
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen storage efficiency of Fe doped carbon nanotubes: molecular simulation study†
Bita Baghaia and
Sepideh Ketabi *b
*b
aDepartment of Applied Chemistry, Faculty of Pharmaceutical Chemistry, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran
bDepartment of Chemistry, Faculty of Pharmaceutical Chemistry, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran. E-mail: sepidehketabi@yahoo.com; s.ketabi@iautmu.ac.ir
First published on 22nd March 2024
Abstract
Given that adsorption is widely regarded as a favorable technique for hydrogen storage, it is appropriate to pursue the development of suitable adsorbent materials for industrial storage. This study aimed to assess the potential of Fe-doped carbon nanotubes (FeCNT) as a remarkable material for hydrogen storage. The structures of pure and Fe-doped CNTs were optimized based on quantum mechanical calculations using density functional theory (DFT) with the Perdew–Burke–Ernzerhof (PBE) method. To gain a comprehensive understanding of the adsorption behavior, Monte Carlo simulation was employed to investigate the adsorption of hydrogen molecules on FeCNT. The study specifically examined the impact of temperature, pressure, and hydrogen mole percentage on the adsorption capacity of FeCNT. The findings indicated that the uptake of hydrogen increased as the pressure increased, and when the pressure exceeded 5 MPa, FeCNT reached a state of near saturation. At room temperature and pressures of 1 and 5 MPa, the hydrogen capacities of FeCNT were determined to be 1.53 and 6.92 wt%, respectively. The radial distribution function diagrams confirmed the formation of a one-layer adsorption phase at pressures below 5 MPa. A comparison of the temperature dependence of hydrogen adsorption between FeCNT and pure CNT confirmed the effectiveness of Fe doping in hydrogen storage up to room temperature. FeCNT exhibited a greater reduction in initial hydrogen capacity at temperatures above room temperature. To evaluate the safety of the system, the use of N2 as a dilution agent was investigated by examining the hydrogen uptake of FeCNT from pure and H2/N2 mixtures at 300 K. The results showed that the addition of N2 to the environment had no significant effect on FeCNT hydrogen storage at pressures below 4 MPa. Furthermore, the study of H2 selectivity from the H2/N2 mixture indicated that FeCNT demonstrated a preference for adsorbing H2 over a wide range of bulk mole fractions at pressures of 4 and 5 MPa, suggesting that these pressures could be considered optimal. Under these conditions, Fe doping can offer an efficient and selective adsorption surface for hydrogen storage.
1. Introduction
Due to excessive consumption of non-renewable energy sources such as fossil fuels in the past few decades, this energy source is expected to end abruptly. Since fossil fuels cause excessive emission of greenhouse gases, they have a large contribution to the problem of global warming and climate change.1 Therefore, due to these problems and the reduction of non-renewable energy reserves, it is necessary to improve new technologies for renewable energy sources. One of the newest renewable energy sources is the technology based on hydrogen energy.Hydrogen is a useful clean energy carrier and has attracted much attention in science and technology.2 Therefore, an effective, safe, and inexpensive system with high hydrogen storage capacity, at temperatures from ambient to 100 °C and pressures below 100 bar is fundamental in practical applications.3–6 To achieve these conditions, it is better to use the interaction of hydrogen with solids instead of compressed or liquid hydrogen (which needs high pressures or low temperatures).2,6
Suitable experimental and theoretical investigations have been stated to attain extremely effective hydrogen storage materials, containing several nanotubes,7 metals, and organic functional groups.8 The hydrogen storage ability of carbon nanotubes (CNTs) is a vital research topic that has attracted worldwide attention.9
Among several potential solid adsorbents, CNTs, which are one-dimensional tubular carbon allotropes, were considered due to their significant surface area, porous structure, and tremendous thermal and chemical stability.10 CNTs, originally developed and reported by S. Iijima,11 are made of rolled graphene sheets.12 Due to their unique structural, electronic, and mechanical properties, CNTs have significant potential in chemical and biological applications.13 Since CNTs have substantial physical and chemical properties, they have numerous applications in nanoelectronic devices and optoelectronics,14 FETs, LEDs,15 polymer composites,16,17 chemical sensors and biosensors,18,19 etc.
The famous properties of CNTs related to binding with hydrogen molecules have proven their good adsorption properties in environmental conditions.
Despite the ability of CNTs in hydrogen energy storage, they are reported to show very low hydrogen uptake capacities,20 which is due to their weak van der Waals interaction with hydrogen.21
Several approaches have been applied to improve low hydrogen adsorption of CNTs. One of the effective methods in this context is doping with other atoms,22,23 which induces defects in the nanotubes. Doping is an approach used to modify the properties of materials and involves introducing impurities or foreign atoms into a material to alter its properties. This technique can be utilized to enhance conductivity, modify band gaps, or induce magnetic properties.24,25
By introducing heteroatoms into the structure of nanotubes, the doping process can modify their properties, enhance their functionality, and induce defects in their structure.
Defects play a significant role in determining the properties and applications of materials. They range from doping in semiconductors to conductivity in mixed ionic–electronic conductors used in batteries, as well as active sites in catalysts.26–28 The disruption of translational symmetry at a defect site induces changes in the local degrees of freedom, which can be classified into configurational, vibrational, spin, and electronic terms. Metal doping affects the structure of carbon nanotubes by introducing changes in their morphological characteristics, compositions, and surface functionality. When doping nanotubes, impurities can be intercalated inside a nanotube, in the space between separate nanotubes, or can substitute one of the atoms of a nanotube. This localization of impurities within the nanotube or between separate nanotubes reduces the likelihood of their migration.
Previous research has also reported the enhancement of H2 absorption and storage in Vanadium oxide nanotube (VONT) by doping some transition metals.23,29 Experimental studies have shown that chemical functionalization and metal doping can improve the hydrogen adsorption capacity of CNTs.30 Defects on the CNT surface have induced the caps to open and thus increase the active sites for the adsorption of hydrogen molecules.31
During the synthesis of CNTs, different factors affect the hydrogen adsorption capacity of CNTs such as synthesis conditions, impurity, and catalyst substances. The metal contaminations in CNT can affect the storage capacity.32 It has been shown that transition metal loading to the carbon structure could improve hydrogen uptake33 of the carbon structures.34 Changes of CNT structures by transition metal doping35–38 and transition metal oxide nanoparticle39–42 have been also reported to enhance H2 adsorption capacity. Numerous studies have demonstrated that the addition of metals such as Ni, Pd, Fe, Pt, Co, and V to carbon structures enhances their hydrogen storage capacity.26,38,39,43–46 Doping carbon nanotubes with Ti at different locations enhances their structural stability, thermodynamic parameters, and hydrogen adsorption.33 Experimental evidence has shown that CNTs grown on Fe, V, Co, and Ni catalysts exhibit H2 adsorption capacities ranging from 2.76 to 5.25 wt%, with CNTs grown on Fe catalyst displaying the highest H2 uptake.47 Recent findings revealed that Fe/CNTs have a higher hydrogen storage capacity at room temperature compared to Co/CNTs.46 Studies on the deposition of Al atoms on Fe-doped CNTs indicated that the Fermi level and energy gap of the CNTs decrease after doping with Fe atoms. Fe doping also disrupts the original electron transfer property of the CNTs, potentially leading to an enhanced interaction with gas molecules.48 As a transition metal, Fe provides active sites that surpass those of C,49 and can form stable chemical structures with a variety of reagents. Furthermore, Fe has a work function similar to that of CNTs, resulting in low contact resistance with CNTs.50 Therefore, it can be concluded that Fe is a suitable metal dopant for modifying and improving the gas adsorption properties of CNTs.51
Moreover, it has indicated that the hydrogen storage of CNTs was influenced by the applied catalyst during the synthesis.47 The CNTs that grow on Fe have shown maximum hydrogen storage capacities.
Recently, effect of Ti and N doping on the hydrogen adsorption abilities of two-dimensional carbon layers have been investigated.52 The outcomes indicated that the hydrogen storage potentials of graphene nano layers can be improved by increasing the concentration of Ti atoms.
Ab initio molecular dynamics (AIMD) using the local density approximation has been employed to simulate the behavior of trimethyl indium (CH3)3In (TMIn) on graphene supported by SiC in an H2 atmosphere and on freestanding graphene. The purpose of this endeavor was to acquire knowledge about the reaction dynamics involved in the supply of In atoms to the graphene surface.53 The focal point of the study was a model that encompassed the zero-layer graphene exposed to TMIn and hydrogen molecules. Through the simulations, the atomistic pathways for TMIn transformations were unveiled, ultimately resulting in the formation of isolated In or InH species on the graphene surface. In particular, it was observed that TMIn exhibited heightened reactivity in the presence of H2, thus confirming the chemical affinity between In and hydrogen species.
In another recent study, Fe-doped carbon aerogels with different percentages of iron atoms were synthesized.54 The results indicated that increasing Fe content induced a decrease in hydrogen adsorption. However, Fe-doped activated carbon aerogels showed enhanced hydrogen storage due to the formation of CNTs in the matrix.
Transition metal (Fe, Ni, and Cu) doping can affect the electrocatalytic activity of the M-Co2P/NCNTs hybrid catalysts for hydrogen evolution reaction. The results have revealed that Fe doping resulting a large adsorption surface and active sites due to the large surface area and greatest partial charge of Fe after doping.55
Os-doping and Fe-doping have been applied to enhance the effectiveness of (9,9) CNT for hydrogen adsorption.56 Electron energy loss functions and the optical adsorption coefficients of the nanotubes have been computed by density functional theory (DFT) method and the result of metal doped CNTs have been compared with pristine CNT. It has been concluded that transition metal doping can increase the hydrogen adsorption ability of CNTs. An enhancement in adsorption efficiency was attained due to the doping of Fe and Os atoms, which overcomes the weak interactions between pristine CNT and hydrogen molecules. In this investigation, quantum mechanical calculations were utilized to compare the adsorption of two H2 molecules on pure and metal-doped CNTs.56 The efficacy of H2 adsorption was examined by analyzing the interactions between light and the studied systems, accomplished by determining the complex dielectric functions. As stated, Fe doping of pristine CNTs enhanced the power efficiency of H2 energy storage in single-walled CNTs. To assess the H2 adsorption capacity of CNTs under different temperature and pressure conditions in an H2 environment (where the number of H2 molecules is proportional to the pressure and temperature), either experimental methods or molecular simulation calculations can be employed.
As previously mentioned, experimental studies on hydrogen adsorption of Fe-decorated CNTs have confirmed an improvement in the hydrogen adsorption of CNTs.38 The hydrogen storage capability of the CNTs has been evaluated using volumetric and electrochemical techniques under ambient pressure. To determine the optimal conditions of temperature and pressure for hydrogen adsorption capacities, computer simulation methods were applied to these CNTs in our study. Particularly, the investigation focused on the hydrogen adsorption capacities from mixtures of gases, as the addition of N2 to the system dilutes the hydrogen environment and enhances safety, which is more favorable for fuel cell applications. Our findings contribute to a better understanding of the fundamental principles and optimal conditions for H2 storage in CNTs, thus shedding light on the rational design of nanoarchitectures for energy storage.
Due to the limitations of experimental conditions, simulation methods are often employed to elucidate microscopic information that cannot be obtained or observed through experiments. By analyzing the acquired information within a wide range that is not easily accessible in practical settings, theoretical calculations have been conducted to investigate the adsorption characteristics of the adsorbent.
The objective of the present research is to assess the impact of Fe doping on the H2 adsorption of carbon nanotubes. Due to the challenges associated with experimentally characterizing the structure properties of the adsorbent, molecular simulation can be employed to determine the optimal adsorption conditions. Molecular simulation allows for the evaluation of selectivity and the relationship between the adsorption properties and the structure. Therefore, our study has organized various relevant variables to predict the optimal conditions for hydrogen storage.
Monte Carlo (MC) simulation method was implemented to compare the absorption capacity of Fe doped CNT (FeCNT) and pristine CNTs. Hydrogen uptake from both pure environments and mixtures of H2 and N2 in Fe-doped CNTs was anticipated. By evaluating the effects of temperature and pressure on the hydrogen adsorption of FeCNT and pristine CNTs, the optimal conditions for hydrogen storage were determined. The radial density profile of the adsorbed phase was evaluated. Additionally, the effects of bulk pressure and composition on hydrogen selectivity from gas mixtures were defined.
2. Computational details
2.1 Quantum mechanics
Optimization of pure and Fe doped CNTs was carried out by quantum mechanical calculations. (9,9) CNT with Fe doped at 20 wt% level was considered as an absorbent in hydrogen adsorption. FeCNT was obtained by replacing the C atom in the CNT structure. Characteristics of studied FeCNT were stoichiometry of Fe14C256, length of 18.51 Å, and a diameter of 14.15 Å.Different percentages of Fe loading have been utilized in several studies. In a recent investigation, Fe7%-CNT has been employed to catalyze the oxygen reduction reaction.57 Similarly, in another study, composites of multi-walled CNTs doped with 8 wt% Fe were extensively used to enhance the electromagnetic interference (EMI) shielding effectiveness (SE) of CNTs. The Fe concentrations in the composites were measured at 0.08 wt%.58 Additionally, Fe-doped carbon aerogels were synthesized with Fe doping at different dopant levels of 1, 5, and 10%. These aerogels exhibited a hydrogen uptake of 3.8 wt%.54 Consequently, considering the existence of prior studies on hydrogen absorption involving lower percentages of Fe doping, our focus was on investigating the synergistic and symmetrical effects surrounding the nanotube. To this end, we conducted hydrogen storage studies on a doped nanotube with a 5 mole percent Fe doping. In future investigations, the authors plan to employ varying metal percentages to elucidate the impact of Fe dopant percentage on the hydrogen storage properties of CNTs.
It is important to acknowledge that the stoichiometry of metal doped CNTs in actual experimental studies might differ from that of modeling studies. Nevertheless, these models should be taken into consideration of other experimental or computational evidence. Moreover, it should be noted that, like our research, doping has been employed in other studies as well.51,56,59
In one particular study, in order to achieve a notable doping effect, Os- or Fe-doped CNT models were obtained by replacing the C atoms in the CNT structure.56 In the study of CO59 and SO2 (ref. 51) adsorption properties of Fe-doped CNTs, the same approach as in our current research was adopted. Fe–CNT was also constructed by replacing an intrinsic CNT with a Fe atom, and the adsorption energies were determined through quantum mechanical calculations.
In the case of such dopants, they constitute an inherent part of the matrix lattice, thereby resolving the issue of dopant clustering. However, in other systems, this doping method may result in clustering or the formation of more intricate systems. This conclusion is illustrated by the storage of Li in graphene, where the separation of Li phases leads to significant capacity limitations.60 Li loading results in a strong repulsion between Li ions at neighboring hexagons.61 Additionally, the positive lithiation energy of graphene implies that the Li adatoms on it should aggregate into clusters and eventually form macroscopic dendrites, instead of forming any stable Li-graphene mixture phase. First-principles computations have shown that the doping of pristine graphene with Li can be stabilized by employing the layered C3B compound, which holds promise as a storage medium at defected adsorption sites. In the case of Li, since it donates its 2s electron to the matrix, an electron-deficient matrix, such as B-substituted C, can better accommodate the extra electrons. However, in the case of transition metals, due to the presence of electrons in the d orbitals, this issue is slightly different. The presence of empty d orbitals enables a direct and stable interaction with C, leading to the creation of defect sites in doping sites.62 It was shown that the CNT doped with Ni resulting in electronic charge transfer and formation of Ni–C bonds at the interface between these two materials.63 In the current study, we employed the generalized-gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) method as the exchange–correlation functional.64,65 To achieve a balance between computational effort and accuracy, the Brillouin zone sampling, 1 × 1 × 3 Monkhorst–Pack mesh was used for the k-point summation. Spin-unrestricted DFT calculations were accomplished for (9,9) CNT and FeCNT. The QUANTUM ESPRESSO package66 was utilized for computations. In the next simulation step, optimized geometries were employed.
2.2 Monte Carlo simulation
Due to the limitation of experimental conditions, simulation methods are usually used for determining microscopic information that cannot be obtained or observed in experiments. By analyzing the adsorption capacities, distribution of gas molecules, selectivity, and other information in a wide range that is not easily accessible in practice, theoretical calculations have been performed to study the adsorption characteristics of adsorbent. To explore the adsorption of H2 molecules in FeCNT, MC simulations were employed at the pressures 0.1–10 MPa and temperatures 75–350 K.The MC simulations were accomplished in a cubic box with the size of 50 Å at the experimental pressures and temperatures applying the Metropolis sampling algorithm.67 The alignment of the nanotube was placed along the z-axis. The Virial equation of state with second and third coefficients was applied to compute gas molecule densities at the studied temperatures and pressures.68 Compression factor (Z) as a function of temperature and pressure is illustrated in the following formula:68
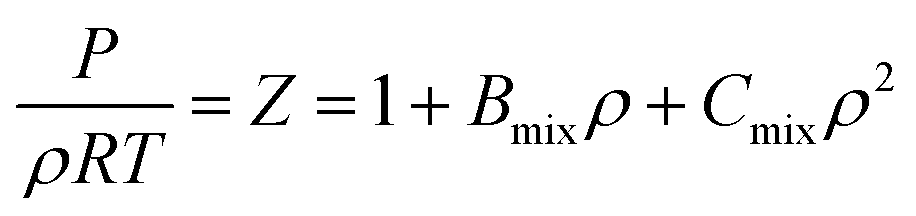 | (1) |
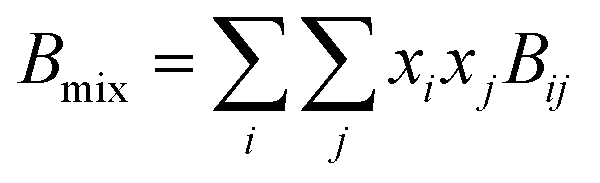 | (2) |
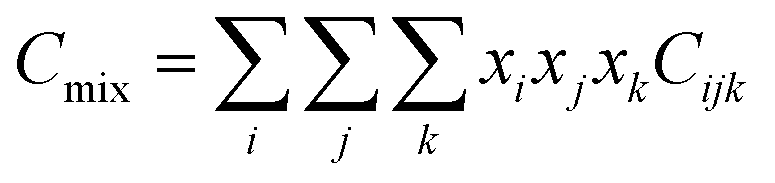 | (3) |
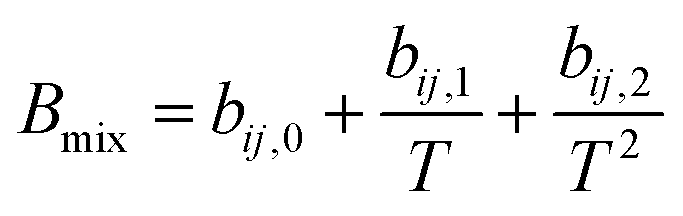 | (4) |
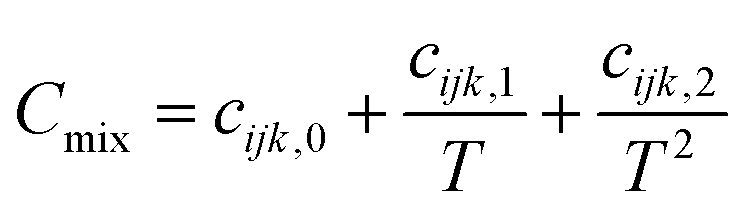 | (5) |
The appropriate coefficients were applied in eqn (4) and (5).68,69
New configurations were created by randomly selecting a molecule and translating it in three dimensions. Periodic boundary conditions were employed in three dimensions.
A reasonable range of translations has been used to achieve a 50% acceptance rate for the produced configurations.
The required configurations of the system were established to calculate the equilibrium properties. Each point is the result of 108 attempts to move. One molecule was picked and displaced randomly in three dimensions x, y, and z. An acceptance rate of 50% for new configurations was achieved by using suitable ranges for translations. The central axis of the nanotube is located along the z-axis.
The initial 5 × 107 cycles were applied for equilibrium and the second 5 × 107 cycles were used to verify ensemble averages. Periodic boundary conditions were applied in all three dimensions and a reflection plane.
The interactions of atoms included bonding parts (between chemically bonded atoms) and non-bonding parts (between any pair of atoms). To calculate the non-bonding interaction energies between the gas–CNT and gas–gas, Lennard-Jones potential function was applied:
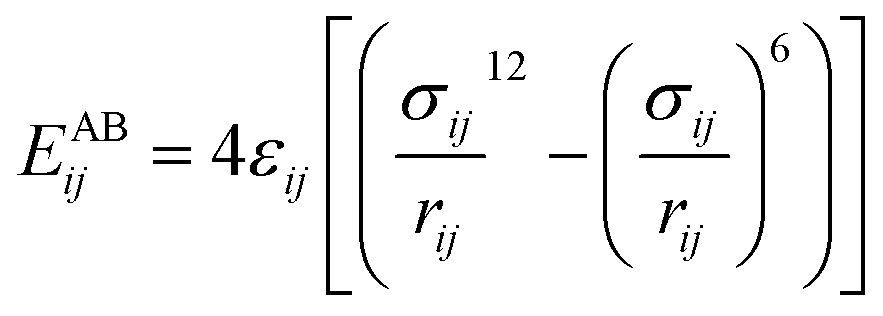 | (6) |
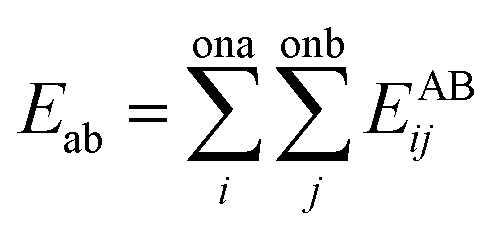 | (7) |
The site–site Lennard-Jones parameters εij and σij for the cross-interaction were obtained according to Lorenz–Berthelot (LB) combining rule75 (the interacting sites i and j are from different species):
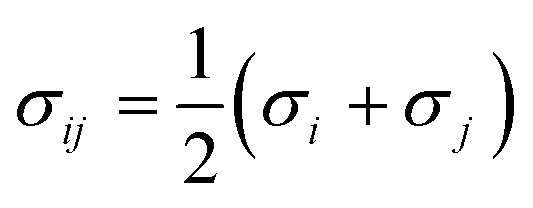 | (8) |
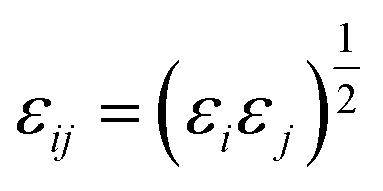 | (9) |
Lennard-Jones cross-interaction parameters are indicated in Table 1.
| Cross interaction | εij (kcal mol−1) | σij (Å) |
|---|---|---|
| H2–Fe | 0.8210 | 2.640 |
| H2–C | 0.0764 | 3.230 |
| N2–Fe | 0.8160 | 2.820 |
| N2–C | 0.0759 | 3.410 |
The Lennard-Jones potential between each pair falls off very rapidly with distance and at 2.5σ the intermolecular interactions have just 1% of its value at σ.76 Therefore, A spherical cut-off distance of 7.5 Å was deployed to the potential interactions between hydrogen molecules.
3. Results and discussion
3.1 Quantum mechanics
After full structural optimizations, Fe-doping causes significantly large distortion in CNT (Fig. 1). Fe atoms move towards the outside of the tube diameter and visible local geometric distortion occurs around the Fe atoms after substituting the Fe atom for the C atom. This is mainly attributed to the difference between the atomic radius of Fe and C. The average Fe–C bond lengths in FeCNT are 1.96 Å, which are significantly larger than the C–C bonds (1.420 Å) of pristine CNT. The increase in the bond lengths combined with the difference in bond angles forces the Fe atoms to protrude outwardly from the nanotube surface.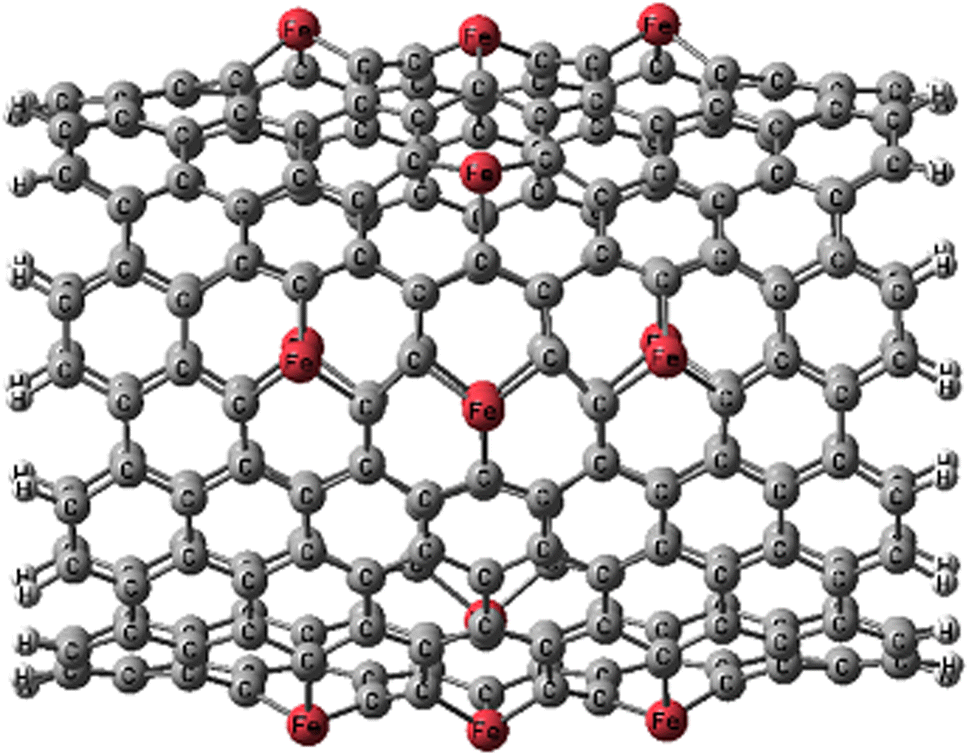 | ||
| Fig. 1 Optimized structure of FeCNT. | ||
The formation energy for Fe-doping into the CNT was defined as
| EF = (EFeCNT − ECNT) − n × (EFe − EC) | (10) |
3.2 Monte Carlo simulation
It has been verified that metal doping can modify the electronic structure, adsorption properties, and activity of surfaces.82 To evaluate the effect of Fe doping on the adsorption properties of CNT, H2 adsorption capacity and the adsorption isotherms of FeCNT were determined by the Monte Carlo simulation method.To evaluate nanotube gas uptake, the adsorption capacity can be an important consideration. Gas adsorption can be assessed in terms of gravimetric adsorption capacity at different temperatures and pressures.
The gravimetric adsorption capacity of pure gases in a nanotube (ρw or wt) is determined by the following equation:83,84
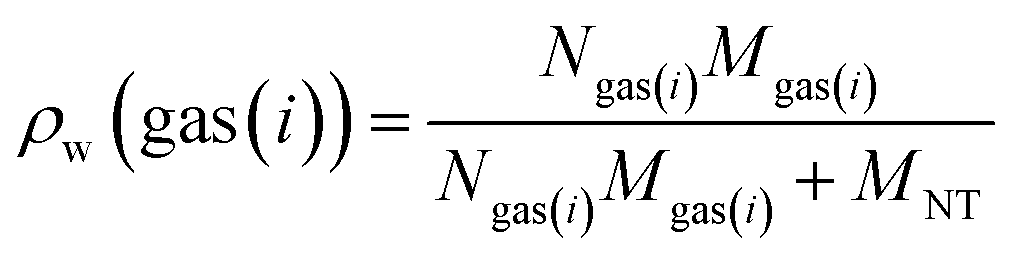 | (11) |
The gravimetric gas (i) adsorption capacity from a mixture of gases are determined by the following equation:
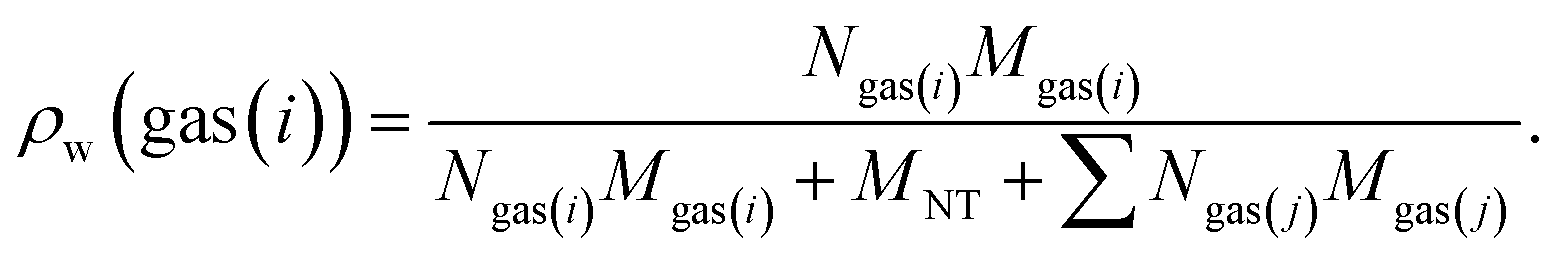 | (12) |
Adsorption capacities of FeCNT from pure and mixture of H2/N2 were determined in different temperatures and pressures. The outcomes have been described in percent adsorption capacity (wt%).
The H2 adsorption of FeCNT was calculated up to 10 MPa at different temperatures. Fig. 2 compares the gravimetric adsorption isotherms of H2 on FeCNT under different pressures within the temperature range of 175–350 K. As can be seen, hydrogen adsorption capacity is a function of the pressure and temperature. At lower pressures, hydrogen uptakes are similar at different temperatures. As anticipated, hydrogen adsorption capacities reduced with increasing temperature, while an increase in adsorptions was observed followed by a pressure increase. As the pressure increases, the FeCNT can adsorb significantly higher amounts of H2. At 175 and 200 K, and pressures larger than 5 MPa, hydrogen capacity decreases (and becomes constant) due to the high gas density in low temperatures and high pressures which produce a saturated phase. As the bulk pressure increases, adsorption (at low temperatures) is not favorable due to the endothermic process (which leads to particle–particle repulsion) and entropy reduction. Hence, a reduction in adsorption capacity is observed.
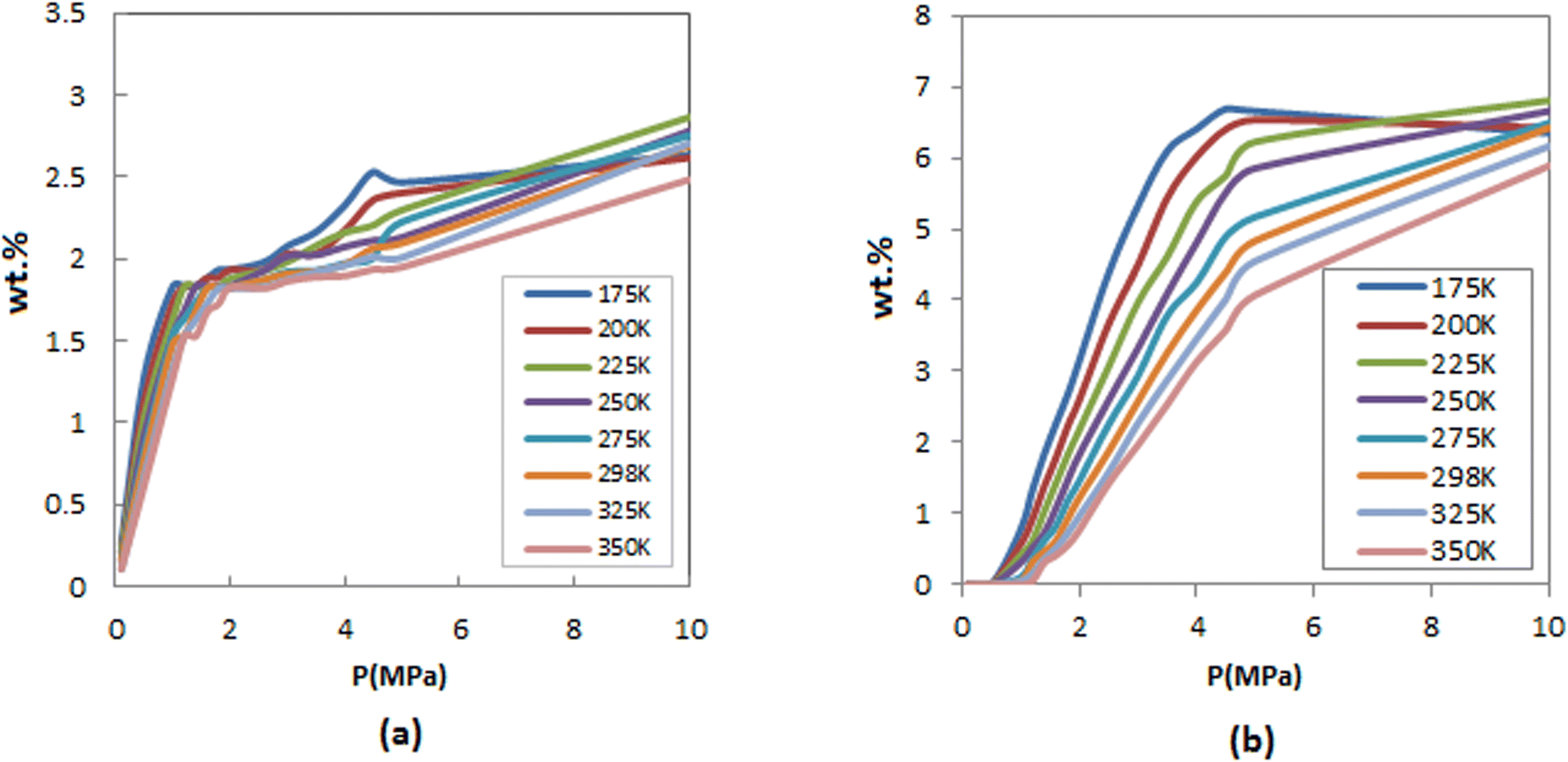 | ||
| Fig. 2 Comparison of H2 adsorption isotherms of FeCNT (a) inside (b) outside the nanotubes at different temperatures. | ||
Hydrogen adsorption isotherms inside and outside the FeCNT are indicated in Fig. 2. It can be seen that the isotherms exhibit similar trends. Nonetheless, due to the steric restriction, hydrogen adsorption inside the FeCNT was lower than outside.
Fig. 3 presents the total capacity for H2 sorption at different pressures and three temperatures. As observed, at pressures greater than 5 MPa, FeCNT is nearly saturated at all three temperatures, with a maximum adsorption capacity of about 9%. To determine the exact H2 storage of FeCNT, the total H2 uptakes at different pressures and temperatures are tabulated in Table 2.
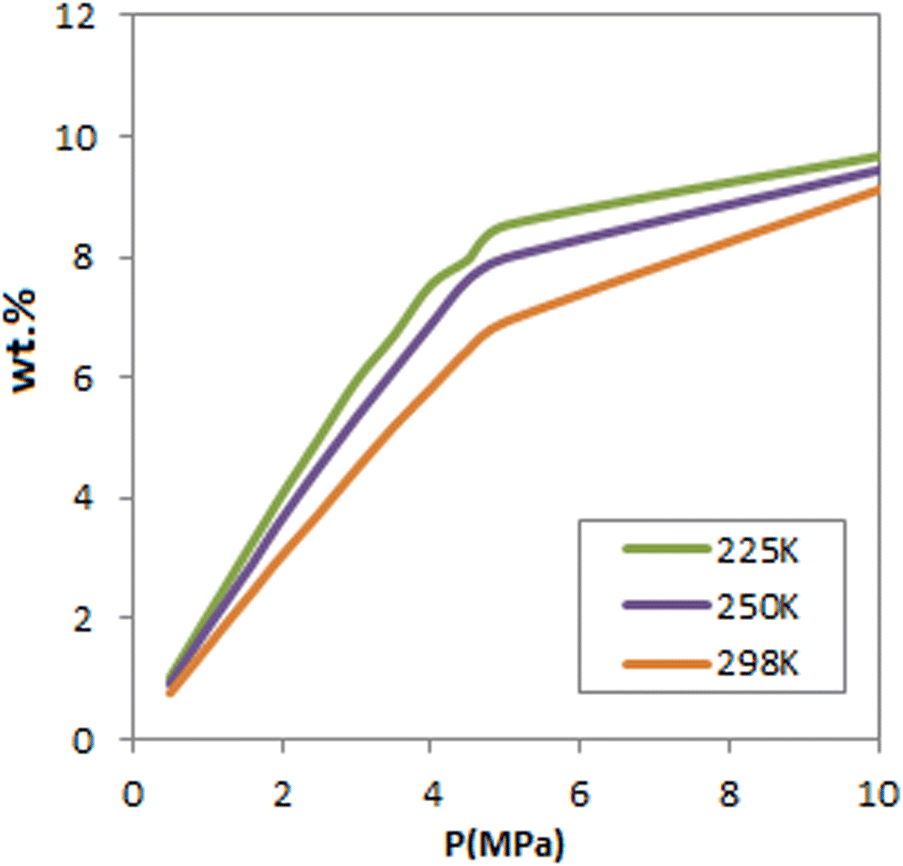 | ||
| Fig. 3 Total H2 adsorption isotherms of FeCNT at 225, 250 and 298 K. | ||
| P (MPa) | Total wt% | |||
|---|---|---|---|---|
| 75 K | 175 K | 250 K | 298 K | |
| 0.1 | 0.5673 | 0.2587 | 0.1554 | 0.1554 |
| 0.5 | 2.9280 | 1.2801 | 0.9250 | 0.7720 |
| 1 | 5.5341 | 2.6040 | 1.8417 | 1.5323 |
| 2 | 9.4252 | 5.0890 | 3.6632 | 3.0504 |
| 4.5 | 10.1821 | 9.2211 | 7.6277 | 6.4612 |
| 5 | 10.2993 | 9.1235 | 7.9834 | 6.9294 |
| 10 | 10.0417 | 10.0861 | 9.4369 | 9.1130 |
As can be seen, of all reported temperatures, the highest amount of H2 uptake by FeCNT falls within the range of 9–10%. The adsorption values at 10 MPa and, 175, 250 and 298 K were 10.086%, 9.4369%, and 9.1130% respectively. Therefore, it can be concluded that FeCNT can enhance the hydrogen adsorption potential of CNTs and exceed the target of the United States Department of Energy (DOE) (6.5 wt%).85,86
Many researchers have investigated the doping effect on the hydrogen adsorption of nanotubes. It has revealed that hydrogen storage of CNT and B doped CNT was 0.02 and 0.157 wt% at 303 K and 10 bar.87 Volumetric measurements of H2 storage of Pd and V doped CNT have confirmed the enhancement of the adsorption capacity of CNTs upon metal doping.39 The results have indicated that H2 uptake of Pd and V doped CNT at 298 K and 65 bar have been 0.125 and 0.1% respectively which were higher than hydrogen storage of CNT (<0.01%). Their results are comparable with our outcome of FeCNT adsorption capacity at 298 K and 0.5 MPa which was 0.77%. The higher H2 adsorption capacity of FeCNT concerning V doped CNT and pure CNT can be also attributed to the εij values of H2–V,29 H2–Fe, and H2–C (see Table 1). DFT investigations88 have also revealed that the enhancement of hydrogen storage by metal doping is due to the electron transfer from metal atoms to carbon.
Many other studies have shown that hydrogen adsorption capacity is increased by metal doping.89–95 The hydrogen adsorptions of the multi-wall CNT and Fe-multi-wall CNT which have been studied by volumetric and electrochemical methods in environmental conditions, were determined to be 0.3% and 0.75%, respectively.38 The results revealed that H2 molecules are adsorbed on the defect sites and pass into the spaces between neighboring carbon layers. The outcomes are compatible with our foundation at 298 K (Table 2), the differences are due to the single wall nanotube applied in the current study. In a recent study, Fe-doped carbon aerogels have been applied for hydrogen uptake at 77 K at pressures up to 30 atm.54 Hydrogen adsorption capacities for 1, 5, and 10% Fe doped carbon aerogels at 25 atm and 77 K were 1.47, 1.38, and 1.28 wt%, respectively. Although the adsorption properties of Fe doped carbon aerogels are different from FeCNT, the results can be comparable with our outcomes. As can be seen in Table 2, FeCNT could adsorb 9.4252 wt% of H2 at 2 MPa and 77 K and was almost saturated at larger pressures.
One of the objectives of the present study is to compare the hydrogen adsorption of FeCNT and pure CNT, and to confirm the efficiency of Fe doping in hydrogen storage of CNTs. Fig. 4 presents a comparison of the H2 adsorption isotherms of FeCNT and pure CNT as a function of pressure at three different temperatures: 170 K, 250 K, and 298 K. At lower pressures, both nanotubes exhibit similar storage capacities. However, as the pressure increases (specifically, pressures exceeding 4.5 MPa), the hydrogen storage of FeCNT surpasses that of pure CNT. Comparison of the H2 adsorption capacities of CNT and FeCNT can also be seen in Table S1.† These findings indicate that at pressures below 4 MPa, the Fe doping did not have a significant impact on the hydrogen adsorption of CNT, as evidenced by Fig. 4. However, at higher pressures, there is a slight improvement in H2 adsorption with FeCNT. Nevertheless, under certain conditions, there are notable changes in the adsorption capacities of FeCNT. For example, at 4.5 MPa and 175 K, the H2 adsorption of CNT and FeCNT were 8.0780% and 9.2211% respectively, representing an approximately 14% increase in adsorption. Similarly, at 5 MPa and 250 K, a 7% improvement was observed, and at 10 MPa and 250 K, a 17% enhancement in H2 storage was observed due to Fe doping of CNT. However, one advantage of Fe doping on the hydrogen storage performance of CNT is the improved H2 selectivity from an H2/N2 mixture environment, which will be discussed in Section 3.2.6. These findings reveal that the H2 storage behaviors of CNT and FeCNT are comparable across all three temperatures but are enhanced by Fe doping at pressures above 4.5 MPa. Consequently, Fe-doping of CNT can be considered a viable strategy for achieving efficient H2 storage at elevated pressures.
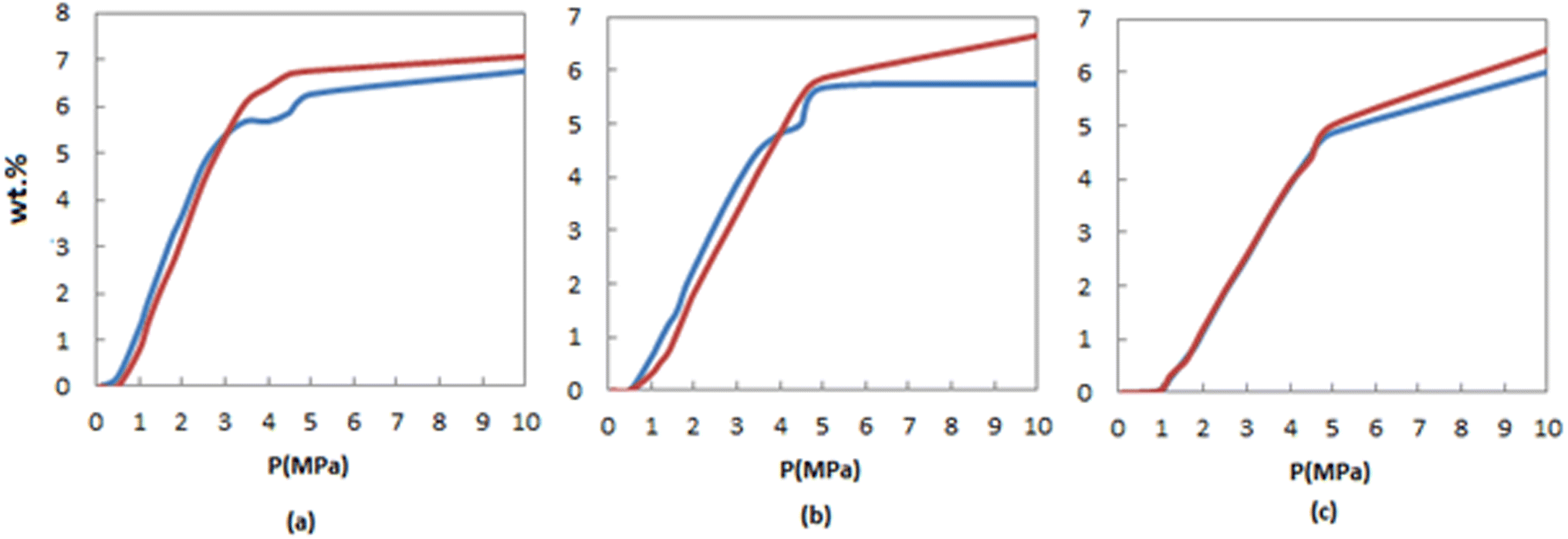 | ||
| Fig. 4 Comparison of H2 adsorption isotherms of FeCNT (red line) and pure CNT (blue line) at (a) 175, (b) 250 and (c) 298 K. | ||
The highest hydrogen adsorption capacity that could be obtained for pure CNT was 4.84 wt% at 10 MPa and 77 K,96 which has been determined by the volumetric method. By acid treatment of CNTs and removal of metal catalysts, a higher storage capacity of CNTs has been observed. The reason was that acid treatment damages the structure of CNTs, and as a result, structural defects with high energy sites can enhance hydrogen adsorption. Our results for FeCNT showed that the H2 adsorption capacity is higher than pure CNTs.
A recent investigation that confirms the effectiveness of transition metal doping on the hydrogen uptake of CNTs has applied optical adsorption spectra analysis in the framework of DFT calculations.56 As mentioned earlier, Fe and Os doping have been shown to overpower the weak interactions between CNT and hydrogen, thus affording a strong surface for hydrogen adsorption.
Of course, the utilization of molecular dynamics methods in investigating the absorption process in nanostructures should not be disregarded in terms of efficiency and accuracy. For instance, the study of high deposition temperatures, which ensures sufficient surface diffusion of Al and Ga adatoms, has been conducted using AIMD simulation to enhance material quality.97 The results suggest that the decomposition of graphene layers is attributed to the surface dissociation of (CH3)3Al and (CH3)3Ga, combined with elevated deposition temperatures. Additionally, the behavior of carbon–carbon bonds can be modeled using interatomic potentials such as original Tersoff98–100 and optimized Tersoff.101 To predict the mechanical behavior of graphene sheets at the atomic scale, MD simulations have applied an interatomic potential with a modified cutoff function for the Tersoff potential.102 The use of this modified Tersoff potential has successfully predicted failure stress and strain values that align well with experimentally estimated values.
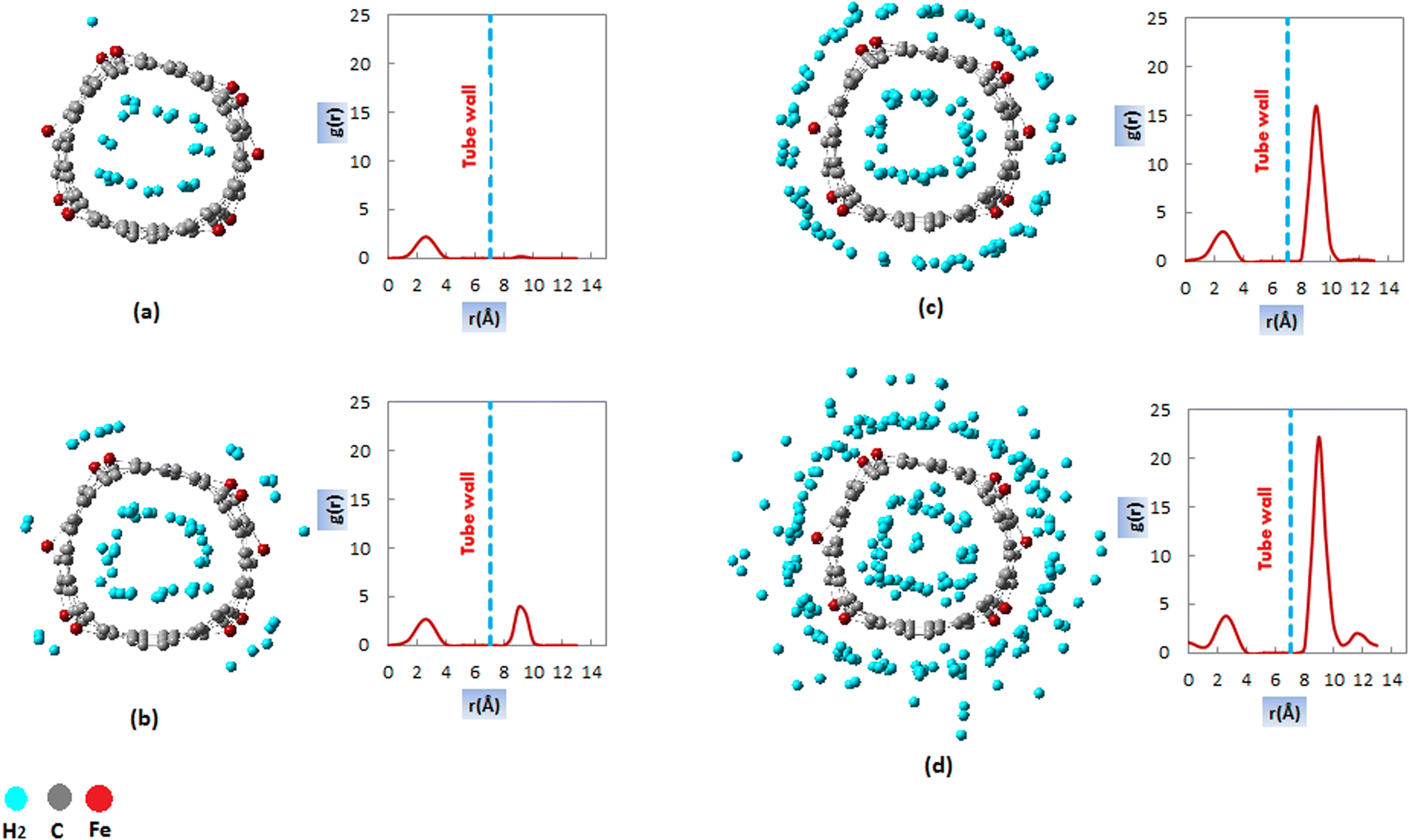 | ||
| Fig. 5 Radial distribution functions of H2 molecules inside and outside the FeCNT versus distance from the central axis of the nanotube and Snapshot generated by the simulation of hydrogen storage at 298 K and (a) 1 MPa (b) 2 MPa (c) 5 MPa (d) 10 MPa. | ||
At lower pressures, 1 MPa (Fig. 5a), the RDFs plots contained only one peak inside the nanotube at about 3 Å from the central axis of CNT. Consequently, at lower pressures, H2 is adsorbed in a cylindrical layer inside the CNT, which is visible in the simulation snapshot at P = 1 MPa. As the pressure increased, an additional peak appeared outside the CNT, which was related to the formation of an adsorbed layer outside the nanotube. The specific trend in the RDF diagrams was comparable to the equilibrium snapshot generated from the FeCNT simulation.
As seen in Fig. 5b, the outside adsorbed H2 molecules prefer to adsorb on the doped Fe atoms (defect sites) due to the larger interaction energies between the Fe atoms and H2. This evidence is justified by comparing the Lennard-Jones cross-interaction parameters of Fe–H2, and C–H2 (Table 1). Since εFe–H2 (0.821 kcal mol−1) is larger than εC–H2 (0.0764 kcal mol−1), therefore, doped Fe atoms are stronger adsorption sites for H2 uptake. It has been proven that defect sites in CNTs can absorb hydrogen molecules, which is associated with a significant increase in the interaction energy and affects the storage capacity of CNTs.103,104
By comparing RDFs at pressures of 2, 5, and 10 MPa, it is clear that the outer peak increased with increasing pressure. This result was due to the greater adsorption of hydrogen molecules at higher pressures. However, the peak height of inside adsorption remained almost constant with increasing pressure. These results are consistent with the H2 adsorption isotherms of FeCNT in Fig. 2. It can be observed that the adsorption capacity within the nanotube becomes almost constant at pressures larger than 2 MPa, while the adsorption capacity outside the nanotube exhibits an increasing trend, superior to the inside capacities. This result is due to the repulsion of H2 molecules at higher pressures and entropy decrease inside the CNT, which are unfavorable conditions for hydrogen adsorption. The outside peak was situated at about 9.5 Å (about 2.45 Å from the CNT wall). There was a minimum potential interaction energy between H2 molecules and FeCNT at this distance. The location of the outside adsorption layer of H2 could be determined by the Lennard-Jones parameters, σFe–H2 = 2.640 Å (see Table 1).
By comparing the RDF profiles, it was found that at 10 MPa, another adsorption layer was formed outside the CNT. As can be seen in Fig. 5d, the second adsorption layer occurred in about 12 Å (2.5 Å from the first layer and about 5 Å from the CNT wall). The result was consistent with the isotherm plots which fitted to the Freundlich isotherm model and confirmed the presence of H2 multi-layer adsorption (see Section 3-2-3). The formation of the second adsorption layer can be recognized in the snapshot generated by the simulation of hydrogen storage at 10 MPa (Fig. 5d).
The equation of Langmuir isotherm is as follows:106,107
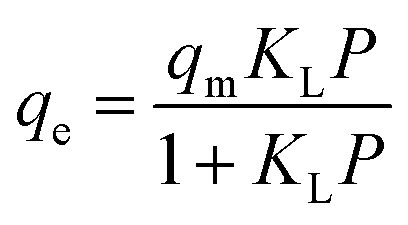 | (13) |
Freundlich isotherm can be employed for multilayer adsorption on heterogeneous adsorbent surfaces. This model assumes that the adsorbent surface is heterogeneous, and active sites and their energies are exponentially distributed.108 In Freundlich isotherm, stronger binding sites are first occupied, until the adsorption energy decreases exponentially after the completion of the adsorption process.109 The presentation of Freundlich isotherm is considered as following equation:110
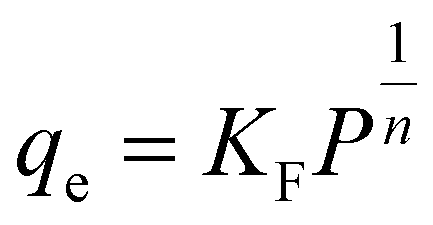 | (14) |
H2 adsorption data on FeCNT were successfully subjected to curve fitting using Langmuir and Freundlich adsorption isotherm model (Fig. 6). The values of Freundlich and Langmuir adsorption isotherm model parameters fitting to the obtained adsorption data are presented in Table 3. The quality of fitting the isotherm models with the obtained adsorption capacities is estimated by the correlation coefficient R2. As can be seen, using the linear regression method, there is a good matching between the simulation data and the predicted values of the isotherm model with R2 close to 1. It can be observed that the isotherm at 175 K shows a better fit with the Freundlich model (Fig. 6b, blue line) with R2 = 0.9999 compared to the isotherm at 298 K with R2 = 0.9897, which is an indication of the heterogeneous nature of the adsorption surface (FeCNT). However, the isotherm at 298 K follows the Langmuir model with R2 = 0.9991 better than at 175 K. This evidence is due to Langmuir mono-layer adsorption at higher temperatures. The results are consistent with the isotherm analysis data of the hydrogen storage of CNTs at 303 K and 10 bar87 which have concluded CNT adsorption data were best fitted by Langmuir isotherm model for monolayer physical adsorption. The maximum adsorption capacity (qm) values obtained from the Langmuir equation for CNT and B doped CNT were 0.033 and 0.414 wt%, and based on our results, qm was 0.6339% at 298 K. As can be seen in Table 3, the Langmuir constant (KL) of FeCNT at 298 K was 0.0248, which is larger than that of CNT (0.0070),87 confirming that Fe doping plays a major role in hydrogen uptake.
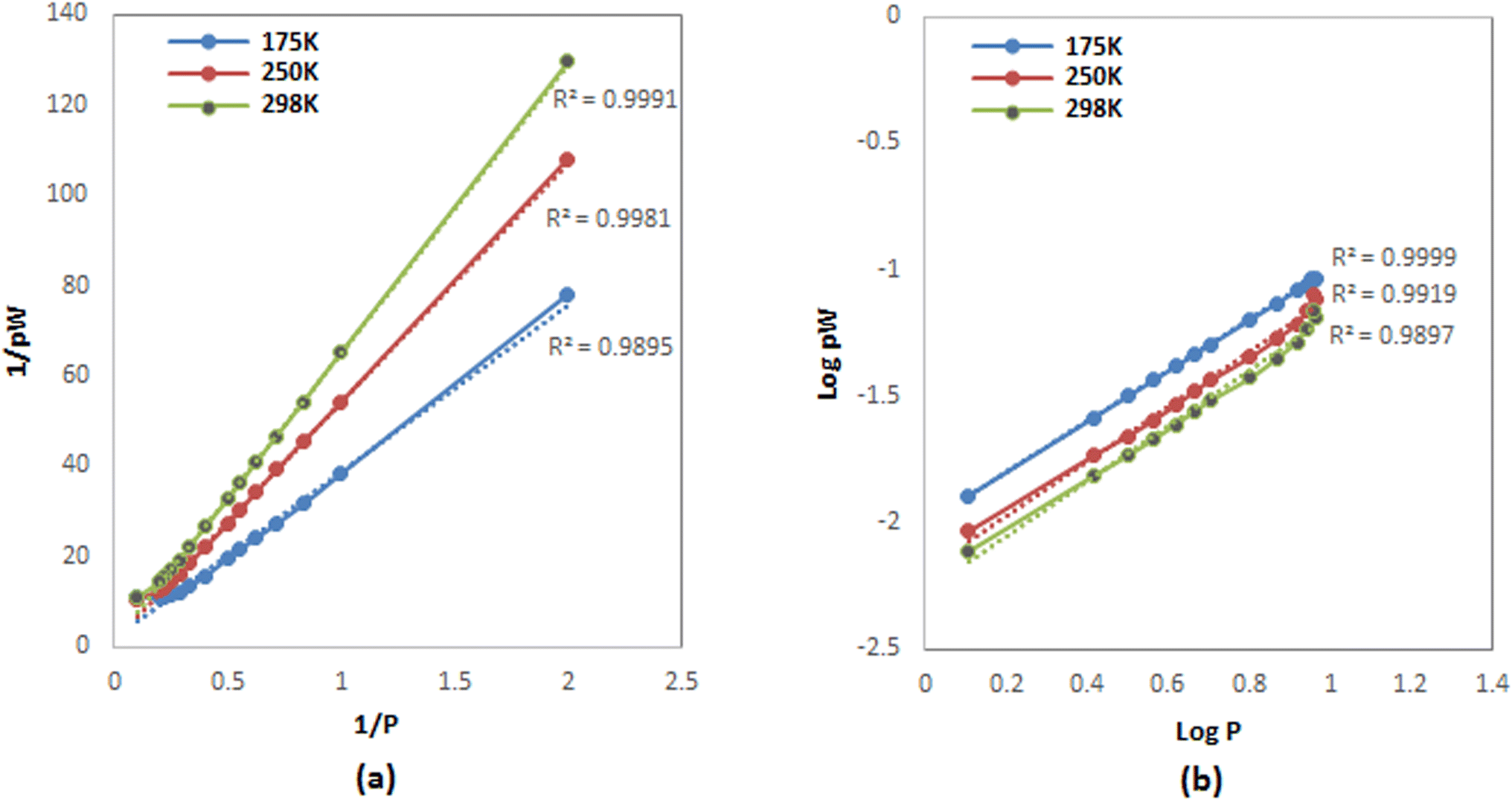 | ||
| Fig. 6 Fitting isotherms plots for H2 adsorption at different temperatures (a) Langmuir (b) Freundlich. | ||
| T (K) | Langmuir parameters | Freundlich parameters | ||
|---|---|---|---|---|
| qm | KL | KF | 1/n | |
| 175 | 0.4452 | 0.0610 | 0.0100 | 1.0031 |
| 250 | 0.5831 | 0.0325 | 0.0065 | 1.0743 |
| 298 | 0.6339 | 0.0248 | 0.0054 | 1.0845 |
Multilayer H2 adsorption can occur at lower temperatures due to the high hydrogen storage that causes the formation of a second adsorption layer. Therefore, it is anticipated that the isotherm follows the Freundlich model, and the existence of multi-layer adsorption is proved. This evidence was confirmed by the RDF diagram at 10 MPa, which showed the second adsorption layer outside the nanotube (see Fig. 5d).
Fig. 7 shows the reduction of total adsorption capacity percentage at 5 MPa with raising temperature. As it is seen, pure CNT and FeCNT exhibited the same behavior with increasing temperature. By increasing the temperature from 175 to 298 K, the hydrogen capacity of pure CNT was reduced from 6.75% to 4.84% (storage decrease about 28% of initial capacity). While the hydrogen capacity of FeCNT was reduced from 6.66% to 5.02% (storage decrease about 24% of initial capacity). These results indicated that hydrogen uptake of FeCNT up to 298 K was better than CNT, and therefore hydrogen storage by FeCNT was maintained up to room temperature higher than pure CNT. It is well inferred from Fig. 7 that at temperatures higher than 298 K, the slope of the plot of CNT was lower than that of FeCNT, which indicates its lesser desorption. As the temperature increased from 298 to 350 K, pure CNT and FeCNT desorbed 7% and 19% of their hydrogen content, respectively. Therefore, it is concluded that with increasing temperature (at temperatures above room temperature), FeCNT shows better reversibility than pure CNT.
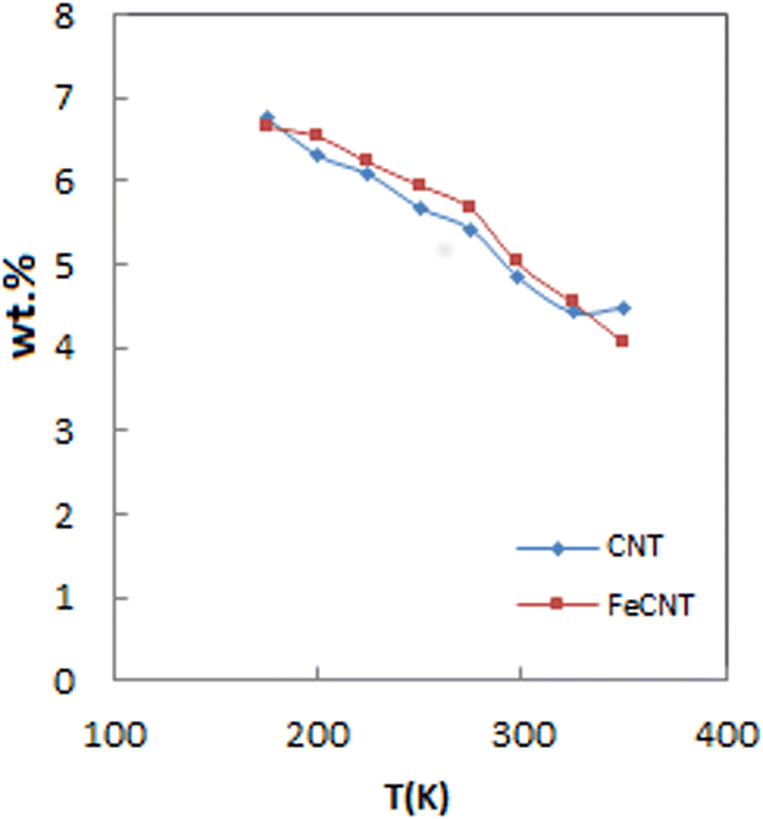 | ||
| Fig. 7 Variation of H2 adsorption capacity of FeCNT and pure CNT upon the temperature increment at 5 MPa. | ||
Fig. 8 shows the dependency of adsorption of H2/N2 mixtures on pressure at 298 K. As can be seen, adsorption isotherms for gas mixtures have similar behavior as pure ones. This figure also shows the effect of mole percentage on FeCNT adsorption capacity. As the mole percentage of H2 in the environment increases, the adsorption capacity increases.
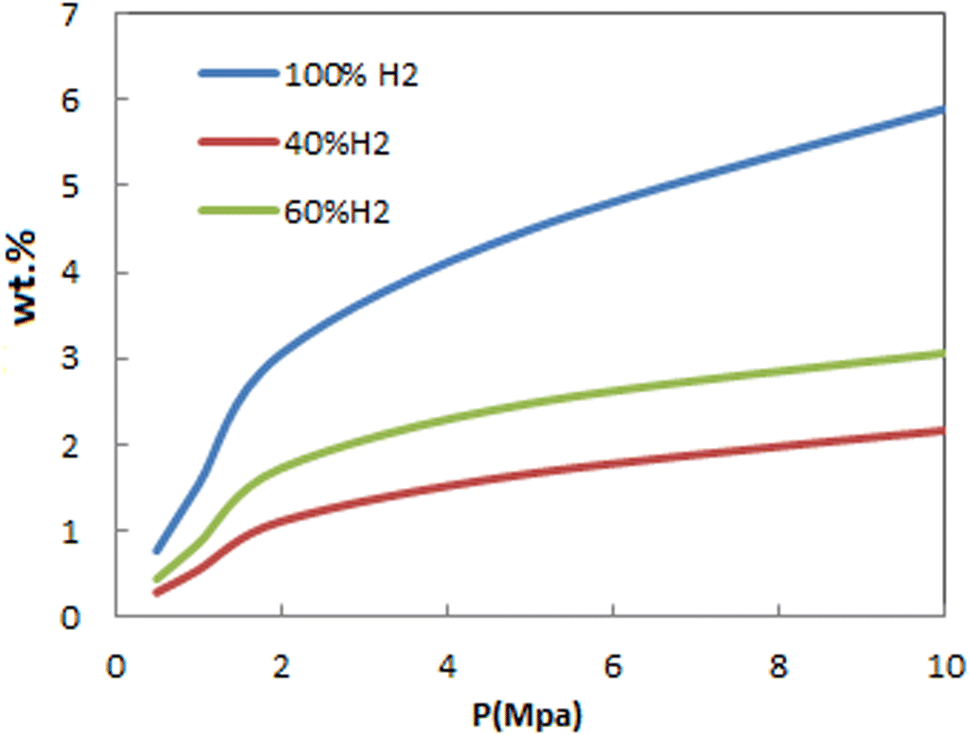 | ||
| Fig. 8 Comparison of hydrogen adsorption isotherms of FeCNT at 298 K from pure H2 and H2/N2 mixture. | ||
To evaluate the influence of adding N2 on FeCNT hydrogen storage, adsorption isotherms for H2 in a gas mixture with N2 and in the absence of N2 were investigated. The H2 adsorption capacities of FeCNT in the gas mixture with N2 and in the absence of N2 at 298 K and at selected pressures is tabulated in Table 4. The adsorption capacity of H2 below 4 MPa in pure and mixture environments was less than 10%, and therefore adding N2 did not affect the H2 uptake of FeCNT. Two factors affecting the adsorption capacity are effective pressure and free adsorbent surface. Since the Lennard-Jones parameter ε of N2 is approximately the same as that of H2, at lower pressures, adding N2 to the environment did not change the interactions between H2 molecules, and therefore did not affect H2 adsorption. As previously mentioned, N2 was a diluting agent, and at pressures below 4 MPa, the addition of N2 to the medium did not significantly affect the hydrogen storage of FeCNT.
| P (MPa) | 40% H2 | 60% H2 | ||
|---|---|---|---|---|
| wt% (H2) in pure H2 | wt% (H2) in H2/N2 mixture | wt% (H2) in pure H2 | wt% (H2) in H2/N2 mixture | |
| 0.5 | 0.310 | 0.2909 | 0.4646 | 0.4453 |
| 1 | 0.6185 | 0.5480 | 0.9247 | 0.8538 |
| 2 | 1.2277 | 1.1131 | 1.8396 | 1.7280 |
| 3 | 1.8367 | 1.6652 | 2.7405 | 2.4799 |
| 4 | 2.4263 | 2.1657 | 3.6339 | 3.0612 |
| 5 | 3.0477 | 2.4214 | 4.4780 | 3.6780 |
| 10 | 5.6833 | 4.0827 | 7.6962 | 5.2460 |
At higher pressures, the restriction of the absorption sites because of the absorption of other gases (i.e., N2) reduces H2 uptake from the gas mixture. In fact, at higher pressures, absorption depends on the free surface area of the adsorbent, and as a result, adding N2 to the environment leads to a decrease in the absorption capacity. As a result, pressures below 4 MPa are appropriate for FeCNT hydrogen storage at 298 K from mixed H2/N2 systems than higher pressures.
The selectivities from the isotherms were calculated by the Ideal Adsorption Solution Theory (IAST),111 which calculate the gas selectivity of adsorbents from a gas mixture through modeling methods. The selectivity SA/B from a binary gas mixture, A and B, is find out by the following equation:112,113
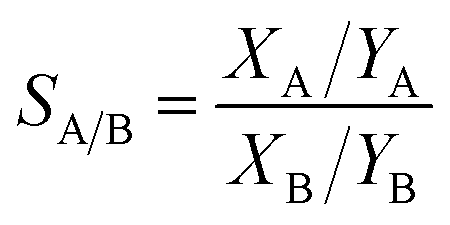 | (15) |
Selectivity depends on the strength of adsorbate–adsorbent interactions, pressure, temperature, and gas phase composition. To achieve optimal conditions for H2 storage from H2/N2 mixture, the effects of pressure and composition on the selectivity of FeCNT were investigated.
To determine the H2/N2 selectivity of FeCNT, the compositions of 0.1–0.9 mole fraction of H2/N2 mixtures at 298 K and pressures up to 10 MPa were considered. The results revealed that H2 selectivity enhanced with the increase of total bulk pressure for all compositions of H2 and N2 mixtures. For instance, the dependence of H2 selectivity of FeCNT versus bulk pressure at 298 K from a H2/N2 gas mixture with 60 mol% of H2 is shown in Fig. 9. As can be seen at lower pressures, SH2/N2 is lower than 1, and therefore H2 is not favorably adsorbed.
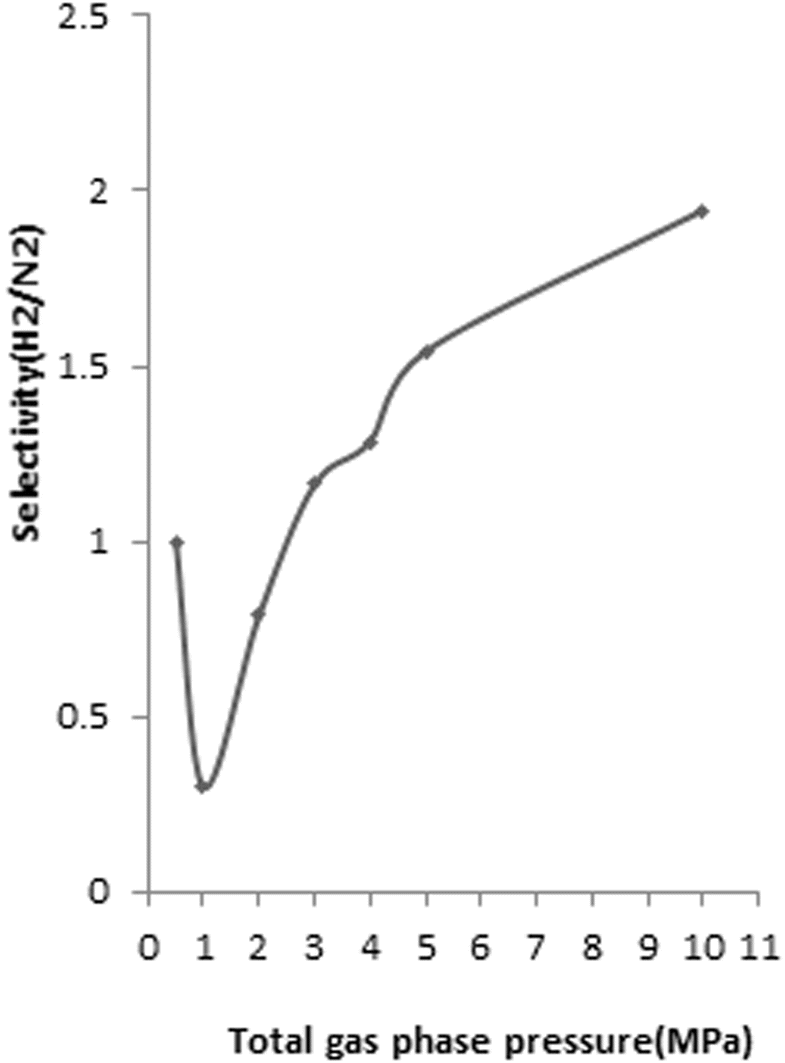 | ||
| Fig. 9 H2 selectivity of FeCNT from mixture of 60 mol% H2/N2 vs. pressure at 298 K (mole fraction of 0.6 H2 and 0.4 N2 mixtures). | ||
As seen in Fig. 9, at pressures 2–3 MPa, SH2/N2 was around 1, which meant that there was no competition. Interactions between gas molecules and NT affect selectivity values.114 The slightly larger H2–Fe and Fe–C interactions compared to N2–Fe and N2–C (see Table 1), leads to nearly identical selectivity of H2 and N2 at lower pressures. At pressures greater than 3 MPa, H2/N2 selectivity tends to be larger than 1, and therefore H2 is efficiently adsorbed. This selectivity is due to the different affinity of the H2 and N2 for adsorbing on the CNT.
Furthermore, it is expected that smaller molecules can be better packed into the CNTs at higher pressures. This fact has been also found in carbon membranes.70 An effective factor in increasing the adsorption capacity with increasing pressure is the packing effect.115
Indeed, in the present study, the smaller kinetic diameter of H2 (0.24 A) compared to N2 (0.36 A)116 leads to higher diffusion and higher selectivity of H2 over N2 in FeCNT, especially at higher pressures. Accordingly, due to superior hydrogen uptake from H2/N2 gas mixtures, pressures larger than 3 MPa are convenient. As can be seen in Fig. 10, at low pressures, the selectivity of H2 is smaller than that of N2 and then enhances along with increasing pressure. The effect of pressure on the selectivity of CH4 over H2 on SiNTs has displayed the same trend.117 As mentioned, smaller molecules are more appropriate for packing in the CNT, the selectivity of CH4/H2 decreases with increasing pressure.
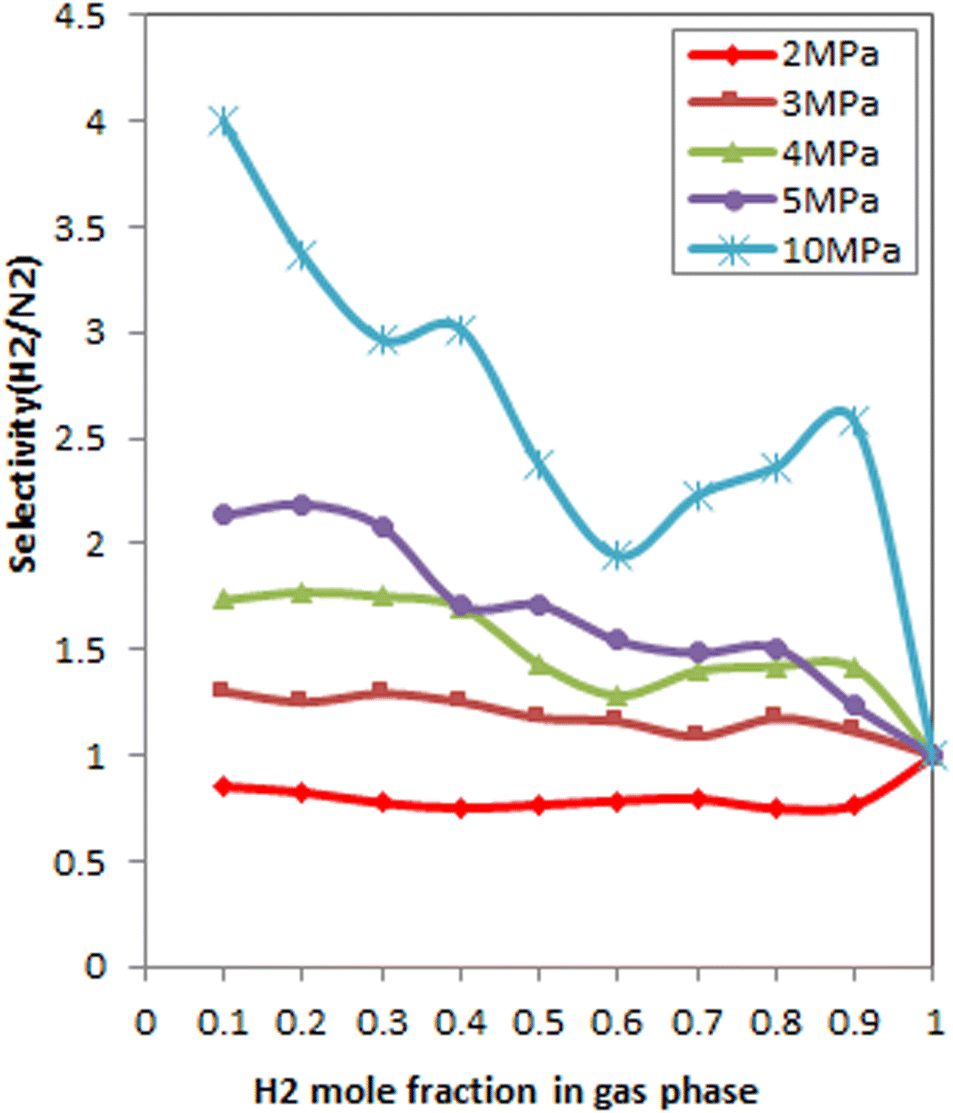 | ||
| Fig. 10 Selectivity of H2/N2 as a function of H2 bulk composition at 298 K and different pressures. | ||
As it is noticed, in addition to the effect of the relative diameters of the two gases, H2/N2 selectivity depends on the structure of the adsorbent and the strength of the interaction of H2 and N2 with the adsorption sites. In a recent study, adsorption of pure H2 or N2 and H2/N2 mixture in SIFSIX-2-Cu-I has been investigated through Monte Carlo simulation using the polarizable force field118 SIFSIX-2-Cu-I is synthesized by organic ligands, metal nodes, and hexafluorosilicate (SiF62−) anions. Since N2 molecules have a denser electron cloud distribution (than H2), F atoms have a higher affinity for N2 molecules. Therefore, selectivity for N2 over H2 has been observed in SIFSIX-2-Cu-i.
In another investigation, polysulfone membrane provided H2/N2 selectivity of 0.76, and mixing it with low concentrations of carbon nanotubes led to an increase in H2/N2 selectivity up to 3.95. These remarkable results are due to the increase in free volume, molecular space, and H2 absorption.119 The trend of H2/N2 selectivity of CNT/polysulfone membrane versus pressure was consistent with our outcomes.
The primary objective of this research was to investigate the efficiency of Fe doping in the hydrogen adsorption properties of CNTs. In this regard, we have also compared FeCNT and CNT by evaluating their H2 selectivity in an H2/N2 mixture environment.
To assess the selectivity performance of CNT and FeCNT, H2/N2 selectivity at different bulk compositions was determined at 5 and 298 K, as shown in Table 5. Considering that the adsorption capacity of FeCNT is greater than that of CNT at pressures larger than 4.5 MPa, the selectivity performance of FeCNT relative to CNT was examined at 5 MPa.
| H2 mole fraction in gas phase | Selectivity (H2/N2) | |
|---|---|---|
| FeCNT | CNT | |
| 0.2 | 2.19 | 0.32 |
| 0.3 | 2.08 | 0.50 |
| 0.4 | 1.71 | 0.73 |
| 0.5 | 1.71 | 0.91 |
As seen in Table 5, the H2/N2 selectivity of FeCNT is greater than CNT and greater than 1, indicating efficient H2 adsorption by FeCNT at 5 and 298 K. On the other hand, the H2/N2 selectivity of CNT is smaller than 1 in almost all studied bulk compositions (except X(H2) = 0.6), indicating that CNT cannot preferentially adsorb H2 over N2.
Fig. 10 denotes the effect of the bulk mole fraction of H2 on the selectivity of H2/N2 at 298 K and different pressures. As can be seen, selectivity improves with increasing pressure. This result is due to the packing effect, which prevails with increasing pressure. Since the packing effect of H2 is greater than that of N2 due to its smaller kinetic diameter,116 as the pressure changes, the selectivity is enhanced significantly. The combination of increasing H2 mole fraction in the gas phase and the adsorption sieve effect increases H2 selectivity.
At 3 MPa, SH2/N2 was equal to 1 in all mole fractions, and it did not change much with the bulk phase composition change. This is because there was no competition, and thus the bulk composition did not affect the H2 selectivity at 3 MPa.
Thus, the pressure of 4 and 5 MPa in which the FeCNT shows an adsorption preference for H2 over the wide range of bulk mole fractions can be considered as an optimum pressure.
As mentioned earlier, at pressures greater than 5 MPa, FeCNT was approximately saturated. Therefore, at 10 MPa, it is reasonable to see a decrease in selectivity with an increasing mole fraction of H2.
Fig. 10 shows that the maximum selectivity of H2/N2 was stated at 0.3 bulk mole fraction of H2 at 5 MPa. However, as previously mentioned, the selectivity increases with increasing pressure and at 10 MPa, FeCNT has the highest values for H2/N2 selectivity. Therefore based on the obtained results, the proposed optimum conditions for H2 selectivity in H2/N2 mixtures were X(H2) = 0.3 at 5 MPa, X(H2) = 0.4 at 4 MPa, and all mole fractions at 10 MPa.
Consequently, considering the result of Section 3.2, which mentioned that adding N2 to the environment does not have a significant effect on FeCNT hydrogen adsorption at pressures below 4 MPa, the composition of X(H2) = 0.4 and pressure of 4 MPa is suggested as the most effective adsorption condition.
4. Conclusion
Considering the ability of metals to absorb hydrogen, it was valuable to study the absorption capacity of new nanomaterials created by metal doping on the surface of other nanomaterials. Consequently, this research focused on evaluating the potential of FeCNT for H2 storage. Monte Carlo simulation was performed to compare the adsorption behavior of FeCNT and investigate the effects of temperature, pressure, and bulk mole fraction of H2 on FeCNT adsorption capacity. Temperature range of 75–350 K and pressure range of 0.5–10 MPa were considered. The simulation results showed that all adsorption isotherms exhibited a similar trend, and at pressures of about 5 MPa and above, FeCNT showed maximum hydrogen capacity.At a constant bulk pressure, increasing the mole fraction of H2 in the gas phase increased the adsorption capacity. The Comparison of hydrogen capacities at increasing temperatures showed that the reversibility of FeCNT was more efficient than pure CNT above ambient conditions.
The results of adding N2 to the medium as a diluent for system safety revealed that hydrogen uptake of FeCNT did not change considerably under 4 MPa, and it has the potential to efficiently adsorb H2 molecules at 298 K from H2/N2 mixture. Since both H2 and N2 are non-polar small molecules, the selectivity of H2 from the H2/N2 mixture was of great interest in this research.
The outcomes of the current research revealed that the H2 storage of CNT is enhanced by Fe doping at pressures above 4.5 MPa, and Fe-doping of CNT can be considered a viable strategy for achieving efficient H2 storage at larger pressures. Moreover, the comparison of H2/N2 selectivity performance of FeCNT and CNT at different bulk compositions and pressures larger than 4.5 MPa indicated that the selectivity of FeCNT is greater than CNT and greater than 1 which confirms FeCNT can preferentially adsorb H2 over N2. As a result, FeCNT is more suitable than pure CNT for hydrogen storage applications because of its improved hydrogen adsorption capacity and can also be used as a potential candidate for efficient H2/N2 adsorption.
Author contributions
Sepideh Ketabi: supervised, data analysis, visualization, writing (original draft & editing), Bita Baghai: performing simulation, methodology, data collection, the whole manuscript was approved by all authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- M. Höök and X. Tang, Depletion of fossil fuels and anthropogenic climate change—A review, Energy Policy, 2013, 52, 797–809 CrossRef.
- L. Schlapbach and A. Züttel, Hydrogen-storage materials for mobile applications, nature, 2001, 414(6861), 353–358 CrossRef CAS PubMed.
- G. Mpourmpakis, E. Tylianakis and G. E. Froudakis, Carbon nanoscrolls: a promising material for hydrogen storage, Nano Lett., 2007, 7(7), 1893–1897 CrossRef CAS PubMed.
- A. Bilic and J. D. Gale, Chemisorption of molecular hydrogen on carbon nanotubes: a route to effective hydrogen storage?, J. Phys. Chem. C, 2008, 112(32), 12568–12575 CrossRef CAS.
- B. Fang, H. Zhou and I. Honma, Ordered porous carbon with tailored pore size for electrochemical hydrogen storage application, J. Phys. Chem. B, 2006, 110(10), 4875–4880 CrossRef CAS PubMed.
- J. Vajo, F. Pinkerton and N. Stetson, Nanoscale phenomena in hydrogen storage, Nanotechnology, 2009, 20(20), 200201 CrossRef PubMed.
- M. Zafar, T. Iqbal, S. Fatima, Q. Sanaullah and S. Aman, Carbon nanotubes for production and storage of hydrogen: challenges and development, Chem. Pap., 2021, 1–17 Search PubMed.
- Y. Cheng, X. Ma, C. Huang, G. Yuan and J. Liao, The effect of functional groups (O, F, or OH) on reversible hydrogen storage properties of Ti2X (X= C or N) monolayer, Int. J. Hydrogen Energy, 2022, 47(67), 28969–28977 CrossRef CAS.
- K. Shivaprasad, S. Raviteja, P. Chitragar and G. Kumar, Experimental investigation of the effect of hydrogen addition on combustion performance and emissions characteristics of a spark ignition high speed gasoline engine, Proc. Technol., 2014, 14, 141–148 CrossRef.
- P. M. Ajayan, Nanotubes from carbon, Chem. Rev., 1999, 99, 1787–1799 CrossRef CAS PubMed.
- S. Iijima, Growth of carbon nanotubes, Mater. Sci. Eng. B, 1993, 19(1–2), 172–180 CrossRef.
- G. Korneva, Functionalization of Carbon Nanotubes, Drexel University, 2008 Search PubMed.
- A. Amiri, M. Maghrebi, M. Baniadam and S. Z. Heris, One-pot, efficient functionalization of multi-walled carbon nanotubes with diamines by microwave method, Appl. Surf. Sci., 2011, 257(23), 10261–10266 CrossRef CAS.
- Y. Itas, C. Ndikilar and T. Zangina, Carbon nanotubes: a review of synthesis and characterization methods/techniques, Int. J. Sci. Technoledge, 2020, 8(2), 43–51 Search PubMed.
- Y. S. Itas, C. E. Ndikilar, T. Zangina, H. Y. Hafeez, A. Safana, M. U. Khandaker, P. Ahmad, I. Abdullahi, B. K. Olawumi and M. A. Babaji, Synthesis of thermally stable h-BN-CNT hetero-structures via microwave heating of ethylene under nickel, iron, and silver catalysts, Crystals, 2021, 11(9), 1097 CrossRef CAS.
- A. Dalton, C. Stephan, J. Coleman, B. McCarthy, P. Ajayan, S. Lefrant, P. Bernier, W. Blau and H. Byrne, Selective interaction of a semiconjugated organic polymer with single-wall nanotubes, J. Phys. Chem. B, 2000, 104(43), 10012–10016 CrossRef CAS.
- T. Ramanathan, F. Fisher, R. Ruoff and L. C. Brinson, Amino-functionalized carbon nanotubes for binding to polymers and biological systems, Chem. Mater., 2005, 17(6), 1290–1295 CrossRef CAS.
- F. Rumiche, H. Wang and J. Indacochea, Development of a fast-response/high-sensitivity double wall carbon nanotube nanostructured hydrogen sensor, Sens. Actuators, B, 2012, 163(1), 97–106 CrossRef CAS.
- J.-M. Tulliani, A. Cavalieri, S. Musso, E. Sardella and F. Geobaldo, Room temperature ammonia sensors based on zinc oxide and functionalized graphite and multi-walled carbon nanotubes, Sens. Actuators, B, 2011, 152(2), 144–154 CrossRef CAS.
- L. Sun, X. Wang, Y. Wang and Q. Zhang, Roles of carbon nanotubes in novel energy storage devices, Carbon, 2017, 122, 462–474 CrossRef CAS.
- S. A. Yamusa, A. Shaari, I. Isah, U. B. Ibrahim, S. I. Kunya, S. Abdulkarim, Y. Itas and M. Alsalamh, Effects of exchange correlation functional (Vwdf3) on the structural, elastic, and electronic properties of transition metal dichalogenides, J. Niger. Soc. Phys. Sci., 2023, 1094 CrossRef.
- J. H. Cho, S. J. Yang, K. Lee and C. R. Park, Si-doping effect on the enhanced hydrogen storage of single walled carbon nanotubes and graphene, Int. J. Hydrogen Energy, 2011, 36(19), 12286–12295 CrossRef CAS.
- A. Salimian, S. Ketabi and H. Aghabozorg, Hydrogen adsorption capacity of vanadium oxide nanotube from pure and mixture gas environment through molecular simulation, Sep. Sci. Technol., 2018, 53(1), 1–12 CrossRef CAS.
- J. Wu, Doping Modification of Carbon Nanotubes and its Applications, Highlights in Science, Engineering and Technology, 2022, 27, 327–333 CrossRef.
- A. Gholizadeh and B. Dehbandi, First-principles study on structural, electronic, and magnetic properties of 3d transition metal-substituted chiral (6, 3) carbon nanotube, Comput. Condens. Matter, 2022, 30, e00621 CrossRef.
- S. Banerjee, K. Dasgupta, A. Kumar, P. Ruz, B. Vishwanadh, J. Joshi and V. Sudarsan, Comparative evaluation of hydrogen storage behavior of Pd doped carbon nanotubes prepared by wet impregnation and polyol methods, Int. J. Hydrogen Energy, 2015, 40(8), 3268–3276 CrossRef CAS.
- C.-H. Chen, T.-Y. Chung, C.-C. Shen, M.-S. Yu, C.-S. Tsao, G.-N. Shi, C.-C. Huang, M.-D. Ger and W.-L. Lee, Hydrogen storage performance in palladium-doped graphene/carbon composites, Int. J. Hydrogen Energy, 2013, 38(9), 3681–3688 CrossRef CAS.
- I. Mosquera-Lois, S. R. Kavanagh, J. Klarbring, K. Tolborg and A. Walsh, Imperfections are not 0 K: free energy of point defects in crystals, Chem. Soc. Rev., 2023, 52, 5812–5826 RSC.
- A. Salimian, S. Ketabi and H. Aghabozorg, Hydrogen storage comparison of M doped vanadium oxide nanotubes (M= Mo, Zr and W): A molecular simulation study, Int. J. Hydrogen Energy, 2018, 43(5), 2831–2839 CrossRef CAS.
- B. I. Kharisov, O. V. Kharissova, U. Ortiz Mendez and I. G. De La Fuente, Decoration of carbon nanotubes with metal nanoparticles: Recent trends, Synthesis and Reactivity in Inorganic, Metal-Organic, and Nano-Metal Chemistry, 2016, 46(1), 55–76 CrossRef CAS.
- F. A. Jibon, M. U. Khandaker, M. H. Miraz, H. Thakur, F. Rabby, N. Tamam, A. Sulieman, Y. S. Itas and H. Osman, Cancerous and non-cancerous brain MRI classification method based on convolutional neural network and log-polar transformation, Healthcare, 2022, 1801 CrossRef PubMed.
- Y. Yürüm, A. Taralp and T. N. Veziroglu, Storage of hydrogen in nanostructured carbon materials, Int. J. Hydrogen Energy, 2009, 34(9), 3784–3798 CrossRef.
- A. Dadgar, A. Maleki, L. Mahdavian and F. Khazali, Computational and structural study of titanium/carbon nanotube nanocomposite, Diamond Relat. Mater., 2022, 126, 109055 CrossRef CAS.
- R. Rakhi, K. Sethupathi and S. Ramaprabhu, Synthesis and hydrogen storage properties of carbon nanotubes, Int. J. Hydrogen Energy, 2008, 33(1), 381–386 CrossRef CAS.
- M. Kumar and Y. Ando, Chemical vapor deposition of carbon nanotubes: a review on growth mechanism and mass production, J. Nanosci. Nanotechnol., 2010, 10(6), 3739–3758 CrossRef CAS PubMed.
- K.-S. Lin, Y.-J. Mai, S.-R. Li, C.-W. Shu and C.-H. Wang, Characterization and hydrogen storage of surface-modified multiwalled carbon nanotubes for fuel cell application, J. Nanomater., 2012, 2012, 13 Search PubMed.
- H.-M. Cheng, Q.-H. Yang and C. Liu, Hydrogen storage in carbon nanotubes, Carbon, 2001, 39(10), 1447–1454 CrossRef CAS.
- A. Reyhani, S. Mortazavi, S. Mirershadi, A. Moshfegh, P. Parvin and A. N. Golikand, Hydrogen storage in decorated multiwalled carbon nanotubes by Ca, Co, Fe, Ni, and Pd nanoparticles under ambient conditions, J. Phys. Chem. C, 2011, 115(14), 6994–7001 CrossRef CAS.
- Y. Suttisawat, P. Rangsunvigit, B. Kitiyanan, M. Williams, P. Ndungu, M. Lototskyy, A. Nechaev, V. Linkov and S. Kulprathipanja, Investigation of hydrogen storage capacity of multi-walled carbon nanotubes deposited with Pd or V, Int. J. Hydrogen Energy, 2009, 34(16), 6669–6675 CrossRef CAS.
- S.-U. Rather, N. Mehraj-ud-din, R. Zacharia, S. W. Hwang, A. R. Kim and K. S. Nahm, Int. J. Hydrogen Energy, 2009, 34, 961–966 CrossRef CAS.
- V. Kiselev and O. Krylov, Adsorption and Catalysis of Transition Metals and their Oxides, Indian J. Phys., 1993, 67, 201–206 Search PubMed.
- A. Lueking and R. T. Yang, Hydrogen spillover from a metal oxide catalyst onto carbon nanotubes—implications for hydrogen storage, J. Catal., 2002, 206(1), 165–168 CrossRef CAS.
- L. Gao, E. Yoo, J. Nakamura, W. Zhang and H. T. Chua, Hydrogen storage in Pd–Ni doped defective carbon nanotubes through the formation of CHx (x= 1, 2), Carbon, 2010, 48(11), 3250–3255 CrossRef CAS.
- A. Sharma, Investigation on platinum loaded multi-walled carbon nanotubes for hydrogen storage applications, Int. J. Hydrogen Energy, 2020, 45(4), 2967–2974 CrossRef CAS.
- M. Aghababaei, A. A. Ghoreyshi and K. Esfandiari, Optimizing the conditions of multi-walled carbon nanotubes surface activation and loading metal nanoparticles for enhanced hydrogen storage, Int. J. Hydrogen Energy, 2020, 45(43), 23112–23121 CrossRef CAS.
- A. Dhanya, N. Ranjan and S. Ramaprabhu, Hydrogen storage studies of Co, Fe, Fe3C nanoparticles encapsulated nitrogen doped carbon nanotubes, Energy Storage, 2023, 5(4), e421 CrossRef CAS.
- E. Dündar-Tekkaya and N. Karatepe, Hydrogen adsorption of carbon nanotubes grown on different catalysts, Int. J. Hydrogen Energy, 2015, 40(24), 7665–7670 CrossRef.
- M. Yoosefian and N. Etminan, Pd-doped single-walled carbon nanotube as a nanobiosensor for histidine amino acid, a DFT study, RSC Adv., 2015, 5(39), 31172–31178 RSC.
- K. Li, W. Wang and D. Cao, Novel chemical sensor for CO and NO: silicon nanotube, J. Phys. Chem. C, 2011, 115(24), 12015–12022 CrossRef CAS.
- S. C. Lim, J. H. Jang, D. J. Bae, G. H. Han, S. Lee, I.-S. Yeo and Y. H. Lee, Contact resistance between metal and carbon nanotube interconnects: Effect of work function and wettability, Appl. Phys. Lett., 2009, 95(26), 264103 CrossRef.
- L. An, X. Jia and Y. Liu, Adsorption of SO2 molecules on Fe-doped carbon nanotubes: The first principles study, Adsorption, 2019, 25, 217–224 CrossRef CAS.
- N. Luhadiya, V. Choyal, S. I. Kundalwal and S. Sahu, Investigation of unified impact of Ti adatom and N doping on hydrogen gas adsorption capabilities of defected graphene sheets, J. Mol. Graph. Model., 2023, 119, 108399 CrossRef CAS PubMed.
- D. G. Sangiovanni, R. Faccio, G. K. Gueorguiev and A. Kakanakova-Georgieva, Discovering atomistic pathways for supply of metal atoms from methyl-based precursors to graphene surface, Phys. Chem. Chem. Phys., 2023, 25(1), 829–837 RSC.
- A. P. Pandey, M. Shaz, V. Sekkar and R. Tiwari, Synergistic effect of CNT bridge formation and spillover mechanism on enhanced hydrogen storage by iron doped carbon aerogel, Int. J. Hydrogen Energy, 2023, 48(56), 21395–21403 CrossRef CAS.
- Y. Pan, Y. Liu, Y. Lin and C. Liu, Metal doping effect of the M–Co2P/Nitrogen-Doped carbon nanotubes (M= Fe, Ni, Cu) hydrogen evolution hybrid catalysts, ACS Appl. Mater. Interfaces, 2016, 8(22), 13890–13901 CrossRef CAS PubMed.
- Y. S. Itas, M. U. Khandaker, A. B. Suleiman, C. E. Ndikilar, A. Lawal, R. Razali, I. I. Idowu, A. M. Danmadami, A. S. Yamusa and H. Osman, Studies of H2 storage efficiency of metal-doped carbon nanotubes by optical adsorption spectra analysis, Diamond Relat. Mater., 2023, 136, 109964 CrossRef CAS.
- F. Tang, W. Xia, H. Zhang, L. Zheng, Y. Zhao, J. Ge and J. Tang, Synthesis of Fe-doped carbon hybrid composed of CNT/flake-like carbon for catalyzing oxygen reduction, Nano Res., 2022, 15(7), 6670–6677 CrossRef CAS.
- Z. Wang, G. Wei and G. L. Zhao, Enhanced electromagnetic wave shielding effectiveness of Fe doped carbon nanotubes/epoxy composites, Appl. Phys. Lett., 2013, 103(18), 183109 CrossRef.
- Y. Liu, H. Zhang, Z. Zhang, X. Jia and L. An, CO adsorption on Fe-doped vacancy-defected CNTs–A DFT study, Chem. Phys. Lett., 2019, 730, 316–320 CrossRef CAS.
- Y. Liu, V. I. Artyukhov, M. Liu, A. R. Harutyunyan and B. I. Yakobson, Feasibility of lithium storage on graphene and its derivatives, J. Phys. Chem. Lett., 2013, 4(10), 1737–1742 CrossRef CAS PubMed.
- K. Persson, Y. Hinuma, Y. S. Meng, A. Van der Ven and G. Ceder, Thermodynamic and kinetic properties of the Li-graphite system from first-principles calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(12), 125416 CrossRef.
- L. Zhao, C. Chai, W. Petz and G. Frenking, Carbones and carbon atom as ligands in transition metal complexes, Molecules, 2020, 25(21), 4943 CrossRef CAS PubMed.
- C. Bittencourt, A. Felten, J. Ghijsen, J.-J. Pireaux, W. Drube, R. Erni and G. Van Tendeloo, Decorating carbon nanotubes with nickel nanoparticles, Chem. Phys. Lett., 2007, 436(4–6), 368–372 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- Q. Wang, Y.-J. Liu and J.-X. Zhao, Theoretical study on the encapsulation of Pd 3-based transition metal clusters inside boron nitride nanotubes, J. Mol. Model., 2013, 19, 1143–1151 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Condens. Matter Phys., 2009, 21(39), 395502 CrossRef PubMed.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, Equation of state calculations by fast computing machines, J. Chem. Phys., 1953, 21(6), 1087–1092 CrossRef CAS.
- J. Estela-Uribe, J. Jaramillo, M. Salazar and J. Trusler, Virial equation of state for natural gas systems, Fluid Ph. Equilib., 2003, 204(2), 169–182 CrossRef CAS.
- M. Jaeschke and A. Humphreys, The GERG Databank of High Accuracy Compressibility Measurements, GERG Tech. Monograph, vol. 4, 1990 Search PubMed.
- Y. Jia, M. Wang, L. Wu and C. Gao, Separation of CO2/N2 gas mixture through carbon membranes: Monte Carlo simulation, Sep. Sci. Technol., 2007, 42(16), 3681–3695 CrossRef CAS.
- A. K. Rappé, C. J. Casewit, K. Colwell, W. A. Goddard III and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114(25), 10024–10035 CrossRef.
- K. Malek and M. Sahimi, Molecular dynamics simulations of adsorption and diffusion of gases in silicon-carbide nanotubes, J. Chem. Phys., 2010, 132(1), 014310 CrossRef PubMed.
- J. I. Paredes, F. Suárez-García, S. Villar-Rodil, A. Martínez-Alonso, J. M. Tascón and E. J. Bottani, N2 physisorption on carbon nanotubes: computer simulation and experimental results, J. Phys. Chem. B, 2003, 107(34), 8905–8916 CrossRef CAS.
- E. Bottani and V. Bakaev, The grand canonical ensemble Monte Carlo simulation of nitrogen on graphite, Langmuir, 1994, 10(5), 1550–1555 CrossRef CAS.
- J.-P. Hansen and I. R. McDonald, Theory of Simple Liquids: with Applications to Soft Matter, Academic press, 2013 Search PubMed.
- A. R. Leach, Molecular Modelling: Principles and Applications, Pearson education, 2001 Search PubMed.
- R. Bian, J. Zhao and H. Fu, Silicon–doping in carbon nanotubes: formation energies, electronic structures, and chemical reactivity, J. Mol. Model., 2013, 19, 1667–1675 CrossRef CAS PubMed.
- T. T. H. Nguyen, C. Le Minh and N. H. Nguyen, A theoretical study of carbon dioxide adsorption and activation on metal-doped (Fe, Co, Ni) carbon nanotube, Comput. Theor. Chem., 2017, 1100, 46–51 CrossRef CAS.
- X.-X. Wang, X.-Y. Li and H.-N. Li, First principles study of Eu doped carbon nanotubes, Phys. Lett. A, 2008, 372(44), 6677–6680 CrossRef CAS.
- Y. Yagi, T. M. Briere, M. H. Sluiter, V. Kumar, A. A. Farajian and Y. Kawazoe, Stable geometries and magnetic properties of single-walled carbon nanotubes doped with 3 d transition metals: A first-principles study, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69(7), 075414 CrossRef.
- W. Zhang, F. Zhang, Z. Zhang, S. Lu and Y. Yang, Electronic structure and magnetism of Fe-doped SiC nanotubes, Sci. China: Phys. Mech. Astron., 2010, 53, 1582–1589 CrossRef CAS.
- T. Yildirim, J. Íñiguez and S. Ciraci, Molecular and dissociative adsorption of multiple hydrogen molecules on transition metal decorated C 60, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(15), 153403 CrossRef.
- J. Toth, Adsorption, CRC Press, 2002 Search PubMed.
- A. A. Rafati, S. M. Hashemianzadeh, Z. B. Nojini and N. Naghshineh, Canonical Monte Carlo simulation of adsorption of O2 and N2 mixture on single walled carbon nanotube at different temperatures and pressures, J. Comput. Chem., 2010, 31(7), 1443–1449 CrossRef CAS PubMed.
- U.D.o. Energy, DOE Technical Targets for Onboard Hydrogen Storage for Light-Duty Vehicles, 2017 Search PubMed.
- P. García-Holley, B. Schweitzer, T. Islamoglu, Y. Liu, L. Lin, S. Rodriguez, M. H. Weston, J. T. Hupp, D. A. Gómez-Gualdrón and T. Yildirim, Benchmark study of hydrogen storage in metal–organic frameworks under temperature and pressure swing conditions, ACS Energy Lett., 2018, 3(3), 748–754 CrossRef.
- S. V. Sawant, S. Banerjee, A. W. Patwardhan, J. B. Joshi and K. Dasgupta, Effect of in-situ boron doping on hydrogen adsorption properties of carbon nanotubes, Int. J. Hydrogen Energy, 2019, 44(33), 18193–18204 CrossRef CAS.
- W. Liu, Y. Zhao, Y. Li, Q. Jiang and E. Lavernia, Enhanced hydrogen storage on Li-dispersed carbon nanotubes, J. Phys. Chem. C, 2009, 113(5), 2028–2033 CrossRef CAS.
- Y.-J. Han and S.-J. Park, Influence of nickel nanoparticles on hydrogen storage behaviors of MWCNTs, Appl. Surf. Sci., 2017, 415, 85–89 CrossRef CAS.
- S. Kaskun and M. Kayfeci, The synthesized nickel-doped multi-walled carbon nanotubes for hydrogen storage under moderate pressures, Int. J. Hydrogen Energy, 2018, 43(23), 10773–10778 CrossRef CAS.
- M. Konni, N. Narayanam, A. S. Dadhich and S. B. Mukkamala, Effect of Reaction Media on Hydrogen Sorption Properties of Mg-Decorated MWCNTs, Fullerenes, Nanotubes and Carbon Nanostructures, 2015, 23(9), 782–787 CrossRef CAS.
- M. Konni, A. S. Dadhich and S. B. Mukkamala, Impact of surface modifications on hydrogen uptake by Fe@ f-MWCNTs and Cu@ f-MWCNTs at non-cryogenic temperatures, Int. J. Hydrogen Energy, 2017, 42(2), 953–959 CrossRef CAS.
- S. ullah Rather, Hydrogen uptake of cobalt and copper oxide-multiwalled carbon nanotube composites, Int. J. Hydrogen Energy, 2017, 42(16), 11553–11559 CrossRef.
- D. Silambarasan, V. Surya, V. Vasu and K. Iyakutti, Investigation of single-walled carbon nanotubes-titanium metal composite as a possible hydrogen storage medium, Int. J. Hydrogen Energy, 2013, 38(34), 14654–14660 CrossRef CAS.
- P. Singh, M. V. Kulkarni, S. P. Gokhale, S. H. Chikkali and C. V. Kulkarni, Enhancing the hydrogen storage capacity of Pd-functionalized multi-walled carbon nanotubes, Appl. Surf. Sci., 2012, 258(8), 3405–3409 CrossRef CAS.
- N. Karatepe and N. Yuca, Hydrogen adsorption on carbon nanotubes purified by different methods, Int. J. Hydrogen Energy, 2011, 36(17), 11467–11473 CrossRef CAS.
- A. Kakanakova-Georgieva, I. G. Ivanov, N. Suwannaharn, C.-W. Hsu, I. Cora, B. Pécz, F. Giannazzo, D. G. Sangiovanni and G. K. Gueorguiev, MOCVD of AlN on epitaxial graphene at extreme temperatures, CrystEngComm, 2021, 23(2), 385–390 RSC.
- A. Kakanakova-Georgieva, G. K. Gueorguiev, D. G. Sangiovanni, N. Suwannaharn, I. G. Ivanov, I. Cora, B. Pécz, G. Nicotra and F. Giannazzo, Nanoscale phenomena ruling deposition and intercalation of AlN at the graphene/SiC interface, Nanoscale, 2020, 12(37), 19470–19476 RSC.
- T. G. Mihopoulos, V. Gupta and K. F. Jensen, A reaction-transport model for AlGaN MOVPE growth, J. Cryst. Growth, 1998, 195(1–4), 733–739 CrossRef CAS.
- R. B. Dos Santos, R. Rivelino, F. de Brito Mota, A. Kakanakova-Georgieva and G. K. Gueorguiev, Feasibility of novel (H 3 C) n X (SiH 3) 3− n compounds (X= B, Al, Ga, In): structure, stability, reactivity, and Raman characterization from ab initio calculations, Dalton Trans., 2015, 44(7), 3356–3366 RSC.
- F. Giannazzo, G. Fisichella, G. Greco, P. Fiorenza and F. Roccaforte, Conductive Atomic Force Microscopy of Two-Dimensional Electron Systems: From AlGaN/GaN Heterostructures to Graphene and MoS2, Conductive Atomic Force Microscopy: Applications in Nanomaterials, 2017, pp. 163–185 Search PubMed.
- G. Rajasekaran, R. Kumar and A. Parashar, Tersoff potential with improved accuracy for simulating graphene in molecular dynamics environment, Mater. Res. Express, 2016, 3(3), 035011 CrossRef.
- H. Wu, D. Wexler, A. R. Ranjbartoreh, H. Liu and G. Wang, Chemical processing of double-walled carbon nanotubes for enhanced hydrogen storage, Int. J. Hydrogen Energy, 2010, 35(12), 6345–6349 CrossRef CAS.
- V. Gayathri and R. Geetha, Hydrogen adsorption in defected carbon nanotubes, Adsorption, 2007, 13, 53–59 CrossRef CAS.
- R. Saadi, Z. Saadi, R. Fazaeli and N. E. Fard, Monolayer and multilayer adsorption isotherm models for sorption from aqueous media, Korean J. Chem. Eng., 2015, 32, 787–799 CrossRef CAS.
- I. Langmuir, The constitution and fundamental properties of solids and liquids. Part I. Solids, J. Am. Chem. Soc., 1916, 38(11), 2221–2295 CrossRef CAS.
- W. Jianlong, Z. Xinmin, D. Decai and Z. Ding, Bioadsorption of lead (II) from aqueous solution by fungal biomass of Aspergillus niger, J. Biotechnol., 2001, 87(3), 273–277 CrossRef CAS PubMed.
- G. Garcıa, A. Faz and M. Cunha, Performance of Piptatherum miliaceum (Smilo grass) in edaphic Pb and Zn phytoremediation over a short growth period, Int. Biodeterior. Biodegrad., 2004, 54(2–3), 245–250 CrossRef.
- J. Zeldowitsch, Adsorption site energy distribution, Acta Physicochim. URSS, 1934, 1(1), 961–973 Search PubMed.
- H. Freundlich, Over the adsorption in solution, J. Phys. Chem., 1906, 57(385471), 1100–1107 Search PubMed.
- A. L. Myers and J. M. Prausnitz, Thermodynamics of mixed-gas adsorption, AIChE J., 1965, 11(1), 121–127 CrossRef CAS.
- B. Van der Bruggen, Membrane Technology, Kirk-Othmer Encyclopedia of Chemical Technology, 2017 Search PubMed.
- M. F. Hasan, E. L. First and C. A. Floudas, Cost-effective CO 2 capture based on in silico screening of zeolites and process optimization, Phys. Chem. Chem. Phys., 2013, 15(40), 17601–17618 RSC.
- L. Huang, L. Zhang, Q. Shao, L. Lu, X. Lu, S. Jiang and W. Shen, Simulations of binary mixture adsorption of carbon dioxide and methane in carbon nanotubes: temperature, pressure, and pore size effects, J. Phys. Chem. C, 2007, 111(32), 11912–11920 CrossRef CAS.
- C. Gu, G.-H. Gao, Y.-X. Yu and T. Nitta, Simulation for separation of hydrogen and carbon monoxide by adsorption on single-walled carbon nanotubes, Fluid Phase Equilib., 2002, 194, 297–307 CrossRef.
- X. Huang, H. Yao and Z. Cheng, Hydrogen separation membranes of polymeric materials, Nanostructured Materials for Next-Generation Energy Storage and Conversion: Hydrogen Production, Storage, and Utilization, 2017, pp. 85–116 Search PubMed.
- S. Balilehvand, S. M. Hashemianzadeh, S. Razavi and H. Karimi, Investigation of hydrogen and methane adsorption/separation on silicon nanotubes: a hierarchical multiscale method from quantum mechanics to molecular simulation, Adsorption, 2012, 18(1), 13–22 CrossRef CAS.
- H. Zhang, Q. Fu, H. Qin, X. Chen, Q. Zhang, Z. Dong, H. Li, S. Wang and M. Wang, An investigation for H2/N2 adsorptive separation in SIFSIX-2-Cu-i, Int. J. Hydrogen Energy, 2023, 48(67), 26251–26259 CrossRef CAS.
- S. Yousef, S. Tuckute, A. Tonkonogovas, A. Stankevičius and A. Mohamed, Ultra-permeable CNTs/PES membranes with a very low CNTs content and high H2/N2 and CH4/N2 selectivity for clean energy extraction applications, J. Mater. Res. Technol., 2021, 15, 5114–5127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08382a |
| This journal is © The Royal Society of Chemistry 2024 |