
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic acceleration of machine learning and molecular docking for prostate-specific antigen ligand design†
Shao-Long Lina,
Yan-Song Chenb,
Ruo-Yu Liua,
Mei-Ying Zhuc,
Tian Zhuc,
Ming-Qi Wang *b and
Bao-Quan Liu*a
*b and
Bao-Quan Liu*a
aDepartment of Bioengineering, College of Life Science, Dalian Minzu University, Dalian 116600, China. E-mail: lbq@dlnu.edu.cn
bSchool of Pharmacy, Jiangsu University, 212013, Zhenjiang, PR China. E-mail: wmq3415@163.com
cBeijing Bionaxin Biotech Co, 1000000, Beijing, China
First published on 13th March 2024
Abstract
Prostate-specific antigen (PSA) serves as a critical biomarker for the early detection and continuous monitoring of prostate cancer. However, commercial PSA detection methods primarily rely on antigen–antibody interactions, leading to issues such as high costs, stringent storage requirements, and potential cross-reactivity due to PSA variant sequence homology. This study is dedicated to the precise design and synthesis of molecular entities tailored for binding with PSA. By employing a million-level virtual screening to obtain potential PSA compounds and effectively guiding the synthesis using machine learning methods, the resulting lead compounds exhibit significantly improved binding affinity compared to those developed before by researchers using high-throughput screening for PSA, substantially reducing screening and development costs. Unlike antibody detection, the design of these small molecules offers promising avenues for advancing prostate cancer diagnostics. Furthermore, this study establishes a systematic framework for the rapid development of customized ligands that precisely target specific protein entities.
Introduction
Prostate cancer ranks as the second most prevalent malignancy among men on a global scale and stands as the fifth leading cause of cancer-related mortality. Timely screening and precise diagnosis are pivotal facets of cancer prevention and therapeutic intervention.1–4 Prostate-specific antigen (PSA) is a pivotal biomarker for both the early diagnosis and ongoing monitoring of prostate cancer, exhibiting an estimated sensitivity of 0.88 for the detection of prostate cancer.5 This biomarker can be divided into various subclasses, including complex PSA (cPSA), free PSA (fPSA), and the PSA homology-specific antigen, specifically the isoform [-2] proprostate-specific antigen (p2PSA). Within the realm of prostate cancer diagnostics, there has been a growing emphasis on exploring and understanding these diverse PSA markers.6,7PSA, an enzyme of the serine protease class, is secreted by prostate epithelial cells and presents as a glycoprotein composed of 237 amino acid residues.4 Prostate-specific antigen is also a natural constituent of semen, where its primary function lies in facilitating the hydrolysis and liquefaction of semen clots, thereby aiding in the release of sperm.8 The Prostate Health Index (PHI) and other composite indices evaluating prostate health, incorporating comprehensive assessments of cPSA, fPSA, and p2PSA, have significantly enhanced the detection rate of prostate cancer.7
Currently, a diverse array of PSA detection chips and assay kits is commercially available, primarily relying on the principle of antigen–antibody specific recognition. However, it is important to note that antibodies used in these assays are associated with considerable costs, necessitate stringent low-temperature storage conditions, and contribute to elevated detection expenses.9 Concurrently, the considerable amino acid sequence homology among cPSA, fPSA, and p2PSA poses a significant challenge, as it can readily lead to antibody cross-reactivity, a phenomenon readily discerned when employing these aforementioned chips and kits.10,11
Interface diversity is crucial for understanding protein functions, designing drug molecules, and developing new antibodies.12,13 It enables proteins to interact with a variety of partners, thus participating in a wide range of biological processes. For instance, ab initio docking methods for antibodies play a significant role in leveraging interface diversity, especially in the recognition of prostate-specific antigens.14 Although antibody recognition is a commonly used approach, exploiting the potential of interface diversity for the combined use of ligands and antibodies in recognizing prostate-specific antigens could offer innovative potential.
Virtual screening has emerged as a pivotal tool within the domain of small molecule development, encompassing applications in drug development and the preparation of molecular probes. This computational approach comprises two fundamental strategies: structure-based design and ligand-based design. Structure-based design capitalizes on the spatial arrangement of proteins and ligands, as well as the interactions with local residues, to construct a lock-and-key model for guiding drug development and molecular probe formulation.15 In contrast, ligand-based design encompasses methodologies such as Quantitative Structure–Activity Relationship (QSAR) and pharmacophore modeling, aiming to identify ligand structures exhibiting structural or functional similarities to known ligands16 The integration of both structure-based and ligand-based virtual screening methodologies has demonstrated notable success in drug development endeavors, thereby enhancing the likelihood of successfully identifying target compounds.17,18 While direct utilization of open source databases like ZINC enables the screening of a vast array of small molecule compounds with binding potential to target proteins, challenges arise when dealing with unknown proteins. In such cases, the establishment of effective pharmacophore models becomes impractical, thereby necessitating substantial efforts for compound property verification. Furthermore, it is worth noting that many compounds within these open databases are characterized by their intricate synthesis requirements and high production costs.19
This research endeavors to elucidate the design and synthesis of a small molecule compound ligand tailored to the distinct binding pocket of prostate-specific antigen (PSA). We employed a comprehensive analytical approach, which incorporated a synergistic integration of Virtual Screening and Machine Learning methodologies, supported by a diverse spectrum of computational and empirical methods, including Surface Plasmon Resonance (SPR), Microscale Thermophoresis (MST), Circular Dichroism (CD), and Molecular Dynamics (MD) simulations, to meticulously evaluate the binding affinity of the synthesized compound with the target protein (refer to Fig. 1).20–23 This endeavor has yielded a promising lead compound characterized by a robust binding affinity, holding significant potential for advancing the diagnosis and treatment of prostate cancer. Moreover, it lays the groundwork for a viable development pathway for designing ligands targeted at specific protein. This multifaceted approach not only facilitates the development of a lead compound for prostate cancer management but also exemplifies a systematic framework for the rational design and synthesis of binding ligands tailored to target proteins.
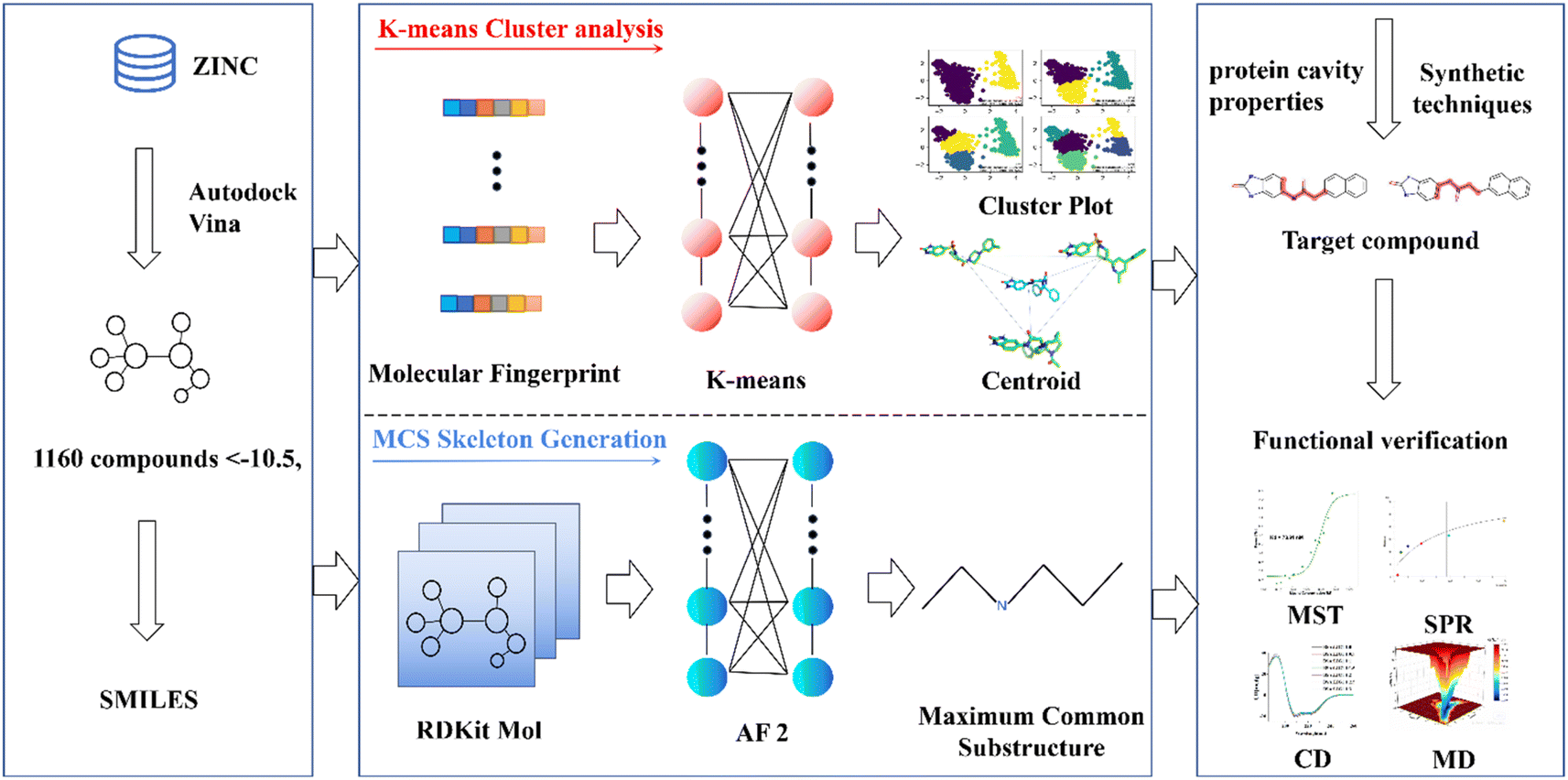 | ||
| Fig. 1 Pipeline for PSA ligand design and validation. This schematic illustrates the comprehensive methodology employed, encompassing highscoring docking experiments, K-means cluster analysis, the extraction of core structural motifs utilizing the Maximum Common Substructure (MCS) approach, exploration of compound databases, and meticulous functional validation assays. These steps collectively facilitate the identification and characterization of specific binding ligands tailored to prostate-specific antigen (PSA). | ||
Materials and methods
Virtual screening
In pursuit of a comprehensive virtual screening of potential ligands, we initiated our investigation by acquiring the prostate-specific antigen (PDB: 3qum) crystal structure, featuring a resolution of 3.2 Å, from the Protein Data Bank (PDB) (https://www.wwpdb.org/).4 Due to the limited availability of crystallized structures of prostate-specific antigen, a key reason for our selection of this protein is that it represents one of the few crystal structures used in market-available reagent kits and chips, employing the antigen–antibody sandwich method. To prepare the protein structure for subsequent analysis, we employed Autodock Tools (ADT), a software suite renowned for its utility in molecular docking studies. ADT was employed to eliminate extraneous water molecules within the protein chain and to introduce hydrogen atoms as required. Subsequently, we accessed the ZINC database (https://zinc.docking.org/) to download a vast collection of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 511709 small molecules, which were then subjected to Autodock (version 2.5) with a grid spacing of 0.375 Å, set between 2.5 Å and 3.5 Å, to prepare the ligands in the Autodock Vina-friendly PDBQT format.24,25
511709 small molecules, which were then subjected to Autodock (version 2.5) with a grid spacing of 0.375 Å, set between 2.5 Å and 3.5 Å, to prepare the ligands in the Autodock Vina-friendly PDBQT format.24,25
For the exploration of potential binding sites within the prostate-specific antigen, we harnessed the capabilities of the PlayMolecule BindScope module. This innovative module leverages voxelization techniques to delineate binding pockets and ligand poses based on distinctive pharmacophore class attributes. Notably, it employs a 3D convolutional neural network (CNN) to predict binding affinities, thus enhancing the precision of our virtual screening efforts.26 Furthermore, we utilized drugs targeting prostate-specific antigen developed by scientists like LeBeau AM for the full protein docking of prostate-specific antigen. This aligns with the cavity prediction by the aforementioned software, therefore, we are confident in designating this cavity as the one for virtual screening.27
In the realm of virtual screening, Autodock Vina emerged as our tool of choice. It capitalizes on a global optimization algorithm, a marked improvement over the genetic algorithm employed by AutoDock 4, thereby bolstering search efficiency and result accuracy. Moreover, Autodock Vina offers support for multithreaded parallel processing, augmenting computational throughput.
Our computational endeavors were executed on a robust 192-core Linux server, enabling the completion of all docking simulations within a time frame of 30 days. The simulation parameters were set with a box size of 60 × 60 × 60 Å3, centered at coordinates X = −30.53, Y = −30.375, and Z = −24.965. A search accuracy level of exhaustiveness = 32 was employed, and the analysis focused on the ten best docking poses selected from the screening results.
K-means clustering
K-means clustering is a prominent technique within the domain of unsupervised machine learning, representing one of the prevailing methods for data clustering in contemporary research. Additionally, k-means is widely applied in pharmaceutical development for drug structure clustering.28,29 This algorithm centers on the identification of cluster centroids, with data points gravitating toward these centroids. Consequently, clusters are formed as a result of this process.30,31 This paper employs the following steps for K-means clustering:Initially, the SMILES representations of the selected compounds postdocking are converted into molecular fingerprints using the RDK software. Additionally, a binary coding or counting vector representation is created for the SMILES notation of chemical and physical properties, en-compassing molecular weight, polarity, charge distribution, stereochemistry, and other relevant features.32 Principal Component Analysis (PCA) is subsequently employed to reduce the dimensionality of the data, ultimately resulting in a 166 bit binary evaluation index serving as the input file for K-means cluster analysis.33
Utilizing the generated algorithm input file, a random function is formulated to select the initial cluster centers for the clustering process. The Euclidean distance metric is then utilized to determine the nearest cluster center for each data point, thereby minimizing the sum of distances between each data point and its respective cluster center. The first iteration produces the initial cluster distribution by assigning data points to the nearest cluster centers. Subsequently, the mean value of data points within each cluster is calculated to establish new cluster centers and minimize intracluster variance. This step is iteratively applied until the cluster distribution stabilizes, indicating the completion of the clustering process.30
In this study, the K-means clustering model adopts the Euclidean distance metric for assigning data points to their respective clusters. The calculation formula is as follows:
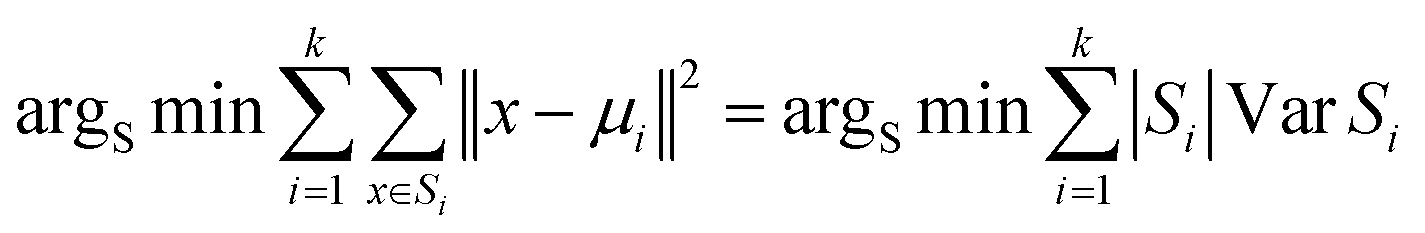 | (1) |
To evaluate the quality of the clustering process and assess the cohesion and separation of the clustering outcomes, the Silhouette Coefficient and Calinski–Harabasz Index are employed as performance indicators.
The Silhouette Coefficient is calculated as follows: For each data point i, the average Euclidean distance between i and other members within the same cluster is computed as a(i). Additionally, b(i) is determined, representing the average Euclidean distance from data point i to all members within other clusters. Subsequently, the silhouette coefficient for each data point i is calculated as follows:
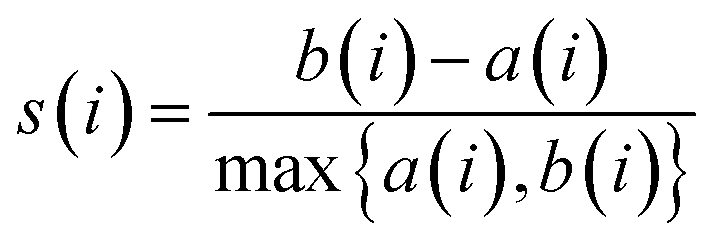 | (2) |
For the entire cluster, the average silhouette coefficient is computed, with n signifying the total number of cluster members:
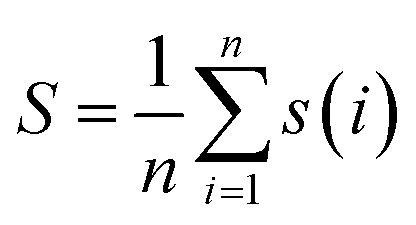 | (3) |
The Silhouette Coefficient yields values within the range [−1, 1]. A higher Silhouette Coefficient indicates superior clustering results, implying that data points are tightly clustered within their respective clusters while maintaining clear separation from other clusters. A coefficient closer to 1 signifies optimal clustering outcomes, while a value near 0 suggests potential overlap among clusters. Conversely, a coefficient near −1 indicates potential misclustering of data points.31
The Calinski–Harabasz Index is computed as follows:
The inter-class dispersion B of the clusters is evaluated. Inter-class dispersion measures the difference between each cluster and the sum of squared distances between all cluster members and the overall average
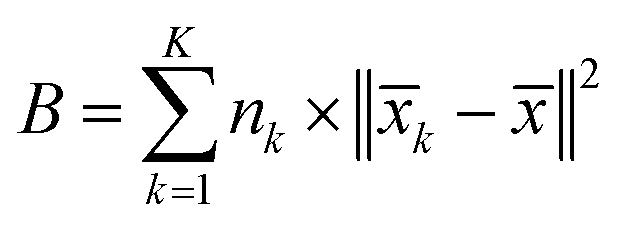 | (4) |
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) k signifies the average of all members in the cluster, and represents the overall average across all clusters.
k signifies the average of all members in the cluster, and represents the overall average across all clusters.
Maximum common substructure identifying
The Rdkit package, available at https://www.rdkit.org/ and specifically the version 2019.03.4.0, plays a crucial role in transforming SMILES notation into graph representations.32 Within this framework, the rdFMCS module is harnessed to identify the Maximum Common Substructure (MCS) within a set of diverse molecules. In the pursuit of this goal, a pairwise comparison strategy is employed, leveraging the graph matching algorithm VF2 to pinpoint the Common Subgraph (CSG) shared between each pair of molecules.To iteratively expand the subgraph, all CSGs are amalgamated, adhering to the constraint of preserving the molecular connectivity. The parameters -atomcompare- and -bondCompare- serve as pivotal tools for evaluating atom and bond equivalences, respectively. Through the meticulous application of these parameters, the framework succeeds in extracting the Maximum Common Subgraph (MCSG). Ultimately, this process culminates in the extraction of the compound's skeletal characteristics, thereby facilitating in-depth analysis and further scientific inquiry.34
Surface plasmon resonance (SPR)
Surface Plasmon Resonance (SPR) is a sensitive and widely employed optical technique for realtime monitoring of biomolecular interactions occurring at the surface of a sensor chip. This study employed the Biacore T200 system, manufactured by GE Healthcare Life Sciences in Uppsala, Sweden, to rigorously quantify the interaction dynamics between the test compound and the designated target protein. To initiate the analysis, the purified target protein was immobilized directly onto a Carboxymethylation 5 (CM5) sensor chip. Subsequently, analytes consisting of small molecules at varying concentrations were introduced into the system for multicycle kinetic assessment.20To prepare the CM5 chip for target protein immobilization, the carboxylic acid groups on the chip's surface (Cytiva) were initially evaluated using a solution comprising a mixture of EDC and NHS (Cytiva) at a temperature of 25 °C and a flow rate of 10 μL min−1. This activation process spanned a duration of 7 minutes. Following activation, the chip was subjected to an injection of a sodium acetate soluble buffer (10 mM; pH 4.5) containing the target protein until the protein content immobilized on the chip reached a level of 2500 resonance units (RU). Ultimately, the multicycle kinetics of the chip were quenched using ethanolamine.35
Subsequently, the target protein was securely anchored to the CM5 chip, and the gradient-diluted analytes were consecutively introduced, with each concentration examined during a distinct cycle. During these experiments, solvent corrections were diligently applied using DMSO to ensure precision in the evaluation of affinity and kinetics. The concentration series of small molecules tested included the following values: 0, 1.25, 2.5, 5, 10, and 20 nM. The experimental conditions were maintained at a temperature of 25 °C, a flow rate of 30 μL min−1, a binding duration of 90 seconds, a dissociation duration of 60 seconds, and a running buffer comprising 5% DMSO in PBS-PSA.
Micro thermal diffusion (MST)
MST (Micro Thermal Diffusion) is an optical method used to characterize the properties of biological molecules, specifically the directed motion of particles within microscopic temperature gradients. This technique involves the labeling of one of the interacting molecules, typically proteins, with fluorescent dyes or the fusion of a GFP (Green Fluorescent Protein) tag. The labeled proteins and ligand molecules are placed in a capillary with specific concentration gradients. A microscale temperature gradient is generated by infrared laser heating, causing thermophoretic motion. This results in changes in the molecular properties such as hydration shell, molecular size, and charge, which subsequently lead to variations in the fluorescence distribution within the reaction system. The MST instrument records changes in fluorescence within the infrared laser-illuminated region inside the sample during the laser's on/off states, allowing for the determination of binding affinities within a relatively short period of time.21 In this experiment, the Monolith NT.115 instrument was employed to detect interactions between compounds and target proteins. Initially, purified proteins were labeled using the MonolithTM RED-NHS second-generation protein labeling kit. The RED dye carried by the NHS ester in the labeling rea-gent can covalently bind to primary amino groups (lysine residues) on the target protein. Subsequently, solutions of the test compounds at 16 gradient concentrations were prepared, and 20 μL volumes of each were mixed thoroughly with the RED-NHS-labeled target proteins.36 The mixed samples were then drawn into a capillary for MST experiments. The MO.Affinity Analysis X86 software was used to fit the MST curve and obtain the binding constant (Kd) values.21Circular dichroism
Circular Dichroism (CD) spectroscopy is a powerful analytical technique employed in the field of structural biology and biophysics. It is used to investigate the secondary structure of biomolecules, particularly proteins and nucleic acids, by studying their differential absorption of left and right circularly polarized light. CD spectroscopy provides valuable insights into the conformational characteristics, stability, and folding of biomolecules, making it an essential tool for researchers in understanding their structural and functional properties.23The CD experiment was conducted using the Chirascan circular dichroism spectrometer produced by Applied Photophysics. The cuvette had a volume of 400 μL, with a PSA concentration of 2 μM. The buffer solution used was PBS with a concentration of 10 mM and a pH of 7.4. During the experiment, a scanning wavelength range of 180–260 nm was employed. In the cuvette, 1.5 μL of a 100 μM small molecule was added sequentially with thorough mixing between additions. Circular dichroism spectra were recorded after each compound addition to investigate the impact of compound titration on the protein.
The obtained results were subjected to analysis using the Circular Dichroism by Neural Networks (CDNN) software. CDNN utilizes neural networks to predict the secondary structure of proteins based on CD spectroscopy data.37
Molecular dynamics simulations
The molecular dynamics simulations described in this study were conducted using the Gromacs 2022.3 software version.38,39 Small molecules were prepared with the General Amber Force Field (GAFF) using AmberTools22, and hydrogen atoms were added and RESP charges were computed using Gaussian 16W.34,35 The calculated potential energy parameters were subsequently integrated into the molecular dynamics system topology file. Simulations were carried out under static conditions at a temperature of 300 K and atmospheric pressure (1 Bar). The Amber 99SB force field was employed, and the solvent consisted of water molecules (Tip3p water model). Sodium ions (Na+) were introduced to neutralize the overall charge of the simulated system. The system underwent initial energy minimization using the steepest descent method, followed by separate equilibration phases: 100000 steps of isothermal-isochoric ensemble (NVT) equilibration and 100000 steps of isothermal-isobaric ensemble (NPT) equilibration, with a coupling constant of 0.1 ps and a duration of 100 ps. Subsequently, a production molecular dynamics simulation was performed, comprising a total of 15000000 steps with a time step of 2 fs, resulting in a total simulation time of 300 ns. After completing the simulation, trajectory analysis was performed using builtin tools in the software, including root mean square deviation (RMSD), root mean square fluctuation (RMSF), and Molecular Mechanics-Generalized Born Surface Area (MMGBSA) calculations, as well as the generation of free energy landscapes.
The Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) method is widely employed for the analysis of binding affinities between proteins and ligands.40 Its binding free energy calculation formula includes contributions from van der Waals interactions, electrostatic interactions, polar solvation, and solvent-accessible surface area (SASA). Free energy landscapes provide insights into the interactions and energy distribution among molecules within the system, aiding in the understanding of molecular interactions, conformations, and stability characteristics. Typically, free energy values are represented using color intensity and contour lines, with the X-axis representing RMSD and the Y-axis representing R(g) (Radius of Gyration). R(g) is a measure of molecular volume and compactness, where smaller R(g) values indicate a compact molecular structure and larger R(g) values indicate a more extended or loose molecular structure.
Results
Molecular selection
In order to identify potential ligand molecules specific to prostate cancer antigens, this study employed virtual screening using Autodock Vina. A total of 1511709 small molecules from the ZINC database were screened. In ESI Table S1,† we have submitted docking scores for 1160 molecules. The selected compounds demonstrate docking scores within the controllable error margin of Autodock Vina (less than 1.5 kcal mol−1) and are superior to the docking score of −8.9 kcal mol−1 for the already discovered drug, Drug 1.27,41 The extensive data available in the ZINC database offers a diverse range of compounds, increasing the possibilities for selecting target molecules. However, two main challenges were encountered: first, conducting property studies and synthesis for all 1160 screened compounds would be excessively demanding in terms of resources; second, many of these compounds were difficult to synthesize.
To streamline the characterization and synthesis efforts following virtual screening, K-means clustering analysis was applied to the 1160 compounds with different cluster numbers (K = 2, 3, 4, 5). The results, as depicted in the Fig. 2, show Silhouette Coefficient values ranging from −1 to 1, with larger values indicating better clustering performance. The Silhouette Coefficient was highest at 0.60 for K = 2, making it the optimal choice among the various clustering options. Subsequently, the compounds of Cluster I, identified through this analysis, were used for further investigation.
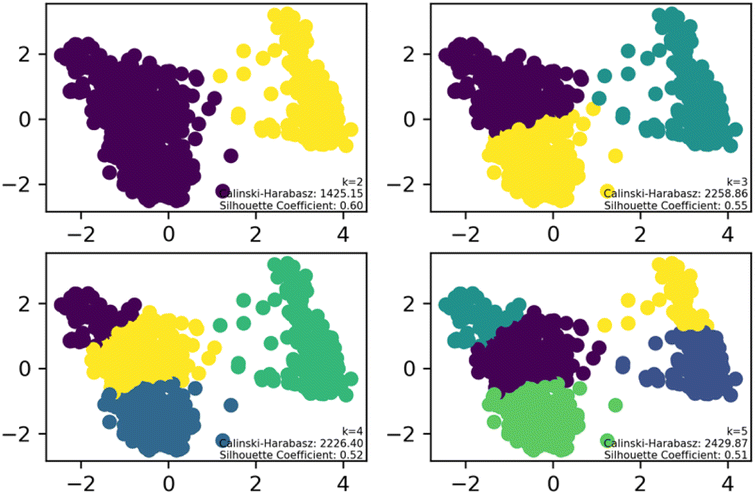 | ||
| Fig. 2 K-means clustering analysis results for small molecules. | ||
To characterize the central compounds within the clusters, the Euclidean distance formula was utilized to calculate the cluster center for Cluster I (ZINC000013683205) and its surrounding compounds, namely ZINC000028333181, ZINC000257278584, and ZINC000257297002, as shown in Fig. 3B. Subsequent sections will provide a detailed analysis of these four structures, including molecular similarity and their interactions with protein cavities.
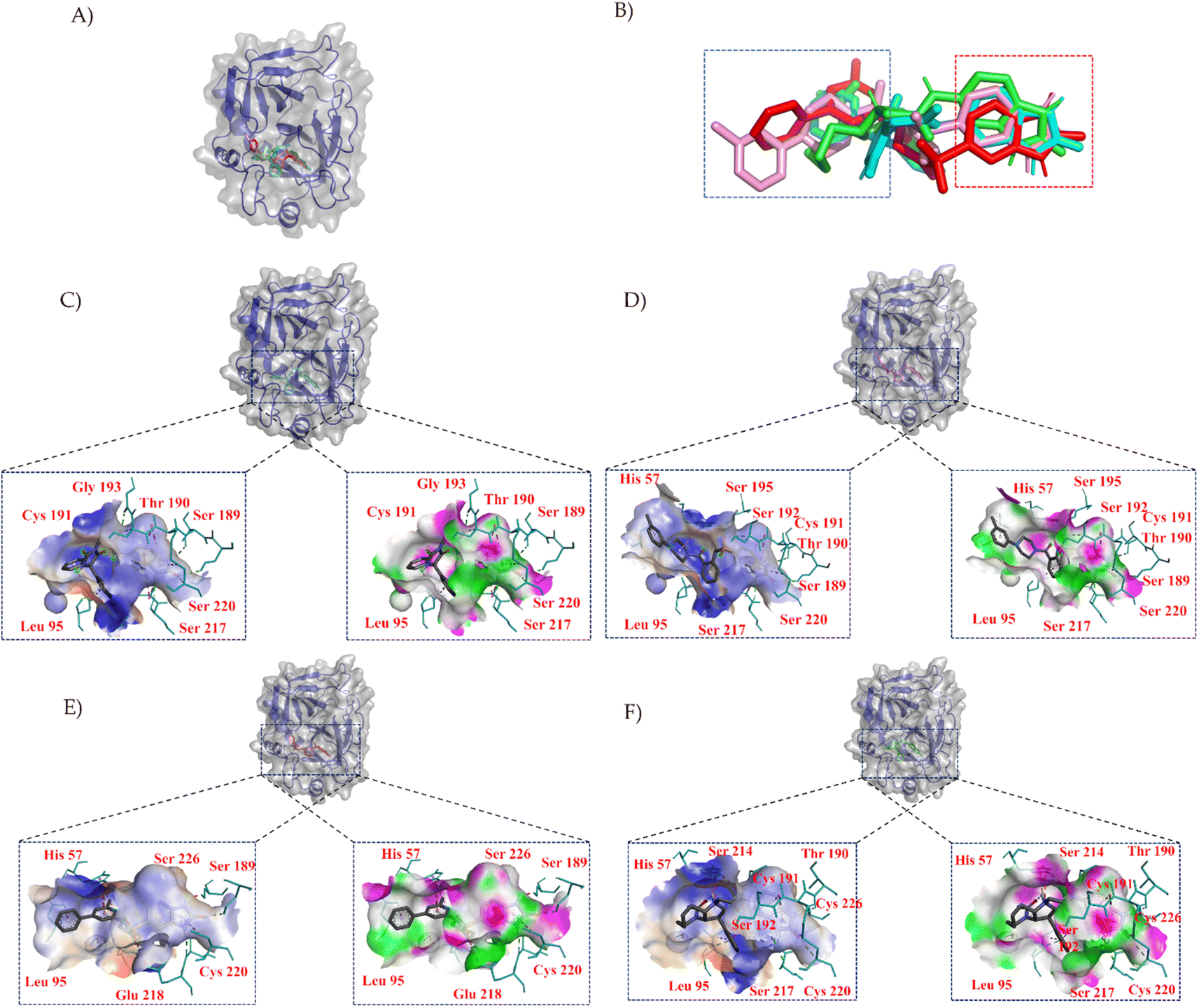 | ||
| Fig. 3 Binding poses of compounds within the protein pocket. (A) Depicts the binding orientations of the central compound and its surrounding compounds in the clustering analysis. (B) Presents the molecular overlay of the compounds. Subfigures (C) to (F) represent the binding interactions of compound 1, compound 2, compound 3, and compound 4 within the pocket, respectively. Hydrophilic residues are shown in blue, while hydrophobic residues are depicted in orange, with color intensity indicating the strength of interaction. Hydrogen bond acceptors are indicated in green, and hydrogen bond donors are represented in pink. Cyan highlights the key amino acid residues involved in the interaction between compounds and the protein. Key chemical interactions are denoted by green (hydrogen bonds) and pink (hydrophobic interactions). | ||
In Fig. 3, we employed PYMOL for visualization.42 It can be observed that these four compounds bind to the same cavity. The compounds ZINC000013683205, ZINC000028333181, ZINC000257278584, and ZINC000257297002, enclosed in the red box, share similar structural motifs, benzimidazolone. This region is primarily involved in hydrogen bond interactions, driven by the structural similarities of these molecules. The structures enclosed in the blue box exhibit greater structural diversity and pose greater synthesis challenges. The blue box primarily consists of hydrophobic groups based on benzene rings, with hydrophobic interactions being the dominant mode of interaction, as indicated in the accompanying table. The central part of the structure includes amide and other structural elements contributing to fewer interactions, suggesting its role as a linker between hydrogen bonding moieties and variable hydrophobic groups.
Due to the sensitivity of the K-means algorithm to initial centroid selection, there is a risk of converging to local optima. This study combined the features of central compounds and their surrounding compounds, along with insights into the cavity's structural characteristics, to further deduce the skeletons of the 1160 compounds, aiding in guiding compound synthesis. The maximum common substructure (MCS) for this cluster was established by employing rdkit software and the VF2 graph matching algorithm on the results of the K-means clustering (K = 2). Ultimately, two maximum common substructures were identified (Fig. S1A and B†).
Compound synthesis and binding affinity validation
In accordance with critical residues, binding cavity characteristics, and structural features, we designed new molecules with potent binding affinities and relatively low synthetic complexities for PSA, as depicted in Fig. 4A and B. The molecular skeleton of compound 1 was retained, along with the preservation of groups that form hydrogen bonds similar to those in the central compound. For the variable hydrophobic regions, a simpler substitution with biphenyl groups was employed, resulting in LIG 1 and 2 as illustrated in Fig. 4A and B.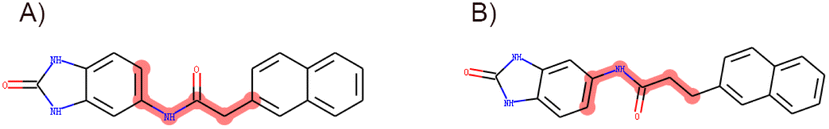 | ||
| Fig. 4 Maximum common substructures selected by K-means clustering and newly designed molecules, with (A) and (B) showcasing the newly designed compounds LIG1 and LIG2, respectively. | ||
The synthesized compounds conform to the key functional group requirements, skeleton criteria, binding cavity specifications, and exhibit lower synthetic complexities, as depicted in Fig. S2.† The nuclear magnetic resonance spectra of these molecules are provided in ESI Fig. S3.†
In the SPR experiment, all obtained data underwent kinetics/affinity fitting analysis, employing a 1:1 binding model. Data analysis was conducted utilizing Biacore evaluation software (version T200 2.0). The results revealed a binding affinity of 59.33 nM between LIG 1 and PSA, whereas the binding affinity between LIG2 and PSA was determined to be 144 nM (Fig. 5A–D). This observation signifies that LIG 1 demonstrates a notably higher binding affinity towards PSA.
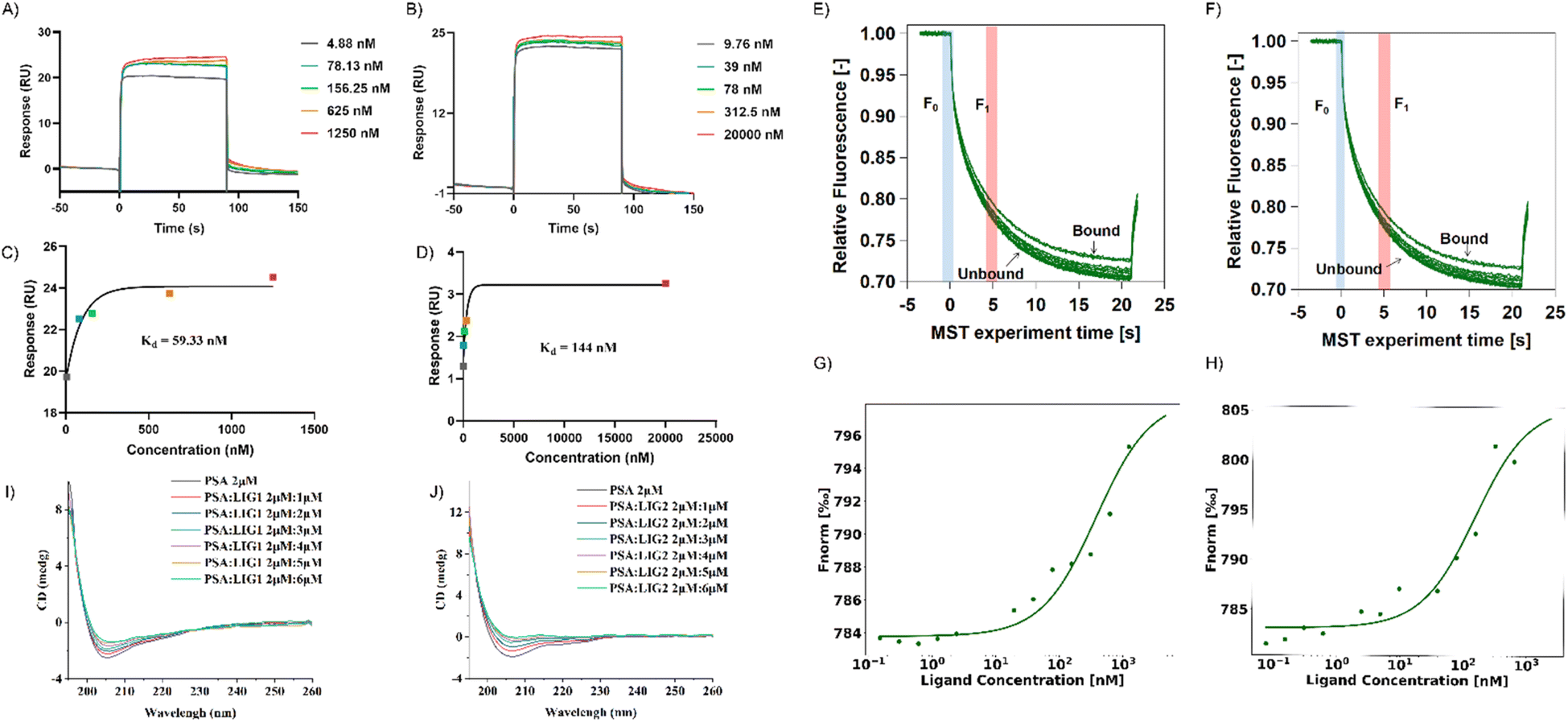 | ||
| Fig. 5 Validation experiments of protein-molecule affinity. (A–D) Surface plasmon resonance experiments. (E–H) Micro thermal diffusion experiments. (I and J) Circular dichroism titrations of LIG 1 and 2 with PSA. | ||
In contrast to SPR principles, MST relies on the influence of temperature gradients on the diffusion behavior of molecules in solution. When two molecules bind or dissociate, their diffusion behavior in a temperature gradient undergoes changes. In this study, we validated the binding of PSA with its ligands in a multisystem approach, providing more comprehensive and reliable results. MST results showed that between concentrations of 0.15 nM to 5 μM, as the concentrations of LIG 1 and LIG 2 increased, the fluorescence signal gradually intensified, indicating that both LIG 1 and LIG 2 could bind to PSA (Fig. 5G and H). The binding affinity between LIG 1 and PSA was determined to be 78.91 nM, while the bind-ing affinity between LIG 2 and PSA was 106.31 nM, indicating that both LIG 1 and LIG 2 possess strong binding affinities, with LIG1 exhibiting stronger binding to PSA.
To further explore the impact of synthesized compounds on the spatial structure of PSA, circular dichroism (CD) titration experiments were conducted. As shown in Fig. 5I and J, it can be observed that upon addition of the compounds, the negative peak height of PSA in the range of 200 nm to 210 nm decreased with increasing compound concentration. This change was further analyzed using CDNN software to assess the impact of compound addition on protein structure. As depicted in Fig. S4,† in the absence of compounds, beta sheets constituted the major part of the protein, accounting for approximately 44% ± 1%, while alpha helices were the least prevalent, comprising approximately 23%. Upon the addition of LIG 1, there was a slight decrease in beta sheet structure by approximately 1%, and when the ratio of alpha helix to PSA and LIG 1 was 1:3, there was a reduction of 3%, resulting in an alpha helix composition of approximately 15%, along with an increase in random coil structure by 5%, accounting for approximately 13.5% of the final structure (as shown in S4A†). In the case of LIG 2 addition, there was a slight increase of approximately 1% in beta sheet structure, and when the ratio of alpha helix to PSA and LIG 2 was 1:3, there was a reduction of 5%, resulting in an alpha helix composition of approximately 29%, along with an increase in random coil structure by 5%, accounting for approximately 12.8% of the final structure (as shown in Fig. S4B†). In summary, given the structural similarity between LIG 1 and LIG 2, their impact on protein structure exhibited a consistent trend, as both compounds significantly altered the PSA structure by converting alpha helices into random coil configurations upon binding with PSA.
Molecular docking and molecular dynamics simulation
LeBeau AM and other scientists used high-throughput screening methods to select some compounds at the same site of prostate-specific antigen. We analyzed two of these compounds that demonstrated better binding effects, and their 2D structures are included in Fig. S5.† Furthermore, in our opinion, the pathways for synthesizing these compounds are challenging. Therefore, we utilized molecular docking with the same criteria as in virtual screening as a more convenient method for comparing binding affinities. We also used molecular dynamics simulations to evaluate the stability of the compounds' binding with PSA. In order to investigate the distinctions in the interaction forces and modes of action between the synthesized compounds 1 (LIG 1), compounds 2 (LIG 2), and the Drug 1 and Drug 2 as reported in the literature with respect to PSA (Prostate-specific antigen), molecular docking was employed for comparative analysis in this study. The docking results are summarized in Table 1. The docking poses of the four compounds are depicted in Fig. 6A–D. Specifically, LIG 1 forms hydrogen bonds with Thr 190, Ser 226, and Ser 217, with bond distances of 2.88 Å, 3.17 Å, and 3.01 Å, respectively. Additionally, it exhibits hydrophobic interactions with His 57, Leu 95, Cys 191, Ser 192, Gly 216, Cys 220, and others. LIG 2 forms hydrogen bonds with Thr 190, Gly 193, and Ser 217, with bond distances of 3.14 Å, 3.26 Å, and 3.20 Å, respectively. It also engages in hydrophobic interactions with His 57, Leu 95, Asp 102, Cys 191, Cys 220, Gly 216, and others. Drug 1 forms hydrogen bonds with His 57, Thr 190, Gly 193, Ser 195, and Ser 226, with bond distances of 3.19 Å, 2.98 Å, 3.13 Å, 2.78 Å, and 2.84 Å, respectively. Furthermore, it exhibits hydrophobic interactions with Leu 95, Cys 191, Ser 192, Cys 220, Trp 215, and others. Drug 2 forms hydrogen bonds with Ala 39, Tyr 94, Ser 195, and Ser 226, with bond distances of 3.12 Å, 3.09 Å, 3.28 Å, and 3.04 Å, respectively. It also engages in hydrophobic interactions with Leu 95, Thr 190, Tyr 215, Ser 217, Glu 218, etc. Based on the docking scores, the synthesized LIG 1 and 2 exhibit scores of −10.8 and −10.3, respectively, which are higher than those of the literature-reported Drug 1 and Drug 2 with scores of −8.9 and −8.1, indicating a stronger binding affinit.43–45| ID | Docking score (kcal mol−1) | Hydrophobic residues | Hydrogen bond residues |
|---|---|---|---|
| LIG 1 | −10.8 | His 57, Leu 95, Cys 191, Ser 192, Gly 216, Cys 220 | Ser 189, Thr 190, Ser 217 |
| LIG 2 | −10.3 | His 57, Leu 95, Asp 102, Cys 191, Cys 220, Gly 216 | Thr 190, Ser 193, Ser 217 |
| Drug 1 | −8.9 | Leu 95, Cys 191, Trp 215 | His 57, Thr 190, Ser 192, Gly 193, Ser 195, Cys 220, Ser 226 |
| Drug 2 | −8.1 | Leu 95, Thr 190, Try 215 | ALA39, His 57, Ser 99, Thr 190, Ser 192, Gly 216 |
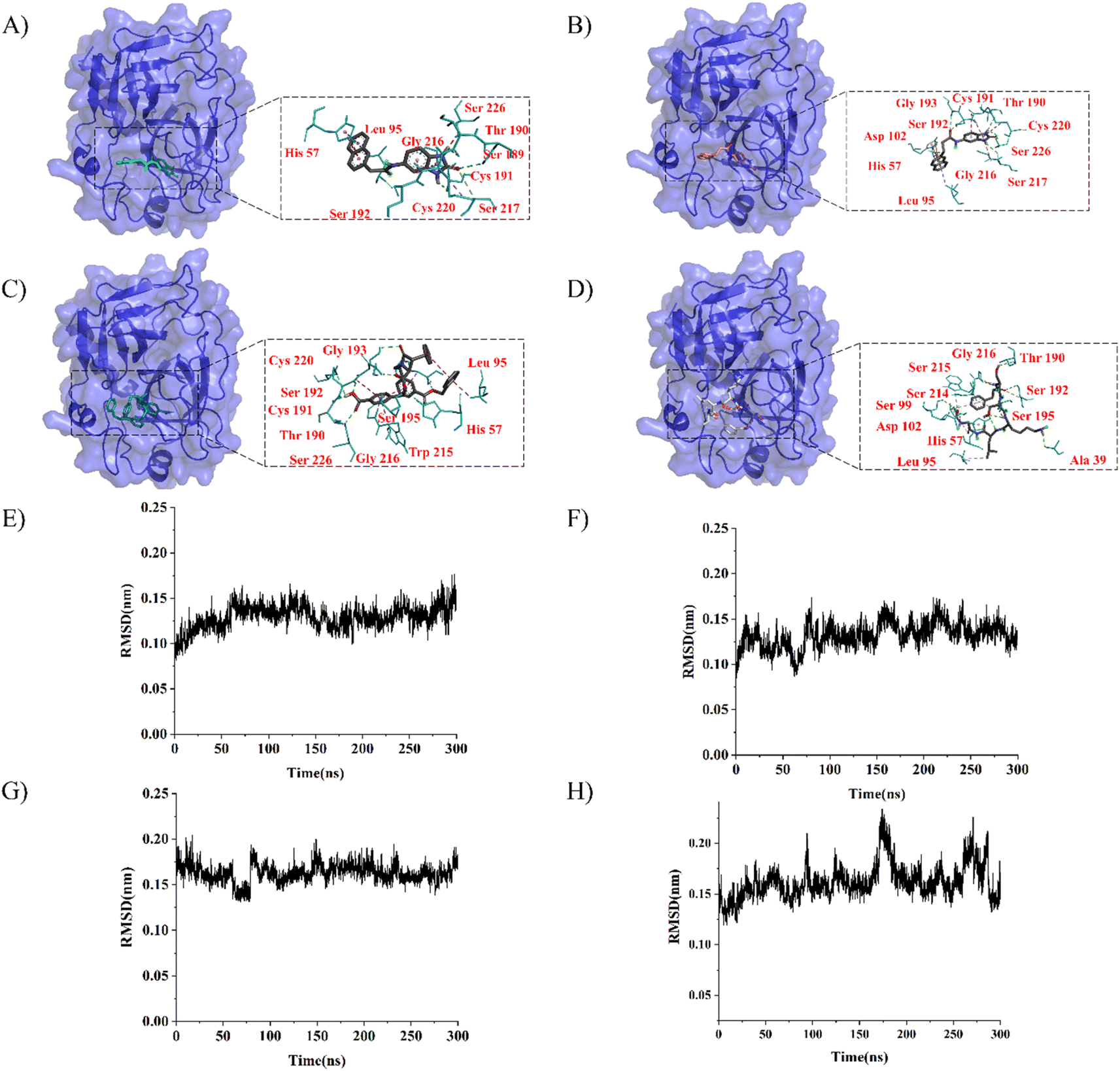 | ||
| Fig. 6 The docking and molecular dynamics simulation results of four compounds were visualized using PYMOL. (A)–(D) Docking poses of LIG 1, LIG 2, Drug 1 and Drug 2. (E)–(H) Represent the Root Mean Square Deviation (RMSD) values during a 300 nanosecond molecular dynamics simulation of the complex structures of LIG 1, LIG 2, Drug 1 and Drug 2, respectively. | ||
To further evaluate the stability of compound-PSA-ligand complexes and ligand conformations, various indicators including RMSD (Root Mean Square Deviation), RMSF (Root Mean Square Fluctuation), and MMPBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) were assessed in this study (Fig. 6E–H). Simulations for all structures were carried out over a 300 ns timescale. The RMSD data reveals that the average RMSD values for LIG1 and LIG2 are 0.1293 nm and 0.1321 nm, with standard deviations of 0.0132 nm and 0.0144 nm, respectively. For Drug 1 and Drug 2, the average RMSD values are 0.1628 nm and 0.1621 nm, with standard deviations of 0.0165 nm and 0.0170 nm, respectively. Lower RMSD values indicate greater system stability, and both the average and variance values for LIG 1 and 2 are lower than those for Drug 1 and Drug 2, suggesting better stability. Overall, all RMSD values are below 0.3 nm, indicating the stability of the four compound structures and validating the rationality of the system setup for further in-depth analysis.
MMPBSA results are presented in Table 2. Among the four systems, the PSA-LIG1 complex exhibits the lowest binding free energy of −36.60 kcal mol−1. Although the free energy scores for LIG2 and Drug 1 are relatively close, with LIG2 scoring −35.36 kcal mol−1 compared to Drug 1 scoring 33.89 kcal mol−1, LIG2 still maintains a lower free energy score. To enhance visualization, MMPBSA results were visualized using the builtin plugins of Gromacs 2022.3.38,39
| Complex | Binding free (kcal mol−1) | Van der Waals (kcal mol−1) | Electrostatic (kcal mol−1) | Polar solvation (kcal mol−1) | SASA (kcal mol−1) |
|---|---|---|---|---|---|
| PSA-LIG1 | −36.60 | −46.53 | −23.87 | 38.75 | −4.95 |
| PSA-LIG2 | −35.36 | −34.97 | −36.75 | 48.87 | −12.51 |
| PSA-Drug1 | −33.89 | −54.72 | −51.95 | 80.02 | −7.24 |
| PSA-Drug2 | −30.59 | −54.33 | −31.80 | 63.41 | −7.87 |
In this study, 2D and 3D free energy landscape plots were constructed. The rmsd_gyrate.log, bindex.ndx, and rmsd_gyrate.log files recorded the relationship between indices and energies. By analyzing these files, the conformations corresponding to the lowest energy for each system were identified at 266 ns, 85 ns, 114 ns, and 71 ns for LIG1, LIG2, Drug1, and Drug2, respectively (as shown in Fig. S6†). The RMSD values for these conformations are close to the average, and R(g) values are relatively low, indicating structural similarity to the initial structures and compactness in the low-energy region. This suggests that during the simulation, molecules may remain in this state for an extended period, demonstrating stability and further confirming the reliability of the binding free energy calculations.
Discussion
In summary, our study encompassed a multifaceted approach to identify specific binding compounds for prostate-specific antigen (PSA) cavities. We employed Autodock Vina for high-throughput docking experiments, K-means clustering analysis, and the determination of maximum common substructures (MCS) through rdFMCS.24,30,32 These methods culminated in the identification and characterization of specific binding ligands with promising affinities for the target protein. Furthermore, we integrated compound and synthesis technique databases to assess cost-effectiveness and synthesis routes. Our rigorous functional validation analyses confirmed the potential of these compounds, offering valuable insights into PSA recognition by small molecules.High-throughput screening techniques enable the detection of alterations in enzyme or receptor function, the interaction between probes and proteins, as well as the kinetic properties of protein-ligand binding.46 Nevertheless, the scarcity of compound samples for high-throughput screening poses a challenge: a substantial number of compound samples are required, but obtaining these samples can be difficult.27,43–45 For instance, compounds targeting prostate-specific antigen, designed by previous researchers using high-throughput screening, entail considerable experimental costs. In contrast, this study employs an integrated approach, combining computer aided drug design and machine learning techniques, to rapidly and cost-effectively identify and synthesize ligands with high binding affinity for prostate-specific antigen within the diverse ZINC database.
In this study, a comprehensive evaluation of compound-protein binding affinity is conducted using four techniques: molecular docking, molecular dynamics simulations, surface plasmon resonance (SPR), and microscale thermophoresis (MST). SPR is an optical phenomenon that arises when incident light at the interface between a metal and a dielectric medium meets specific energy and momentum matching conditions, leading to the excitation of coherent oscillations of free electrons on the metal surface.20 MST employs infrared lasers for localized heating of molecules, inducing directional movement, and subsequently analyzing the molecular distribution ratio within the temperature gradient field via fluorescence analysis.21 Despite the distinct principles underlying the two systems, the experimental values obtained are comparable. Although the data for LIG 1 and LIG 2 are relatively similar, both systems demonstrate that LIG 1 exhibits a higher binding affinity for prostate-specific antigen than LIG 2, corroborating the molecular docking and molecular dynamics simulation data. This, to some extent, validates the reliability of the computational data and ultimately establishes a dependable, rapid, and cost-effective ligand screening approach for specific proteins.
However, our work also revealed certain challenges. Conventional antibody-based prostate cancer detection methods exhibit limitations related to temperature sensitivity, cost, and cross-reactivity. While our synthesized compounds exhibit lower binding affinities, they address cost and storage issues. Nevertheless, there remains room for optimization. Future research directions could include enhancing compound binding affinities through fragment-based drug design and designing residues for multiple PSA cavities.47 Additionally, cost-effective fluorescent labeling methods, such as incorporating Fmoc groups, could be explored to improve PSA detection.48
Conclusions
In conclusion, our study provides a foundation for the development of lead compounds for prostate cancer treatment and molecular probe design. Our comprehensive screening approach, integrating various techniques and databases, streamlines compound selection and synthesis. As we look ahead, further research can refine our compounds, making them more potent, cost-effective, and applicable in prostate cancer diagnostics. This work exemplifies the potential of a multidisciplinary approach to target-specific protein pocket, offering a promising avenue for future drug development and molecular probe design endeavors.Author contributions
Shao-Long Lin: conceptualization writing, methodology, data curation. Yan-Song Chen: data curation. Ruo-Yu Liu: methodology. Mei-Ying Zhu: data curation. Tian Zhu: supervision. Ming-Qi Wang: supervision, writing – review & editing. Bao-Quan Liu Wang: supervision, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Department of Education of Liaoning Province, grant number LNYJG2022508.Notes and references
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, Ca-Cancer J. Clin., 2023, 73, 17–48 CrossRef PubMed
.
- C. Fitzmaurice, Global Burden of Disease Cancer Collaboration, JCO, 2018, 36, 1568 Search PubMed
- S. A. Khan, K. V. Hernandez-Villafuerte, M. T. Muchadeyi and M. Schlander, Int. J. Cancer, 2021, 149, 790–810 CrossRef CAS PubMed
- R. Ménez, S. Michel, B. H. Muller, M. Bossus, F. Ducancel, C. Jolivet-Reynaud and E. A. Stura, J. Mol. Biol., 2008, 376, 1021–1033 CrossRef PubMed
- S. W. D. Merriel, L. Pocock, E. Gilbert, S. Creavin, F. M. Walter, A. Spencer and W. Hamilton, BMC Med., 2022, 20, 54 CrossRef CAS PubMed
- X. Filella, E. Fernández-Galan, R. Fernández Bonifacio and L. Foj, Pharmacogenomics Pers. Med., 2018, 11, 83–94 CAS
- P.-J. Lamy, Y. Allory, A.-S. Gauchez, B. Asselain, P. Beuzeboc, P. de Cremoux, J. Fontugne, A. Georges, C. Hennequin, J. Lehmann-Che, C. Massard, I. Millet, T. Murez, M.-H. Schlageter, O. Rouvière, D. Kassab-Chahmi, F. Rozet, J.-L. Descotes and X. Rébillard, Eur. Urol. Focus, 2018, 4, 790–803 CrossRef PubMed
- S. P. Balk, Y.-J. Ko and G. J. Bubley, J. Clin. Oncol., 2003, 21, 383–391 CrossRef CAS PubMed
- R. C. Ladner, A. K. Sato, J. Gorzelany and M. de Souza, Drug Discovery Today, 2004, 9, 525–529 CrossRef CAS PubMed
- A. I. Barbosa, A. P. Castanheira, A. D. Edwards and N. M. Reis, Lab Chip, 2014, 14, 2918–2928 RSC
- J. L. Garcia-Cordero and S. J. Maerkl, Lab Chip, 2014, 14, 2642–2650 RSC
- A. Singh, T. Dauzhenka, P. J. Kundrotas, M. J. E. Sternberg and I. A. Vakser, Proteins: Struct., Funct., Bioinf., 2020, 88, 1180–1188 CrossRef CAS PubMed
- A. Morin, J. Meiler and L. S. Mizoue, Trends Biotechnol., 2011, 29, 159–166 CrossRef CAS PubMed
- X. Xu, C. Yan and X. Zou, J. Comput. Chem., 2018, 39, 2409–2413 CrossRef CAS PubMed
- N. Kumar, R. Srivastava, A. Prakash and A. M. Lynn, J. Biomol. Struct. Dyn., 2020, 38, 3396–3410 CrossRef CAS PubMed
- R. Singh, A. V. Pokle, P. Ghosh, A. Ganeshpurkar, R. Swetha, S. K. Singh and A. Kumar, J. Biomol. Struct. Dyn., 2023, 41, 6089–6103 CrossRef CAS PubMed
- S. Kumar, I. Ali, F. Abbas, N. Khan, M. K. Gupta, M. Garg, S. Kumar and D. Kumar, In silico pharmacol., 2023, 11, 20 CrossRef PubMed
- A. Biswas and V. Jayaprakash, in CADD and Informatics in Drug Discovery, ed. M. Rudrapal and J. Khan, Springer Nature, Singapore, 2023, pp. 283–311 Search PubMed
- S. Wang, G. Dong and C. Sheng, Chem. Rev., 2019, 119, 4180–4220 CrossRef CAS PubMed
- J. Homola, S. S. Yee and G. Gauglitz, Sens. Actuators, B, 1999, 54, 3–15 CrossRef CAS
- M. Jerabek-Willemsen, T. André, R. Wanner, H. M. Roth, S. Duhr, P. Baaske and D. Breitsprecher, J. Mol. Struct., 2014, 1077, 101–113 CrossRef CAS
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed
- D. Lu, H. Wang, M. Chen, L. Lin, R. Car, W. E., W. Jia and L. Zhang, Comput. Phys. Commun., 2021, 259, 107624 CrossRef CAS
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed
- M. Skalic, G. Martínez-Rosell, J. Jiménez and G. De Fabritiis, Bioinformatics, 2019, 35, 1237–1238 CrossRef CAS PubMed
- A. M. LeBeau, M. Kostova, C. S. Craik and S. R. Denmeade, Biol. Chem., 2010, 391, 333–343 CAS
- J. Lu, L. Chen, J. Yin, T. Huang, Y. Bi, X. Kong, M. Zheng and Y.-D. Cai, J. Biomol. Struct. Dyn., 2016, 34, 906–917 CrossRef CAS PubMed
- A. Belkadi, S. Kenouche, N. Melkemi, I. Daoud and R. Djebaili, Struct. Chem., 2021, 32, 2235–2249 CrossRef CAS
- T. Caliński and J. Harabasz, Commun. Stat., 1974, 3, 1–27 Search PubMed
- S. Aranganayagi and K. Thangavel, in International Conference on Computational Intelligence and Multimedia Applications (ICCIMA 2007), 2007, vol. 2, pp. 13–17 Search PubMed
- RDKit, https://www.rdkit.org/, accessed August 22, 2023 Search PubMed
- N. Salem and S. Hussein, Procedia Comput. Sci., 2019, 163, 292–299 CrossRef
- Y. Cao, T. Jiang and T. Girke, Bioinformatics, 2008, 24, i366–i374 CrossRef CAS PubMed
- H. Du, L. Guo, F. Fang, D. Chen, A. A Sosunov, G. M McKhann, Y. Yan, C. Wang, H. Zhang, J. D. Molkentin, F. J. Gunn-Moore, J. P. Vonsattel, O. Arancio, J. X. Chen and S. D. Yan, Nat. Med., 2008, 14, 1097–1105 CrossRef CAS PubMed
- K. L. Holmes and L. M. Lantz, in Methods in Cell Biology, Academic Press, 2001, vol. 63, pp. 185–204 Search PubMed
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed
- J. E. Smith, R. D. Jones, M. L. Braun, A. G. Walker, L. M. Hall, D. A. Kozakov and S. Vajda, J. Chem. Inf. Model., 2019, 59, 2041–2052 CrossRef PubMed
- D. Seeliger and B. L. de Groot, J. Comput.-Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed
- R. M. Adlington, J. E. Baldwin, G. W. Becker, B. Chen, L. Cheng, S. L. Cooper, R. B. Hermann, T. J. Howe, W. McCoull, A. M. McNulty, B. L. Neubauer and G. J. Pritchard, J. Med. Chem., 2001, 44, 1491–1508 CrossRef CAS PubMed
- P. Singh, S. A. Williams, M. H. Shah, T. Lectka, G. J. Pritchard, J. T. Isaacs and S. R. Denmeade, Proteins, 2008, 70, 1416–1428 CrossRef CAS PubMed
- H. Koistinen, G. Wohlfahrt, J. M. Mattsson, P. Wu, J. Lahdenperä and U.-H. Stenman, Prostate, 2008, 68, 1143–1151 CrossRef CAS PubMed
- V. Blay, B. Tolani, S. P. Ho and M. R. Arkin, Drug Discovery Today, 2020, 25, 1807–1821 CrossRef CAS PubMed
- N. Astrain-Redin, C. Sanmartin, A. K. Sharma and D. Plano, J. Med. Chem., 2023, 66, 3703–3731 CrossRef CAS PubMed
- D. Liu, D. Fu, L. Zhang and L. Sun, Chin. Chem. Lett., 2021, 32, 1066–1070 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08550c |
| This journal is © The Royal Society of Chemistry 2024 |