
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-induced microfluidic production of ultrasmall platinum nanoparticles†
Marcello
Marelli
 *a,
Patricia
Perez Schmidt
a,
Xuan Trung
Nguyen
b,
Emanuela
Pitzalis
b,
Lorenzo
Poggini
cd,
Laura
Ragona
e,
Katiuscia
Pagano
e,
Laura Antonella
Aronica
f,
Laura
Polito
a and
Claudio
Evangelisti
*b
*a,
Patricia
Perez Schmidt
a,
Xuan Trung
Nguyen
b,
Emanuela
Pitzalis
b,
Lorenzo
Poggini
cd,
Laura
Ragona
e,
Katiuscia
Pagano
e,
Laura Antonella
Aronica
f,
Laura
Polito
a and
Claudio
Evangelisti
*b
aCNR-SCITEC, Institute of Science and Chemical Technologies “Giulio Natta”, Via Fantoli 16/15, 20138 Milano, Italy. E-mail: marcello.marelli@cnr.it
bCNR-ICCOM, Institute of Chemistry of OrganoMetallic Compounds, Via G. Moruzzi 1, 56124 Pisa, Italy. E-mail: claudio.evangelisti@cnr.it
cCNR-ICCOM, Institute of Chemistry of OrganoMetallic Compounds, Via Madonna del Piano 10, 50019 Sesto Fiorentino, Italy
dDepartment of Chemistry “U. Schiff” – DICUS – and INSTM Research Unit, University of Florence, Via della Lastruccia 3-13, 50019 Sesto Fiorentino, FI, Italy
eCNR-SCITEC, Institute of Science and Chemical Technologies “Giulio Natta”, Via Corti 12, 20133 Milano, Italy
fDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy
First published on 30th September 2024
Abstract
We describe here the synthesis of ultrasmall Pt nanoparticles (NPs) obtained by a robust and reliable protocol using UV-Vis photoreduction of a platinum salt precursor, under continuous flow conditions. These ligand-free Pt NPs were rapidly dispersed onto a solid support or stabilized towards aggregation as a colloidal solution by the addition of an appropriate ligand in the reaction mixture. The proposed protocol exploits a microfluidic platform where the Pt4+ precursor is photo-reduced to small Pt0 NPs (1.3 nm) at room temperature in the presence of ethanol, without any additional reducing agent. We apply the protocol to prepare Pt NPs highly dispersed on carbon support (Pt/C) proven to be a very efficient heterogeneous catalyst for both the hydrosilylation of terminal alkynes and hydrogenation of nitroaromatic compounds, selected as model reactions. Furthermore, we exploit the versatility of this microfluidic approach to produce stabilized aqueous/ethanol colloidal solutions of Pt NPs, employing a ligand of choice (e.g., PVP or a thiol-ligand). These colloids offer long-term storage and further ligand modification. We showcase the synthesis of biocompatible glycol-stabilized Pt nanoparticles as an exemplary application.
Noble metal nanoparticles (NPs), particularly platinum NPs, are extensively studied for academic and industrial purposes.1,2 The peculiar properties endowed by the size and shape are transformed to unique optical and electronic characteristics.3 Thanks to that, several fields of work benefit from their application spanning from catalysis, optoelectronics, sensors, energy production, and environmental remediation to nano-medicine and biotechnologies.4,5
Heterogeneous Pt catalysts containing small Pt NPs finely dispersed on a high-surface-area support have been used for more than 100 years to promote a large variety of reactions for the production of fine and specialty chemicals (e.g. hydrogenations, hydrosilylations, oxidations, dehydrogenations, hydrogenolysis)6–8 and for thermo- and electrocatalytic processes in the field of hydrogen production (e.g. reforming of small organic molecules, water electrolysis).9–11 Small Pt NPs have also been largely studied in photocatalysis12,13 as electron-trapping agents and in several fuel cell technologies14 such as Polymer Electrolyte Fuel Cells (PEMFCs), or Direct Methanol Fuel Cells (DMFCs), where they are a fundamental component. The control of the size and shape of the supported NPs plays a crucial role in their catalytic performances controlling activity, selectivity, and stability.15
In the biomedical field, the physicochemical properties of Pt NPs could be exploited in diagnostics and treatments. Noteworthily, modulation of the Pt NPs’ size, shape, and surface functionalization can determine their biocompatibility. For instance, functionalization with peptides, DNA, or low molecular weight ligands among others can increase Pt NPs’ specificity and targeting efficacy, being in this way excellent candidates for biomedical applications such as for cancer treatment.16–18 Platinum is involved in the design of synthetic enzymes (nanozymes) or is used as a radical scavenger in therapies for oxidative stress diseases.5,19–22 Moreover, despite some misgivings about nanotoxicity, Pt NPs showed good stability in acidic cellular vesicle environments, promising cytocompatibility in vivo.23
All these applications and capabilities are strongly entangled with the fine control of NP structure and chemical surroundings in turn expressed in the different properties and activities. An effective design requires a carefully planned production route and fine control of synthesis parameters. Among the plethora of synthetic routes available, our focus lies on colloidal synthesis.24,25 This approach offers simplicity and scalability: typically, a metal–organic precursor is dissolved in a suitable solvent, subjected to reduction to yield metal NPs (via chemical, thermal, or light-induced processes), and stabilized using a surfactant or a coordinating additive. The last step is generally useful to obtain a stable colloidal solution; however, the stabilizers sometimes need to be removed to activate the NPs or, conversely, are necessary to bring the desired function to the metal (e.g. specific steric hindrance, active tags, or enhanced bio-compatibility). In conventional batch reactors, these methods could suffer from inhomogeneous mixing of reactants. As a result, the nucleation and growth steps are not properly separated in time, leading to polydispersity in NPs’ size and shape and poor batch-to-batch reproducibility and thus difficulty in producing these materials on an industrial scale. Micro- and milli-fluidic continuous flow setups have been recently identified as optimal methods to address this scaling issue. These systems offer enhanced mixing, allowing precise control over reaction conditions. By spatially and temporally separating the nucleation and growth steps, they enable the synthesis of size-controlled metal NPs with narrower size distributions. Simply by using multiple flow reactors operating simultaneously (i.e. numbering up), continuous flow setups can circumvent the issues related to the scaling up of the NP production.26
This work aims to propose a novel, more sustainable, microfluidic strategy for the synthesis of ultrasmall Pt NPs that can be effectively used to obtain supported Pt-based heterogeneous catalysts or ligand-stabilized Pt NPs as innovative bio-active nanomaterials. Central to this approach is the exploitation of continuous-flow microfluidics combined with UV-photoreduction of Pt atoms, offering distinct advantages over conventional batch methods. These include enhanced control over the reaction parameters, fast and precise reagent mixing, and higher process reproducibility.27–30 Particularly, the microfluidic process offers exclusive benefits such as more efficient light exposure, reduced material consumption (particularly during the optimization phase), and the potential for easy scale up, all of which enhance the overall sustainability of the system. Moreover, the UV-light exploitation to promote the synthesis avoids the use of harsh chemical reductants and allows the synthesis at room temperature for a greener process. Pt photoreduction under UV light was deeply studied by Harada and co-workers disclosing the NPs’ growth mechanism and the influences of the stabilizer under batch conditions.31–34 They observed a fast reduction step favouring the formation of a large amount of nucleation sites (that is a good prerequisite to obtain small NPs) but poor stability in aqueous media and particle precipitation. In this work, we overcame this challenge through microfluidic implementation, facilitating the synthesis of ultrasmall Pt NPs yet stable enough to be highly dispersed onto a solid support (i.e. carbon).
The synthesis was carried out at room temperature (25 °C) in a water–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture, without the use of additional reducing agents (Fig. 1). The setup used has been designed by our group and recently successfully employed for the synthesis of ultrasmall Au NPs.35 The procedure is facile, highly reproducible, flexible, robust, and reliable. In detail, the as-prepared Pt NPs without any additional ligand were immediately dispersed on carbon as the support leading to Pt/C catalysts with different Pt loadings (i.e. 1 wt% and 0.1 wt%). The unsupported ligand-free Pt NPs were not stable against agglomeration at room temperature for long times (i.e. after ca. 30 minutes the formation of a precipitate was observed).
:1) mixture, without the use of additional reducing agents (Fig. 1). The setup used has been designed by our group and recently successfully employed for the synthesis of ultrasmall Au NPs.35 The procedure is facile, highly reproducible, flexible, robust, and reliable. In detail, the as-prepared Pt NPs without any additional ligand were immediately dispersed on carbon as the support leading to Pt/C catalysts with different Pt loadings (i.e. 1 wt% and 0.1 wt%). The unsupported ligand-free Pt NPs were not stable against agglomeration at room temperature for long times (i.e. after ca. 30 minutes the formation of a precipitate was observed).
 | ||
| Fig. 1 Microfluidic reactor sketch: (1) reagent inlet, (2) pump, (3) reactor coiled around the UV lamp, (4) circuit loop, (5) direct carbon impregnation and/or (6) product outlet. | ||
By flowing the reaction mixture through a FEP (fluorinated ethylene propylene) tubing setup, we achieve Pt NP formation without the need for harsh reducing agents at room temperature. UV light initiates solvent decomposition, leading to the generation of hydroxyl radicals and solvated electrons, which efficiently reduce Pt4+ to form Pt0 NPs via Pt2+ ions following the mechanism (Scheme S1†) previously described by Harada et al.33 We assume that the overall reaction mechanism and kinetics proceed through a two-step reduction process, followed by nucleation and aggregative growth. We hypothesize that the spatial confinement of the microfluidic system limits further NP accretions and so limits their size.31 UV-vis spectra of the Pt colloidal solution (Fig. S2† – obtained at the beginning of each 20-minute cycle) substantially retrace the profile obtained by Harada.31 The characteristic smooth band in the range of 300–400 nm, from the Pt4+ precursor, is barely detectable already after 20 minutes (first cycle). Signals stabilize after 80 minutes, indicating that the precursor was consumed obtaining a clear yellow-brownish solution.
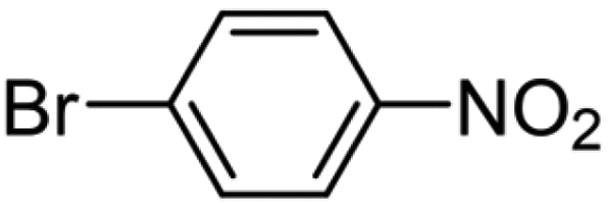
The as-prepared Pt NPs showed a very narrow NP size distribution with a mean diameter (dm) of 1.3 nm (standard deviation (SD) = 0.3 nm) and almost 80% of them were below 1.5 nm (SD 0.3 nm) in size (Fig. 2, above). The mean size observed in the as-prepared Pt NPs agrees with the formation of FCC-derived truncated octahedral shape Ptn nanoparticles (n = 116).36 This size allows a relative stability of the Pt NPs dominated by their electronic shell structure occurring as a result of their geometric structure. On the other hand, after their immobilization on the carbon support TEM analysis showed that a thermodynamically favorable increase of the particle size, corresponding to 0.6 nm (dm = 1.9 nm), occurred. This value is in agreement with the addition of one Pt layer on the top of the just obtained Pt NPs, leading to truncated octahedron NPs containing 260 Pt atoms. Anyways, a narrow size distribution was observed (SD = 0.3 nm) together with the absence of segregated Pt NP agglomerates (Fig. 2, down). X-ray Photoelectron Spectroscopy (XPS) has been employed to further study the Pt0 NPs and Fig. S3† shows the survey scan. Furthermore, we acquired in detail the Pt 4f region, revealing an almost complete reduction to Pt0 (85.1%) as reported in Fig. 2, with a component located at 70.2 eV. A minor amount of Pt(IV) at 74.1 eV (4.4%) and Pt(II) at 72.1 eV (10.5%) were revealed according to the reported mechanism29,31 that counts a slow conversion of PtCl62− precursor to the PtCl42− ionic complex, followed by a fast reduction of the latter to obtain Pt0 metal clusters.20,22 In light of the possible application of the carbon-supported Pt NPs as heterogeneous catalysts, two classes of reactions were selected: hydrogenation of halonitroaromatics34,35 and hydrosilylation of terminal alkynes.36 The catalytic results obtained in both reactions carried out under very mild conditions (i.e. 70 °C, solvent-less, for hydrosilylation and 25 °C, 1.0 bar H2 for hydrogenation), are reported in Table 1. In the hydrosilylation of 1-hexyne with dimethylphenylsilane, our Pt catalysts with two different loadings (1 wt% and 0.1 wt%) were tested (entries 1 and 2). Both catalysts exhibited good catalytic activity with >95% conversion after 2 h of reaction time. 1H NMR analysis revealed that there were only β-(E) isomer ((E)-hex-1-en-1-yldimethyl(phenyl)silane) and α product (hex-1-en-2-yldimethyl(phenyl)silane). The β-(E)/α selectivity was found to be 75%/25%. Notably, the Pt loading did not affect selectivity and activity, as both loadings demonstrated similar β-(E)/α selectivity and turnover frequencies (TOFs) of 3840 h−1 and 3520 h−1, respectively. The Pt-based catalysts showed high efficiency in the other model reaction: chemo-selective hydrogenation, particularly of p- and o-chloronitrobenzene and p-bromonitrobenzene (entries 3–5). Carbon-supported Pt catalysts were extensively studied in this process and a strong correlation between the Pt NP sizes (either an electronic or geometric effect) and the chemo-selectivity was reported.37,38 Particularly, according to the above reported results, we previously reported that under similar reaction conditions Pt NPs measuring 2 nm diameter obtained by a different approach and dispersed on the same carbon support were very selective to the formation of haloanilines, limiting the hydro-dehalogenation reaction.39
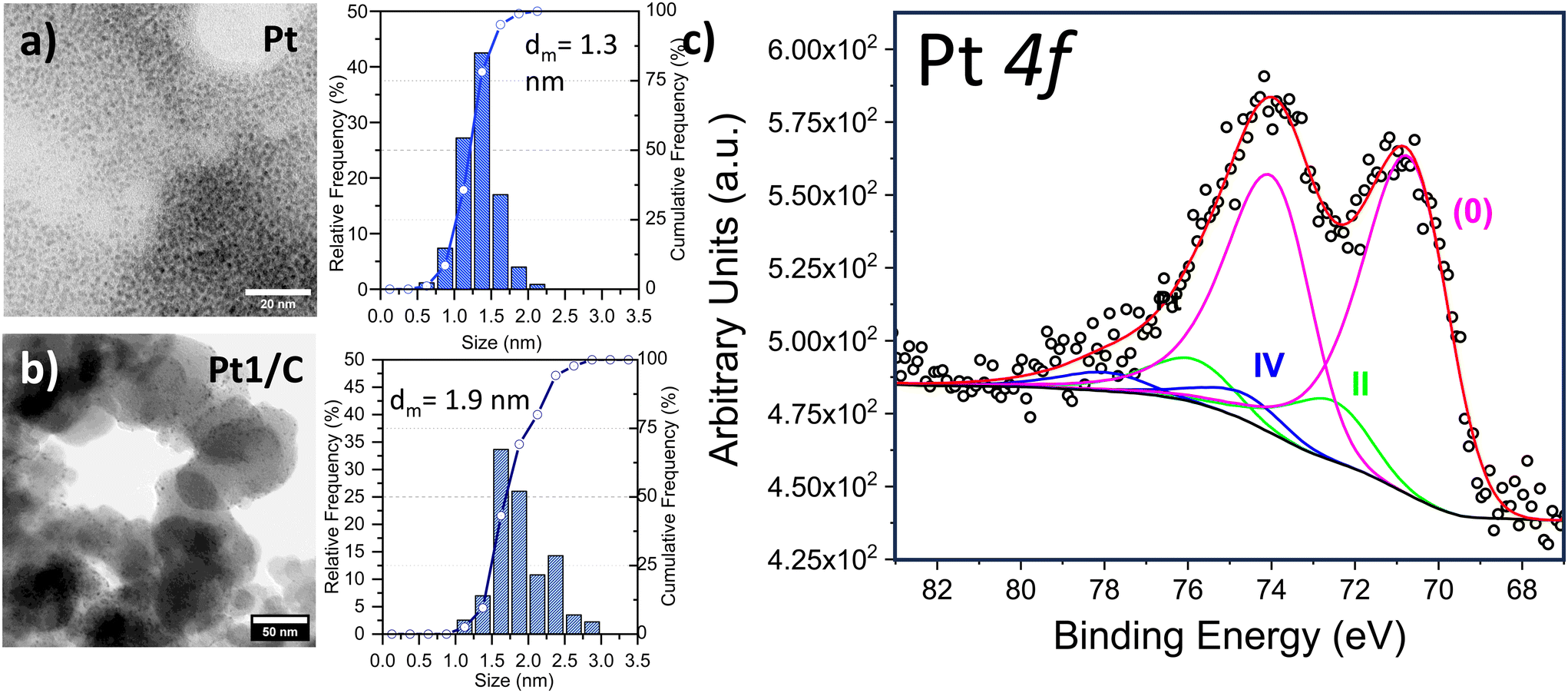 | ||
| Fig. 2 Left side, TEM micrographs and related NP size distribution of (a) freshly prepared Pt0 NPs and (b) derived Pt0 NPs supported on carbon (Pt/C), metal loading 1 Pt wt.% alongside the (c) corresponding XPS spectrum at the Pt 4f edge (right side). | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Time (min) | Conv. (%) | Products sel. (%) | TOFc (h−1) |
| a Reaction conditions: Pt/C (5 × 10−4 mmol Pt), 1-hexyne (8 mmol), Me2PhSiH (2 mmol), T = 70 °C. b Reaction conditions: Pt/C (5 × 10−4 mmol Pt), halonitrobenzene (1.28 mmol), T = 25 °C, P(H2) = 0.1 MPa, MeOH (5 mL). c Calculated as mol converted substrate × mol. Pt−1 × min−1 at lower than 50% of reaction conversion. d Isomeric ratio derived from 1H-NMR integrals of the olefinic protons. e Chemoselectivity calculated by GC-FID. | ||||||
| Hydrosilylation of 1-hexynea | ||||||
| 1 |

|
Pt/C 0.1 wt% | 120 | 100 | 75 β-(E); 25(α)d | 3840 |
| 2 | Pt/C 1.0 wt% | 120 | 95 | 73 β-(E); 27(α)d | 3520 | |
| Hydrogenation of haloarenesb | ||||||
| 3 |

|
Pt/C 1.0 wt% | 50 | 98 | 99 XAN; 1 ANe | 3957 |
| 4 |

|
Pt/C 1.0 wt% | 135 | 96 | 99 XAN; 1 ANe | 1041 |
| 5 |
|
Pt/C 1.0 wt% | 90 | 100 | > 99 ![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) |
1673 |

The UV-derived Pt NPs showed excellent catalytic activity in these reactions, with exceptionally high selectivity for halo-aminoaromatic products (≥99% selectivity in all cases). As expected, on increasing the steric hindrance (from p- to o-) a slight decrease of reaction rate was observed (TOF = 3957 and 1041 h−1, for p- and o-chloronitrobenzene, respectively).40 Interestingly, the catalyst displayed negligible dehalogenation activity, even with bromo-substituted substrates (see entry 5, Table 1), which is typically challenging to achieve with other Pt catalysts.41–43
The high versatility of the protocol can allow obtaining Pt NP colloidal solution stable in time (avoiding NP aggregation and/or precipitation) at room temperature by the addition of an appropriate ligand (e.g. a polymer or a molecular ligand) to the reagent mixture and endow a new functionality to the ultra-small Pt NPs. With this aim, we added polyvinylpyrrolidone (PVP) (0.1 wt%) to the starting mixture obtaining PVP-stabilized Pt NPs at room temperature. However, they can be stored for a long time (months) at room temperature without the formation of any agglomerate, due to the control of size exerted by PVP in the photoreduction process.33 (See Fig. S4† for the reaction scheme.) The PVP-Pt NPs are slightly larger than the as prepared ligand-free ones (dm = 1.9 nm; SD = 0.3 nm, Fig. 3), and in agreement with the above discussion about the possible formation of more stable truncated octahedron Pt NPs containing 260 Pt atoms. We then synthesized supported catalysts by impregnation of PVP-stabilized Pt NPs on carbon (Pt(PVP)/C; Pt loading of 0.1 wt% and 1.0 wt%). The Pt(PVP)/C catalysts showed that Pt NPs retain the sizes without particle growth or agglomeration during the deposition step. The presence of PVP at the surface of the Pt NPs leads to a negative effect on the catalytic activity of the catalyst in the hydrogenation of haloarenes (TOF = 3957 and 2413 h−1 for Pt/C and Pt(PVP)/C, respectively) that can be ascribed to the steric hindrance of the ligand at the Pt NP surface. On the other hand, only a slightly negative effect on the selectivity (98% vs. 99% to XAN) was observed. Overall, the above reported results demonstrated that the Pt(PVP)/C catalyst exhibited a notable catalytic efficiency (i.e. activity and selectivity), provided the possibility for long term storage of the PVP-stabilized Pt NPs. The proposed synthetic approach can also be exploited to confer innovative properties to the Pt NPs. By utilizing a specific stabilizer in the reaction mixture, we can indeed engineer the NP surface, introducing unique functionalities. As a proof of concept of the extreme process flexibility, a glycan derivative of lactose (3-Lac) was added to the reaction mixture. The (3-Lac) ligand, namely the (29-thio-[(3,6,9,12,15,18-hexaoxaundecanyl-β-D-galactopyranosyl)(1 → 4)-β-D-glucopyranoside]), has an amphiphilic linker with a terminal thiol moiety able to anchor the particle's surface and was previously employed in our laboratory to decorate analogous Au-Glyco NPs.33 Pt(3-Lac) NP synthesis followed the same steps (Fig. S05†), leading to a clear yellow stable colloidal solution that was purified by ultrafiltration and centrifugation. Pt(3-Lac) NPs have a mean diameter of 2.1 nm (SD 0.4 nm) with almost 90% of the NPs < 2.5 nm, without the formation of any NP agglomerates (Fig. 3). The strong broadening of 1H NMR resonances in the Pt(3-Lac) 1D spectrum, compared to those of free 3-Lac, together with the disappearance of resonances of chemical groups in the direct surroundings of thiol (Hβ of 3-Lac) confirmed the binding of 3-Lac to the Pt NPs (Fig. 3). NMR-DOSY analysis (Fig. S06†) highlighted a single main diffusion coefficient, confirming a narrow size distribution of NPs. An estimated hydrodynamic radius of 3.7 ± 0.2 nm was derived from the measured diffusion coefficient D = 5.08 ± 0.4 × 10−11 m2 s−1 from eqn (2) in the Materials and methods section.†
 | ||
| Fig. 3 Representative TEM micrographs and related size distributions of Pt PVP (left side above) and Pt(3-LAC) NP colloidal solution (left side below). Overlay of 1D 1H NMR spectra of the 3-Lac free ligand (black) and Pt(3-Lac) NPs (red). The red arrow highlights the resonances of chemical groups in the direct surroundings of thiol groups (2.636 ppm for Hβ). | ||
Conclusions
In summary, we developed an optimized, robust, and reliable protocol combining the photo-induced UV-reduction of a Pt4+ precursor to size-controlled Pt0 NPs with a microfluidic platform, carried out at room temperature. The approach allows the continuous flow synthesis of ligand-free Pt NPs (mean diameter of 1.3 nm) that can be rapidly immobilized on a heterogeneous support (i.e. carbon), minimizing their over-growth and agglomeration. The derived Pt/C supported system has been proven to be a very efficient heterogeneous catalyst for both the hydrogenation of nitroaromatic compounds and hydrosilylation of terminal alkynes, selected as model reactions. Interestingly, PVP-stabilized Pt NPs obtained with the same approach and deposited onto carbon Pt(PVP)/C still maintained a notable catalytic activity and selectivity in the reported reactions. The high flexibility of the approach was exploited to achieve quantitative collection of ultrasmall Pt NPs, stabilized towards the agglomeration by the presence of additional ligands in the reaction medium (i.e. PVP or a thiol functionalized glycan ligand, 3-Lac). The direct synthesis of glyco-Pt nano-systems has never been reported and an optimized protocol for Glyco-NP preparation is of relevance as they emerged as excellent platforms for many bio-applications.2,44,45 In addition, Pt(3-Lac) NMR characterization allowed us to estimate the ligand concentration on metal surfaces and the size of the decorated Pt NP particles. The proposed photo-induced microfluidic production of ultrasmall Pt NP opens the way to efficiently produce supported or unsupported Pt nanoparticles having very reproducible structural and morphological features to be exploited in catalysis and/or in other fields, such as biomedicine. Thanks to the ease of scale-up, the relatively low cost, and simplicity compared to other competing continuous flow technologies, the proposed approach can be applied in a larger scale production of a wide range of metal nanoparticles, decorated with different functional groups at their surface.Author contributions
M. M.: conceptualization, writing – original draft, writing – review & editing, methodology, and investigation, P. P. S.: writing – review & editing and investigation, X. T. N.: writing – review & editing and investigation, E. P.: writing – review & editing and investigation, L. POG.: writing – review & editing and investigation, L. R.: writing – review & editing and investigation, K. P.: writing – review & editing and investigation, L. A. A.: writing – review & editing and investigation, L. POL.: conceptualization, writing – review & editing, methodology, and investigation, C. E.: conceptualization, writing – original draft, writing – review & editing, and investigation.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mr Brunetto Cortigiani for his assistance in using the MatchLab (University of Florence) platform.References
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chemistry and properties of nanocrystals of different shapes, 2005, vol. 105 Search PubMed.
- M. A. R. Khan, M. S. Al Mamun and M. H. Ara, Microchem. J., 2021, 171, 106840 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- G. J. Leong, M. C. Schulze, M. B. Strand, D. Maloney, S. L. Frisco, H. N. Dinh, B. Pivovar and R. M. Richards, Appl. Organomet. Chem., 2014, 28, 1–17 CrossRef CAS.
- M. Jeyaraj, S. Gurunathan, M. Qasim, M.-H. Kang and J.-H. Kim, Nanomaterials, 2019, 9, 1719 CrossRef CAS PubMed.
- Chemistry of the Platinum Group Metals Recent Developments, ed. F. R. Hartley, Elsevier Science, 1991 Search PubMed.
- H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed.
- N. Jung, D. Y. Chung, J. Ryu, S. J. Yoo and Y. E. Sung, Nano Today, 2014, 9, 433–456 CrossRef CAS.
- M. A. Khan, H. Zhao, W. Zou, Z. Chen, W. Cao, J. Fang, J. Xu, L. Zhang and J. Zhang, Recent Progresses in Electrocatalysts for Water Electrolysis, Springer Singapore, 2018, vol. 1 Search PubMed.
- A. Chen and P. Holt-hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- Z. Wei, M. Janczarek, K. Wang, S. Zheng and E. Kowalska, Catalysts, 2020, 10, 1070 CrossRef CAS.
- K. Wang and E. Kowalska, Front. Chem., 2022, 10 DOI:10.3389/fchem.2022.972494.
- G. Ran, Q. Fu and W. Xu, RSC Adv., 2015, 5, 14740–14746 RSC.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- S. Y. Gutiérrez de la Rosa, R. Muñiz Diaz, P. T. Villalobos Gutiérrez, R. Patakfalvi and Ó. Gutiérrez Coronado, Int. J. Mol. Sci., 2022, 23, 9404 CrossRef.
- D. Guarnieri, P. Melone, M. Moglianetti, R. Marotta, P. A. Netti and P. P. Pompa, Nanoscale, 2017, 9, 11288–11296 RSC.
- Y. Teow and S. Valiyaveettil, Nanoscale, 2010, 2, 2607–2613 RSC.
- N. Fakhri, S. Abarghoei, M. Dadmehr, M. Hosseini, H. Sabahi and M. R. Ganjali, Spectrochim. Acta, Part A, 2020, 227, 117529 CrossRef CAS PubMed.
- A. Naseer, A. Ali, S. Ali, A. Mahmood, H. S. Kusuma, A. Nazir, M. Yaseen, M. I. Khan, A. Ghaffar, M. Abbas and M. Iqbal, J. Mater. Res. Technol., 2020, 9, 9093–9107 CrossRef CAS.
- H. Khan, H. R. Mirzaei, A. Amiri, E. Kupeli Akkol, S. M. Ashhad Halimi and H. Mirzaei, Semin. Cancer Biol., 2021, 69, 24–42 CrossRef CAS.
- D. Pedone, M. Moglianetti, E. De Luca, G. Bardi and P. P. Pompa, Chem. Soc. Rev., 2017, 46, 4951–4975 RSC.
- Y. Bendale, V. Bendale, R. Natu and S. Paul, J. Pharmacopunct., 2016, 19, 114–121 CrossRef PubMed.
- M. V. Danilenko, V. E. Guterman, I. N. Novomlinskiy and I. V. Pankov, Colloid Polym. Sci., 2023, 301, 433–443 CrossRef CAS.
- J. Quinson and K. M. Ø. Jensen, Adv. Colloid Interface Sci., 2020, 286, 102300 CrossRef CAS.
- E. J. Roberts, L. R. Karadaghi, L. Wang, N. Malmstadt and R. L. Brutchey, ACS Appl. Mater. Interfaces, 2019, 11, 27479–27502 CrossRef CAS PubMed.
- A. Singh and K. Miyabayashi, RSC Adv., 2019, 10, 362–366 RSC.
- M. J. Hossain, M. S. Rahman, M. S. Rahman, M. A. Ali, N. C. Nandi, P. Noor, K. N. Ahmed and S. Akhter, J. Nanostruct. Chem., 2016, 6, 49–56 CrossRef CAS.
- S. Gimondi, H. Ferreira, R. L. Reis and N. M. Neves, ACS Nano, 2023, 17, 14205–14228 CrossRef CAS PubMed.
- U. Laura, M. Arruebo and V. Sebastian, Dalton Trans., 2018, 47, 1693–1702 RSC.
- M. Harada and Y. Kamigaito, Langmuir, 2012, 28, 2415–2428 CrossRef CAS.
- M. Harada, K. Okamoto and M. Terazima, Langmuir, 2006, 22, 9142–9149 CrossRef CAS PubMed.
- M. Harada and H. Einaga, Langmuir, 2006, 22, 2371–2377 CrossRef CAS PubMed.
- M. Sakamoto, M. Fujistuka and T. Majima, J. Photochem. Photobiol., C, 2009, 10, 33–56 CrossRef CAS.
- P. Perez Schmidt, K. Pagano, C. Lenardi, M. Penconi, R. M. Ferrando, C. Evangelisti, L. Lay, L. Ragona, M. Marelli and L. Polito, Angew. Chem., 2023, 62, e202210140 CrossRef CAS.
- R. Al-Shareef, M. Harb, Y. Saih, S. Ould-Chikh, D. H. Anjum, J.-P. Candy and J.-M. Basset, J. Phys. Chem. C, 2018, 122, 23451–23459 CrossRef CAS.
- Z. Wang, C. Wang, S. Mao, B. Lu, Y. Chen, X. Zhang, Z. Chen and Y. Wang, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- Q. Guan, C. Zhu, Y. Lin, E. I. Vovk, X. Zhou, Y. Yang, H. Yu, L. Cao, H. Wang, X. Zhang, X. Liu, M. Zhang, S. Wei, W.-X. Li and J. Lu, Nat. Catal., 2021, 4, 840–849 CrossRef CAS.
- W. Oberhauser, C. Evangelisti, C. Tiozzo, M. Bartoli, M. Frediani, E. Passaglia and L. Rosi, Appl. Catal., A, 2017, 537, 50–58 CrossRef CAS.
- P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445–2465 RSC.
- C. Evangelisti, L. A. Aronica, M. Botavina, G. Martra, C. Battocchio and G. Polzonetti, J. Mol. Catal. A: Chem., 2013, 366, 288–293 CrossRef CAS.
- X. Cui, K. Junge, X. Dai, C. Kreyenschulte, M. M. Pohl, S. Wohlrab, F. Shi, A. Brückner and M. Beller, ACS Cent. Sci., 2017, 3, 580–585 CrossRef CAS.
- M. Takasaki, Y. Motoyama, K. Higashi, S. H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2008, 10, 1601–1604 CrossRef CAS.
- E. Dosekova, J. Filip, T. Bertok, P. Both, P. Kasak and J. Tkac, Med. Res. Rev., 2017, 37, 514–626 CrossRef.
- A. Dag, M. Callari, H. Lu and M. H. Stenzel, Polym. Chem., 2016, 7, 1031–1036 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02971b |
| This journal is © The Royal Society of Chemistry 2024 |