
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of super reductive EDA photocatalyst for challenging reactions: a theoretical and experimental study†
Yi-Hui Deng‡
bc,
Qini Li‡a,
Manhong Li‡a,
Leifeng Wang *a and
Tian-Yu Sun*b
*a and
Tian-Yu Sun*b
aSchool of Pharmaceutical Sciences (Shenzhen), Sun Yat-Sen University, No.66, Gongchang Road, Shenzhen 518107, P. R. China. E-mail: wanglf33@mail.sysu.edu.cn
bShenzhen Bay Laboratory, Shenzhen 518132, P. R. China. E-mail: Tian-Yu_Sun@pku.edu.cn
cLab of Computational Chemistry and Drug Design, State Key Laboratory of Chemical Oncogenomics, Peking University Shenzhen Graduate School, Shenzhen 518055, P. R. China
First published on 8th January 2024
Abstract
We reported a novel electron-donor-acceptor (EDA) photocatalyst formed in situ from isoquinoline, a diboron reagent, and a weak base. To further optimize the efficiency of this photocatalyst, Density Functional Theory (DFT) calculations were conducted to investigate the substituent effects on the properties of vertical excitation energy and redox potential. Subsequently, we experimentally validated these effects using a broader range of substituents and varying substitution positions. Notably, the 4-NH2 EDA complex derived from 4-NH2-isoquinoline exhibits the highest photocatalytic efficiency, enabling feasible metal free borylation of aromatic C–H bond and detosylaion of Ts-anilines under green and super mild conditions. These experimental results demonstrate the effectiveness of our strategy for photocatalyst optimization.
Introduction
Photocatalysis1–9 using electron-donor-acceptor (EDA) complexes10–12 has gained significant attention as it provides novel pathways for radical generation. Diboron reagents exhibit high reactivity, rendering them excellent candidates for EDA complex construction when paired with electron donor or acceptor partners (Scheme 1). Recently, Glorius13 disclosed a photoinduced decarboxylative borylation of aryl N-hydroxyphthalimide esters via the use of Katritzky salts as an acceptor in EDA complex I. Aggarwal14 proposed an EDA complex II formed by 2-iodophrnyl thiocarbonate, bis(pinacolato)diboron (B2pin2) and N,N-dimethylformamide, revealing boronic esters with high stereocontrol. König15 realized an ipso-borylation of substituted arenes utilizing EDA complex III, generated between thiolate, B2pin2 and boryl anion activated substrates. Very recently, Procter16 found that heterocyclic sulfonium moieties could be exploited as redox-active labels to form visible light-absorbing EDA complexes IV in combination with triarylamines, offering a mild, metal free and broadly applicable strategy for the formation of aryl radicals. While substantial efforts have been devoted to catalytic systems based on EDA complexes under relatively mild reaction conditions, it should be pointed out that the chemistry related to EDA complexes in this regard remains largely unexplored because of the countless random combinations of electron donors and electron acceptors.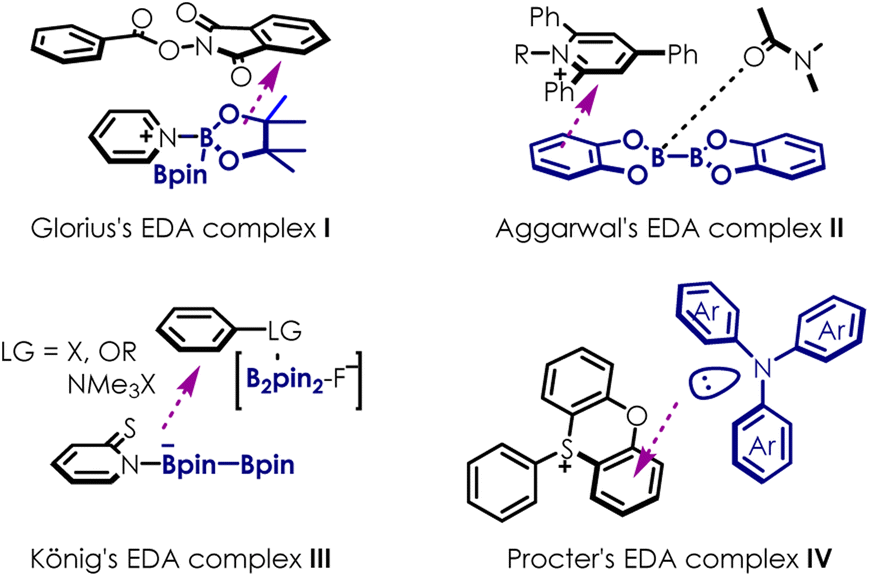 | ||
| Scheme 1 Typical EDA complexes. | ||
Although diborons have been employed as hydrogen or hydride equivalents in several reductive transformations,17,18 they have rarely served as precursors for highly reductive EDA complexes, which act as single electron donors in radical mediated transformations.19–21 Moreover, diboron compounds are capable of undergoing reductive addition with N-containing heterocycles (NCHs), such as pyridine and isoquinoline derivatives. Most reported methods rely on strong bases (e.g., MeOK/MeONa) to facilitate the formation of boronic ions, which can coordinate with potential electron acceptors. Considering that the strong bases could complicate purification and limit substrate scope, we wonder whether highly reductive excited EDA complexes could be generated using simple NCHs/diboron as precursors for electron acceptors and weak base as electron donors under visible light irradiation.
Motivated by this thought, we developed a new photocatalytic system using isoquinoline as the NCH instead of the commonly used pyridine for activation of diboron reagent22 (Scheme 2). Almost quantitatively and exclusively, DL-N,N-diborate dimer A was obtained through the reductive coupling of isoquinoline and B2pin2. Dimer A could coordinate with a weak base (triethylamine, NEt3, ΔG = 11.6 kcal mol−1) in situ, forming EDA complex B. EDA complex B was then excited by 390 nm light, resulting in a highly reducing excited state B* 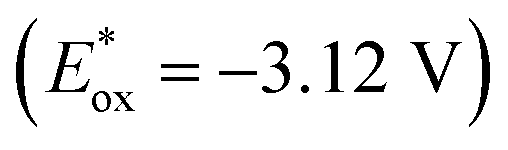 . EDA complex B* spontaneously underwent single electron transfer (SET) with electron deficient substrates such as aryl halides (e.g., for PhCl, Ered = −3.04 V vs. SCE),23 yielding radical intermediates that can proceed further transformations. The electronic structure of EDA complex is the key factor dictating the reactivity of a photocatalytic system. Although substituent groups are known to impact the electronic structure, there is currently limited research on the substituent effects on this complex. Therefore, investigating substituents for EDA complex photocatalysts derived from NCHs may offer a route to fine-tuning redox potentials, enabling access to more difficult reduction processes with higher efficiencies and chemoselectivities.
. EDA complex B* spontaneously underwent single electron transfer (SET) with electron deficient substrates such as aryl halides (e.g., for PhCl, Ered = −3.04 V vs. SCE),23 yielding radical intermediates that can proceed further transformations. The electronic structure of EDA complex is the key factor dictating the reactivity of a photocatalytic system. Although substituent groups are known to impact the electronic structure, there is currently limited research on the substituent effects on this complex. Therefore, investigating substituents for EDA complex photocatalysts derived from NCHs may offer a route to fine-tuning redox potentials, enabling access to more difficult reduction processes with higher efficiencies and chemoselectivities.
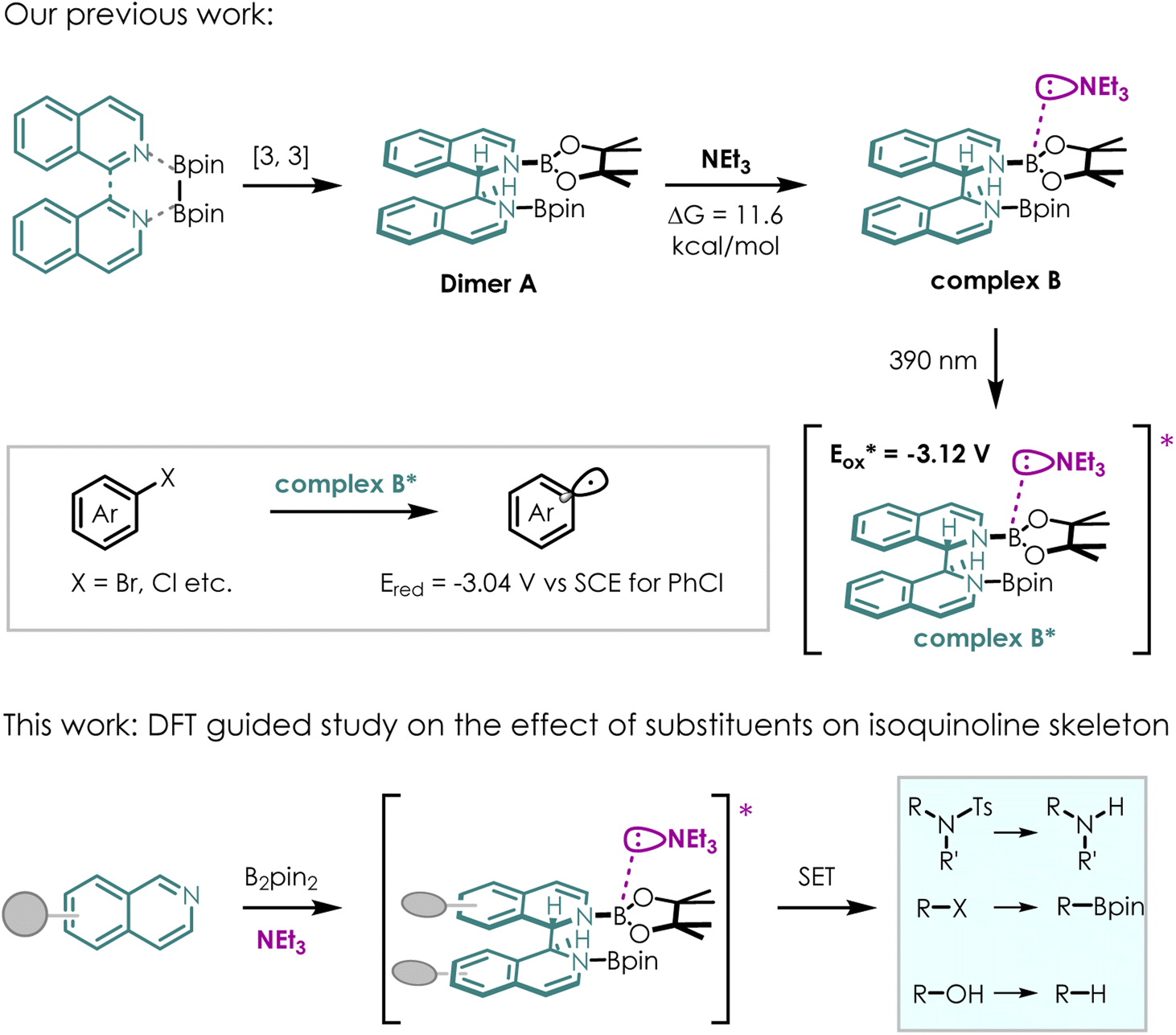 | ||
| Scheme 2 Our EDA complex proposal. | ||
Preliminary computational design and experimental validation
In this study, we employed Density Functional Theory (DFT) to elucidate the substituent effects on the properties of vertical excitation energy and redox potential of the photocatalyst. First, the original structure of the photocatalyst was computationally modified by introducing electron donating group –NH2 and electron withdrawing group –CN at the 6-position of isoquinoline—a strategically chosen site with minimized steric effects (Scheme 3). We speculated that as the vertical excitation energy of the photocatalyst increases, it becomes less prone to excitation, leading to a decrease in reaction efficiency. However, once the photocatalyst is excited, due to its higher energy state, it will exhibit enhanced reducing capability, thereby resulting in an increase in reaction efficiency. Theoretically, the photocatalytic efficiency should be jointly determined by the vertical excitation energy and redox potential.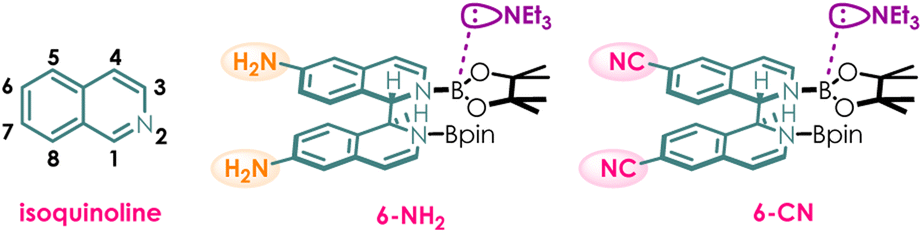 | ||
| Scheme 3 The atomic numbering of isoquinoline and the structures of 6-NH2 and 6-CN. | ||
The vertical excitation energies (Evex) and redox potentials (E0S1) of the original photocatalyst complex B and its 6-substituted amino and cyano derivatives (6-NH2 and 6-CN) were calculated using time-dependent density functional theory24–26 (TDDFT) and corresponding experiments were conducted to validate our computational predictions.
As shown in Table 1, the introduction of the electron-donating amino group (6-NH2) resulted in a minor increase of 0.05 eV in the excited state energy (4.00 eV) compared to the original complex B (3.95 eV), which means a mere ∼4 nm blue-shift in the absorption peak. Based on this result, it can be inferred that if the visible light used is capable of exciting complex B, it is highly likely to also excite 6-NH2.
|
|||
|---|---|---|---|
| Substituent | Evex_calc. (eV) | E0S1,calc.(2PC˙+/1PC*) (V vs. SCE) | Experimental yields |
| 6-NH2 | 4.00 | −3.26 | 64% |
| Complex B | 3.95 | −3.12 | 40% |
| 6-CN | 3.58 | −2.51 | 32% |
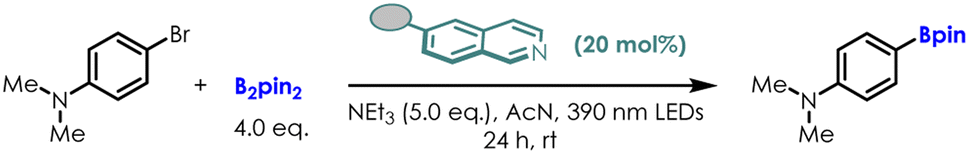
Notably, the 6-NH2 lowers the redox potential (−3.26 V) according to computational findings. Therefore, it is speculated that the 6-NH2 photocatalyst could enable more difficult reductions. In contrast, computational results indicate that although the electron withdrawing cyano substituent (6-CN) decreases the excitation energy significantly, it concurrently increases the redox potential (−2.51 V), which is unfavorable for the reduction reactions.
The experimental results with different 6-substituted isoquinoline were also shown in Table 1. The variation in experimental yields of the borylation of 4-bromo-dimethylaniline correlates with the trends in the reduction potentials of these isoquinoline derivatives. 6-NH2, with the lowest reduction potential (−3.26 V), showed the highest experimental yield (64%), surpassing that of the original photocatalyst complex B (40%). For 6-CN, which has the highest reduction potential (−2.51 V), its yield is only 32%. These experimental findings validate the trends predicted by our calculations and demonstrate the effectiveness of our strategy for photocatalyst optimization.
Experimental screening and theoretical interpretation for the high efficiency of 4-NH2
Based on the above results, we further examined the substituent effects on isoquinoline by experiments, including a wider range of substituents (–NH2, –OMe, –Me, –OH, –F, –COOMe and –CN) and different substitution positions (3-, 4-, 5-, 6-, 7-, and 8-positions) (Table 2). It can be observed that electron rich substituents generally increase the yield, while electron poor substituents tend to decrease it. The highest yield was achieved with 4-substituted amino isoquinoline (4-NH2), reaching 70%, which is 75% higher than the yield of non-substituted isoquinoline (40%). To gain deeper insights into the reasons for the superior performance of 4-NH2, natural transition orbitals27 (NTOs) for the S1 excited state of each complex were computed using Multiwfn program (version 3.8)28 and visualized through VMD 1.93.29
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
Electron rich substituents | Electron poor substituents | ||||||
| –H | –NH2 | –OMe | –Me | –OH | –F | –COOMe | –CN | |
| a Reactions were run on 0.2 mmol scales, 1H-NMR yield using pyrazine as internal standard. | ||||||||
| 3 | 40% | 57% | 56% | 51% | 50% | 27% | ||
| 4 | 70% | 57% | 32% | 48% | 48% | 24% | ||
| 5 | 60% | 58% | 50% | 27% | ||||
| 6 | 64% | 63% | 65% | 62% | 37% | 32% | ||
| 7 | 60% | 64% | 49% | 53% | 33% | |||
| 8 | 63% | 57% | 45% | 31% | ||||
NTOs provide visual representations of the hole and electron density distributions involved in an electronic transition. The hole NTOs represent where electron density originates, corresponding to the occupied orbitals involved in the transition. The electron NTOs represent where electron density migrates to, corresponding to the virtual orbitals involved. Analyzing the hole and electron NTO pairs provides insight into the electron flow during the excitation.
As shown in Fig. 1, the NTOs analysis reveal that the 4-NH2 lone pair directly participate in the hole NTO. It is a well-established principle that electron donating groups typically raise orbital energies.30–32 Therefore, the presence of 4-NH2 leads to an increase in the energy of the hole NTO. However, the 4-NH2 lone pair do not participate in the electron NTO, implying its minimal impact on the electron NTO. Therefore, when considering the combined influence of 4-NH2 on both hole NTO and electron NTO, it leads to a reduction in excitation energy (3.60 eV) compared to that of the 6-NH2 (4.00 eV). Furthermore, the 4-NH2 complex has a lower redox potential (−3.45 V) than the 6-NH2 complex of (−3.26 V). To further analyze the reason why the 4-NH2 complex has an even lower redox potential, spin population analysis was conducted.
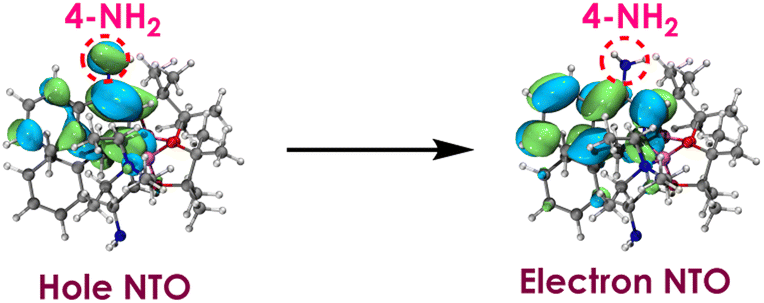 | ||
| Fig. 1 Natural transitions orbital (NTO) pair for S1 excited state for 4-amino derivative. | ||
As shown in Fig. 2, the spin of the 4-amino radical cation primarily distributes on the 4-position, whereas in the 6-H cation radical, the spin population exhibits greater delocalization. Consequently, the presence of the 4-NH2 can enhance the stabilization of the cationic species.
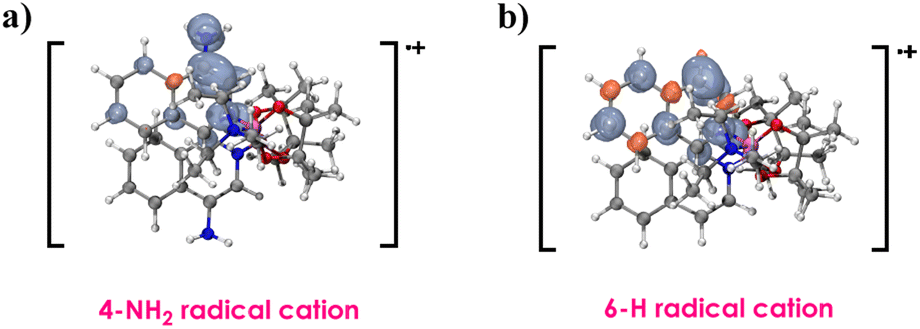 | ||
| Fig. 2 Spin density distribution of the radical cation of (a) 4-amino derivative and (b) original complex B. | ||
The 4-NH2 complex has lower excitation energies (3.60 eV), concurrently, it also has a lower redox potential (−3.45 V) since the 4-NH2 can enhance the stabilization of the cationic species. Therefore, the 4-NH2 complex exhibits the highest photocatalytic efficiency.
Borylation of aromatic C–H bond
After screening and identifying 4-NH2 isoquinoline as an excellent photocatalyst, we applied it to more challenging aromatic C–H bond borylation reactions (ESI Table S1†). A selection of electron rich aromatic substrates was submitted to the C–H borylation sequence using B2pin2 as the aryl radical trap for the photochemical progress (Table 3). Electron donating/withdrawing group substituted anilines and phenol (1a–1n) were smoothly converted to the corresponding arylboronates in moderate to high yields (34–82%) and excellent regioselectivity (rr > 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to single isomers), showcasing the compatibility of our method with a broad array of functionality (e.g., difluoromethoxyl-, trifluoromethyl-, trifluoromethoxy-, cyano-, ester-, alkyl-, fluoride- and naked hydroxyl/amino). The regioselectivity of the borylation reaction is governed by both electronic and steric effects of the substituents, in a similar manner to the triptycenyl sulfide-induced nucleophilic substitution reactions reported by Miura and co-workers.33 Heterocycles such as morpholine (2o) and pyrrolidine (2p) were also competent substrates. Medicinally relevant fluorine-containing anilines afforded C–H borylation products with high efficiencies (2b–2e, 2j, 2k). Of note is the ability of this method to effectively construct C–B bond from hindered aryl C–H bond, as shown by the reaction of substrates 1h–1n, yielding the desired borylation products in good yield and excellent chemoselectivities. Furthermore, the borylation reaction with bicyclic aromatic substrates, such as 1,2,3,4-tetrahydroisoquinoline derivative (1q), benzomorpholine (1r), 2,3-dihydrobenzofuran (1s) and chromane (1t), all proceeded well and afforded the corresponding borylation products as single regio-isomers in accordance with the high reactivity of these positions for electrophilic substitution. Remarkably, reductive cleavages of unusual C–O bonds (1u)/detosylation (1v) followed with direct C–H bond borylation under standard conditions afforded the borylated phenol (2u) and aniline (2v) derivatives effectively. Both of these transformations are highly important because cascade site selective transformation of aromatic C–H bond remains a major challenge.
:1 to single isomers), showcasing the compatibility of our method with a broad array of functionality (e.g., difluoromethoxyl-, trifluoromethyl-, trifluoromethoxy-, cyano-, ester-, alkyl-, fluoride- and naked hydroxyl/amino). The regioselectivity of the borylation reaction is governed by both electronic and steric effects of the substituents, in a similar manner to the triptycenyl sulfide-induced nucleophilic substitution reactions reported by Miura and co-workers.33 Heterocycles such as morpholine (2o) and pyrrolidine (2p) were also competent substrates. Medicinally relevant fluorine-containing anilines afforded C–H borylation products with high efficiencies (2b–2e, 2j, 2k). Of note is the ability of this method to effectively construct C–B bond from hindered aryl C–H bond, as shown by the reaction of substrates 1h–1n, yielding the desired borylation products in good yield and excellent chemoselectivities. Furthermore, the borylation reaction with bicyclic aromatic substrates, such as 1,2,3,4-tetrahydroisoquinoline derivative (1q), benzomorpholine (1r), 2,3-dihydrobenzofuran (1s) and chromane (1t), all proceeded well and afforded the corresponding borylation products as single regio-isomers in accordance with the high reactivity of these positions for electrophilic substitution. Remarkably, reductive cleavages of unusual C–O bonds (1u)/detosylation (1v) followed with direct C–H bond borylation under standard conditions afforded the borylated phenol (2u) and aniline (2v) derivatives effectively. Both of these transformations are highly important because cascade site selective transformation of aromatic C–H bond remains a major challenge.
| a Reactions were run on 0.2 mmol scales, isolated yield. |
|---|
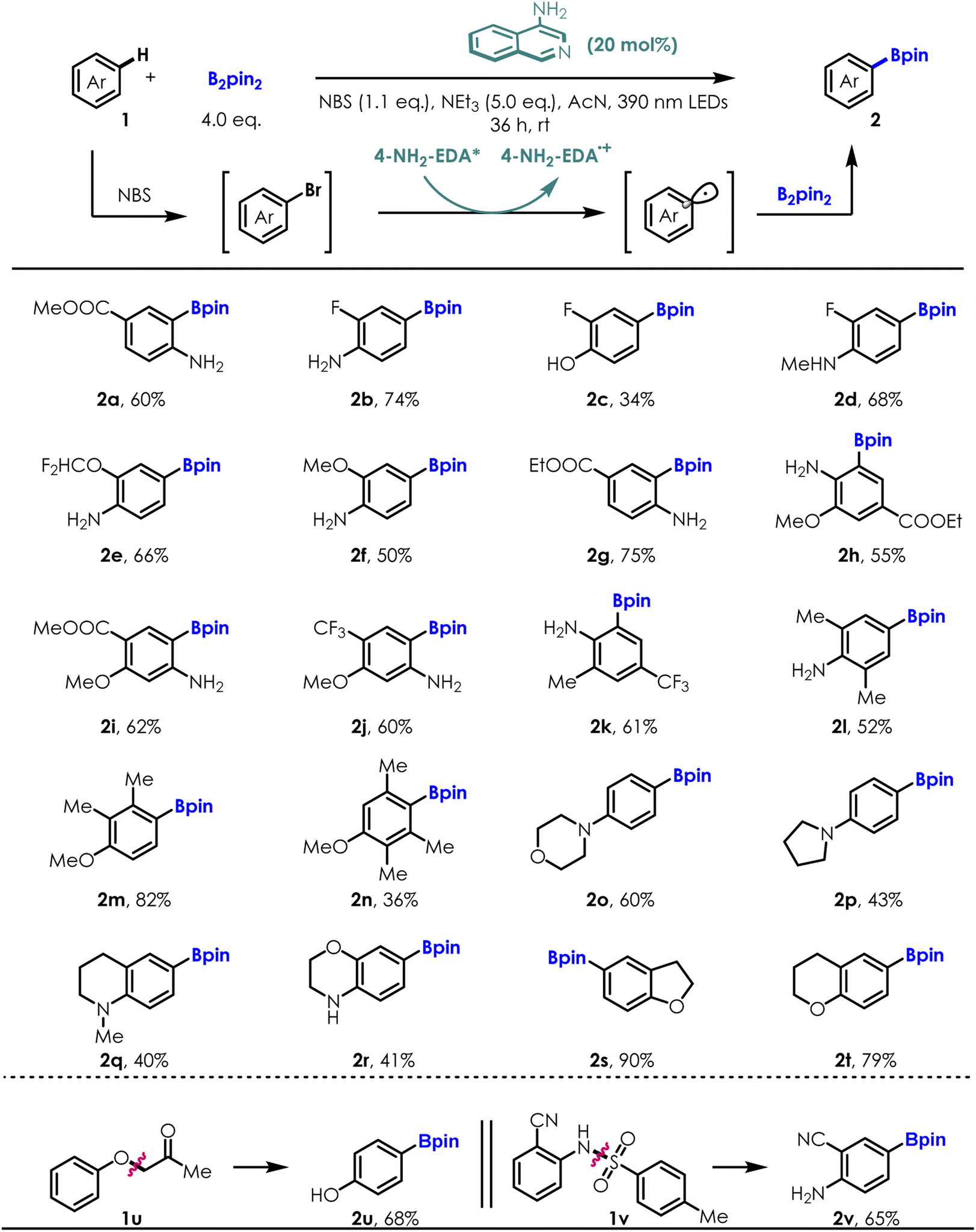 |
Detosylaion of Ts-anilines
To further demonstrate the utility of this novel reductive protocol with 4-NH2 isoquinoline, challenging detosylation34–37 without any metal additives was examined (Table 4). A variety of both electron-rich (3a, 3f), electron-deficient (3c–3e) and more hindered (3f) tosylated primary anilines afforded the desired detosylated products in good to excellent yields (NMR, yields of products were taken using pyrazine as an internal standard) under standard conditions. As a contrast, the detosyaltion yield was improved remarkably using 4-amino-isoquinoline instead of isoquinoline (4a; 88% vs. 60%). Secondary amines (3h, 3i) were efficient substrates for this transformation as well. Several heterocyclic compounds containing N-Ts groups, such as pyridine, pyrrole, 7-azaindole and indole, are also proven to be amenable (3j–3m). Surprisingly, reductive deoxygenation38–40 of fluorenone (3o) and fluorenol (3n) was accomplished with high efficiency (75–79%, 4n). Unfortunately, this protocol is not suitable for alkyl amine compounds.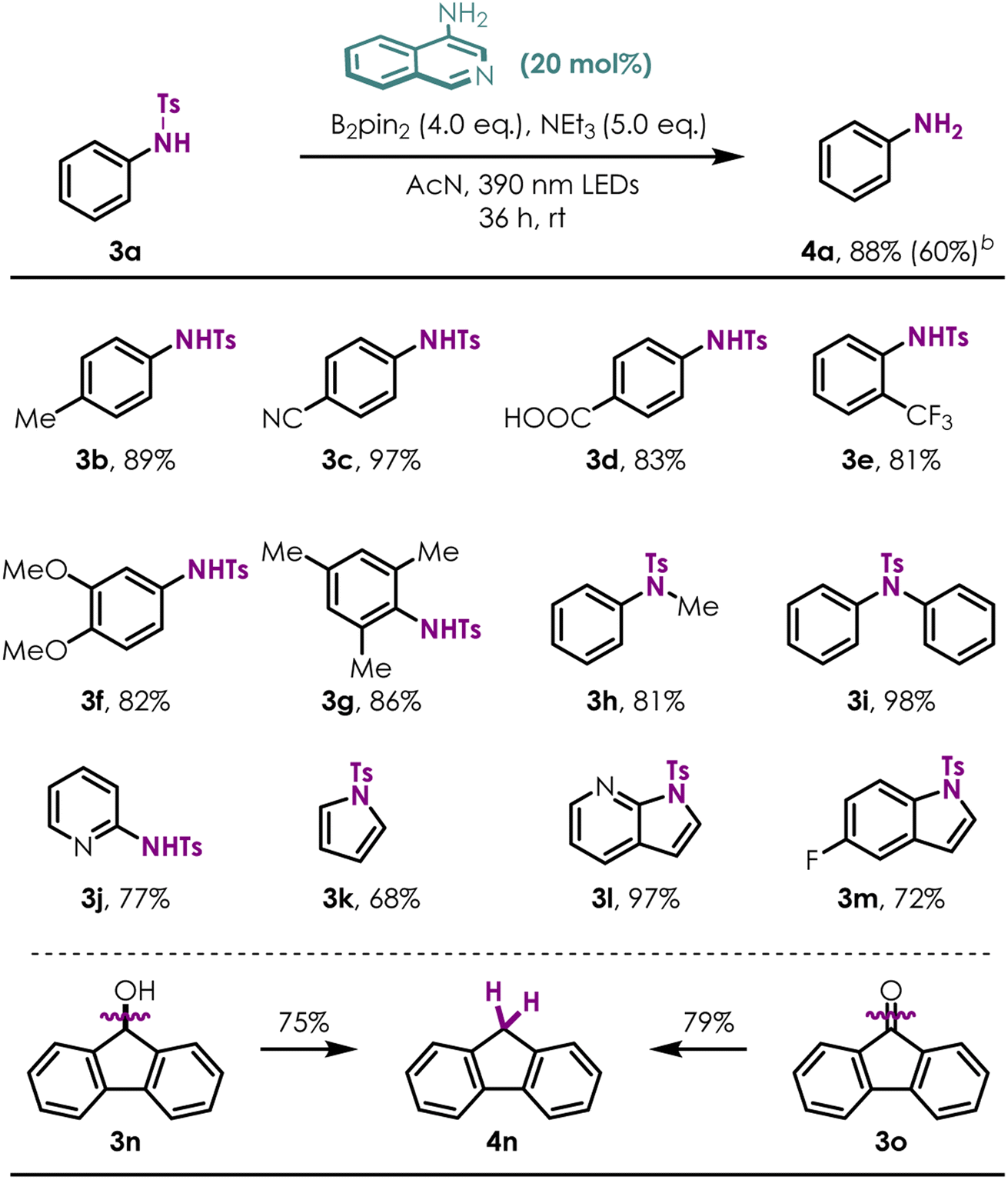
Conclusions
In conclusion, the substituent effects on the properties of vertical excitation energy and redox potential of the in situ formed EDA photocatalyst has been investigated. The 4-NH2 complex has lower excitation energies (3.60 eV) and a lower redox potential (−3.45 V), exhibiting the highest photocatalytic efficiency. Several important reductive transformations, such as aromatic C–H bond borylation, detosylation as well as deoxygenation, has been achieved with high functional groups tolerance and good to excellent yields.Author contributions
Yi-Hui Deng: data curation, formal analysis, investigation, software, visualization, writing – original draft; Qini Li: data curation, formal analysis, investigation, writing – original draft; Manhong Li: data curation, formal analysis, investigation, writing – original draft; Leifeng Wang: conceptualization, supervision, writing – review & editing; Tian-Yu Sun: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of this research by the National Natural Science Foundation of China (No. 22271316), the Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515011994), the Key-Area Research and Development Program of Guangdong Province (2020B010188001), the Technology & Innovation Commission of Shenzhen Municipality, the Fundamental Research Funds for the Central Universities (No. 23ykbj010), and the Sun Yat-sen University Startup Fund. The calculations were carried out at the Shenzhen Bay Laboratory Supercomputing Center.Notes and references
- H. M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC.
- X. Yu, J. Chen and W. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- N. L. Reed and T. P. Yoon, Chem. Soc. Rev., 2021, 50, 2954–2967 RSC.
- M. S. Galliher, B. J. Roldan and C. R. J. Stephenson, Chem. Soc. Rev., 2021, 50, 10044–10057 RSC.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed.
- K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed.
- N. Lalloo, C. E. Brigham and M. S. Sanford, Acc. Chem. Res., 2022, 55, 3430–3444 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- Y. Yuan, S. Majumder, M. Yang and S. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS.
- Z. Yang, Y. Liu, K. Cao, X. Zhang, H. Jiang and J. Li, Beilstein J. Org. Chem., 2021, 17, 771–799 CrossRef CAS PubMed.
- F. Sandfort, F. Strieth-Kalthoff, F. J. R. Klauck, M. J. James and F. Glorius, Chem.–Eur. J., 2018, 24, 17210–17214 CrossRef CAS PubMed.
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed.
- S. Wang, H. Wang and B. König, Chem, 2021, 7, 1653–1665 CAS.
- A. Dewanji, L. van Dalsen, J. A. Rossi-Ashton, E. Gasson, G. E. M. Crisenza and D. J. J. Procter, Nat. Chem., 2023, 15, 43–52 CrossRef CAS PubMed.
- B. Wang, J. Ma, H. Ren, S. Lu, J. Xu, Y. Liang, C. Lu and H. Yan, Chin. Chem. Lett., 2022, 33, 2420–2424 CrossRef CAS.
- M. Jang, T. Lim, B. Y. Park and M. S. Han, J. Org. Chem., 2022, 87, 910–919 CrossRef CAS PubMed.
- L. Bai and L. Jiao, Chem, 2023, 9, 1–23 CAS.
- F. Zhou and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202201102 CrossRef CAS PubMed.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124–9128 CrossRef CAS PubMed.
- M. Li, S. Liu, H. Bao, Q. Li, Y. Deng, T. Sun and L. Wang, Chem. Sci., 2022, 13, 4909–4914 RSC.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 94107 CrossRef.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
- G. L. Eakins, J. S. Alford, B. J. Tiegs, B. E. Breyfogle and C. J. Stearman, J. Phys. Org. Chem., 2011, 24, 1119–1128 CrossRef CAS.
- J. T. E. Quinn, J. X. Zhu, X. Li, J. L. Wang and Y. N. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- C. P. Hsieh, H. P. Lu, C. L. Chiu, C. W. Lee, S. H. Chuang, C. L. Mai, W. N. Yen, S. J. Hsu, E. W. G. Diau and C. Y. Yeh, J. Mater. Chem., 2010, 20, 1127–1134 RSC.
- Y. Nishii, M. Ikeda, Y. Hayashi, S. Kawauchi and M. Miura, J. Am. Chem. Soc., 2020, 142, 1621–1629 CrossRef CAS PubMed.
- I. A. MacKenzie, L. F. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- M. Schmalzbauer, M. Marcon and B. König, Angew. Chem., Int. Ed., 2021, 60, 6270–6292 CrossRef CAS PubMed.
- F. Glaser, C. Kerzig and O. S. Wenger, Chem. Sci., 2021, 12, 9922–9933 RSC.
- R. Miyajima, Y. Ooe, T. Miura, T. Ikoma, H. Iwamoto, S. Takizawa and E. Hasegawa, J. Am. Chem. Soc., 2023, 145, 10236–10248 CrossRef CAS PubMed.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- H. Fang and M. Oestreich, Chem. Sci., 2020, 11, 12604–12615 RSC.
- O. P. Williams, A. F. Chmiel, M. Mikhael, D. M. Bates, C. S. Yeung and Z. K. Wickens, Angew. Chem., Int. Ed., 2023, 62, e2023001 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07558c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |