DON (60 μg mL−1) at different time intervals. Utilizing the annexin V-FITC/PI kit, we examined the apoptosis pathway in the tumor spheroids following incubation with HACD–TMAV-DON. The results indicated that exposure to HACD–TMAV-DON for 24–72 h led to a range of 25–70% cell deaths, as evident by the substantial population in Q4 and Q2 quadrants, indicating early and late apoptosis (annexin V/FITC positive), respectively (Fig. S18, ESI†). In contrast, the untreated control group of spheroids resulted in the population of the Q3 quadrant, signifying viable A549 cells (Fig. S18, ESI†). This study thus reaffirmed the increased apoptotic efficacy of HACD–TMAV-DON on tumor spheroids over time. The apoptotic effects of anticancer agents are often associated with the intratumoral accumulation of ROS, surpassing a critical threshold and inducing cellular toxicity.51 To validate the role of intratumoral ROS in the apoptotic efficacy of HACD–TMAV-DON, we treated tumor spheroids with HACD–TMAV-DON (60 μg mL−1), followed by staining with DCFH-DA dye and subjected them to flow cytometric analysis. Observations made from this study revealed a noteworthy increase in the fluorescence intensity of DCF over time, indicating a significant rise in intratumoral ROS levels (Fig. S19, ESI†). The mean fluorescence intensity of DCF increased from 896 to 2487 within 6 h–48 h (Fig. S19, ESI†). This enhanced intratumoral ROS was believed to be the primary factor responsible for cell apoptosis triggered by HACD–TMAV-DON.Conclusion
In summary, a HACD–TMAV nano-conjugate was engineered by leveraging electrostatic interactions between a positively charged TMAV and negatively charged HACDs, harnessing the advantageous features of both the vesicle and carbon dots. Taking advantage of hyaluronate's high affinity for the CD44 receptor, we designed the HACD–TMAV conjugate for targeted drug delivery and selective bioimaging of A549 cancer cells overexpressing CD44. Encapsulation of the glutamine antagonist DON within the HACD–TMAV conjugate showed superior drug loading efficiency compared to individual carriers (HACDs and TMAV). The CD44-mediated, target-specific delivery of DON by HACD–TMAV exhibited ∼6.0-fold higher anticancer efficacy against A549 cells (CD44+) than HepG2 cells (CD44−) and normal HEK 293 cells. The enhanced anticancer effectiveness of DON-loaded HACD–TMAV resulted from simultaneous inhibition of intracellular glutamine and SLC1A5, a glutamine transporter, effectively starving cancer cells of their essential nutrient. The simultaneous inhibition of intracellular glutamine and SLC1A5 leads to cancer cell starvation, chemo-sensitization, and ultimately, ROS-mediated apoptosis via the downregulation of the PI3K/AKT/mTOR signaling cascade. Moreover, the HACD–TMAV-DON formulation showed ∼2.0-fold and ∼4.5-fold higher cytotoxicity against A549 cells compared to individual carriers and free DON, respectively. Moreover, HACD–TMAV-DON induced a ∼3.5-fold reduction in tumor size in a 3D tumor spheroid model of A549 cells. The dual-targeting strategy presented here offers a glimpse into a promising era of nanomedicine, where cancer therapies are tailored to individual molecular profiles, enhancing efficacy and minimizing side effects. Hence, the DON-encapsulated HACD–TMAV conjugate holds promise as a theranostic tool for treating lung adenocarcinoma.Author contributions
Afreen Zaman: conceptualization, methodology, investigation, formal analysis, analysis and interpretation of data, software, and writing – original draft. Aparajita Ghosh: investigation, data curation, validation, formal analysis, and editing. Anup Kumar Ghosh: methodology, investigation, validation, formal analysis, and editing. Prasanta Kumar Das: supervision, design of the study, analysis and interpretation of data, project administration, funding acquisition, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
P. K. D. is thankful to the Science and Engineering Research Board, DST, India, for financial assistance (No. CRG/2021/000235). A. Z., A. G. and A. K. G. acknowledge UGC, India, and SERB-DST, India, and IACS, India, for research fellowships, respectively. The authors thank Gopal Manna for assisting in preparing the TOC.References
- A. Sharma, D. Shambhwani, S. Pandey, J. Singh, H. Lalhlenmawia, M. Kumarasamy, S. K. Singh, D. K. Chellappan, G. Gupta, P. Prasher, K. Dua and D. Kumar, ACS Omega, 2023, 8, 10–41 CrossRef CAS PubMed.
- A. M. Cryer and A. J. Thorley, Pharm. Ther., 2019, 198, 189–205 CrossRef CAS PubMed.
- A. Crintea, A. G. Dutu, G. Samasca, I. A. Florian, I. Lupan and A. M. Craciun, Life, 2021, 11, 682 CrossRef CAS PubMed.
- A. Babu, A. K. Templeton, A. Munshi and R. Ramesh, J. Nanomater., 2013, 2013, 863951 CrossRef.
- J. Wang, T. Zhou, Y. Liu, S. Chen and Z. Yu, Front. Pharmacol., 2022, 12, 781425 CrossRef PubMed.
- M. K. Alshammari, E. Y. Almomen, K. F. Alshahrani, S. F. Altwalah, M. Kamal, M. F. Al-Twallah, S. H. Alsanad, M. H. Al-Batti, F. J. Al-Rasheed, A. Y. Alsalamah, M. B. Alhazza, F. A. Alasmari, Abida and M. Imran, Biomedicines, 2023, 11, 473 CrossRef CAS PubMed.
- U. K. Sukumar, B. Bhushan, P. Dubey, I. Matai, A. Sachdev and G. Packirisamy, Int. Nano Lett., 2013, 3, 45 CrossRef.
- K. Bukowski, M. Kciuk and R. Kontek, Int. J. Mol. Sci., 2020, 21, 3233 CrossRef CAS PubMed.
- R. Thapa and G. D. Wilson, Stem Cells Int., 2016, 2016, 2087204 CrossRef.
- C. Chen, S. Zhao, A. Karnad and J. W. Freeman, J. Hematol. Oncol., 2018, 11, 64 CrossRef.
- Y. Yu, C. Huang, F. Chen, W. Pan and L. Zhang, Mater. Des., 2023, 225, 111551 CrossRef CAS.
- S. Y. Lee, M. S. Kang, W. Y. Jeong, D. W. Han and K. S. Kim, Cancers, 2020, 12, 940 CrossRef CAS PubMed.
- J. Jiang, S. Srivastava and J. Zhang, Cancers, 2019, 11, 804 CrossRef CAS PubMed.
- L. Chen and H. Cui, Int. J. Mol. Sci., 2015, 16, 22830–22855 CrossRef CAS PubMed.
- S. Dinda, M. Ghosh and P. K. Das, Langmuir, 2016, 32, 6701–6712 CrossRef CAS PubMed.
- D. Sarkar, M. Chowdhury and P. K. Das, Langmuir, 2022, 38, 3480–3492 CrossRef CAS PubMed.
- M. Chowdhury and P. K. Das, ACS Appl. Bio Mater., 2021, 4, 5132–5144 CrossRef CAS PubMed.
- G. Nocito, G. Calabrese, S. Forte, S. Petralia, C. Puglisi, M. Campolo, E. Esposito and S. Conoci, Cancers, 2021, 13, 1991 CrossRef CAS PubMed.
- V. Mishra, A. Patil, S. Thakur and P. Kesharwani, Drug Discovery Today, 2018, 23, 1219–1232 CrossRef CAS PubMed.
- M. Zheng, S. Ruan, S. Liu, T. Sun, D. Qu, H. Zhao, Z. Xie, H. Gao, X. Jing and Z. Sun, ACS Nano, 2015, 9, 11455–11461 CrossRef CAS PubMed.
- A. Truskewycz, H. Yin, N. Halberg, D. T. H. Lai, A. S. Ball, V. K. Truong, A. M. Rybicka and I. Cole, Small, 2022, 18, 2106342 CrossRef CAS PubMed.
- S. Dinda, D. Mandal, S. Sarkar and P. K. Das, Chem. – Eur. J., 2017, 23, 15194–15202 CrossRef CAS PubMed.
- P. Zou, G. Y. Shi and C. Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3669–3679 CrossRef CAS.
- A. Ghosh, A. K. Ghosh, A. Zaman and P. K. Das, Mol. Pharmaceutics, 2023, 20, 6391–6406 CrossRef CAS PubMed.
- L. Zhang, Z. Lin, Y. X. Yu, B. P. Jiang and X. C. Shen, J. Mater. Chem. B, 2018, 6, 6534–6543 RSC.
- H. A. Crosby, M. Ihnat and K. E. Miller, MOJ Toxicol., 2015, 1, 30–38 Search PubMed.
- L. Tenora, J. Alt, R. P. Dash, A. J. Gadiano, K. Novotna, V. Veeravalli, J. Lam, Q. R. Kirkpatrick, K. M. Lemberg, P. Majer, R. Rais and B. S. Slusher, J. Med. Chem., 2019, 62, 3524–3538 CrossRef CAS.
- F. Wu, A. Lukinius, M. Bergström, B. Eriksson, Y. Watanabe and B. Långström, Eur. J. Cancer, 1999, 35, 1155–1161 CrossRef CAS PubMed.
- H. C. Yoo, S. J. Park, M. Nam, J. Kang, K. Kim, J. H. Yeo, J. K. Kim, Y. Heo, H. S. Lee, M. Y. Lee, C. W. Lee, J. S. Kang, Y. H. Kim, J. Lee, J. Choi, G. S. Hwang, S. Bang and J. M. Han, Cell Metab., 2020, 31, 267–283 CrossRef CAS.
- M. Hassanein, M. D. Hoeksema, M. Shiota, J. Qian, B. K. Harris, H. Chen, J. E. Clark, W. E. Alborn, R. Eisenberg and P. P. Massion, Clin. Cancer Res., 2013, 19, 560–570 CrossRef CAS PubMed.
- J. Huang, L. Chen, J. Wu, D. Ai, J. Q. Zhang, T. G. Chen and L. Wang, J. Med. Chem., 2022, 65, 16033–16061 CrossRef CAS PubMed.
- N. H. Park, W. Cheng, F. Lai, C. Yang, P. Florez de Sessions, B. Periaswamy, C. W. Chu, S. Bianco, S. Liu, S. Venkataraman, Q. Chen, Y. Y. Yang and J. L. Hedrick, J. Am. Chem. Soc., 2018, 140, 4244–4252 CrossRef CAS PubMed.
- S. Herath, R. H. Sadeghi, P. Radfar, R. Ladwa, M. Warkiani, K. O'Byrne and A. Kulasinghe, Front. Oncol., 2022, 11, 801269 CrossRef PubMed.
- C. M. Leenders, L. Albertazzi, T. Mes, M. M. Koenigs, A. R. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- S. Cantekin, T. F. A. de Greefab and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- P. Kesharwani, R. Chadar, A. Sheikh, W. Y. Rizg and A. Y. Safhi, Front. Pharmacol., 2022, 12, 800481 CrossRef PubMed.
- Y. S. Jian, C. W. Chen, C. A. Lin, H. P. Yu, H. Y. Lin, M. Y. Liao, S. H. Wu, Y. F. Lin and P. S. Lai, Int. J. Nanomed., 2017, 12, 2315–2333 CrossRef CAS PubMed.
- L. Zhong, L. Xu, Y. Liu, Q. Li, D. Zhao, Z. Li, H. Zhang, H. Zhang, Q. Kan, Y. Wang, J. Sun and Z. He, Acta Pharm. Sin. B, 2019, 9, 397–409 CrossRef PubMed.
- H. Y. Seok, N. S. Rejinold, K. M. Lekshmi, K. Cherukula, I. K. Park and Y. C. Kim, J. Controlled Release, 2018, 280, 20–30 CrossRef CAS PubMed.
- J. Li, M. Li, L. Tian, Y. Qiu, Q. Yu, X. Wang, R. Guo and Q. He, Int. J. Pharm., 2020, 578, 119122 CrossRef CAS PubMed.
- M. Kumar and S. J. George, Nanoscale, 2011, 3, 2130–2133 RSC.
- K. M. Lemberg, J. J. Vornov, R. Rais and B. S. Slusher, Mol. Cancer Ther., 2018, 17, 1824–1832 CrossRef CAS PubMed.
- J. Alt, M. C. Potter, C. Rojas and B. S. Slusher, Anal. Biochem., 2015, 474, 28–34 CrossRef CAS PubMed.
- B. He, N. Lu and Z. Zhou, Curr. Opin. Cell Biol., 2009, 21, 900–912 CrossRef CAS PubMed.
- M. Spada, C. Piras, G. Diana, V. P. Leoni, D. V. Frau, G. Serreli, G. Simbula, R. Loi, A. Noto, F. Murgia, P. Caria and L. Atzori, Antioxidants, 2023, 12, 683 CrossRef CAS PubMed.
- M. Butler, L. T. van der Meer and F. N. van Leeuwen, Trends Endocrinol. Metab., 2021, 32, 367–381 CrossRef CAS PubMed.
- Z. Wang, F. Liu, N. Fan, C. Zhou, D. Li, T. Macvicar, Q. Dong, C. J. Bruns and Y. Zhao, Front. Oncol., 2020, 10, 589508 CrossRef PubMed.
- J. Jin, J. K. Byun, Y. K. Choi and K. G. Park, Exp. Mol. Med., 2023, 55, 706–715 CrossRef CAS PubMed.
- M. Hassanein, J. Qian, M. D. Hoeksema, J. Wang, M. Jacobovitz, J. Xiangming, F. T. Harris, B. K. Harris, K. L. Boyd, H. Chen, R. Eisenberg and P. P. Massion, Int. J. Cancer, 2015, 137, 1587–1597 CrossRef CAS PubMed.
- H. Yang, R. M. Villani, H. Wang, M. J. Simpson, M. S. Roberts, M. Tang and X. Liang, J. Exp. Clin. Cancer Res., 2018, 37, 266 CrossRef CAS PubMed.
- E. Panieri and M. M. Santoro, Cell Death Dis., 2016, 7, e2253 CrossRef CAS PubMed.
- A. Ghoneum, A. Y. Abdulfattah, B. O. Warren, J. Shu and N. Said, Int. J. Mol. Sci., 2020, 21, 3100 CrossRef CAS PubMed.
- M. R. Dutordoir and D. A. A. Bates, Biochim. Biophys. Acta, 2016, 1863, 2977–2992 CrossRef PubMed.
- L. Y. Tian, D. J. Smit and M. Jücker, Int. J. Mol. Sci., 2023, 24, 2652 CrossRef CAS PubMed.
- D. Miricescu, A. Totan, I. I. Stanescu-Spinu, S. C. Badoiu, C. Stefani and M. Greabu, Int. J. Mol. Sci., 2021, 22, 173 CrossRef CAS PubMed.
- C. Porta, C. Paglino and A. Mosca, Front. Oncol., 2014, 4, 64 Search PubMed.
- D. G. Hardie, Cell Metab., 2014, 20, 939–952 CrossRef CAS PubMed.
- O. Trédan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef PubMed.
- I. W. Y. Mak, N. Evaniew and M. Ghert, Am. J. Transl. Res., 2014, 6, 114–118 Search PubMed.
- J. H. Véchot, A. Rafii, C. Touboul and J. Pasquier, Int. J. Mol. Sci., 2018, 19, 181 CrossRef PubMed.
- N. Manduca, E. Maccafeo, R. De Maria, A. Sistigu and M. Musella, Front. Immunol., 2023, 14, 1175503 CrossRef CAS PubMed.
- G. Gamerith, J. Rainer, J. M. Huber, H. Hackl, Z. Trajanoski, S. Koeck, E. Lorenz, J. Kern, R. Kofler, J. M. Kelm, H. Zwierzina and A. Amann, Oncotarget, 2017, 68, 112647–112661 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic scheme, experimental details, characterization data, 1H NMR and mass spectra of TMAVs, DLS correlogram of TMAVs, DLS study of HACD–TMAV, calibration curve of DON, UV-vis spectra of individual DON-loaded nano-vehicles, FTIR spectra of HACD–TMAV-DON, cytocompatibility of HACD–TMAV against A549, HepG2 and HEK293 cells, flow cytometric analysis of apoptosis in A549 cells using HACD–TMAV-DON, nuclear staining of A549 cells with Hoechst 33342 dye, calibration curve of glutamine, comparative analysis of the intracellular ROS levels, formation of 3D tumor spheroids, flow cytometry analysis of apoptosis and ROS generation in 3D tumor spheroids. See DOI: https://doi.org/10.1039/d4nr00426d |
| This journal is © The Royal Society of Chemistry 2024 |