
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the effect of acid and alkali treatment on a halloysite-based catalyst for dry reforming of methane†
Ahmed Abotaleba,
Dema Al-Masri ab,
Alaa Alkhateba,
Kamal Mrouec,
Atef Zekric,
Yasmin Mashhourd and
Alessandro Sinopoli*a
ab,
Alaa Alkhateba,
Kamal Mrouec,
Atef Zekric,
Yasmin Mashhourd and
Alessandro Sinopoli*a
aQatar Environment and Energy Research Institute, Hamad Bin Khalifa University, P.O. Box 34110, Doha, Qatar. E-mail: asinopoli@hbku.edu.qa
bEarthna Center for a Sustainable Future, Qatar Foundation, Doha, Qatar
cHBKU Core Labs, Hamad Bin Khalifa University, P.O. Box 34110, Doha, Qatar
dDepartment of Chemistry and Earth Sciences, College of Arts and Sciences, Qatar University, Doha P.O. Box 2713, Qatar
First published on 5th February 2024
Abstract
Dry reforming of methane (DRM) has recently received wide attention owing to its outstanding performance in the reduction and conversion of CH4 and CO2 to syngas (H2 and CO). From an industrial perspective, nickel (Ni)-supported catalysts have been deemed among the most suitable catalysts for DRM owing to their low cost and high activity compared to noble metals. However, a downside of nickel catalysts is their high susceptibility to deactivation due to coke formation and sintering at high temperatures. Using appropriate supports and preparation methods plays a major role in improving the activity and stability of Ni-supported catalysts. Halloysite nanotubes (HNTs) are largely utilized in catalysis as a support for Ni owing to their abundance, low cost, and ease of preparation. The treatment of HNTs (chemical or physical) prior to doping with Ni is considered a suitable method for increasing the overall performance of the catalyst. In this study, the surface of HNTs was activated with acids (HNO3 and H2SO4) and alkalis (NaOH and Na2CO3 + NaNO3) prior to Ni doping to assess the effects of support treatment on the stability, activity, and longevity of the catalyst. Nickel catalysts on raw HNT, acid-treated HNT, and alkali-treated HNT supports were prepared via wet impregnation. A detailed characterization of the catalysts was conducted using X-ray diffraction (XRD), BET surface area analysis, scanning electron microscopy (SEM), transmission electron microscopy (TEM), solid-state nuclear magnetic resonance (ssNMR), H2-temperature programmed reduction, (H2-TPR), CO2-temperature programmed desorption (CO2-TPD), and Ni-dispersion via H2-pulse chemisorption. Our results reveal a clear alteration in the structure of HNTs after treatment, while elemental mapping shows a uniform distribution of Ni throughout all the different supports. Moreover, the supports treated with a molten salt method resulted in the overall highest CO2 and CH4 conversion among the studied catalysts and exhibited high stability over 24 hours testing.
1. Introduction
Carbon dioxide (CO2) and methane (CH4) have been identified by the scientific community as two of the main greenhouse gases responsible for the global climate change. With the global average temperature increasing and the frequent occurrence of environmental disasters, it has become urgent to mitigate greenhouse gases and greenhouse gas effects on a global scale. In this regard, dry reforming of methane (DRM) represents a valuable process for the transformation of methane and carbon dioxide into valuable synthetic gas (syngas) comprising H2 and CO, with an H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO molar ratio close to unity. The produced syngas can be used as a feedstock in the synthesis of higher hydrocarbons in the Fischer–Tropsch process.1–4 The DRM reaction requires copious amounts of energy, with reaction temperatures in the range 550–900 °C. However, side reactions such as methane decomposition, Boudouard, and reverse water–gas shift (RWGS) reactions, are responsible for coke deposition (hence catalyst deactivation) and low product selectivity. To overcome these side reactions, noble metals such as Rh, Ru, Pt and Pd have been largely utilized as catalysts for DRM,5–8 although the rarity and high cost of noble metals impose some limitations on their adoption at a large scale.9–11 Consequently, over the last decade, nickel has attracted significant attention in the catalysis community as an ideal candidate for replacing noble metals in the industry owing to its high activity, good C–H bond breaking ability, and cost-effectiveness.12–16 Although nickel may solve some cost and material availability issues, Ni-based catalysts are prone to coke formation and sintering at high temperatures.17
:CO molar ratio close to unity. The produced syngas can be used as a feedstock in the synthesis of higher hydrocarbons in the Fischer–Tropsch process.1–4 The DRM reaction requires copious amounts of energy, with reaction temperatures in the range 550–900 °C. However, side reactions such as methane decomposition, Boudouard, and reverse water–gas shift (RWGS) reactions, are responsible for coke deposition (hence catalyst deactivation) and low product selectivity. To overcome these side reactions, noble metals such as Rh, Ru, Pt and Pd have been largely utilized as catalysts for DRM,5–8 although the rarity and high cost of noble metals impose some limitations on their adoption at a large scale.9–11 Consequently, over the last decade, nickel has attracted significant attention in the catalysis community as an ideal candidate for replacing noble metals in the industry owing to its high activity, good C–H bond breaking ability, and cost-effectiveness.12–16 Although nickel may solve some cost and material availability issues, Ni-based catalysts are prone to coke formation and sintering at high temperatures.17
Various strategies have been investigated to enhance the stability of nickel catalysts by tuning their morphology and electronic properties.5,18 For example, using nickel in the presence of other co-catalysts/promoters (typically transition metals) has been investigated by Li et al. by doping silica substrate with La3+ ions,19–22 resulting in improved activity and stability of Ni-based catalysts. Similarly, Alotaibi et al. evaluated the effect of La and Ca as promoters for Ni-based zeolite catalysts for DRM, demonstrating a higher stability for the La-containing catalyst.23 Developing core–shell structured Ni-based catalysts is another strategy for improving catalytic performance, attributed to the confinement effect and the high dispersion of the catalyst. In fact, the presence of an outer layer shell can prevent coke deposition on top of the active metal phase.20,24 An elegant example that succeeded in confining the active nickel into the channels of mesoporous silica has been reported by Xie et al.25–31 Core–shell structure catalysts have been gradually developed, and nickel has been encapsulated, or confined, in well-defined structures, such as perovskites,32,33 pyrochlores,34 fluorites,35 hexaaluminates,36 spinels,37 and sandwich-like structures.38,39 Additionally, the size of a nickel particles was found to strongly influence the coke formation, as reported by Gould et al., who demonstrated that 5 nm is the ideal size to enhance the coking-resistance performance of a nickel catalyst.40–43 Despite the large number of approaches and investigations, coke formation and sintering still hinder the adoption of Ni-based catalysts in practical and industrial applications.20,24
The development of novel catalytic supports has always become more relevant within the context of sustainable development and cyclic economy. In this regard, natural clays have recently been exploited in various catalytic applications owing to their abundancy, high thermal conductivity, good surface area, and low cost.44–51 Among the natural clays, halloysites (HNT), characterized by the molecular formula Al2SiO2(OH)4·nH2O and a two-layer structure, have been extensively studied. The outer layer of HNTs comprises a tetrahedral silicon-oxygen network, whereas the inner layer is represented by an octahedral aluminium–oxygen unit. HNTs have been largely adopted as materials for environmental and catalytic applications; however, they possess a natural inert character,52–54 and their surface modification is needed to strengthen the interaction between catalyst and support.55–57 Furthermore, the morphology of the catalytic support may influence the diffusion of active metal precursor in the impregnation step.58
In the literature, several examples of chemical HNT surface modification methods have been reported. Generally, they can be divided into acid treatment and alkali treatment based on the nature of the used chemical. The ultimate goal of both acid and alkali treatments is to create defects on the HNT surface or to unzip the typical spiral structure of the HNT. A similar approach was applied to different bulk materials (e.g., boron nitride and TiO2), as an effective way to expose more active sites and to improve the catalytic activity of those materials.59–63 Acid treatment with HNO3 has been reported to increase the catalytic performance of nickel on HNT for steam–CO2 dual reforming of methane.55 A strategy to increase the surface area of HNT was represented by etching with sulphuric acid through the selective etching of Al–OH on the surface of HNT.64 In studies using alkali treatment, reacting HNTs with a low concentration of sodium hydroxide (NaOH) resulted in the formation of hydroxyl groups on the surface of HNTs, allowing for a higher dispersion of the treated HNT in water and polar solvents.65 An interesting approach towards HNT alkali treatment is represented by the use of molten salts. Such an approach was reported by Chen et al. for the fabrication of Pt@HNT catalysts, resulting in improved activity and selectivity for the hydrogenation of cinnamaldehyde.56 In a similar fashion, Zhang and his group selectively etched the outer silica layer of halloysite by ball milling with molten salts, leading to the formation of defects, where the resulting Ni-based modified HNT catalyst exhibited enhanced catalytic performance, compared to the grinding method.66
Over the past decade, various treatment methods have been applied to HNT supports for catalytic methane conversion reactions. In this work, we present a comprehensive comparative study on the effect of acid versus alkali treatment on raw halloysite clay together with the catalytic activities of the corresponding nickel-based catalysts. Specifically, raw HNT (S1) was modified by treatment with HNO3 (S3), H2SO4 (S5), ball-milled molten salt (S7), NaOH (S9), and ground molten salt (S11), and its textural properties were investigated. The six HNT samples were doped with nickel salt using the wet impregnation method. The resulting doped catalysts S2 (Ni@S1), S4 (Ni@S3), S6 (Ni@S5), S8 (Ni@S7), S10 (Ni@S9), and S12 (Ni@S11) were extensively characterized and tested in a DRM reaction. Various characterization techniques, including TEM, BET, ssNMR, XRD, CO2-TPD, TGA, and H2-TPR, were employed to investigate the effects of the different support treatments. Our findings indicate that the activity and stability of the final catalysts were strongly affected by the type of treatment owing to different Ni dispersion, reducibility, basicity, and different numbers of active sites. In general, alkali treatment of the HNT support yielded more stable catalysts in the DRM, with an overall higher CH4 and CO2 conversion than those recorded for supports treated with acids.
2. Methodology
2.1 Catalyst preparation
A series of Ni-based halloysite catalysts were synthesized by acid treatment (HNO3 and H2SO4) and by alkali treatment (NaOH and Na2CO3–NaNO3 molten salt), followed by wet impregnation. Nano-halloysite clay (Al2Si2O5(OH)4·2H2O) was purchased from Sigma-Aldrich and used as support. Nitric acid (HNO3 ≥69.0%), sulphuric acid (H2SO4 95–97%), nickel chloride hexahydrate (NiCl2·6H2O), sodium hydroxide pellets (NaOH), sodium nitrate (Na2NO3), and sodium carbonate (Na2CO3) were all supplied from Sigma-Aldrich and used in the catalyst synthesis process.The main HNT treatment procedures were adapted from the literature. Specifically, for the acid HNO3 (ref. 67) and H2SO4 (ref. 64) treatments, halloysite (2 g) was refluxed in 120 mL of 3 M HNO3 or H2SO4 solution for 8 hours. The resulting solid was washed several times with deionized (DI) water until it reached pH 4 to 5; the samples were then dried at 105 °C for 12 hours, followed by calcination at 1000 °C for 6 hours. In the alkali (NaOH) treatment, halloysite (10 g) was mixed in 100 mL of 5 M NaOH solution and heated to reflux for 8 hours. The obtained solid was then washed several times with DI water until a pH of 6 to 7 was achieved; the samples were then dried at 105 °C for 12 hours.65 For the molten salt treatment of HNT nanotubes, a mixture of HNT (2 g), Na2CO3 (0.6 g) and NaNO3 (2 g) was milled in a ceramic ball mill device at a speed of 300 rpm (ball-to-sample mass ratio ≃ 10:1) for an hour and then calcined at 350 °C for 2 h. Next, the resultant sample was washed several times with deionized water to remove soluble salts and impurities and then dried in a vacuum oven for 12 hours. The effect of grinding the solid mixture rather than using ball milling was also explored.66 Raw halloysite nanotubes were used as a reference to evaluate the effect of the treatments.
The impregnation method was used to synthesize Ni-based halloysite nanotubes. For this purpose, a certain amount of nickel chloride, with a nominal content of 10%, was dissolved in deionized water, after which the treated HNT nanotubes were added to the prepared aqueous solution at room temperature. The obtained slurry was accurately mixed for 1 hour, after which urea was added to the mixture in excess with respect to nickel salt weight. The mixture was stirred for a further 60 minutes at room temperature prior to refluxing at 80 °C for 10 hours. In the next step, the resulting solid was washed and dried in an oven for 12 hours. Finally, all the samples were calcined at 800 °C for 6 hours. Throughout this paper, the samples were labelled as follows: raw HNT (S1); Ni@raw HNT (S2); HNO3-HNT (S3); Ni@HNO3-HNT (S4); H2SO4-HNT (S5); Ni@H2SO4-HNT (S6); ball-milled molten salt-HNT (S7); Ni@ball-milled molten salt-HNT (S8); NaOH-HNT (S9); Ni@NaOH-HNT (S10); ground molten salt-HNT (S11); and Ni@grinded molten salt-HNT (S12).
2.2 Catalyst characterization
All solid-state NMR experiments were carried out at 14.1 T using a Bruker AVANCE III 600 MHz wide bore spectrometer operating at Larmor frequencies of 600.13 MHz for 1H and 119.2 MHz for 29Si, equipped with a Bruker 3.2 mm triple-resonance low-temperature magic angle spinning (LT-MAS) NMR probe. Samples were ground into fine powders and packed into 3.2 mm outer diameter zirconia rotors. The 29Si CP-MAS (cross-polarization magic angle spinning) spectra were acquired at room temperature with a 10 kHz MAS rate using a ramped-amplitude cross-polarization pulse sequence with a 1H π/2 excitation pulse of 3.0 μs, a recycle delay of 3 s, and cross-polarization contact times ranging from 5.0 to 8.0 ms. SPINAL-64 heteronuclear decoupling with ca. 90–100 kHz RF field strength was applied to decouple protons during signal acquisition,68 along with the collection ranging from 1280 to 2048 FIDs to obtain reliable signal-to-noise ratios. Additionally, 29Si single pulse (SP) experiments with high-power proton decoupling were performed on some samples at a 10 kHz MAS rate using a 1.5 μs (π/6 flip angle) 29Si excitation pulse and a 15 s recycle delay with the accumulation ranging from 1120 to 2000 transients. All 29Si NMR spectra were externally referenced with respect to liquid tetramethylsilane (TMS) at 0 ppm.Specific BET surface areas of the as-prepared catalysts were measured by a surface area analyser (ASAP 2420, Micromeritics) at 77 K. Before measurements, samples were dried at 90 °C for 30 min, followed by degassing at 350 °C for 6 h under vacuum to remove contaminates and water. The specific surface area was calculated using the BET equation and BJH desorption for pore size analysis.
The X-ray powder diffraction (XRD) patterns of the prepared catalysts were conducted using an X-ray diffractor (XRD-6100x, Shimadzu) with Cu-Kα (λ = 1.5406 Å) radiation. The measurement was conditioned at 40 kV and 30 mA, with a scanning speed of 7° min−1 ranging from 10° to 70°.
The TEM investigations were performed on a field emission gun TALOS (FEI, Hillsboro, Oregon, USA) operated at 200 kV and equipped with an FEI Energy Dispersive X-ray (EDX) detector and a high-angle annular dark-field, HAADF detector. Esprit software from Bruker (Billerica, Massachusetts, USA) was used to obtain the qualitative element analyses. Specimens were prepared by dispersing samples in isopropanol and droplets of the suspension were deposited on a thin copper grid. After the isopropanol was vaporized, the thin copper grid was mounted in a TEM holder and then inserted into the sample chamber of the transmission electron microscope. Moreover, actual Ni contents on the as-prepared catalysts were performed by SEM-EDS FEI Quanta650FEG for imaging and equipped with a Bruker XFlash 6I60 detector for EDS analysis.
H2 temperature-programmed reduction (H2-TPR) of the as-prepared catalysts was performed by applying an AutoChem 2950 (Micromeritics) apparatus equipped with thermal conductivity. Prior to the experiments, 50 mg of catalyst was pretreated in N2 (100 mL min−1) at 300 °C for 30 min to remove impurities, followed by cooling to ambient temperature in N2 stream. Catalysts were reduced with a 10 vol% H2–Ar mixture (100 mL min−1) by heating up to 800 °C, at a ramp rate of 10 °C min−1.
Catalyst basic sites were identified through CO2-temperature programmed desorption (CO2-TPD) analysis using AutoChem 2950 apparatus, where 500 mg of catalyst was pretreated in N2 (100 mL min−1) at 300 °C for 30 min to remove impurities. After cooling to ambient temperature in N2, the catalyst was saturated with CO2 gas (100 mL min−1), followed by flushing the sample with Ar (100 mL min−1) to remove excess CO2. Finally, samples were heated up to 800 °C at a ramp rate of 10 °C min−1 while detecting the CO2-desorption.
Pulse chemisorption was also performed by an AutoChemII 2950 station. The nickel-doped samples (S2, S4, S6, S8, S10, and S12) were placed in a U-shaped stainless reactor to quantify the Ni-dispersion. Prior to the experiments, 50 mg of catalyst was reduced by 10 vol% H2–Ar mixture (50 mL min−1) and heated up to 750 °C, at a ramp rate of 10 °C min−1. Then, Ar gas was purged to remove the excess H2, at 775 °C for 60 minutes, followed by cooling down to ambient temperature in Ar. Pulse chemisorption was performed at 35 °C, where a cryo-cooler from micromeritics was used to inject liquid nitrogen to cool and maintain the sample at the desired temperature. The volume of the injection loop was 0.5 cm3. Pulses of H2 (10 vol% H2 in Ar) were injected into the catalytic reactor, and Argos was used as the carrier gas. H2 consumption, TPR and CO2-TPD were all measured using a thermal conductivity TCD detector, equipped with a water trap. Pulse injection, sample temperature and TCD signals were controlled and monitored by Micromeritics AutoChem software.
Thermogravimetric analysis (TGA) of both the reduced and spent catalysts was performed using a Discovery TGA from TA Instruments. The catalysts (∼10 mg) were heated up to 800 °C at a rate of 10 °C min−1 with air flow (20 mL min−1).
2.3 Catalytic activity and stability tests
Catalytic activity measurements of the DRM were performed using a MicroEffi reactor (EMP) equipped with an SS310 fixed-bed reactor (internal diameter 9.1 mm and length 304.8 mm). Approximately 200 mg of catalyst was loaded and reduced by 5% H2/Ar (100 mL min−1) at 800 °C. The reactor was then cooled to 750 °C by N2 flow (50 mL min−1). A mixture of equimolar CH4/CO2 (50 mL min−1) passed through the catalyst with a gas hourly space velocity (GHSV) of 30000 mL g−1 h−1. Catalytic performance was evaluated at 750 °C for 24 hours. Gaseous products (e.g., CO, H2, CH4 and CO2) were analysed by applying an on-line mass spectrometer mks-CIRRUS™ 2 atmospheric pressure gas monitoring. Conversions of CH4 and CO2, as well as the molar ratio of H2/CO, were determined from the corresponding difference between the outlet and inlet flow rates. The CH4 and CO2 conversions (%) as well as H2/CO molar ratio were calculated using the following equations: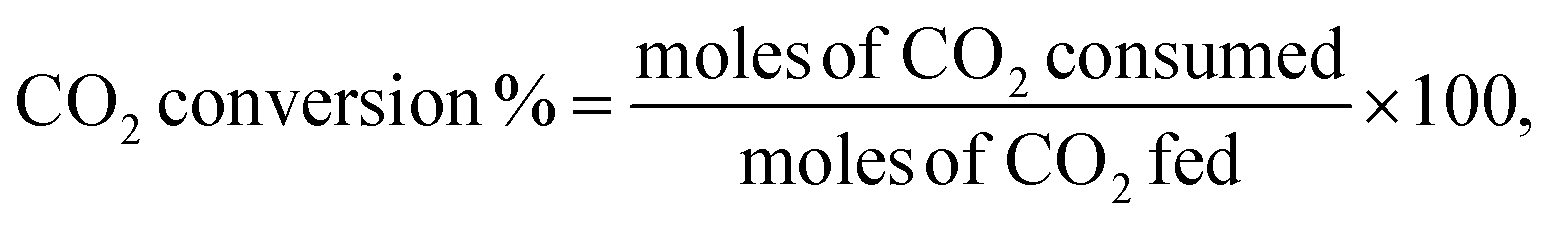
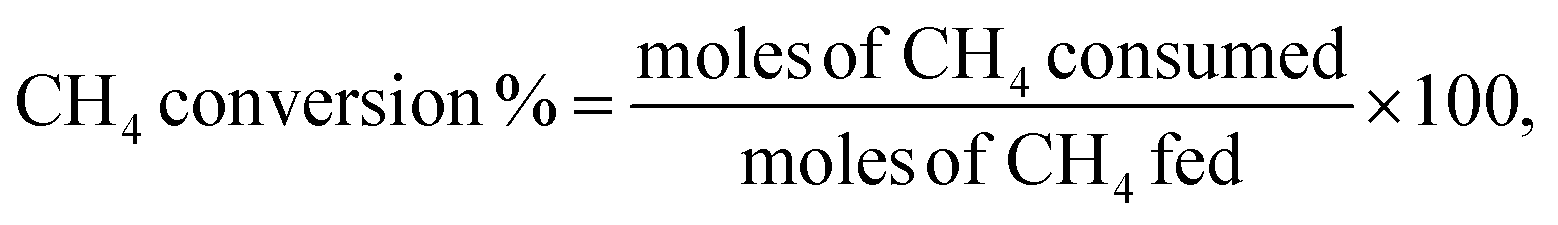
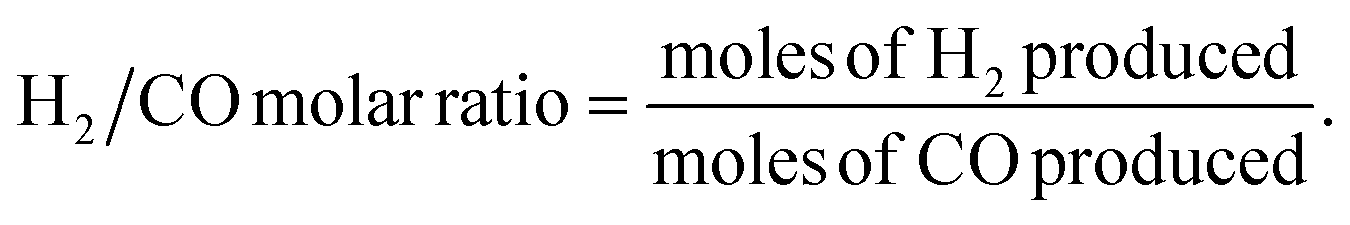
3. Results and discussion
Aluminosilicate nanotubular clay is selected because of its low cost, natural availability, chemical/thermal stability and relatively high surface area (approx. 60–80 m2 g−1).51,69–71 The acid treatment of HNT is a common strategy used by the materials community that involves the use of strong acids, such as sulfuric acid or nitric acid, to modify the surface of the nanotubes.64,72,73 This process leads to the removal of impurities and amorphous materials, resulting in the exposure of a higher proportion of the tubular structure, by etching the inner alumina layer (and in a minor percentage the silica layer) and thus enhancing the material's ability to interact with the metal catalyst. The acid treatment process also affects the porosity and active sites on the surface of the HNT, making it more accessible for reactant molecules to interact with the substrate. These pores and active sites provide sites for catalysis, increasing the material's reactivity and efficiency. The availability of pores and active sites upon acid treatment makes HNT a more effective adsorbent and catalyst support. This allows for higher loading of active species and facilitates the diffusion of reactants into the inner structure of the nanotubes.Recently, alkali-treated HNTs have attracted wide attention as a valuable alternative to acid-treated HNT.65,74,75 Alkali treatment involves the use of alkaline solutions, such as sodium hydroxide (NaOH) or Na2CO3–NaNO3 molten salts, to modify the surface of the HNT. This process can create different functional groups (e.g., –OH) on the nanotube surface, which can enhance its interaction with carbon dioxide (CO2) molecules, leading to a higher CO2 adsorption capacity. Furthermore, alkaline treatment can increase the resistance of HNT to coke deposition, which is the accumulation of carbonaceous materials on the catalyst surface during reactions, prolonging the catalyst's lifespan and maintaining its activity over extended reaction periods. Additionally, alkali treatment may unzip the tubular structure of the HNT, exposing more of its surface.
Both acid and alkaline treatments of HNT offer distinct advantages. Acid treatment increases the surface area and availability of pores and active sites, while alkaline treatment enhances CO2 adsorption, resistance to coke deposition, catalyst activity, and stability. These treatments make HNT a versatile material with valuable applications in adsorption and catalysis. The choice between acid and alkaline treatments depends on the specific needs and intended applications of the modified HNT.
3.1 Solid-state NMR analysis
Solid-state nuclear magnetic resonance (ssNMR) spectroscopy was used to better demonstrate the effects of the different treatments on the silica outer layer of HNT, and the spectra are shown in Fig. 1. The 29Si CP-MAS NMR spectrum of raw HNT (S1) shows a single sharp signal at −91.8 ppm (from TMS) corresponding to the crystallographically equivalent Si atoms of the tetrahedral SiO4 layers on the HNT outer surface in accordance with reported values in the literature.76 Moreover, the observed chemical shift indicates a Q3-type halloysite silicon site, Si(OSi)3(OAl2), in which each silicon atom of the SiO4 tetrahedron is bridged by one oxygen atom to two aluminium atoms from the adjacent AlO6 octahedral layer in its second coordination sphere.76–78 It is worth noting that the Qn assignments used herein are consistent with the notations used by Lippmaa et al. and Smith et al. for silicates and layered aluminosilicates.79,80 According to this notation, all silicate anions can be described as a combination of Qn units, where the Q symbol represents a silicon atom bonded to four oxygen atoms (Q = SiO4) and the superscript shows the number of other Q units (Si–O tetrahedra) attached to the silicon tetrahedron under study.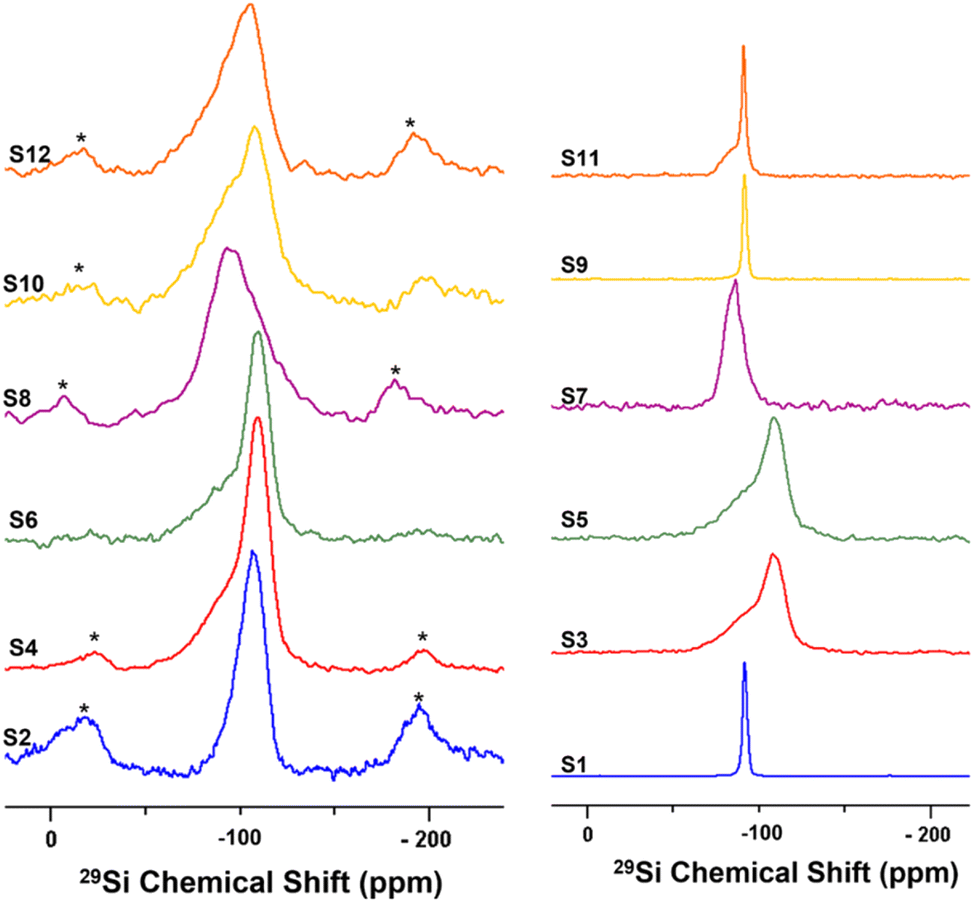 | ||
| Fig. 1 Solid-state 29Si NMR spectra at 10 kHz MAS for undoped samples (right) and nickel-doped catalysts (left). | ||
For S3, a broad silicon signal centred at −109 ppm is observed; this peak can be attributed to the formation of a Q4-type Si(OSi)4 amorphous three-dimensional silica site as a result of the destruction of the HNT crystal lattice by acid treatment. The downfield (high-frequency) shoulder might be attributed to some silicon-rich nanoparticles or minor Q3-type sites from unreacted HNT. Similarly, S5 shows a broad silicon signal at −110 ppm. Ni-doped catalysts S2, S4, and S6 show a broad silicon signal at −110 ppm, similar to the acid-treated HNTs (S3 and S5, respectively), attributed to the common calcination step after doping. The HNT treated using the ball-milled molten salt method (S7) shows a broad silicon signal at −86.6 ppm. This signal could be assigned to Q2-type silicon environments, such as Si(OSi)2(OH)2 or Si(OSi)2(OH)(OAl) sites. Interestingly, its Ni-doped counterpart, S8, shows a broad and featureless silicon signal centred around −93 ppm. S9 shows a silicon signal identical to that of the unreacted raw HNT at −92 ppm. Here, it is important to highlight that NaOH treatment is the only method not comprising a calcination or heating step at the end (other than the drying step). In contrast, S10 shows a broad featureless silicon signal centred at −108 ppm. S11 shows a signal at −91.3 ppm, similar to the one in S1, and a downfield shoulder that might correspond to minor Q2-type environments, such as Si(OSi)2(OH)2 or Si(OSi)2(OH)(OAl), suggesting the partial efficacy of the treatment and the presence of unreacted raw HNTs. Instead, S12 shows a broad and featureless silicon peak centred at −106 ppm. In general, the broad and almost featureless 29Si NMR line shapes observed for some of the samples are likely due to the presence of disordered and/or amorphous electronic environments around the silicon atoms in these solids as a consequence of both chemical and thermal treatments. This structural disorder is caused by variations in local geometry around a certain nucleus, such as changes in the number and type of coordinating atoms, the nature of atoms on the next nearest neighbouring sites, and variations in bond lengths and angles. From an NMR perspective, it has been demonstrated that such a disorder in the structure around a nucleus causes a distribution in the NMR chemical shift of that nucleus, which results in such broadened line shapes.81
3.2 XRD investigation
The XRD patterns of all samples are shown in Fig. 2. For raw halloysite (S1), the XRD pattern showed the characteristic peaks of pristine halloysite at 2θ = 11.78°, 19.9°, 24.8°, 35.2°, 54.5°, 62.1°, and 63.5 according to JCPDS#29-1487.82 A small amount of SiO2 was also observed, which agrees with the chemical analysis of halloysite.83 Amongst the undoped samples, the non-calcinated ones (S1 and S9) retained their typical HNT peaks. However, all the other undoped samples (S3, S5, S7, and S11), whose preparation procedures included a calcination step after treatment, did not show the characteristic peaks of raw halloysite, revealing the formation of amorphous halloysite.84 Similarly, for all the impregnated samples, the HNT peaks disappeared after calcination at 800 °C. Furthermore, the relative intensity of the SiO2 peak to the structural peaks of the halloysite phase increased for the modified samples, indicating the extraction of silica from the halloysite structure.67,83,85 In the impregnated samples, no peaks were observed for NiO, demonstrating an effective reduction to metallic nickel in the presence of urea. Peaks for metallic nickel were observed for samples S6, S8, and S10, and nickel aluminium oxide spinel peaks were observed for S8 only.67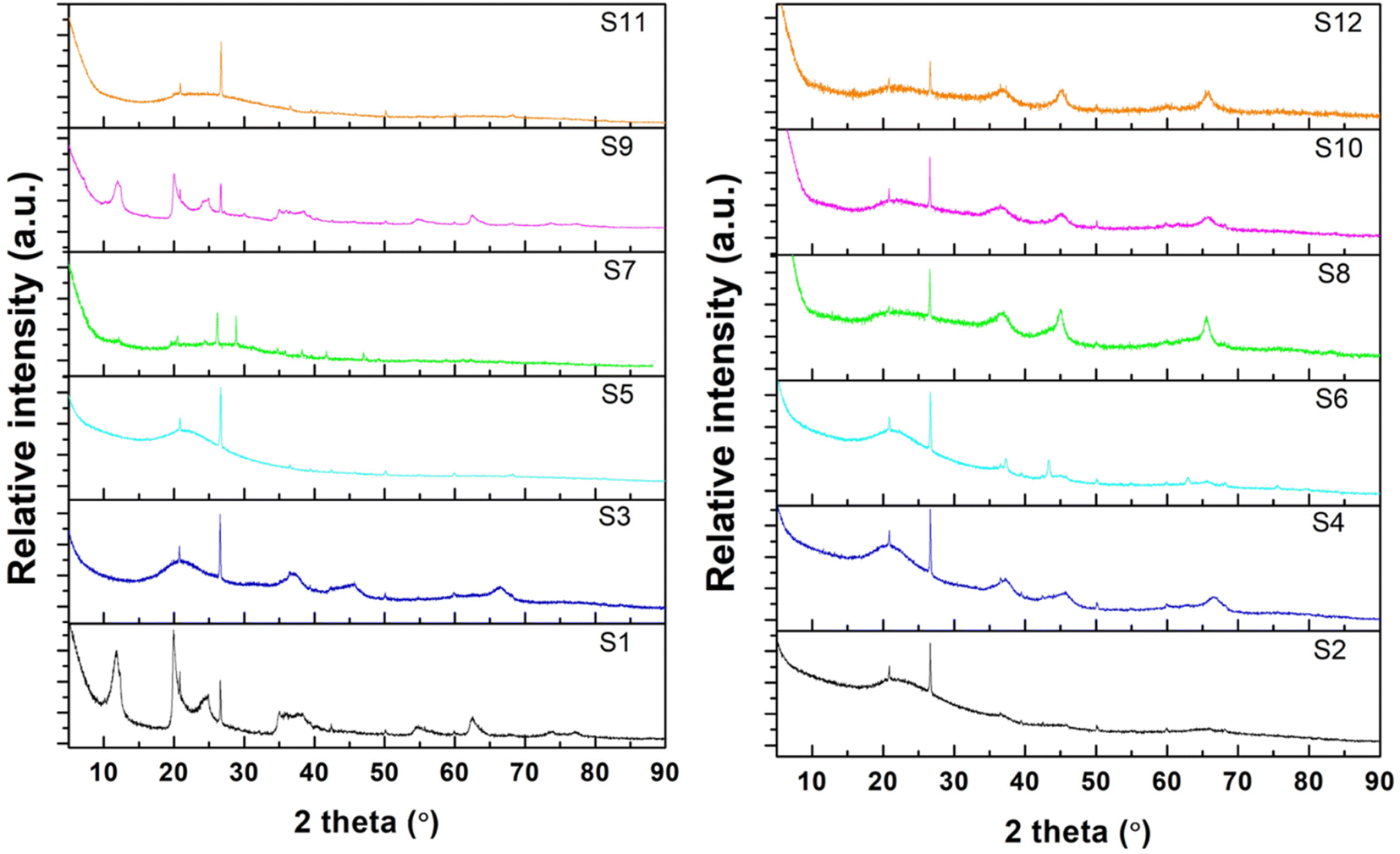 | ||
| Fig. 2 XRD patterns of the synthesized HNTs: undoped samples (left) and impregnated samples (right). | ||
3.3 Morphology investigation
Scanning electron microscopy and energy-dispersive X-ray spectroscopy (SEM-EDS) were conducted for the doped catalyst to evaluate the amount of loaded nickel. Despite the initial loading of 10 wt% nickel, the final percentage of nickel ranged from 3.6 to 11%, with S10 and S2 having the lowest loadings (see Fig. S1†).To better understand the changes in the morphology of the HNTs upon treatment, transmission electron microscopy (TEM) was conducted on the raw HNT support and the treated HNT supports (Fig. 3). Fig. 3a illustrates the structure of the raw HNTs, depicting a nanotubular structure with an inner lumen diameter of about 11.5 nm. Fig. 3b and c show the microscopic images of HNO3 and H2SO4 treated HNT, respectively. Here, the HNT walls and edges appear smoother and more rounded hypothetically owing to acid treatment, whereby the acid attacks the walls of the HNT, particularly the alumina layer, as alumina is more soluble in acid than silica.64,67 The etching of alumina via acid could also be the reason why the inner lumen area (consisting of alumina) of acid-treated samples is less vivid.
 | ||
| Fig. 3 TEM images of the treated supports: (a) S1, (b) S3, (c) S5, (d) S7, (e) S9, and (f) S11. | ||
However, HNTs subjected to alkaline treatment with NaOH and the molten salt methods do not show similar smooth edges throughout the HNT, as depicted in Fig. 3d and e, respectively. However, the lumen area is not very visible. Alkaline treatment dissolves silica, which could have caused the collapse of the HNT walls, destroying the lumen area. Specifically, for the HNT treated with the ball-milled molten salt method (Fig. 3e), the HNTs adopt a more plate-like structure rather than their original tubular shape. On the contrary, no such changes are observed in the molten salt grinding method, where the HNTs retain their tubular morphology. In general, all the HNT samples treated with alkalis resulted in tubular shapes that were shorter and smaller in diameter than those of their acid-treated counterparts.
TEM characterization was conducted on the Ni metal-doped supports, and the results are presented in Fig. 4. Visible nickel particles were not observed in the TEM image for Ni-raw HNT (Fig. 4a); instead, Ni was found to be uniformly distributed through the support in the STEM-EDX elemental mapping (Fig. S2†). Interestingly, the presence of nanosheets was observed on most of the treated supports with finer nanosheets growing on the alkali-treated supports (Fig. 4d–f). The Ni particle size was calculated using TEM images and image processing software (see Table S2 and Fig. S6†), revealing smaller particles for S2, S4, and S6 in the range of 7–9 nm, and larger particles for the alkali-treated samples S8, S10, and S12, in the range of 13–30 nm.
 | ||
| Fig. 4 TEM images of Ni loaded on HNT supports: (a) S2, (b) S4, (c) S6, (d) S8, (e) S10, and (f) S12. | ||
3.4 Textural properties
The nitrogen adsorption–desorption isotherms and pore size analyses of the samples are reported in Fig. S3 and S4,† respectively. All samples showed a type IV isotherm with a hysteresis loop, demonstrating the existence of mesopores according to the IUPAC classification.86 The corresponding BET surface areas and pore volumes are summarized in Table S1.† The BET surface area of the alkali-treated samples generally decreased from 65 to 19, 50, and 47 m2 g−1 (for S7, S9, and S11, respectively), while the total pore volume changed from 0.275 to 0.105, 0.321, and 0.277 cm3 g−1 (for S7, S9, and S11, respectively), when compared to the raw halloysite, owing to the effect of silica etching. Additionally, in the acid-treated HNT, a general decrease in BET surface area and a similar pore volume, respectively, was observed, for the HNO3-treated sample (40 m2 g−1 and 0.230 cm3 g−1) and for the H2SO4-treated sample (62 m2 g−1 and 0.354 cm3 g−1) compared to raw HNT. This behaviour can be attributed to the favourable etching of alumina instead of the silica layer when using acid treatment, resulting in better preservation of the original structure, as observed in the TEM images.After decorating all the samples with Ni, the BET surface area and pore volume of the metal-containing samples were higher than those of the corresponding undoped halloysite samples (see Table 1). Therefore, the resulting pore structure was found to be sufficient to allow for the sufficient diffusion of methane molecules and products.
| Sample | SBET [m2 g−1] | Micropore area [m2 g−1] | Vt [cm3 g−1] | Pore size [nm] | Si/Al ratio |
|---|---|---|---|---|---|
| S2: Ni-raw HNT | 53 | 12 | 0.281 | 13.9 | 1 |
| S4: Ni-AHNT, HNO3 | 55 | 13 | 0.269 | 12 | 1.5 |
| S6: Ni-AHNT, H2SO4 | 66 | 10 | 0.298 | 12.4 | 1.3 |
| S8: Ni-AlHNT, NaNO3 ball milled | 73.88 | 5.05 | 0.290 | 8.82 | 0.94 |
| S10: Ni-AlHNT NaOH | 50.9 | 3.14 | 0.271 | 16.38 | 1.03 |
| S12: Ni-AlHNT, NaNO3 grinded | 52 | 0 | 0.244 | 7.7 | 1.01 |
H2-TPR experiments were performed to explore the reduction behaviour of the investigated catalysts. H2-TPR profiles of the raw and modified halloysite samples are shown in Fig. 5. In general, peaks at temperatures lower than 400 °C indicate the reduction of free NiO. Peaks at temperatures below 600 °C are attributed to metal oxides, such as NiO·Al2O3 (surface NiAl2O4 spinel or defeated NiAl2O4 spinel) or NiO, having a weak interaction with the support. Peaks at temperatures above 600 °C are assigned to the reduction of complex-bonded metal oxide species, such as crystalline NiAl2O4 spinel, or to the reduction of NiO species strongly interacting with the support.67,87,88 Nickel spinels can easily be formed from a reaction between Ni and the Al2O3 layer. However, it is believed that the NiAl2O4 spinel cannot be easily reduced to a Ni0 species; thus, incomplete reduction of NiAl2O4 produces Ni sites that interact strongly with the support. Such strong interaction plays an important role in stabilizing the catalytic activity of the Ni during DRM.88 Nickel loading for all the prepared catalysts calculated by H2-TPR is reported, as shown in Table 2.
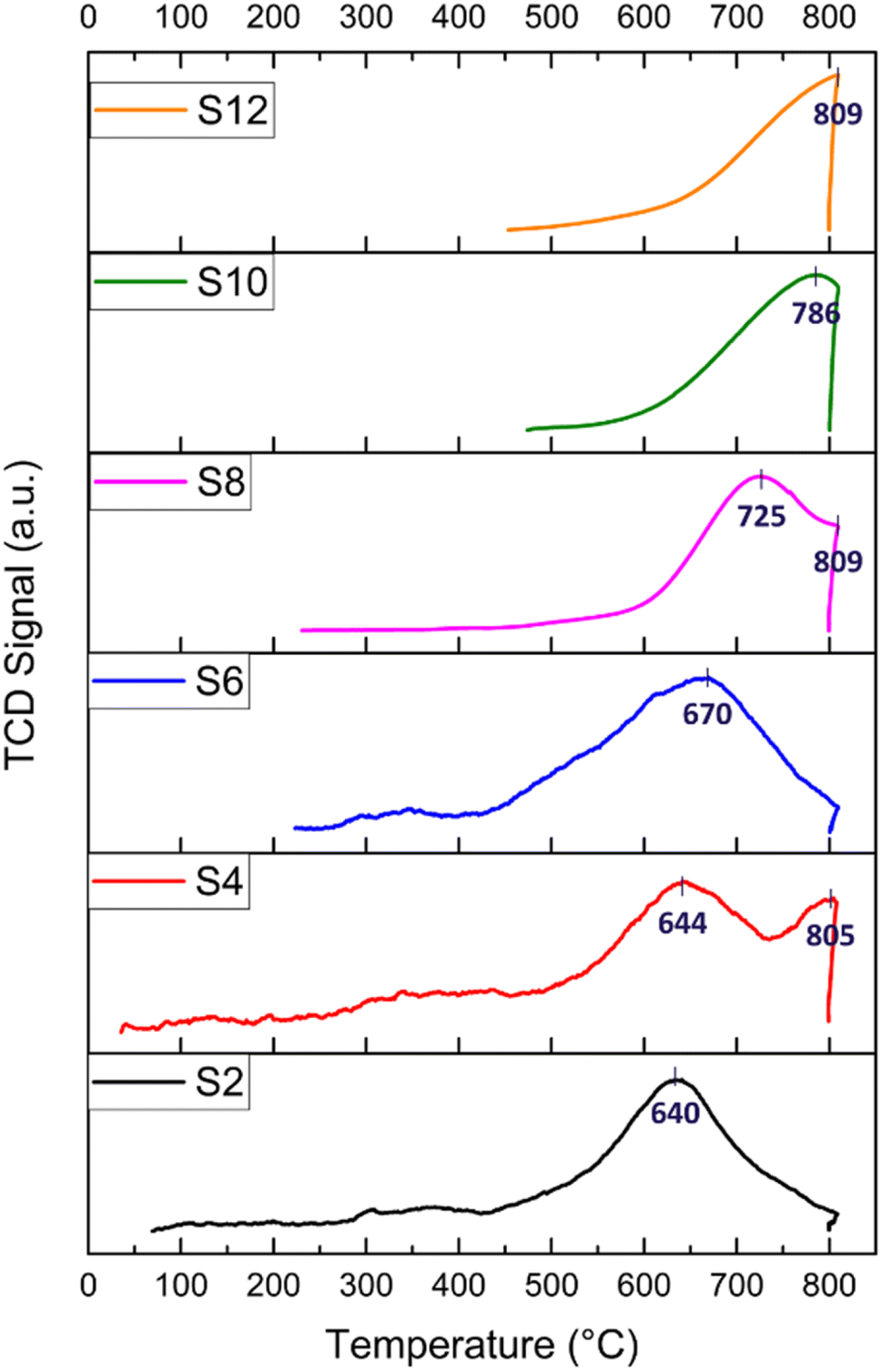 | ||
| Fig. 5 H2-TPR profiles of the HNT-supported Ni catalysts; numbers indicate reduction temperatures. | ||
| Sample | Ni loading [wt%] from SEM | H2-pulse chemisorption | H2-TPR | CO2-TPD | ||||
|---|---|---|---|---|---|---|---|---|
| Ni dispersion [%] | Metallic surface area [m2 per g metal] | Peaks [°C] | Ni loading [wt%] | H2 consumption (cm3 per g STP) | Peaks [°C] | CO2 adsorption (cm3 per g STP) | ||
| S2 | 5.86 | 0.21 | 1.38 | 640 | 8.82 | 26.45 | 73, 394 | 3.75 |
| S4 | 7.63 | 1.29 | 8.55 | 644, 805 | 11.45 | 34.34 | 102, 424, 795 | 8.42 |
| S6 | 11.01 | 0.20 | 1.34 | 670 | 11.02 | 33.07 | 79 | 0.09 |
| S8 | 9.09 | 0.31 | 2.03 | 725, 809 | 9.85 | 29.55 | 86, 401 | 186.24 |
| S10 | 3.63 | 0.74 | 4.89 | 786 | 8.14 | 24.42 | 89, 402 | 555.04 |
| S12 | 9.57 | 2.74 | 18.19 | 809 | 6.01% | 18.04 | 77, 395 | 225.70 |
For the S2 catalyst, reduction was performed at 640 °C, and the peaks indicated a good interaction between the support and the active metal. Similarly, the acid-treated samples S4 and S6 exhibited a reduction peak at 644 and 670 °C, respectively, in addition to a peak at 805 °C for S4, which could be attributed to the potential dissolution of SiO2 by HNO3. All the alkali-treated samples (S8, S10, and S12) exhibited reduction peaks at higher temperatures (725, 786, and 809 °C, respectively) compared to the acid-treated catalysts. The reduction of NiO at higher temperatures in the alkali-treated catalysts and in the S4 (805 °C peak) could be attributed to the effect of the alkaline treatment on the morphology of HNT, specifically leading to the formation of sheets (such as with S8) and to the creation of defects on the silica layer. Both two-dimensional nanosheet structures and defect formation strengthened the interaction between NiO and the substrate as well as caused an increased interfacial area. It has been demonstrated that the etching of silica in the outer layer of the halloysite causes some defects and cavities that can confine Ni particles, which, consequently, leads to the formation of a strong metal–support interaction.18,66
The H2 consumption is calculated for all the catalysts, and it ranges from 18 to 34 cm3 per g STP (see Table 2). The highest H2 consumption is recorded for S4, which also has the highest Ni loading (11.45 wt%) calculated from the H2-TPR. This high Ni loading is responsible for the high initial activity in relation to catalyst performance. Following this, S6 exhibited 33.06 cm3 per g STP H2 consumption and a Ni loading of 11 wt%, recording the second-best initial conversion. In general, the alkali-treated catalysts exhibited an H2 consumption lower than that of the acid-treated samples. In particular, S8 exhibited a reasonable H2 consumption of 29.55 cm3 per g STP with 9.85 wt% Ni, demonstrating its acceptable catalytic activity with adequate Ni loading owing to the size reduction and good surface area induced by the ball milling process. Both S2 and S10 displayed average H2 consumption and Ni loading compared to the other samples, with approximately 8 wt% Ni. Conversely, S12 reported the lowest H2 consumption of 18 cm3 per g STP, with a total Ni loading of 6 wt%. Notably, S12 underwent a grinding process only, resulting in relatively larger particles compared to the ball-milled technique, thus leading to the lowest final Ni loading among the samples.
Before moving to the CO2-TPD analysis, it is important to add a note about the quantification of nickel loading. Despite the nominal 10 wt% loadings discussed in the Catalyst preparation section, the resulting Ni loadings, estimated by both SEM EDS and H2-TPR and reported in Table 2, have values that differ from the expected 10%. During the synthesis of the catalyst, the Ni2+ ion dissolved in the aqueous media is adsorbed on the surface of the HNT. Based on the previous chemical treatment, the presence of terminal groups, cavities, and other cations, as well as different surface areas, porosity, and dispersion in water, the treated halloysite supports are prone to adsorb nickel ions differently. The resulting reduced nickel catalyst is then estimated by SEM EDS and H2-TPR. The values obtained by applying the two methods can be different because the SEM EDS provides a semi-quantitative elemental analysis, and the results are affected by background and matrix effects. Although the metal loading obtained by H2-TPR comes from an indirect calculation based on the H2 consumption, it is generally considered more accurate than SEM EDS analysis. In general, inductively coupled plasma mass spectrometry (ICP-MS) represents the most accurate technique for measuring catalyst loading on a support, but this analysis is beyond the scope of our work.
The basicity of the samples was examined by CO2-TPD, and the results are shown in Fig. 6 (the CO2 desorption peak values are shown in Table 2). In general, CO2 desorption peaks indicate the presence of weak basic sites when T < 150 °C (attributed to OH− groups), medium basic sites at T = 150–300 °C (attributed to metal–O2− pairs), and strong basic sites when T > 300 °C (attributed to isolated O2− anions).18
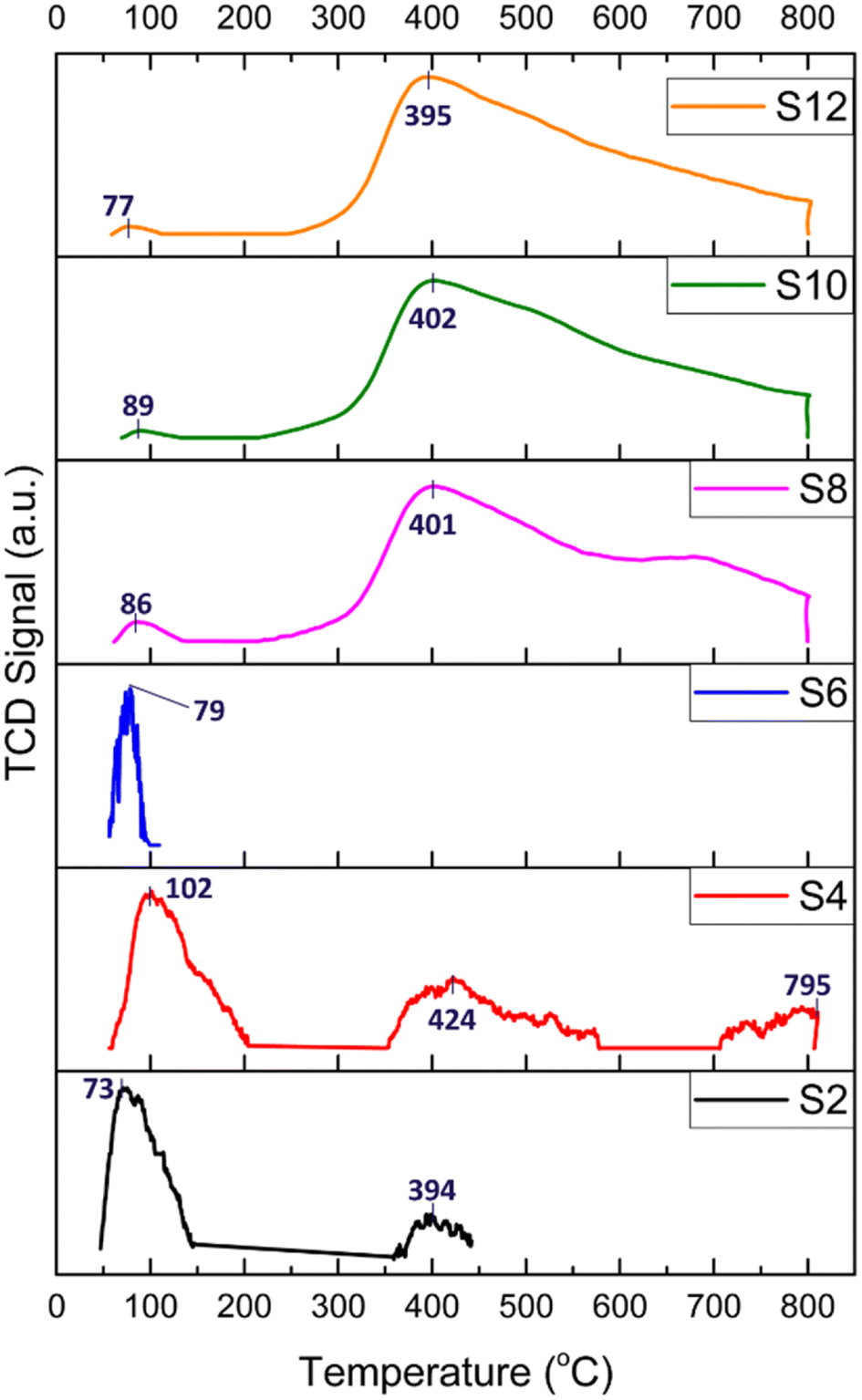 | ||
| Fig. 6 CO2-TPD profiles of the HNT-supported Ni catalysts; numbers indicate desorption temperatures. | ||
The TPD profiles depicted in Fig. 6 of the acid-treated catalysts (S2, S4, and S6) generally present peaks attributed to a very weak basic strength and a main peak below 102 °C, resulting from reactive bicarbonates sourced from the interactions of CO2 with hydroxyl groups on the support surfaces. Interestingly, for S4, three CO2 desorption peaks were detected at 102, 424 and 795 °C. This pattern was attributed to the formation of defects and cavities on both the silica and alumina layers of the HNT caused by HNO3 treatment, thereby producing a more stable space for the confinement of CO2 molecules.
For the alkali-treated catalysts (S8, S10, and S12), the CO2 desorption peaks are observed at higher temperatures, which typically indicate a relatively stronger basic site. The desorption peak was around 400 °C and could be linked to more stable carbonates stemming from the interactions of CO2 with the surface oxygen atoms of Ni. Moreover, the area of the desorption peaks reflects the number of basic sites on the surface of the catalysts. As depicted in Fig. 6, the area of the desorption peaks of the alkali-treated catalysts was much larger than that of the acid-treated catalysts.
The CO2 consumption of the samples is obtained by CO2-TPD, as detailed in Table 2. The raw HNT and acid-treated samples, S2, S4, and S6, exhibited CO2 consumptions of 3.75, 8.42, and 0.09 cm3 per g STP, respectively. These findings confirm their low basicity and subsequent lack of stability, as observed in the performance test (Fig. 7). In contrast, the alkali-treated samples, S8, S10, and S12, demonstrated significantly higher values at 186.24, 555.04, and 225.70 cm3 per g STP, respectively. These results indicate the elevated basicity of these samples owing to the formation of hydroxyl terminations, resulting in higher CO2 adsorption, reduced coke formation, and, consequently, a more stable catalyst, as corroborated by the stability performance test.
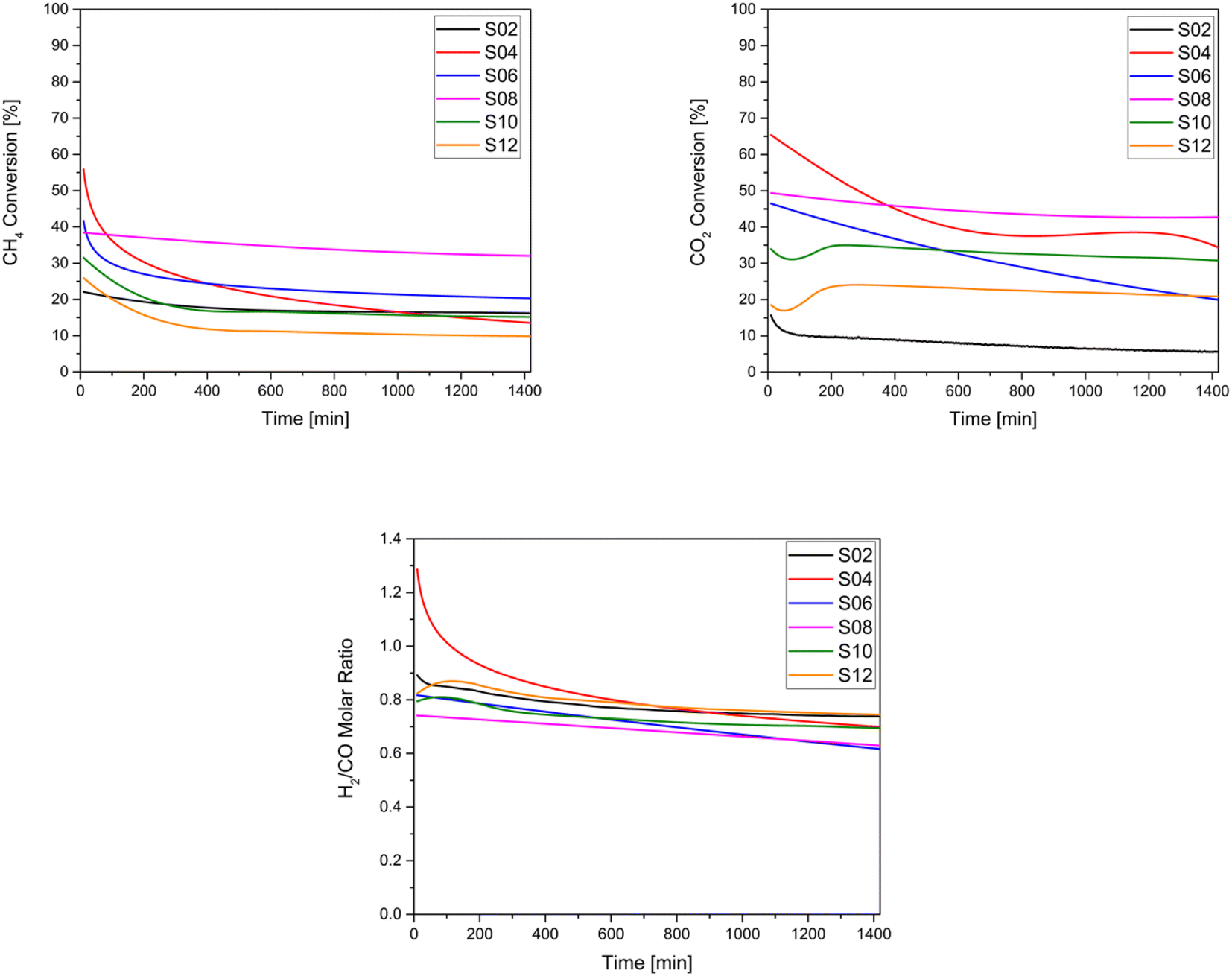 | ||
| Fig. 7 Catalytic performances of the studied catalysts: (a) CH4 conversion (b) CO2 conversion and (c) H2/CO molar ratio. | ||
The Ni dispersion and the corresponding Ni metallic surface area, as derived from the H2-pulse chemisorption, are presented in Table 2. The Ni dispersion values range from 0.2% to 2.7%, with corresponding Ni metallic surface areas ranging from 1.3 to 18.2 m2 gNi−1.
Similar low dispersion values in clay-based catalysts, as observed here, have been commonly reported in the literature. For example, Zhao et al. reported comparable values between 0.17 and 0.6 for a similar acid-treated catalyst.55 Moreover, the pulse chemisorption experiments suggest that the Ni metallic surface area might not be the sole factor governing the catalytic reaction. In fact, catalyst S12, with the highest Ni surface area of 18.2 m2 gNi−1, demonstrated poor catalytic performance and stability.
3.5 Catalyst activity and stability evaluation
Because DRM is an endothermic reaction, based on the Gibbs free energy, it requires a temperature in the range of 627–1000 °C and atmospheric pressure to approach equilibrium conversion, a reduction of carbon deposition, and stability.89 Here, the catalytic activity of the samples was evaluated at a temperature of 750 °C. For these tests, the conversion of CH4 and CO2 as well as the measurement of the H2/CO ratio were conducted. As illustrated in Fig. 7, it can be observed that CO2 conversion was generally higher than CH4 conversion for all samples, except for the untreated Ni-HNT (S2) and for S6 owing to their low basicity. Thus, the reverse water-gas shift reaction becomes favourable according to the following equation: CO2 + H2 ↔ H2O + CO. Amongst the catalysts, the overall highest CO2 and CH4 conversions were observed for S8 (ball-milled molten salt method), which also depicted remarkable stability over 24 hours of testing and was associated with negligible coke deposition. However, S12 (ground molten salt method) depicted the lowest CH4 conversion (10%) and the second lowest CO2 conversion (12%) after S2 owing to the lowest Ni loading (6 wt%, see Table 2) and potentially in the absence of micropores (see Table 1), which leads to a shorter residence time for the reactants. In general, all the treated catalysts outperformed the CO2 conversion recorded for untreated HNT S2. All the analysed catalysts resulted in an average value of 0.75 in terms of the H2/CO ratio (Fig. 7), as a consequence of the reverse water–gas shift reaction.According to Fig. 7, which presents the stability of the samples indicated by the decrease in CH4 and CO2 conversion, no apparent deactivation of S2 and S8 over a period of 24 hours was observed, as the catalysts maintained a stable CH4 and CO2 conversion. In contrast, a significant deactivation was largely observed for S4 and S6 (both acid treated), where the conversion dropped from 56% to 14% (CH4) and 65% to 34% (CO2) for S4, and from 42% to 20% (CH4) and 46% to 20% (CO2) for S6, after 24 h. The high initial conversion exhibited by the acid-treated samples S4 and S6 can be attributed to their relatively higher H2 consumption indicated by the H2-TPR, along with their overall higher Ni content. Conversely, samples S2 and S12, with lower Ni loading, showed the opposite trend. In general, samples with higher basicity, namely S8, S10, and S12, demonstrated significantly higher CO2 adsorption (see Table 2). Thus, they exhibited a negligible CO2 conversion drop (see Table 3), corresponding to −7%, −7%, and −0.04%, respectively. To summarise, all the alkali-treated materials appeared to be stable and superior to those supports treated with acids.
| Sample | CH4 conversion [%] | CO2 conversion [%] | Coke formation [%] | Average Ni particle size increases owing to sintering [nm] | ||
|---|---|---|---|---|---|---|
| t = 10 [min] | t = 1440 [min] | t = 10 [min] | t = 1440 [min] | |||
| S2 | 22.22 | 15.83 | 16.4 | 6.36 | 4.71 | +42 |
| S4 | 55.90 | 13.55 | 65.39 | 34.31 | 33.38 | +22 |
| S6 | 41.68 | 20.32 | 46.47 | 19.96 | 21.45 | +33 |
| S8 | 38.42 | 32.02 | 49.39 | 42.74 | 2.67 | +17 |
| S10 | 33.93 | 15.58 | 37.77 | 30.55 | 13.30 | +29 |
| S12 | 25.89 | 9.98 | 21.58 | 21.54 | 11.72 | +54 |
The performance testing also revealed that simple mechanical processes, such as ball-milling and grinding, have a strong influence on the textural properties and catalytic conversion of the final catalysts. In this regard, S8 (ball-milling process) outperformed S12 (grinding process) in terms of CO2 and CH4 conversion, and coke formation. In the future, the modification of catalytic supports, such as halloysites, should not be limited to only chemical treatments but also mechanical processes will certainly play a role in improving the catalytic performances.
3.6 Characterization of the spent catalysts
TGA and TEM analyses were performed to investigate the type and quantity of coke deposited on the catalysts, and the results are shown in Fig. 8 and 9, respectively. The weight % from the TGA analysis of the spent catalysts after 24 hours of DRM reaction at 750 °C is also reported in Table 3 in terms of coke formation %. No significant weight loss (>5%) was recorded for the fresh samples (Fig. S8†). For all the spent catalysts, only one significant drop in weight % was observed in all TGA profiles in the temperature range of 500–650 °C, indicating only one main type of carbon coke morphology deposited on the catalyst. In general, the number of weight losses in the TGA profile indicates the type of carbon, while the intensity of these signals shows the amount of formed coke. The temperature range at which the main loss occurs is associated with the temperature at which filamentous carbon forms. The weight loss percentages depicted in Fig. 8 are consistent with the stability exhibited by the catalysts in terms of CH4 and CO2 conversion drops. Samples showing negligible deactivation over 24 hours of time-on-stream are also those presenting the least coke formation, as highlighted by the minimum weight loss in the TGA profile (e.g., S2 and S8).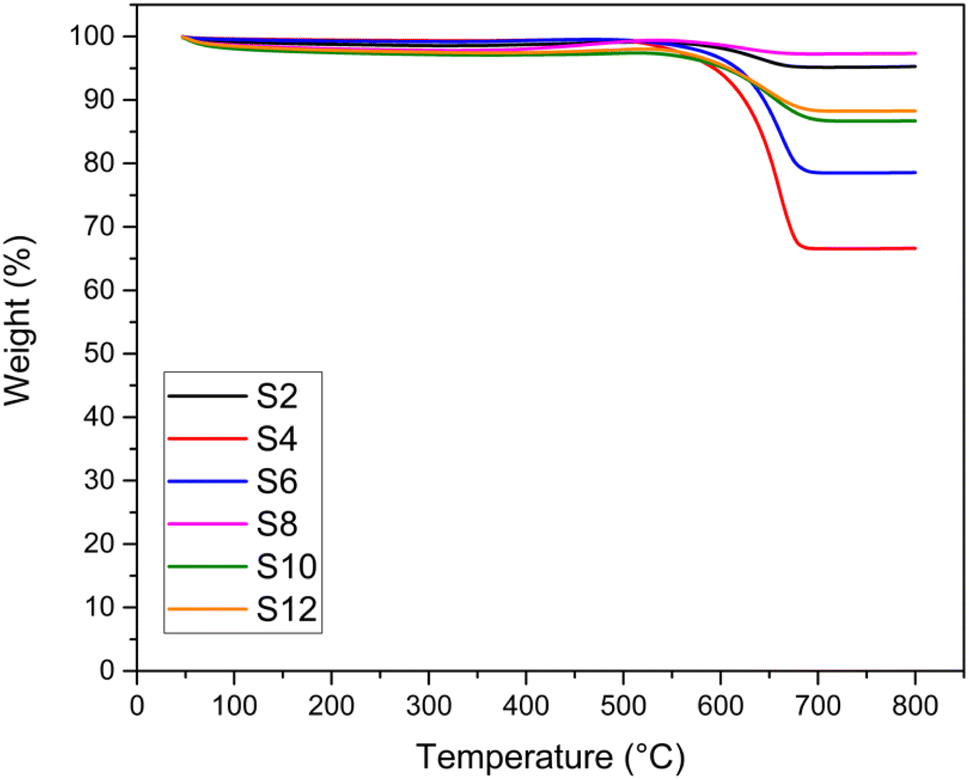 | ||
| Fig. 8 TGA analysis of the spent catalysts. | ||
 | ||
| Fig. 9 TEM images of spent catalysts: (a) S2 – Ni-raw HNT, (b) S4 – Ni-AHNT (HNO3), (c) S6 – Ni-AHNT (H2SO4), (d) S8 – Ni-AlHNT (ball milled), (e) S10 – Ni-AlHNT (NaOH), and (f) S12 – Ni-AlHNT (ground). | ||
TEM images (Fig. 9) of the spent catalysts revealed the formation of carbon filaments. Such filaments gradually accumulate on the support surface by the transformation of active surface carbons within the reaction time, eventually leading to catalyst deactivation. From the figures, it is difficult to estimate the amount of coke formed. However, the general trend seems more in favour of alkali-treated catalysts that show a lower amount of carbon deposited. Interestingly, the acid-treated samples (S4 and S6), with the presence of carbon filaments, also exhibit some amorphous carbon formations. This can also explain the drop in the corresponding TGA profile (Fig. 8) occurring at a temperature lower than that of the other catalysts.
TEM analysis also revealed a typical sintering effect on nickel (Table 3 and Fig. 9). All the spent catalysts showed the formation of large nickel nanoparticles up to 130 nm (Fig. S5†). From the corresponding particle size distribution (Table S2 and Fig. S7†), it can be observed that the mean particle size for spent catalysts ranged from 30 to 67 nm, with the alkali-treated samples characterized by a broader distribution.
The average Ni particle size analysis reveals that although S8 had the highest Ni particle size among the fresh samples, it exhibited the lowest nickel sintering, followed by S10. Conversely, the acid-treated samples S4 and S6, followed by the alkali ground sample, S12, showed a higher Ni sintering effect with respect to the original particle size. This further confirmed that grinding the HNT support after the molten salt treatment did not result in the desired effect otherwise demonstrated by the ball-milling process, which yielded a catalyst with superior resistance against coke formation and sintering. As expected, the raw HNT support reported the highest Ni sintering.
4. Conclusions
This work explored the use of aluminosilicate nanotubular clay for catalytic applications, with a focus on the dry reforming of methane. The study highlights the effect of different acid and alkali treatments on HNTs, revealing how these treatment methods affect the material's structure and properties. The reported results showed an enhanced surface area and more accessible active sites in acid-treated HNTs, resulting in higher Ni loading with a smaller particle size, which makes them versatile catalyst supports with higher initial activity. Moreover, alkali-treated HNTs exhibited enhanced CO2 adsorption capacity, resistance to coke deposition and to Ni sintering, and overall catalyst stability. This study employed various analytical techniques to characterize the treated HNTs, shedding light on their structural changes. The research also assesses their textural properties, reduction behaviour, basicity, and catalytic performance in DRM reactions. Ultimately, this work provides valuable insights into the potential applications of aluminosilicate nanotubular clay in catalysis and underlines the significance of treatment methods in tailoring its performance for specific applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge Qatar Environment and Energy Research Institute (QEERI, at Hamad Bin Khalifa University) for supporting this research. The authors would like also to thank the core lab staff at Hamad Bin Khalifa University (HBKU), namely Dr Akshath Raghu Shetty, Mr Janarthanan Ponraj, and Dr Ayman Samara for their material characterization support. The authors thank Ms Aliya Banu, Ms Mona Sawali, and Mr Ahmadyar Qureshi for their contribution as interns during the preliminary stages of the project. Open Access funding provided by the Qatar National Library.References
- H. M. Torres Galvis, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Iron particle size effects for direct production of lower olefins from synthesis gas, J. Am. Chem. Soc., 2012, 134(39), 16207–16215 CrossRef CAS PubMed
.
- R. M. Navarro, M. Pena and J. Fierro, Hydrogen production reactions from carbon feedstocks: fossil fuels and biomass, Chem. Rev., 2007, 107(10), 3952–3991 CrossRef CAS PubMed
- G. A. Olah, A. Goeppert, M. Czaun and G. S. Prakash, Bi-reforming of methane from any source with steam and carbon dioxide exclusively to metgas (CO–2H2) for methanol and hydrocarbon synthesis, J. Am. Chem. Soc., 2013, 135(2), 648–650 CrossRef CAS PubMed
- L. Mo, K. K. M. Leong and S. Kawi, A highly dispersed and anti-coking Ni–La 2 O 3/SiO 2 catalyst for syngas production from dry carbon dioxide reforming of methane, Catal. Sci. Technol., 2014, 4(7), 2107–2114 RSC
- J. Kehres, J. G. Jakobsen, J. W. Andreasen, J. B. Wagner, H. Liu, A. Molenbroek, J. Sehested, I. Chorkendorff and T. Vegge, Dynamical properties of a Ru/MgAl2O4 catalyst during reduction and dry methane reforming, J. Phys. Chem. C, 2012, 116(40), 21407–21415 CrossRef CAS
- J. F. Múnera, L. M. Cornaglia, D. V. Cesar, M. Schmal and E. A. Lombardo, Kinetic studies of the dry reforming of methane over the Rh/La2O3− SiO2 catalyst, Ind. Eng. Chem. Res., 2007, 46(23), 7543–7549 CrossRef
- J. Niu, X. Du, J. Ran and R. Wang, Dry (CO2) reforming of methane over Pt catalysts studied by DFT and kinetic modeling, Appl. Surf. Sci., 2016, 376, 79–90 CrossRef CAS
- L. Foppa, M.-C. Silaghi, K. Larmier and A. Comas-Vives, Intrinsic reactivity of Ni, Pd and Pt surfaces in dry reforming and competitive reactions: Insights from first principles calculations and microkinetic modeling simulations, J. Catal., 2016, 343, 196–207 CrossRef CAS
- B. Pawelec, S. Damyanova, K. Arishtirova, J. G. Fierro and L. Petrov, Structural and surface features of PtNi catalysts for reforming of methane with CO2, Appl. Catal., A, 2007, 323, 188–201 CrossRef CAS
- J. Guo, C. Xie, K. Lee, N. Guo, J. T. Miller, M. J. Janik and C. Song, Improving the Carbon Resistance of Ni-Based Steam Reforming Catalyst by Alloying with Rh: A Computational Study Coupled with Reforming Experiments and EXAFS Characterization, ACS Catal., 2011, 1(6), 574–582 CrossRef CAS
- R. Duarte, O. Safonova, F. Krumeich, M. Makosch and J. A. van Bokhoven, Oxidation state of Ce in CeO2-promoted Rh/Al2O3 catalysts during methane steam reforming: H2O activation and alumina stabilization, ACS Catal., 2013, 3(9), 1956–1964 CrossRef CAS
- L. Xu, H. Song and L. Chou, One-Pot Synthesis of Ordered Mesoporous NiO–CaO–Al2O3 Composite Oxides for Catalyzing CO2 Reforming of CH4, ACS Catal., 2012, 2(7), 1331–1342 CrossRef CAS
- J. L. Rogers, M. C. Mangarella, A. D. D'Amico, J. R. Gallagher, M. R. Dutzer, E. Stavitski, J. T. Miller and C. Sievers, Differences in the nature of active sites for methane dry reforming and methane steam reforming over nickel aluminate catalysts, ACS Catal., 2016, 6(9), 5873–5886 CrossRef CAS
- W. Tu, M. Ghoussoub, C. V. Singh and Y.-H. C. Chin, Consequences of surface oxophilicity of Ni, Ni-Co, and Co clusters on methane activation, J. Am. Chem. Soc., 2017, 139(20), 6928–6945 CrossRef CAS PubMed
- S. E. Evans, J. Z. Staniforth, R. J. Darton and R. M. Ormerod, A nickel doped perovskite catalyst for reforming methane rich biogas with minimal carbon deposition, Green Chem., 2014, 16(10), 4587–4594 RSC
- Y. Guo, J. Zou, X. Shi, P. Rukundo, Z.-j. Wang and A. Ni, CeO 2–CDC-SiC catalyst with improved coke resistance in CO 2 reforming of methane, ACS Sustainable Chem. Eng., 2017, 5(3), 2330–2338 CrossRef CAS
- N. Wang, W. Qian, W. Chu and F. Wei, Crystal-plane effect of nanoscale CeO 2 on the catalytic performance of Ni/CeO 2 catalysts for methane dry reforming, Catal. Sci. Technol., 2016, 6(10), 3594–3605 RSC
- K. Bakhtiari, A. S. Kootenaei, S. Maghsoodi, S. Azizi and S. M. T. Ghomsheh, Synthesis of high sintering-resistant Ni-modified halloysite based catalysts containing La, Ce, and Co for dry reforming of methane, Ceram. Int., 2022, 48(24), 37394–37402 CrossRef CAS
- T. Margossian, K. Larmier, S. M. Kim, F. Krumeich, A. Fedorov, P. Chen, C. R. Müller and C. Copéret, Molecularly Tailored Nickel Precursor and Support Yield a Stable Methane Dry Reforming Catalyst with Superior Metal Utilization, J. Am. Chem. Soc., 2017, 139(20), 6919–6927 CrossRef CAS PubMed
- R. Chai, G. Zhao, Z. Zhang, P. Chen, Y. Liu and Y. Lu, High sintering-/coke-resistance Ni@SiO2/Al2O3/FeCrAl-fiber catalyst for dry reforming of methane: one-step, macro-to-nano organization via cross-linking molecules, Catal. Sci. Technol., 2017, 7(23), 5500–5504 RSC
- X. Li, D. Li, H. Tian, L. Zeng, Z.-J. Zhao and J. Gong, Dry reforming of methane over Ni/La2O3 nanorod catalysts with stabilized Ni nanoparticles, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS
- H. Arandiyan, H. Dai, J. Deng, Y. Liu, B. Bai, Y. Wang, X. Li, S. Xie and J. Li, Three-dimensionally ordered macroporous La0.6Sr0.4MnO3 with high surface areas: Active catalysts for the combustion of methane, J. Catal., 2013, 307, 327–339 CrossRef CAS
- R. Alotaibi, Ni catalysts with different promoters supported on zeolite for dry reforming of methane., Appl Petrochem Res, 2015 DOI:10.1007/s13203-015-0117-y
- C. Wang, X. Jie, Y. Qiu, Y. Zhao, H. A. Al-Megren, S. Alshihri, P. P. Edwards and T. Xiao, The importance of inner cavity space within Ni@ SiO2 nanocapsule catalysts for excellent coking resistance in the high-space-velocity dry reforming of methane, Appl. Catal., B, 2019, 259, 118019 CrossRef CAS
- L. Li, S. He, Y. Song, J. Zhao, W. Ji and C.-T. Au, Fine-tunable Ni@porous silica core–shell nanocatalysts: Synthesis, characterization, and catalytic properties in partial oxidation of methane to syngas, J. Catal., 2012, 288, 54–64 CrossRef CAS
- S. Li, C. Zhang, Z. Huang, G. Wu and J. Gong, A Ni@ZrO2 nanocomposite for ethanol steam reforming: enhanced stability via strong metal–oxide interaction, Chem. Commun., 2013, 49(39), 4226–4228 RSC
- T. Xie, L. Shi, J. Zhang and D. Zhang, Immobilizing Ni nanoparticles to mesoporous silica with size and location control via a polyol-assisted route for coking- and sintering-resistant dry reforming of methane, Chem. Commun., 2014, 50(55), 7250–7253 RSC
- A. Rodriguez-Gomez, R. Pereñiguez and A. Caballero, Nickel Particles Selectively Confined in the Mesoporous Channels of SBA-15 Yielding a Very Stable Catalyst for DRM Reaction, J. Phys. Chem. B, 2018, 122(2), 500–510 CrossRef CAS PubMed
- Z. Bian, I. Y. Suryawinata and S. Kawi, Highly carbon resistant multicore-shell catalyst derived from Ni-Mg phyllosilicate nanotubes@silica for dry reforming of methane, Appl. Catal., B, 2016, 195, 1–8 CrossRef CAS
- C. Zhang, W. Zhu, S. Li, G. Wu, X. Ma, X. Wang and J. Gong, Sintering-resistant Ni-based reforming catalysts obtained via the nanoconfinement effect, Chem. Commun., 2013, 49(82), 9383–9385 RSC
- B. B. Lynch, B. D. Anderson, W. J. Kennedy and J. B. Tracy, Synthesis and chemical transformation of Ni nanoparticles embedded in silica, Nanoscale, 2017, 9(47), 18959–18965 RSC
- Y. Chai, Y. Fu, H. Feng, W. Kong, C. Yuan, B. Pan, J. Zhang and Y. Sun, A Nickel-Based Perovskite Catalyst with a Bimodal Size Distribution of Nickel Particles for Dry Reforming of Methane, ChemCatChem, 2018, 10(9), 2078–2086 CrossRef CAS
- G. Valderrama, A. Kiennemann, C. U. de Navarro and M. R. Goldwasser, LaNi1-xMnxO3 perovskite-type oxides as catalysts precursors for dry reforming of methane, Appl. Catal., A, 2018, 565, 26–33 CrossRef CAS
- S. Bhattar, A. Krishnakumar, S. Kanitkar, A. Abedin, D. Shekhawat, D. J. Haynes and J. J. Spivey, 110th Anniversary: Dry reforming of methane over Ni-and Sr-substituted lanthanum zirconate pyrochlore catalysts: Effect of Ni loading, Ind. Eng. Chem. Res., 2019, 58(42), 19386–19396 CrossRef CAS
- M. A. Naeem, P. M. Abdala, A. Armutlulu, S. M. Kim, A. Fedorov and C. R. Müller, Exsolution of Metallic Ru Nanoparticles from Defective, Fluorite-Type Solid Solutions Sm2RuxCe2–xO7 To Impart Stability on Dry Reforming Catalysts, ACS Catal., 2020, 10(3), 1923–1937 CrossRef CAS
- Y. Zhu, N. Jin, R. Liu, X. Sun, L. Bai, H. Tian, X. Ma and X. Wang, Bimetallic BaFe2MAl9O19 (M = Mn, Ni, and Co) hexaaluminates as oxygen carriers for chemical looping dry reforming of methane, Appl. Energy, 2020, 258, 114070 CrossRef CAS
- W. O. Alabi, K. O. Sulaiman, H. Wang, Y. Hu and C. Patzig, Effect of spinel inversion and metal-support interaction on the site activity of Mg-Al-Ox supported Co catalyst for CO2 reforming of CH4, J. CO2 Util., 2020, 37, 180–187 CrossRef CAS
- S. Das, J. Ashok, Z. Bian, N. Dewangan, M. Wai, Y. Du, A. Borgna, K. Hidajat and S. Kawi, Silica–Ceria sandwiched Ni core–shell catalyst for low temperature dry reforming of biogas: Coke resistance and mechanistic insights, Appl. Catal., B, 2018, 230, 220–236 CrossRef CAS
- J. Dou, R. Zhang, X. Hao, Z. Bao, T. Wu, B. Wang and F. Yu, Sandwiched SiO2@ Ni@ ZrO2 as a coke resistant nanocatalyst for dry reforming of methane, Appl. Catal., B, 2019, 254, 612–623 CrossRef CAS
- T. D. Gould, A. Izar, A. W. Weimer, J. L. Falconer and J. W. Medlin, Stabilizing Ni Catalysts by Molecular Layer Deposition for Harsh, Dry Reforming Conditions, ACS Catal., 2014, 4(8), 2714–2717 CrossRef CAS
- G. Wu, C. Zhang, S. Li, Z. Han, T. Wang, X. Ma and J. Gong, Hydrogen Production via Glycerol Steam Reforming over Ni/Al2O3: Influence of Nickel Precursors, ACS Sustainable Chem. Eng., 2013, 1(8), 1052–1062 CrossRef CAS
- Y. Cao, M. Lu, J. Fang, L. Shi and D. Zhang, Hexagonal boron nitride supported mesoSiO2-confined Ni catalysts for dry reforming of methane, Chem. Commun., 2017, 53(54), 7549–7552 RSC
- X. Zhao, M. Lu, H. Li, J. Fang, L. Shi and D. Zhang, In situ preparation of Ni nanoparticles in cerium-modified silica aerogels for coking- and sintering-resistant dry reforming of methane, New J. Chem., 2017, 41(12), 4869–4878 RSC
- J.-J. Shao, K. Raidongia, A. R. Koltonow and J. Huang, Self-assembled two-dimensional nanofluidic proton channels with high thermal stability, Nat. Commun., 2015, 6(1), 7602 CrossRef PubMed
- J. Im, H. Shin, H. Jang, H. Kim and M. Choi, Maximizing the catalytic function of hydrogen spillover in platinum-encapsulated aluminosilicates with controlled nanostructures, Nat. Commun., 2014, 5(1), 3370 CrossRef PubMed
- D.-Y. Kang, N. A. Brunelli, G. I. Yucelen, A. Venkatasubramanian, J. Zang, J. Leisen, P. J. Hesketh, C. W. Jones and S. Nair, Direct synthesis of single-walled aminoaluminosilicate nanotubes with enhanced molecular adsorption selectivity, Nat. Commun., 2014, 5(1), 3342 CrossRef PubMed
- E. Paineau, M.-E. M. Krapf, M.-S. Amara, N. V. Matskova, I. Dozov, S. Rouzière, A. Thill, P. Launois and P. Davidson, A liquid-crystalline hexagonal columnar phase in highly-dilute suspensions of imogolite nanotubes, Nat. Commun., 2016, 7(1), 10271 CrossRef CAS PubMed
- Y. Lin, X. Wang, J. Liu and J. D. Miller, Natural halloysite nano-clay electrolyte for advanced all-solid-state lithium-sulfur batteries, Nano Energy, 2017, 31, 478–485 CrossRef CAS
- L. Yu, H. Wang, Y. Zhang, B. Zhang and J. Liu, Recent advances in halloysite nanotube derived composites for water treatment, Environ. Sci.: Nano, 2016, 3(1), 28–44 RSC
- G. I. Fakhrullina, F. S. Akhatova, Y. M. Lvov and R. F. Fakhrullin, Toxicity of halloysite clay nanotubes in vivo: a Caenorhabditis elegans study, Environ. Sci.: Nano, 2015, 2(1), 54–59 RSC
- H. U. Hambali, A. A. Jalil, A. A. Abdulrasheed, T. J. Siang, Y. Gambo and A. A. Umar, Zeolite and clay based catalysts for CO2 reforming of methane to syngas: A review, Int. J. Hydrogen Energy, 2022, 47(72), 30759–30787 CrossRef CAS
- T. Tsoufis, F. Katsaros, B. J. Kooi, E. Bletsa, S. Papageorgiou, Y. Deligiannakis and I. Panagiotopoulos, Halloysite nanotube-magnetic iron oxide nanoparticle hybrids for the rapid catalytic decomposition of pentachlorophenol, Chem. Eng. J., 2017, 313, 466–474 CrossRef CAS
- X. Zhang, P. Wang, X. Wu, S. Lv and J. Dai, Application of MnOx/HNTs catalysts in low-temperature NO reduction with NH3, Catal. Commun., 2016, 83, 18–21 CrossRef CAS
- H. Dai, P. Yu, H. Liu, S. Xiong, X. Xiao, J. Deng and L. Huang, Ni-Based catalysts supported on natural clay of attapulgite applied in the dry reforming of methane reaction, New J. Chem., 2020, 44(37), 16101–16109 RSC
- Z. Zhao, P. Ren and W. Li, Supported Ni catalyst on a natural halloysite derived silica–alumina composite oxide with unexpected coke-resistant stability for steam-CO2 dual reforming of methane, RSC Adv., 2016, 6(55), 49487–49496 RSC
- Q. Wang, Y. Wang, Y. Zhao, B. Zhang, Y. Niu, X. Xiang and R. Chen, Fabricating roughened surfaces on halloysite nanotubes via alkali etching for deposition of high-efficiency Pt nanocatalysts, CrystEngComm, 2015, 17(16), 3110–3116 RSC
- Y. Wang, C. Liu, Y. Zhang, B. Zhang and J. Liu, Facile Fabrication of Flowerlike Natural Nanotube/Layered Double Hydroxide Composites as Effective Carrier for Lysozyme Immobilization, ACS Sustainable Chem. Eng., 2015, 3(6), 1183–1189 CrossRef CAS
- J. J. González, J. Da Costa-Serra and A. Chica, Biogas dry reforming over Ni–Ce catalyst supported on nanofibered alumina, Int. J. Hydrogen Energy, 2020, 45(40), 20568–20581 CrossRef
- C. Xie, C. Mak, X. Tao and F. Yan, Photodetectors Based on Two-Dimensional Layered Materials Beyond Graphene, Adv. Funct. Mater., 2017, 27(19), 1603886 CrossRef
- Y. Wang, N. Xu, D. Li and J. Zhu, Thermal Properties of Two Dimensional Layered Materials, Adv. Funct. Mater., 2017, 27(19), 1604134 CrossRef
- J. Kang, V. K. Sangwan, J. D. Wood and M. C. Hersam, Solution-Based Processing of Monodisperse Two-Dimensional Nanomaterials, Acc. Chem. Res., 2017, 50(4), 943–951 CrossRef CAS PubMed
- H. Zeng, C. Zhi, Z. Zhang, X. Wei, X. Wang, W. Guo, Y. Bando and D. Golberg, “White Graphenes”: Boron Nitride Nanoribbons via Boron Nitride Nanotube Unwrapping, Nano Lett., 2010, 10(12), 5049–5055 CrossRef CAS PubMed
- Y. Wang, D. Widmann, M. Heenemann, T. Diemant, J. Biskupek, R. Schlögl and R. J. Behm, The role of electronic metal-support interactions and its temperature dependence: CO adsorption and CO oxidation on Au/TiO2 catalysts in the presence of TiO2 bulk defects, J. Catal., 2017, 354, 46–60 CrossRef CAS
- I. Surya, K. Waesateh, A. Masa and N. Hayeemasae, Selectively etched halloysite nanotubes as performance booster of epoxidized natural rubber composites, Polymers, 2021, 13(20), 3536 CrossRef CAS PubMed
- S. Zeng, C. Reyes, J. Liu, P. A. Rodgers, S. H. Wentworth and L. Sun, Facile hydroxylation of halloysite nanotubes for epoxy nanocomposite applications, Polymer, 2014, 55(25), 6519–6528 CrossRef CAS
- M. Lu, J. Fang, L. Han, K. Faungnawakij, H. Li, S. Cai, L. Shi, H. Jiang and D. Zhang, Coke-resistant defect-confined Ni-based nanosheet-like catalysts derived from halloysites for CO 2 reforming of methane, Nanoscale, 2018, 10(22), 10528–10537 RSC
- Z. Zhao, P. Ren and W. Li, Supported Ni catalyst on a natural halloysite derived silica–alumina composite oxide with unexpected coke-resistant stability for steam-CO 2 dual reforming of methane, RSC Adv., 2016, 6(55), 49487–49496 RSC
- B. Fung, A. Khitrin and K. Ermolaev, An improved broadband decoupling sequence for liquid crystals and solids, J. Magn. Reson., 2000, 142(1), 97–101 CrossRef CAS PubMed
- S. Sadjadi, Halloysite-based hybrids/composites in catalysis, Appl. Clay Sci., 2020, 189, 105537 CrossRef CAS
- M. Massaro, C. G. Colletti, G. Lazzara, S. Milioto, R. Noto and S. Riela, Halloysite nanotubes as support for metal-based catalysts, J. Mater. Chem. A, 2017, 5(26), 13276–13293 RSC
- M. Massaro, R. Noto and S. Riela, Halloysite Nanotubes: Smart Nanomaterials in Catalysis, Catalysts, 2022, 12(2), 149 CrossRef CAS
- Z. Asadi, S. Sadjadi, M. Nekoomanesh-Haghighi and N. Bahri-Laleh, Effects of acid-treatment of halloysite on the characteristics and catalytic performance of palladated halloysite in lubricants hydrogenation reaction, Inorg. Chem. Commun., 2022, 140, 109438 CrossRef CAS
- Z. Zhao, J. Ran, Y. Jiao, W. Li and B. Miao, Modified natural halloysite nanotube solely employed as an efficient and low-cost solid acid catalyst for alpha-arylstyrenes production via direct alkenylation, Appl. Catal., A, 2016, 513, 1–8 CrossRef CAS
- M. Lu, J. Fang, L. Han, K. Faungnawakij, H. Li, S. Cai, L. Shi, H. Jiang and D. Zhang, Coke-resistant defect-confined Ni-based nanosheet-like catalysts derived from halloysites for CO2 reforming of methane, Nanoscale, 2018, 10(22), 10528–10537 RSC
- F. Ferrante, M. Bertini, C. Ferlito, L. Lisuzzo, G. Lazzara and D. Duca, A computational and experimental investigation of halloysite silicic surface modifications after alkaline treatment, Appl. Clay Sci., 2023, 232, 106813 CrossRef CAS
- E. Abdullayev, A. Joshi, W. Wei, Y. Zhao and Y. Lvov, Enlargement of halloysite clay nanotube lumen by selective etching of aluminum oxide, ACS Nano, 2012, 6(8), 7216–7226 CrossRef CAS PubMed
- C. Cheng, W. Song, Q. Zhao and H. Zhang, Halloysite nanotubes in polymer science: Purification, characterization, modification and applications, Nanotechnol. Rev., 2020, 9(1), 323–344 CrossRef
- A. F. Peixoto, A. C. Fernandes, C. Pereira, J. Pires and C. Freire, Physicochemical characterization of organosilylated halloysite clay nanotubes, Microporous Mesoporous Mater., 2016, 219, 145–154 CrossRef CAS
- K. A. Smith, R. J. Kirkpatrick, E. Oldfield and D. M. Henderson, High-resolution silicon-29 nuclear magnetic resonance spectroscopic study of rock-forming silicates, Am. Mineral., 1983, 68(11–12), 1206–1215 CAS
- E. Lippmaa, M. Maegi, A. Samoson, G. Engelhardt and A. R. Grimmer, Structural studies of silicates by solid-state high-resolution silicon-29 NMR, J. Am. Chem. Soc., 1980, 102(15), 4889–4893 CrossRef CAS
- R. F. Moran, D. M. Dawson and S. E. Ashbrook, Exploiting NMR spectroscopy for the study of disorder in solids, Int. Rev. Phys. Chem., 2017, 36(1), 39–115 Search PubMed
- J. A. Torres-Luna, G. I. Giraldo-Gómez, N. R. Sanabria-González and J. G. Carriazo, Catalytic degradation of real-textile azo-dyes in aqueous solutions by using Cu–Co/halloysite, Bull. Mater. Sci., 2019, 42(4), 137 Search PubMed
- S. Kadi, S. Lellou, K. Marouf-Khelifa, J. Schott, I. Gener-Batonneau and A. Khelifa, Preparation, characterisation and application of thermally treated Algerian halloysite, Microporous Mesoporous Mater., 2012, 158, 47–54 CrossRef CAS
- H. Wu, J. Zhang and Z. Zhou, in Effect of heat treatment on structure, morphology and surface properties of halloysite, IOP Conference Series: Materials Science and Engineering, IOP Publishing, 2019, p. 012111 Search PubMed
- P. Yuan, D. Tan, F. Aannabi-Bergaya, W. Yan, M. Fan, D. Liu and H. He, Changes in structure, morphology, porosity, and surface activity of mesoporous halloysite nanotubes under heating, Clays Clay Miner., 2012, 60(6), 561–573 CrossRef CAS
- T. Lin, S. Zhao, S. Niu, Z. Lyu, K. Han and X. Hu, Halloysite nanotube functionalized with La-Ca bimetallic oxides as novel transesterification catalyst for biodiesel production with molecular simulation, Energy Convers. Manage., 2020, 220, 113138 CrossRef CAS
- T. Wu, S. Li and Q. Yan, Effect of Interaction between Nickel and Alumina on Coke Deposition on Ni/Al∼ 2O∼ 3 Catalyst for Partial Oxidation of Methane to Syngas, Chin. J. Catal., 2001, 22(5), 504–506 Search PubMed
- L. Zhou, L. Li, N. Wei, J. Li and J. M. Basset, Effect of NiAl2O4 formation on Ni/Al2O3 stability during dry reforming of methane, ChemCatChem, 2015, 7(16), 2508–2516 CrossRef CAS
- F. Sharifianjazi, A. Esmaeilkhanian, L. Bazli, S. Eskandarinezhad, S. Khaksar, P. Shafiee, M. Yusuf, B. Abdullah, P. Salahshour and F. Sadeghi, A review on recent advances in dry reforming of methane over Ni-and Co-based nanocatalysts, Int. J. Hydrogen Energy, 2022, 47(100), 42213–42233 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07990b |
| This journal is © The Royal Society of Chemistry 2024 |