
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel catalyzed C–S cross coupling of sterically hindered substrates enabled by flexible bidentate phosphines†
Ivo H.
Lindenmaier
,
Robert C.
Richter
and
Ivana
Fleischer
 *
*
Institute of Organic Chemistry, Faculty of Science, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: ivana.fleischer@uni-tuebingen.de
First published on 29th February 2024
Abstract
Due to the smaller size of nickel compared to palladium, the C–S cross-coupling of sterically challenging aryl electrophiles with alkyl thiols under nickel catalysis remained elusive. Herein, we report the nickel-catalyzed cross-coupling of alkyl thiols with aryl triflates bearing functional groups in ortho-position relative to the leaving group using Ni(cod)2/DPEphos (L1) or dppbz (L2) as the catalytic system. For substrates featuring non-coordinating ortho-substituents, the reaction operates under mild conditions using L1, while for electrophiles bearing coordinating groups, the ligand L2 and elevated temperatures are required. The synthetic utility could be demonstrated on numerous examples, including biologically relevant compounds, and on larger scale. Instead of Ni(cod)2, more cost-efficient Ni(OAc)2 can also be employed in the presence of zinc as reductant. Furthermore, insights into the reaction mechanism were obtained by competition experiments, isolation of organometallic intermediates and computations.
Introduction
Thioethers are represented in a large range of natural compounds,1 pharmaceuticals,2 agrochemicals3 as well as in functional materials.4 Additionally, compounds containing sulfur in higher oxidation state can be conveniently accessed from the corresponding thioethers.5 Therefore, the efficient and selective construction of the C–S bond has been thoroughly investigated leading to the development of the transition-metal catalyzed Migita reaction as the most prominent procedure.6 Until today, a wide variety of synthetic methods under catalysis of Fe, Ni, Cu and Pd has been established, covering even less activated electrophiles such as aryl chlorides or carbamates while operating under mild conditions.7Despite numerous contributions towards the coupling of electronically deactivated electrophiles, the usage of sterically demanding aryl (pseudo)halides has been much less studied. Compared to other cross-couplings such as the Suzuki–Miyaura-coupling, for which ortho-substituted substrates have been frequently reported,8 in C–S couplings, the low reactivity of the electrophile is more problematic due to the high binding affinity of thiol(ate)s,9 possibly leading to off-cycle species before catalytic turnover can occur. In addition, if neighboring groups contain heteroatoms, catalysis can be hampered by coordination to the catalyst. Whereas aryl thiols seem to be less problematic and have been more frequently reported (Scheme 1A),10 only few protocols operating under palladium catalysis can tolerate sterically demanding electrophiles in couplings with more nucleophilic alkyl thiols.
 | ||
| Scheme 1 Literature overview for the C–S coupling of sterically hindered electrophiles and our objective. | ||
Such systems have been independently published by the groups of Nolan,11 Buchwald12 and Hartwig,13 but all of these protocols still required harsh conditions that are typical for many C–S couplings (Scheme 1B). The only example for Pd-catalyzed coupling of bulky aryl halides with alkyl thiols under mild conditions was presented by Organ.14 However, these reports do not primarily focus on sterically challenging substrates, which is why the scope of ortho-substituted halides is limited to few examples.
Meanwhile, no general efficient procedures exist under nickel catalysis, with the only recently published exceptions requiring photoredox conditions.15 This may owe to the smaller radius of nickel compared to palladium, resulting in higher steric strain around the metal center. Recently, our group reported the coupling of aryl chlorides and triflates with sterically demanding alkyl thiols at room temperature using a nickel precatalyst containing Xantphos ligand (4,5-bis-(diphenylphosphino)-9,9-dimethylxanthen).16 Aryl triflates not only showed superior reactivity, but can also be accessed from the corresponding phenols, a widely available and naturally abundant substance class. Nevertheless, substituents in ortho-position were not tolerated even at higher temperatures with ortho-methyl as the only exception. Consequently, we envisioned the development of a nickel-catalyzed procedure that would allow for the synthesis of sterically hindered aryl alkyl thioethers (Scheme 1B).
Results and discussion
In the beginning of our investigations, 2-biphenyl trifluoromethanesulfonate (2-PhPhOTf, 1a) was chosen as the model substrate in the coupling with n-heptane thiol (2a). In previous projects we found the inorganic base KOAc explicitly suitable for mild C–S couplings and decided to take advantage of this reactivity again. The earlier used precatalyst (Xantphos)Ni(o-tolyl)Cl, as well as a catalytic system consisting of Ni(cod)2 and Xantphos proved ineffective, thus a ligand screening was performed (Scheme 2). It revealed, that bidentate triaryl phosphines with wide bite-angles were necessary for this reaction. Other ligand classes such as N-heterocyclic carbenes, bidentate alkyl-, monodentate aryl and alkyl phosphines or bidentate N-ligands were generally ineffective (for detailed screening information, see ESI, Table S1†). Of all ligands tested, DPEphos (bis[(2-diphenylphosphino)phenyl] ether, L1), as well as ferrocene derivative dppf (1,1′-bis(diphenylphosphino)ferrocene) showed the highest reactivity, while other ligands led to no conversion to the product. For further investigations, DPEphos was chosen for its higher economic efficiency compared to dppf. Since bulky substrates would imply steric strain on the metal center, we hypothesized that the ligand needed to be flexible in the backbone, which was represented by the large difference in reactivity between more rigid Xantphos and more flexible DPEphos.17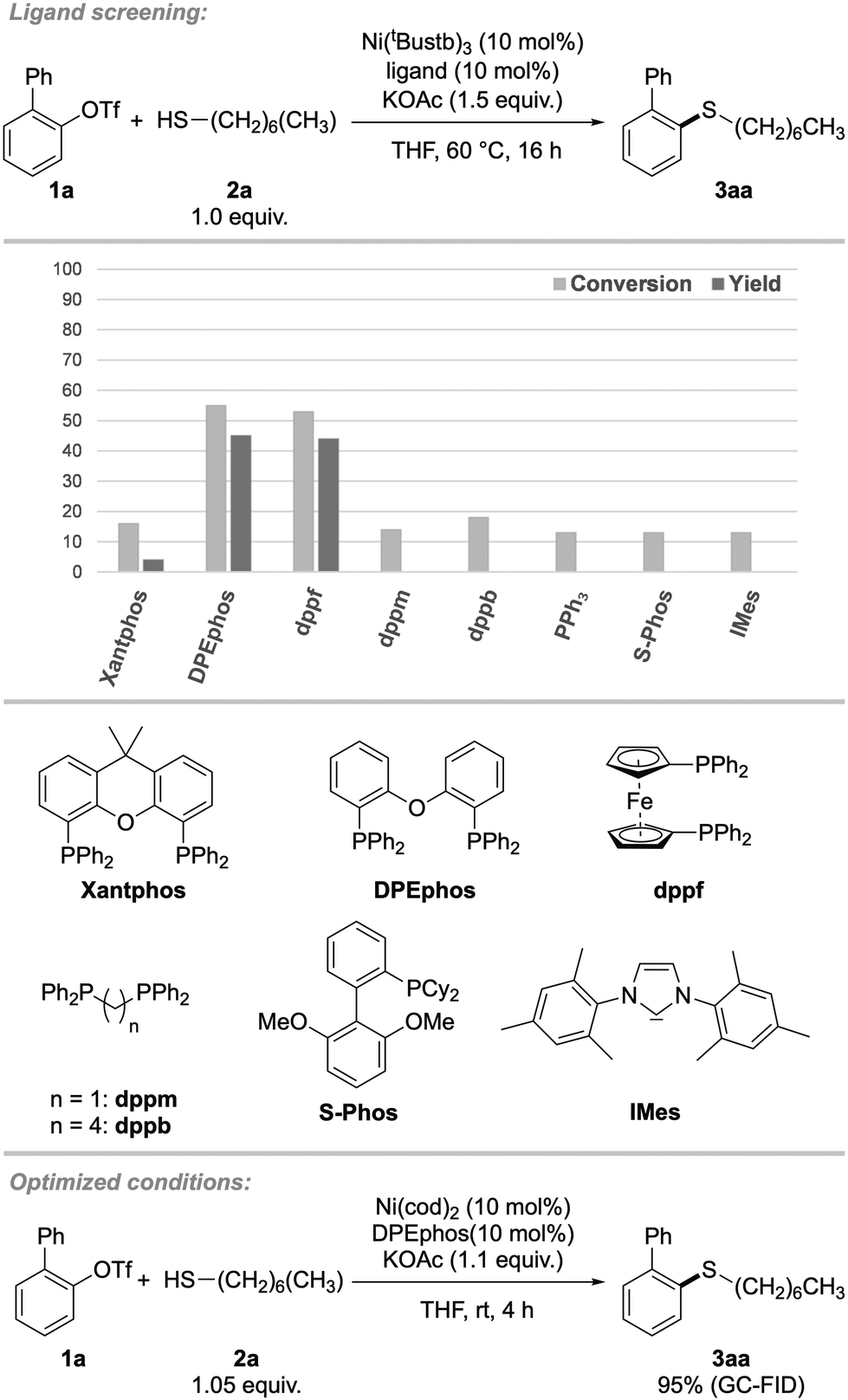 | ||
| Scheme 2 Optimization of reaction parameters. 2-Biphenyl triflate (1a) (0.1 mmol), heptane thiol (2a) (1.0 equiv.), KOAc (1.5 equiv.), Ni(4-tBu-stilbene)3 (10 mol%), ligand (10 mol%), THF (2 mL), 60 °C, 16 h. For monodentate ligands, 20 mol% were used. Yield and conversion were determined by GC–FID using pentadecane as internal standard. | ||
Having identified a suitable catalytic system, other reaction parameters were investigated. Conveniently, lowering the reaction temperature to room temperature did not result in reduced yields. Surprisingly, the Ni(0)-precatalyst Ni(cod)2 performed much better than Ni(0)-stilbene. This can be explained by the challenging oxidative addition that may be more hindered by the coordination of stilbene compared to cod, or the higher molar ratio of stilbene. To our delight, even tertiary adamantane thiol (2b) gave excellent yield at room temperature with a short reaction time of two hours (for kinetic investigation see ESI, Fig. S1†) and therefore this bulky thiol was chosen as model substrate for further studies. Remarkably, conversions were close to yields and unreacted aryl triflate could be recovered. This was generally the case for most substrates presented in both aryl triflate and alkyl thiol scope (vide infra). For ligands that gave no product, the conversion was usually ∼10%, indicating that oxidative addition had occurred, but further catalytic steps were not supported.
With these optimized conditions in hand, the substrate scope with respect to electrophiles was explored (Scheme 3). The isolation of previously screened 2-biphenyl thioether 3ab was successful in excellent yield. Simple alkyl substituents with increasing steric demand showed high yields of 3bb and 3cb, while an ortho-tert-butyl group in 3db proved more challenging, giving poor yield even under harsher conditions. An allyl substituent of 3eb was tolerated in excellent yield, leaving the double bond intact. Despite these promising results, aryl triflate 1f bearing two ortho-methyl groups gave no yield with the catalytic system discussed above. Thus, we further optimized towards the coupling of this highly challenging substrate and found ligand dppbz (1,2-bis(diphenylphosphino)benzene, L2) competent for this transformation, albeit only primary thiols could be used, and higher temperature was necessary. Using these modified conditions, thioether 3fc was formed in modest, but still synthetically useful yield, especially since the starting material can be recovered. The synthesis of disubstituted thioether 3gb derived from 1,2-phenyl ditriflate was possible in high yield without raising the catalyst loading. Next, electron-withdrawing functional groups were investigated. The trifluoromethyl-substituted thioether 3hb was obtained in good yield, while nitrile 3ib gave excellent yield. Remarkably, biselectrophile 3jb with a chloro-substituent gave the desired monosubstituted product in good yield. When we moved towards substrates bearing functional groups with potentially coordinating ability in ortho-position, L1 gave little to no product. To our surprise, L2, which was previously used for the coupling of 2,6-dimethylphenyl triflate (1f), performed well with these substrates. Accordingly, acyl 3kb as well as ester 3lb groups were feasible when using dppbz (L2), although slightly elevated temperatures were necessary. Strikingly, boronic ester substituted triflate 1m was converted in high yield, possibly enabling subsequent functionalization of 3mb by orthogonal cross-coupling protocols. Conveniently, a neighboring methoxy group, which is commonly present in numerous bioactive phenols,18 gave the acetovanillone-derived thioether 3nb in excellent yield. Although heterocycles occasionally needed slightly more forcing conditions (longer reaction times or higher temperatures), thioethers with pyridine (3ob), quinoline (3pb), thiophene (3qb), chromanone (3rb) and benzodioxol (3sb) functionalities were furnished in high yields even when using the corresponding bromide instead of triflate. Notably, amine- and amide-substituted triflates showed no conversion to the products in this reaction (3tb and 3ub), probably due to strong chelating effects. However, suitable protecting groups could render nitrogen-containing groups feasible, but would require additional optimization. Finally, the thioetherification of triflates 1v–1x demonstrated the possibility to employ substrates derived from biologically active phenols or natural products, though higher temperatures and longer reaction times were generally required, which can be explained by the low solubility of such compounds. Synthesis of thioether 3xb also highlights the possibility to obtain sulfur bioisosteres of well-known drug molecules, such as Febuxostat. In addition, 1-pyrene triflate 1y was subjected to the reaction, showcasing the possibility for functionalization of motifs relevant for functional materials.
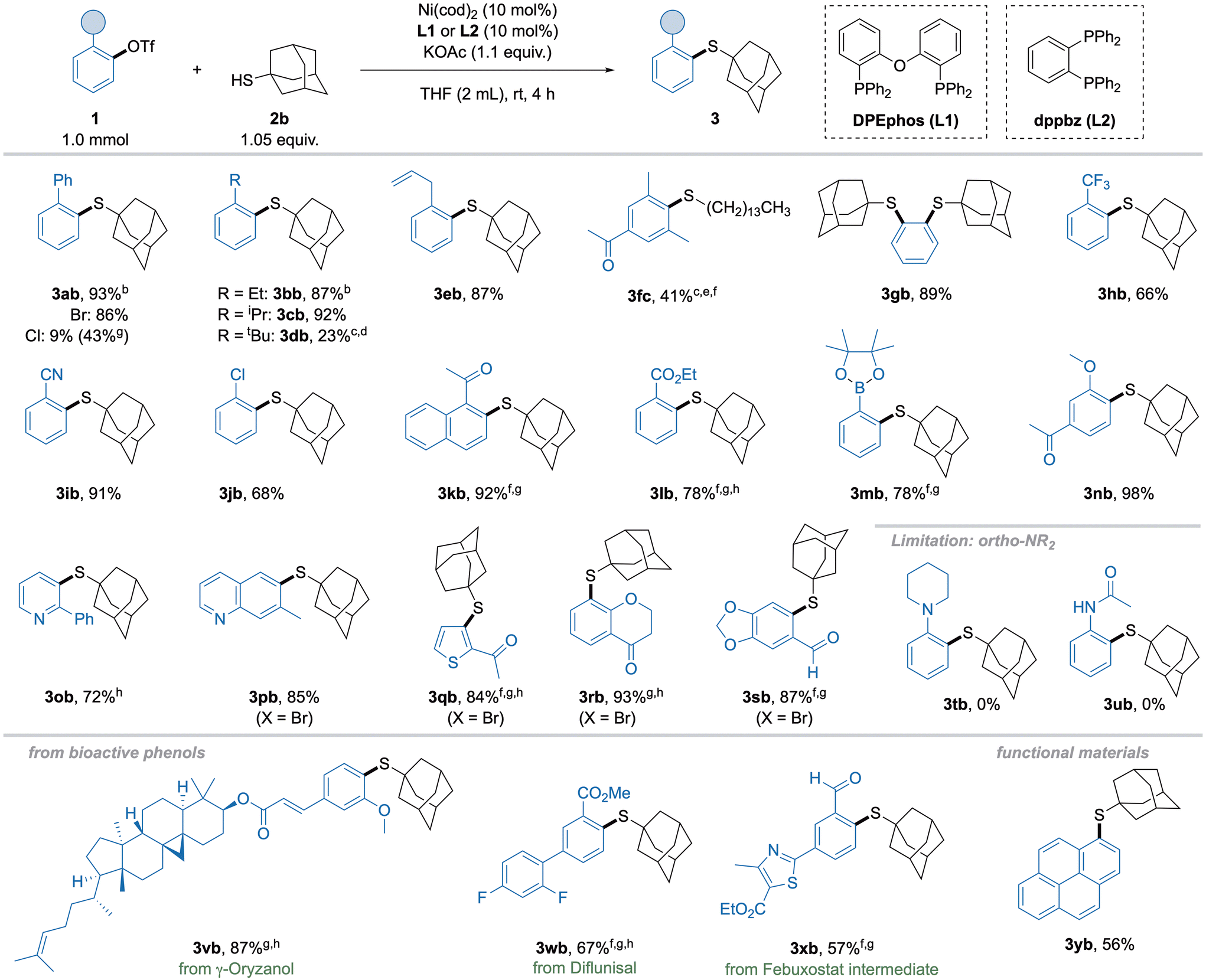 | ||
Scheme 3 Substrate scope with respect to aryl triflates. Standard conditions: 1 (1 mmol), 2b (1.05 equiv.), KOAc (1.1 equiv.), L1 (10 mol%), Ni(cod)2 (10 mol%), THF (2 mL), rt, 4 h. Isolated yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 h. b100 °C, 16 h. cGC-MS. dTetradecane thiol (1.05 equiv.) instead of 2b. eL2 (10 mol%) instead of L1. f60 °C. g6 h. 2 h. b100 °C, 16 h. cGC-MS. dTetradecane thiol (1.05 equiv.) instead of 2b. eL2 (10 mol%) instead of L1. f60 °C. g6 h. | ||
Subsequently, the scope of alkyl thiols was investigated (Scheme 4). Surprisingly, primary and secondary thiols initially gave diminished yields compared to sterically more demanding adamantane thiol (2b), which can be ascribed to the stronger poisoning effect of sterically less hindered thiols. This effect was not present in preliminary screening experiments, probably due to lower concentration of thiol in the reaction mixture. Fortunately, lowering the thiol concentration by adding more solvent combined with slower addition of thiol resulted in the formation of primary alkyl thioether 3ac in 78% isolated yield. Using this slightly modified procedure, simple primary, secondary and tertiary alkyl thiols 2c–e were coupled in high to excellent yields, clearly favoring higher substituted thiols. In the thiol moiety, functional groups such as ester, ketone and cyclic ether were tolerated, although yields of 3af, 3ag and 3aj were lower when the functionality was in close proximity to the thiol group. Contrary to the aryl triflate scope, yields with these thiols could neither be improved by employing L2 nor by higher temperatures. The cyclopropyl group in 3ah was compatible with the method but showed modest yield. Azetidine 3ai only gave poor yield, which could be explained by poisoning by the carbamate. Aryl thiols were not compatible with this method.
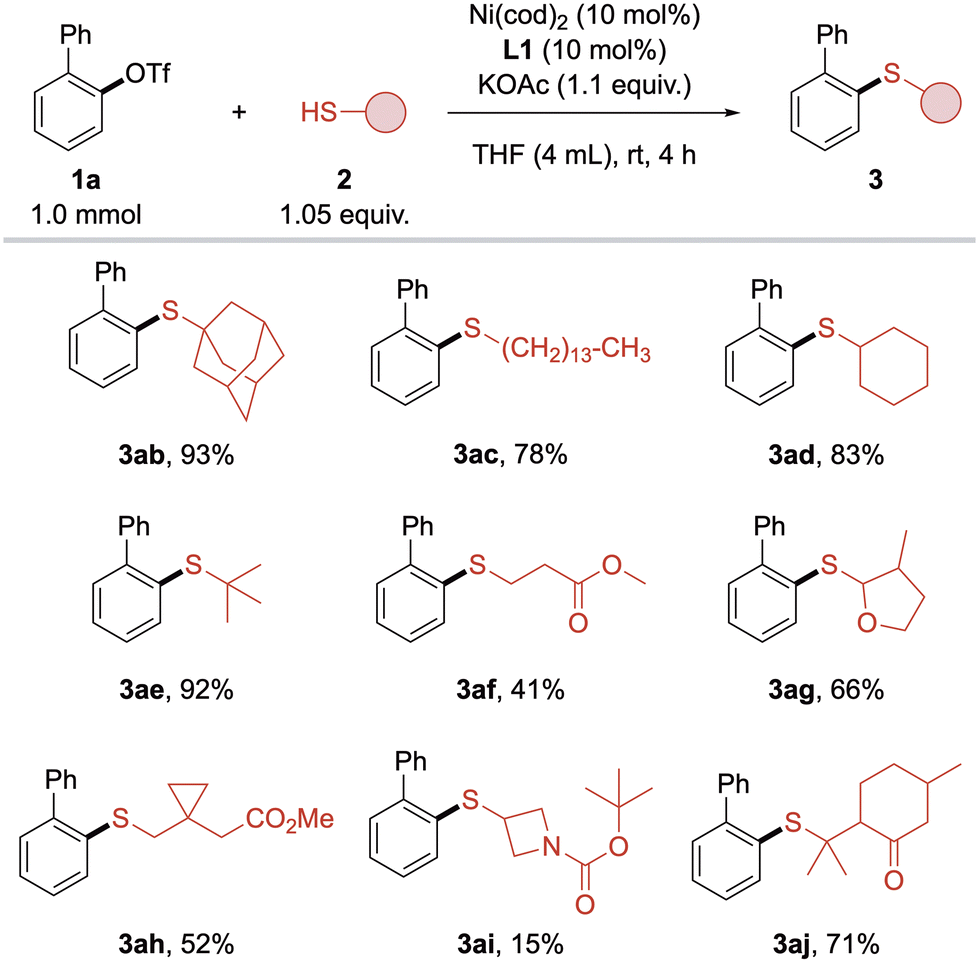 | ||
| Scheme 4 Substrate scope with respect to alkyl thiols. Standard conditions: 1a (1 mmol), 2 (1.05 equiv.), KOAc (1.1 equiv.), L1 (10 mol%), Ni(cod)2 (10 mol%), THF (4 mL), rt, 4 h. Isolated yields. | ||
Aiming for a better understanding of the reactivity in this system, competition experiments were performed.19 Accordingly, the expected reactivity order between halogens OTf ≈ Br > Cl was revealed by intermolecular competition (Scheme 5A). In an intramolecular competition, chemoselectivity between OTf and Br was even less expressed, and the disubstitution product 3zbb was favored. Yet, chemoselective monosubstitution was possible between OTf/Cl yielding thioether 3Ab in good yield (Scheme 5B).
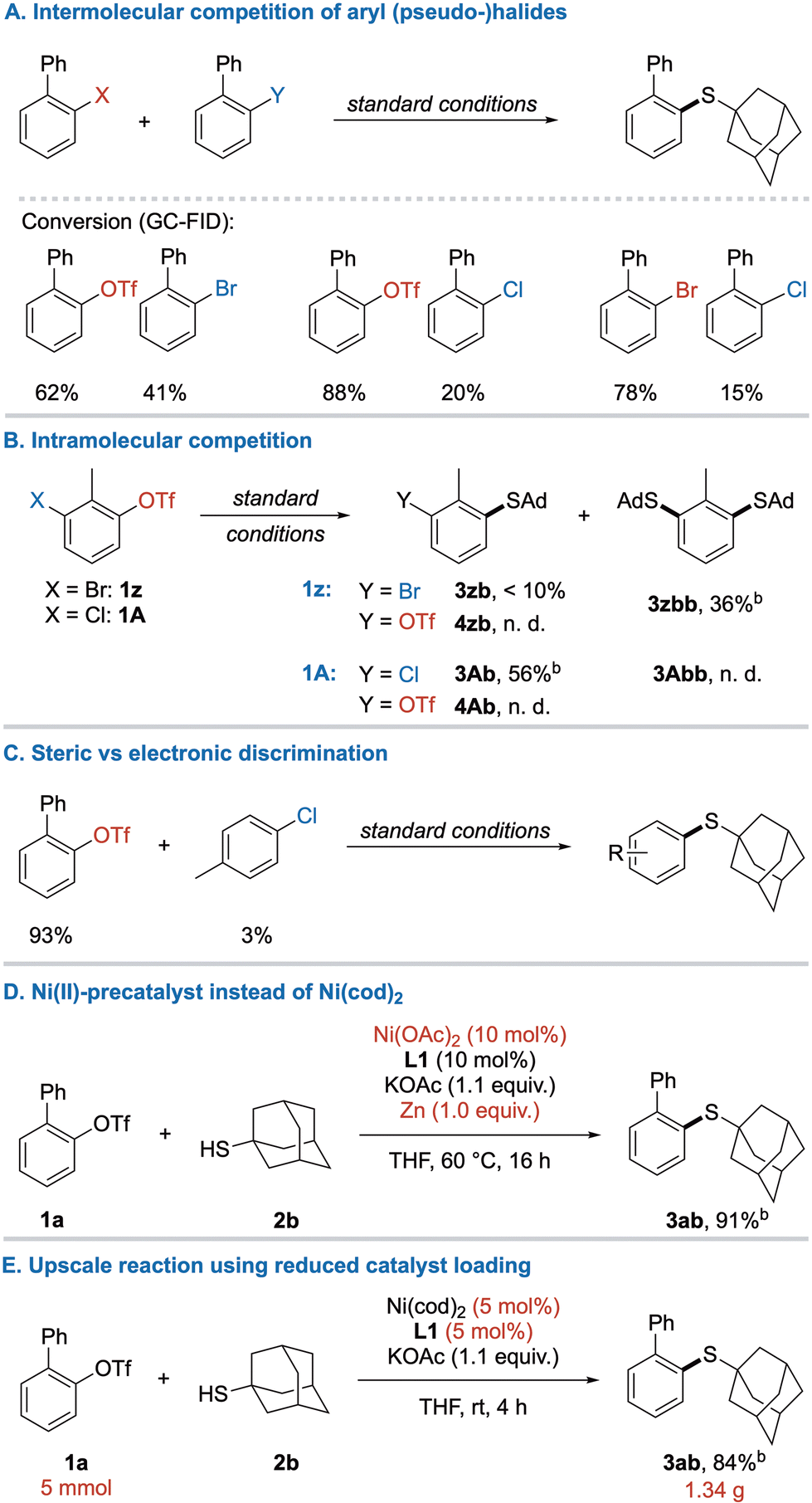 | ||
| Scheme 5 Further exploration of reactivity in the C–S cross-coupling of (pseudo)halides with alkyl thiols. Standard conditions: aryl (pseudo-)halides (each 0.2 mmol), 2b (1.05 equiv.), KOAc (1.1 equiv.), L1 (10 mol%), Ni(cod)2 (10 mol%), THF (2 mL), rt, 4 h. Yields and conversions were determined by quantitative GC-FID unless stated otherwise. aIsolated yield. | ||
Further results of intramolecular competition were previously observed in the investigation of scope for 1,2-biselectrophiles (Scheme 3, 3gb and 3jb). Next, sterically hindered triflate 1a was subjected to coupling in the presence of the electronically less reactive 4-chloro toluene to compare the influence of both factors (Scheme 5C). Interestingly, the higher reactivity of C–OTf bond can overcome the steric hinderance caused by an ortho-phenyl group, leaving 4-chloro toluene almost unreacted. Furthermore, the air-sensitive Ni(cod)2 could be replaced by Ni(OAc)2 under reductive conditions (Scheme 5D), greatly improving the cost-efficiency of this reaction as well as enabling storage of all reagents under benchtop-conditions. However, longer reaction times and heating were required for the activation of the precatalyst. Upscaling of the model reaction to 5 mmol proceeded without issues and delivered 1.34 g of thioether 3ab (Scheme 5E).
Intrigued by the substrate-depending demand for different ligands, we thrived to get a better understanding of the mechanistic steps, especially compared to our previously developed Ni/Xantphos catalytic system.16a,b In these studies, the coordination and deprotonation of the thiol was identified to be the likely slowest step, operating in a concerted-metalation-deprotonation (CMD)-like fashion.20 Since this was only computed for unsubstituted aryl chlorides, we questioned how the mechanism would differ when introducing ortho-substituents. The lower BDE of aryl triflates compared to chlorides and the high steric demand of the herein presented substrates should exhibit opposite effects on the reactivity.
Reductive elimination, though, would be presumably facile in this system as the relief of steric strain acts as the driving force and was therefore not further investigated. Hence, DFT computations were performed to study the steps of oxidative addition (OA) and thiol coordination and deprotonation (TCD, Fig. 1).21 The computed barriers were ΔG‡OA = 4.3 kcal mol−1 and ΔG‡TCD = 9.8 kcal mol−1, indicating that oxidative addition occurs more easily, showcasing the high reactivity of triflates even in a constrained environment. In contrast, the coordination of the bulky thiol required a higher activation barrier. Since we were unable to determine the transition state for the acetate coordination to B due to more complex effects such as salt precipitation and solubility, no rate-determining state could be identified. However, the mechanism of the OA and TCD were studied more detailed to elucidate the proceeding of the reaction despite the high steric strain around the nickel center aiming to rationalize the need for flexible ligands. Generally, oxidative addition of Ni(0) into aryl triflates is computationally poorly investigated with most reports focusing on sulfonamides or esters.22 The reported mechanisms differ between SN2-like, 3- or 5-centered transition states.23 Our computations indicated an SN2-like mechanism for the oxidative addition (TSA–B), while competing pathways showed higher energy barriers. Presumably, the steric pressure would render the SN2-mechanism more favorable in this case. The barrier of the oxidative addition, however, is remarkably low, and the process is strongly exergonic, showcasing the high reactivity of aryl triflates. Nevertheless, we found the initial precoordination of the π-system (A) fairly unfavored, but possible due to the great excess of 1a at the beginning of the reaction (see SI). The product of the oxidative addition is the [(DPEphos)Ni(biphenyl)]+-cation (B), which is in accordance with the obtained crystal structure (vide infra).
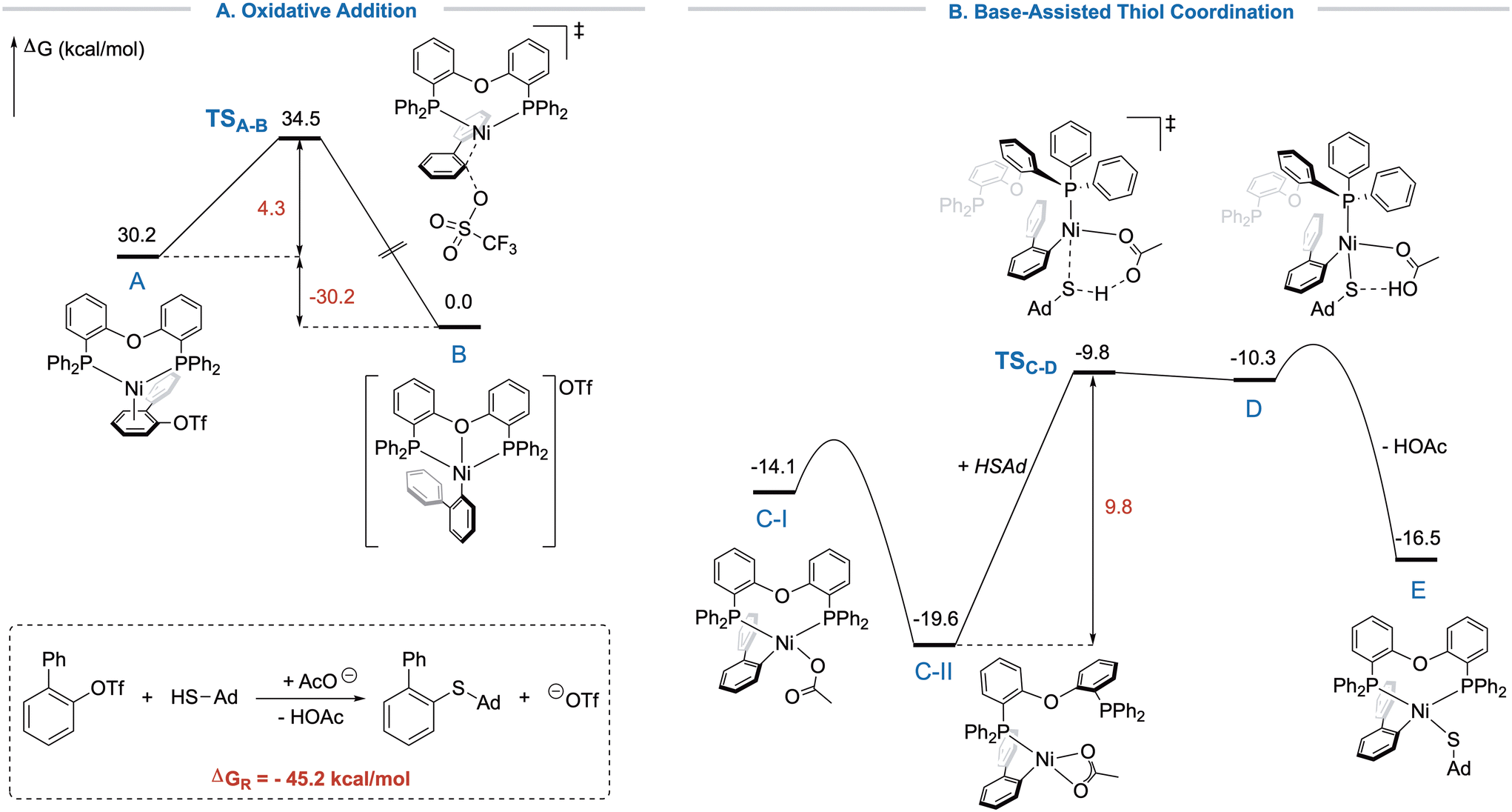 | ||
| Fig. 1 Free energy profile for the oxidative addition and thiol coordination in the cross-coupling of 2-PhPhOTf with HSAd. Structures and energies were calculated at the M06-L(SMD:THF)/def2-QZVPP//B3LYP-D3BJ/def2-SVP level of theory. | ||
Notably, when we moved towards the thiol coordination, computations pointed out a displacement of one of the phosphines from nickel by initial η2-coordination of the acetate, which is energetically more favorable than the corresponding η1-structure, demonstrating a major difference to previous results.16a However, structure C-II could be energetically favored due to reduced steric strain. A similar η1-coordination of DPEphos was previously demonstrated on sterically demanding Cu-complexes.24 Simultaneously to the acetate switching back to η1-coordination, the free coordination site trans to the loosely bound phosphine is occupied by the thiol, followed by proton transfer to acetate. In the transition state, a complete dissociation of the previously displaced phosphine is observed with the now inactive arm of L1 rotated away from nickel (TSC–D). Competing bidentate transition-states could not be located. Essentially, L1 appears to act as monodentate PPh3-like ligand during the step of thiol coordination and deprotonation. This feature of bidentate phosphine ligands was previously proposed from the group of Doyle for the nickel-catalyzed Suzuki–Miyaura coupling of sterically hindered substrates.25 This proposed mechanism could explain the large difference in reactivity between the otherwise similar Xantphos and DPEphos (L1). Analysis of the intrinsic reaction coordinate (IRC) revealed that the reactive mode of the transition state corresponds to a H+-transfer from thiol to acetate while all other bonds remained almost unchanged. We additionally found energy minima shortly after the transition state, where the proton is bound to the acetate, but still exhibits weak H-bond interactions to the sulfur atom. As soon as the H+-transfer from thiol to acetate is complete (D), acetic acid is displaced by the phosphine, leading to structure E, from which product formation will eventually occur via reductive elimination (not shown). This interplay demonstrates the need for a specific ligand that is both bulky and flexible to protect sensitive intermediates by steric repulsion, but allows for the generation of additional coordination sites, when necessary. The Gibbs Energy of the overall reaction was computed to amount to −45.2 kcal mol−1.
Encouraged from these findings, we sought to obtain additional indices for the mechanism, e.g. isolating the product of oxidative addition to Ni(0). It is a known issue that addition of bidentate phosphine ligands to Ni(cod)2 can lead to mixtures of Ni(cod)2, (P∩P)Ni(cod) and (P∩P)2Ni. (P∩P)2Ni is considered a catalytic sink, which will not engage in oxidative addition.26 In accordance with previous reports,26 we only observed NMR-signals for (DPEphos)Ni(cod) upon mixing a 1:1 ratio of DPEphos and Ni(cod)2. However, when adding 1 equiv. of 2-PhPhOTf (1a), no reaction was observed after 1 hour. We speculated that the aryl triflate would initially bind with its π-surface, meaning that it must displace cod in an equilibrium, which could be shifted by using an excess of aryl triflate. Accordingly, complex Ni1 was isolated as a fine orange powder (Scheme 6A), and characterized by NMR, HR-MS and X-Ray analysis. The obtained connectivity of Ni1 showed a distorted square-planar complex that crystallized as a cation due to the moderate coordination ability of the triflate counterion.27 Consequently, the reactivity of complex Ni1 was further investigated. Thus, the reaction of Ni1 with 1.05 equiv. adamantane thiol (2b) in the presence of KOAc yielded coupling product 3ab in 69% yield (Scheme 6B). In absence of base, the reaction gave slightly lower yield, which can be explained by reduced efficiency of the thiol coordination and deprotonation. Furthermore, Ni1 could be efficiently employed as an active catalyst in the standard reaction (Scheme 6C), indicating that Ni1 was indeed an intermediate of the catalytic cycle. We thus propose that the reaction proceeds via the typical Ni0/NiII-cycle that has been previously proposed in our earlier work.16a Although our calculations indicated a flexible η1/η2-ligation state of DPEphos during base-assisted thiol coordination, we were unable to obtain experimental data to support this hypothesis.
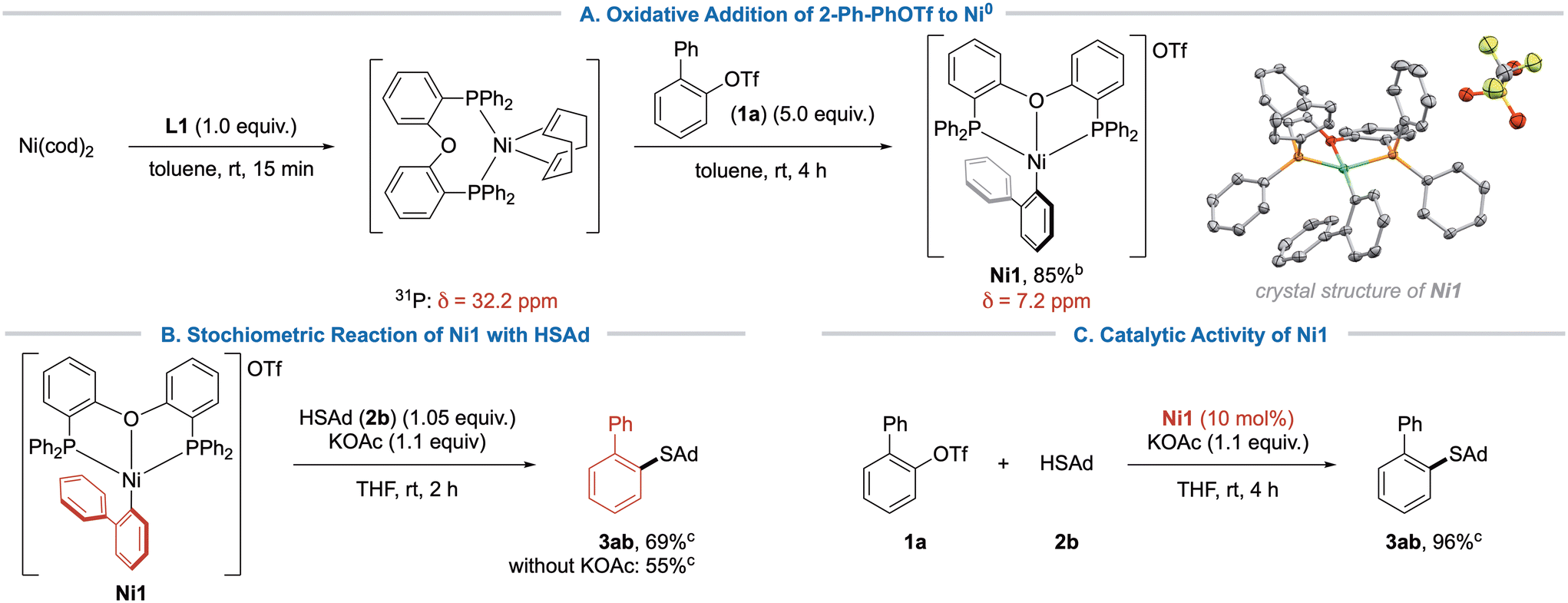 | ||
| Scheme 6 Isolation and reactivity of oxidative addition product Ni1. The ORTEP of Ni1 was obtained by X-Ray diffraction of suitable crystals. Thermal ellipsoids are shown at 50% propability and hydrogen atoms are omitted for clarity. aIsolated yield. bYield determined by quantitative GC-FID using pentadecane as internal standard. | ||
Conclusions
In summary, we developed the first nickel-catalyzed C–S cross coupling for aryl triflates bearing substituents in ortho-position. Although aryl bromides and, to some extent, chlorides were feasible substrates as well, the combination of the superior reactivity of aryl triflates with a flexible ligand (L1) allowed for the coupling of sterically hindered substrates, including heterocycles, in high yields. Additionally, a wide range of functional groups with coordinating abilities, such as the methoxy group, carbonyls and boronic ester, was tolerated when using another ligand (L2). Moreover, the synthetic utility could be demonstrated by the thioetherification of triflates derived from biologically relevant phenols. Experimental and computational mechanistic studies supported a Ni0/NiII catalytic cycle. Furthermore, computations indicated an unusual switch in the ligation state of the bidentate phosphine, rationalizing the need for a flexible ligand.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the analytics department of the University of Tübingen for their excellent work, as well as Dr. C. Maichle-Mössmer for performing X-Ray analysis. The financial support from the University of Tübingen, DFG (grant FL 878/9-1), FCI (scholarship I.H.L.) and Studienstiftung des Deutschen Volkes (scholarship R.C.R.) is gratefully acknowledged.References
- C. Jiménez, Marine sulfur-containing natural products, in Stud. Nat. Prod. Chem, ed. R. Atta ur, Elsevier, 2001, vol. 25, pp. 811–917 Search PubMed.
- (a) K. A. Scott and J. T. Njardarson, Analysis of US FDA-Approved Drugs Containing Sulfur Atoms, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed; (b) M. Feng, B. Tang, H. S. Liang and X. Jiang, Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- (a) P. Devendar and G.-F. Yang, Sulfur-Containing Agrochemicals, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed; (b) H. W. Hilton, N. S. Nomura, W. L. Yauger and S. S. Kameda, Absorption, translocation, and metabolism of metribuzin (BAY-94337) in sugarcane, J. Agric. Food Chem., 1974, 22, 578–582 CrossRef CAS PubMed.
- (a) M. Criado-Gonzalez and D. Mecerreyes, Thioether-based ROS responsive polymers for biomedical applications, J. Mater. Chem. B, 2022, 10, 7206–7221 RSC; (b) W.-J. Wang, J. Liu, Y.-T. Yan, X.-L. Yang, W.-Y. Zhang, G.-P. Yang and Y.-Y. Wang, Uncommon thioether-modified metal–organic frameworks with unique selective CO2 sorption and efficient catalytic conversion, CrystEngComm, 2021, 23, 1447–1454 RSC.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Oxidation of sulfides to sulfoxides and sulfones with 30% hydrogen peroxide under organic solvent- and halogen-free conditions, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
- M. Toshihiko, S. Tomiya, A. Yoriyoshi, S. Jun-ichi, K. Yasuki and K. Masanori, The Palladium Catalyzed Nucleophilic Substitution of Aryl Halides by Thiolate Anions, Bull. Chem. Soc. Jpn., 1980, 53, 1385–1389 CrossRef.
- (a) V. J. Geiger, R. M. Oechsner, P. H. Gehrtz and I. Fleischer, Recent Metal-Catalyzed Methods for Thioether Synthesis, Synthesis, 2022, 54, 5139–5167 CrossRef CAS; (b) F. Abedinifar, S. Bahadorikhalili, B. Larijani, M. Mahdavi and F. Verpoort, A review on the latest progress of C-S cross-coupling in diaryl sulfide synthesis: Update from 2012 to 2021, Appl. Organomet. Chem., 2022, 36, e6482 CrossRef CAS; (c) P. K. Behera, P. Choudhury, P. Behera, A. Swain, A. K. Pradhan and L. Rout, Transition Metal Catalysed C-S Cross-Coupling Reactions at Room Temperature, ChemistrySelect, 2022, 7, e202202919 CrossRef CAS; (d) I. P. Beletskaya and V. P. Ananikov, Transition-Metal-Catalyzed C–S, C–Se, and C–Te Bond Formations via Cross-Coupling and Atom-Economic Addition Reactions. Achievements and Challenges, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed; (e) S. Huang, M. Wang and X. Jiang, Ni-catalyzed C–S bond construction and cleavage, Chem. Soc. Rev., 2022, 51, 8351–8377 RSC.
- (a) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, An N-Heterocyclic Carbene Ligand with Flexible Steric Bulk Allows Suzuki Cross-Coupling of Sterically Hindered Aryl Chlorides at Room Temperature, Angew. Chem., Int. Ed., 2003, 42, 3690–3693 CrossRef CAS PubMed; (b) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, Modified (NHC)Pd(allyl)Cl (NHC = N-Heterocyclic Carbene) Complexes for Room-Temperature Suzuki−Miyaura and Buchwald−Hartwig Reactions, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed; (c) K. L. Billingsley, K. W. Anderson and S. L. Buchwald, A Highly Active Catalyst for Suzuki–Miyaura Cross-Coupling Reactions of Heteroaryl Compounds, Angew. Chem., Int. Ed., 2006, 45, 3484–3488 CrossRef CAS PubMed; (d) J.-S. Ouyang, Y.-F. Li, F.-D. Huang, D.-D. Lu and F.-S. Liu, The Highly Efficient Suzuki–Miyaura Cross-Coupling of (Hetero)aryl Chlorides and (Hetero)arylboronic Acids Catalyzed by “Bulky-yet-Flexible” Palladium–PEPPSI Complexes in Air, ChemCatChem, 2018, 10, 371–375 CrossRef CAS.
- (a) J. Luo, Y. Liang, M. Montag, Y. Diskin-Posner, L. Avram and D. Milstein, Controlled Selectivity through Reversible Inhibition of the Catalyst: Stereodivergent Semihydrogenation of Alkynes, J. Am. Chem. Soc., 2022, 144, 13266–13275 CrossRef CAS PubMed; (b) T. Itoh and T. Mase, Practical Thiol Surrogates and Protective Groups for Arylthiols for Suzuki−Miyaura Conditions, J. Org. Chem., 2006, 71, 2203–2206 CrossRef CAS PubMed; (c) B. Zeysing, C. Gosch and A. Terfort, Protecting Groups for Thiols Suitable for Suzuki Conditions, Org. Lett., 2000, 2, 1843–1845 CrossRef CAS PubMed.
- (a) J. Xu, R. Y. Liu, C. S. Yeung and S. L. Buchwald, Monophosphine Ligands Promote Pd-Catalyzed C–S Cross-Coupling Reactions at Room Temperature with Soluble Bases, ACS Catal., 2019, 9, 6461–6466 CrossRef CAS PubMed; (b) T. Scattolin, E. Senol, G. Yin, Q. Guo and F. Schoenebeck, Site-Selective C−S Bond Formation at C−Br over C−OTf and C−Cl Enabled by an Air-Stable, Easily Recoverable, and Recyclable Palladium(I) Catalyst, Angew. Chem., Int. Ed., 2018, 57, 12425–12429 CrossRef CAS PubMed; (c) A. C. Jones, W. I. Nicholson, H. R. Smallman and D. L. Browne, A Robust Pd-Catalyzed C–S Cross-Coupling Process Enabled by Ball-Milling, Org. Lett., 2020, 22, 7433–7438 CrossRef CAS PubMed; (d) T.-Y. Yu, H. Pang, Y. Cao, F. Gallou and B. H. Lipshutz, Safe, Scalable, Inexpensive, and Mild Nickel-Catalyzed Migita-Like C−S Cross-Couplings in Recyclable Water, Angew. Chem., Int. Ed., 2021, 60, 3708–3713 CrossRef CAS PubMed; (e) R. Sikari, S. Sinha, S. Das, A. Saha, G. Chakraborty, R. Mondal and N. D. Paul, Achieving Nickel Catalyzed C–S Cross-Coupling under Mild Conditions Using Metal–Ligand Cooperativity, J. Org. Chem., 2019, 84, 4072–4085 CrossRef CAS PubMed; (f) C.-W. Chen, Y.-L. Chen, D. M. Reddy, K. Du, C.-E. Li, B.-H. Shih, Y.-J. Xue and C.-F. Lee, CuI/Oxalic Diamide-Catalyzed Cross-Coupling of Thiols with Aryl Bromides and Chlorides, Chem. – Eur. J., 2017, 23, 10087–10091 CrossRef CAS PubMed.
- G. Bastug and S. P. Nolan, Carbon–Sulfur Bond Formation Catalyzed by [Pd(IPr*OMe)(cin)Cl] (cin = cinnamyl), J. Org. Chem., 2013, 78, 9303–9308 CrossRef CAS PubMed.
- M. Murata and S. L. Buchwald, A general and efficient method for the palladium-catalyzed cross-coupling of thiols and secondary phosphines, Tetrahedron, 2004, 60, 7397–7403 CrossRef CAS.
- M. A. Fernández-Rodríguez and J. F. Hartwig, A General, Efficient, and Functional-Group-Tolerant Catalyst System for the Palladium-Catalyzed Thioetherification of Aryl Bromides and Iodides, J. Org. Chem., 2009, 74, 1663–1672 CrossRef PubMed.
- J. L. Farmer, M. Pompeo, A. J. Lough and M. G. Organ, [(IPent)PdCl2(morpholine)]: A Readily Activated Precatalyst for Room-Temperature, Additive-Free Carbon–Sulfur Coupling, Chem. – Eur. J., 2014, 20, 15790–15798 CrossRef CAS PubMed.
- (a) M. Nikitin, F. Babawale, S. Tastekin, M. Antonietti, I. Ghosh and B. König, C(sp2)–S cross-coupling reactions with nickel, visible light, and mesoporous graphitic carbon nitride, Green Chem., 2024 10.1039/D3GC04517J; (b) I. Ghosh, N. Shlapakov, T. A. Karl, J. Düker, M. Nikitin, J. V. Burykina, V. P. Ananikov and B. König, General cross-coupling reactions with adaptive dynamic homogeneous catalysis, Nature, 2023, 619, 87–93 CAS.
- (a) R. M. Oechsner, J. P. Wagner and I. Fleischer, Acetate Facilitated Nickel Catalyzed Coupling of Aryl Chlorides and Alkyl Thiols, ACS Catal., 2022, 12, 2233–2243 CrossRef CAS; (b) R. M. Oechsner, I. H. Lindenmaier and I. Fleischer, Nickel Catalyzed Cross-Coupling of Aryl and Alkenyl Triflates with Alkyl Thiols, Org. Lett., 2023, 25, 1655–1660 CrossRef CAS PubMed; (c) P. H. Gehrtz, V. Geiger, T. Schmidt, L. Sršan and I. Fleischer, Cross-Coupling of Chloro(hetero)arenes with Thiolates Employing a Ni(0)-Precatalyst, Org. Lett., 2019, 21, 50–55 CrossRef CAS PubMed.
- G. M. Adams and A. S. Weller, POP-type ligands: Variable coordination and hemilabile behaviour, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef CAS.
- E. Orlo, C. Russo, R. Nugnes, M. Lavorgna and M. Isidori, Natural Methoxyphenol Compounds: Antimicrobial Activity against Foodborne Pathogens and Food Spoilage Bacteria, and Role in Antioxidant Processes, Foods, 2021, 10, 1807 CrossRef CAS PubMed.
- (a) E. D. Entz, J. E. A. Russell, L. V. Hooker and S. R. Neufeldt, Small Phosphine Ligands Enable Selective Oxidative Addition of Ar–O over Ar–Cl Bonds at Nickel(0), J. Am. Chem. Soc., 2020, 142, 15454–15463 CrossRef CAS PubMed; (b) E. K. Reeves, E. D. Entz and S. R. Neufeldt, Chemodivergence between Electrophiles in Cross-Coupling Reactions, Chem. – Eur. J., 2021, 27, 6161–6177 CrossRef PubMed.
- D. Lapointe and K. Fagnou, Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- (a) S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (b) F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (c) F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC; (d) A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed; (e) Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- (a) K. W. Quasdorf, A. Antoft-Finch, P. Liu, A. L. Silberstein, A. Komaromi, T. Blackburn, S. D. Ramgren, K. N. Houk, V. Snieckus and N. K. Garg, Suzuki−Miyaura Cross-Coupling of Aryl Carbamates and Sulfamates: Experimental and Computational Studies, J. Am. Chem. Soc., 2011, 133, 6352–6363 CrossRef CAS PubMed; (b) L. V. Hooker and S. R. Neufeldt, Ligation state of nickel during C-O bond activation with monodentate phosphines, Tetrahedron, 2018, 74, 6717–6725 CrossRef CAS PubMed; (c) X. Li and X. Hong, Computational studies on Ni-catalyzed C−O bond activation of esters, J. Organomet. Chem., 2018, 864, 68–80 CrossRef CAS.
- (a) R. Y. Liu, J. M. Dennis and S. L. Buchwald, The Quest for the Ideal Base: Rational Design of a Nickel Precatalyst Enables Mild, Homogeneous C–N Cross-Coupling, J. Am. Chem. Soc., 2020, 142, 4500–4507 CrossRef CAS PubMed; (b) R. Tang, J. Petit, Y. Yang, A. Link, F. Bächle, M.-E. L. Perrin, A. Amgoune and A. Tlili, Nickel-Catalyzed Trifluoromethylselenolation of Aryl and Alkenyl-Triflates: Scope and Mechanism, ACS Catal., 2023, 12553–12562 CrossRef CAS.
- R. Venkateswaran, M. S. Balakrishna, S. M. Mobin and H. M. Tuononen, Copper(I) Complexes of Bis(2-(diphenylphosphino)phenyl) Ether: Synthesis, Reactivity, and Theoretical Calculations, Inorg. Chem., 2007, 46, 6535–6541 CrossRef CAS PubMed.
- J. E. Borowski, S. H. Newman-Stonebraker and A. G. Doyle, Comparison of Monophosphine and Bisphosphine Precatalysts for Ni-Catalyzed Suzuki–Miyaura Cross-Coupling: Understanding the Role of the Ligation State in Catalysis, ACS Catal., 2023, 7966–7977 CrossRef CAS PubMed.
- A. L. Clevenger, R. M. Stolley, N. D. Staudaher, N. Al, A. L. Rheingold, R. T. Vanderlinden and J. Louie, Comprehensive Study of the Reactions Between Chelating Phosphines and Ni(cod)2, Organometallics, 2018, 37, 3259–3268 CrossRef CAS.
- T. Hayashida, H. Kondo, J.-i. Terasawa, K. Kirchner, Y. Sunada and H. Nagashima, Trifluoromethanesulfonate (triflate) as a moderately coordinating anion: Studies from chemistry of the cationic coordinatively unsaturated mono- and diruthenium amidinates, J. Organomet. Chem., 2007, 692, 382–394 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo00260a |
| This journal is © the Partner Organisations 2024 |