
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of core–shell magnetic metal–organic framework composites Fe3O4@MIL-101(Fe, Co) for degradation of RhB by efficiently activating PMS†
Huizhong Wu‡
 ,
Qiong Yi‡,
Xiang Li,
Yingxi Wang* and
Ling Li*
,
Qiong Yi‡,
Xiang Li,
Yingxi Wang* and
Ling Li*
Ministry-of-Education Key Laboratory for the Synthesis and Application of Organic Function Molecules, Hubei University, 430062, People's Republic of China. E-mail: lingli@hubu.edu.cn; 16890202@qq.com
First published on 23rd May 2024
Abstract
Low catalytic efficiency and catalyst recovery are the key factors limiting the practical application of advanced oxidation processes. In this work, a core–shell magnetic nanostructure Fe3O4@MIL-101(Fe, Co) was prepared via a simple solvothermal method. The core–shell structure and magnetic recovery performance were characterized by various technologies. The results of dye degradation experiments proved that within 10 minutes, the Fe3O4@MIL-101(Fe, Co)/PMS system can degrade more than 95% of 10 mg per L Rhodamine (RhB) at an initial pH of 7, which possesses higher catalytic activity than the Fe3O4/PMS system and the MIL-101(Fe, Co)/PMS system. The effects of initial solution pH and coexisting anions in water on the degradation of RhB were further discussed. The results showed that Fe3O4@MIL-101(Fe, Co) displayed excellent degradation efficiency in a wide pH range of 3–11 and capability of resisting coexisting anions. It is worth mentioning that after five cycles, the RhB removal rate can still be maintained at over 90% after 10 minutes of reaction. Free radical quenching experiments were further studied, confirming that ˙OH and SO4−˙ were involved in the degradation of RhB, while the dominating active free radical was SO4−˙. The possible reaction mechanism of the RhB degradation process was also inferred.
1. Introduction
With the rapid development of industrial production, the demand for dyes is increasing year by year. Dyes are mainly used in the textile, leather, food, paper, cosmetics, rubber, plastics, and printing industries. The unreasonable discharge of dye wastewater from different industries is the main source of water pollution.1,2 According to reports, dyes are toxic, mutagenic, carcinogenic, and can cause skin irritation. These types of macromolecular organic matter cannot be naturally degraded in water, and excessive exposure to them can cause irreversible harm to aquatic animals, plants, and human beings.3,4 Therefore, it is urgent to develop an economical and efficient dye wastewater treatment technology. At present, various methodologies including physical methods (adsorption, ion exchange, filtration, coagulation, and membrane separation), chemical oxidation (Fenton reagent, ozone oxidation, photocatalysis, and electrocatalysis), and biological methods (aerobic and anaerobic degradation) have been reported for dye wastewater treatment.5–14 However, traditional physical methods cannot completely remove pollutants, and biodegradation is not convenient for large-scale application due to its high implementation cost. Chemical oxidation, also known as advanced oxidation process (AOPs), is considered one of the most effective technologies for producing reactive oxygen species (ROS) to remove organic pollutants in the water environment.15 In recent years, peroxomonosulfate (PMS) has been widely used as a multifunctional activator in AOPs systems due to its easy activation, powerful oxidation ability, and high efficiency. The reactive oxygen species generated by activating PMS, such as SO4−˙, ˙OH, O2˙−, and even singlet oxygen (1O2), can decompose or even completely mineralize macromolecular organic compounds into CO2 and H2.16 Compared with ˙OH (E0 = 1.8–2.7 V) produced by traditional Fenton reaction, SO4−˙ (E0 = 2.5–3.1 V) has a higher oxidation potential, a better pH adaptation range (2.0–8.0), a longer half-life (30–40 μs), and stronger selectivity for organic pollutants.17,18 The combination of transition metal ions and PMS is one of the effective strategies for generating active oxygen species in AOPs.However, in homogeneous reactions, metal ions are difficult to recycle, which can lead to secondary pollution of the aqueous solution The combination of transition metal ions and PMS is one of the effective strategies to generate active oxygen in AOPs. However, in homogeneous reaction, metal ions are difficult to recycle, which will cause secondary pollution to the aqueous solution.19 Recently, the emergence of nanomaterials as heterogeneous catalysts to activate PMS has brought a new dawn to the field of sewage treatment.
As a type of nanomaterials, metal–organic framework materials (MOFs) possess clear structures, diverse types, design flexibility, and modifiability. Their super-large specific surface area and porous structure enable MOFs to expose more active sites, thus providing them with a wide range of applications in wastewater treatment. Recently, the appearance of nanomaterials as heterogeneous catalysts to activate PMS has brought a new dawn to the field of sewage treatment. As a kind of nanomaterials, metal–organic framework materials (MOFs) possess clear structure, rich types, designability and modifiability, the super-large specific surface area and porous structure enable MOFs to expose more active sites, so they are provided with a very broad application prospect in wastewater treatment.20,21 Because iron is environmentally friendly, in Fe-MOFs, Fe(III) can activate PMS or H2O2 to generate free radicals and facilitate the Fe(III)/Fe(II) cycle. Additionally, organic carboxylic acid ligands form strong coordination bonds and stable skeletons. Well-dispersed pores in Fe-MOFs, formed by organic carboxylic acid and Fe(III), not only expose metal sites more effectively but also reduce the chance of Fe leaching.22 Due to their composition and structural characteristics, iron-based MOFs such as MIL-101(Fe), MIL-100(Fe), and MIL-53(Fe) have been widely used in activating PMS in advanced oxidation processes.23,24 However, the circulation of Fe(III)/Fe(II) in a single Fe-MOFs/PMS system is slow, resulting in unsatisfactory efficiency in catalyzing PMS to produce reactive oxygen species.25 Previous studies have shown that the doping of another metal element can increase the number of metal active sites in the material. More importantly, the synergistic effect between metal ions can accelerate the interface electron transfer rate, thereby increasing the rate of ROS production.26 In our previous work, different percentages of Co2+ were doped into MIL-101, and the bimetallic MOFs: MIL-101 (Fe, CO20%) exhibited the best catalytic degradation performance.27
As we all know, magnetic separation is a promising strategy that can meet the current demand for high-performance separation technologies due to its low cost and high efficiency.28 Therefore, the introduction of magnetic Fe3O4 is a common method for convenient catalyst recycling. However, due to the small specific surface area, small pore volume, and poor mass transfer ability of transition metal oxidants such as Fe3O4, the activation efficiency of PMS is not high.29 Consequently, they are often compounded with other materials to enhance the catalytic ability of the catalysts. For example, Wang et al. prepared self-propelled magnetic MnO2@Pollen micromotors, which can serve as efficient catalysts for activating PMS to degrade tetracycline. In this material, Fe3O4 not only facilitates recycling and magnetic control through an external magnetic field but also cooperates with MnO2 to efficiently degrade TC.30 Furthermore, previous research has indicated that the combination of magnetic nanoparticles and nano-MOFs can greatly enhance pollutant degradation.31 Given the presence of Fe(II) in Fe3O4, the catalytic activity of PMS activation can be significantly improved by the synergistic effect between different metal ions. While the MIL series of Fe-MOFs only contain Fe(III) sites with weak Fenton activity, Fe(II) in Fe3O4 can serve as an electron donor to accelerate the cycle of Fe(III)/Fe(II) when combined with Fe-MOFs.32 Lv et al. prepared FeII@MIL-100 by introducing additional Fe(II) into the original Fe-MOFs. The cooperation between Fe(II) and Fe-MOFs facilitated the Fe(III)/Fe(II) cycle, thus enabling the catalyst to exhibit higher catalytic capacity.33
Based on this, Fe3O4 was coated with MIL-101(Fe, Co) to prepare an efficient catalytic heterogeneous catalyst with magnetic recovery, facilitating easy separation and avoiding secondary pollution. Due to the presence of Fe(II), it can promote the transformation between Fe(III) and Fe(II), Co(II) and Co(III), endowing the composite with excellent catalytic degradation performance. The structure and properties of the material were characterized by XRD, FT-IR, UV-vis, SEM, and TEM. RhB was used as a model pollutant in the degradation experiment. Compared with MIL-101(Fe, Co) and Fe3O4, it was evident that the Fe3O4@MIL-101(Fe, Co)/PMS system exhibited better degradation performance, with a RhB removal rate reaching 99% within 10 minutes. Cyclic experiments and ion interference experiments were conducted to evaluate the practicality of the materials. Free radicals in the reaction system were investigated through free radical quenching experiments. Scheme 1 depicts the synthesis of Fe3O4@MIL-101(Fe, Co) and the mechanism of PMS activation.
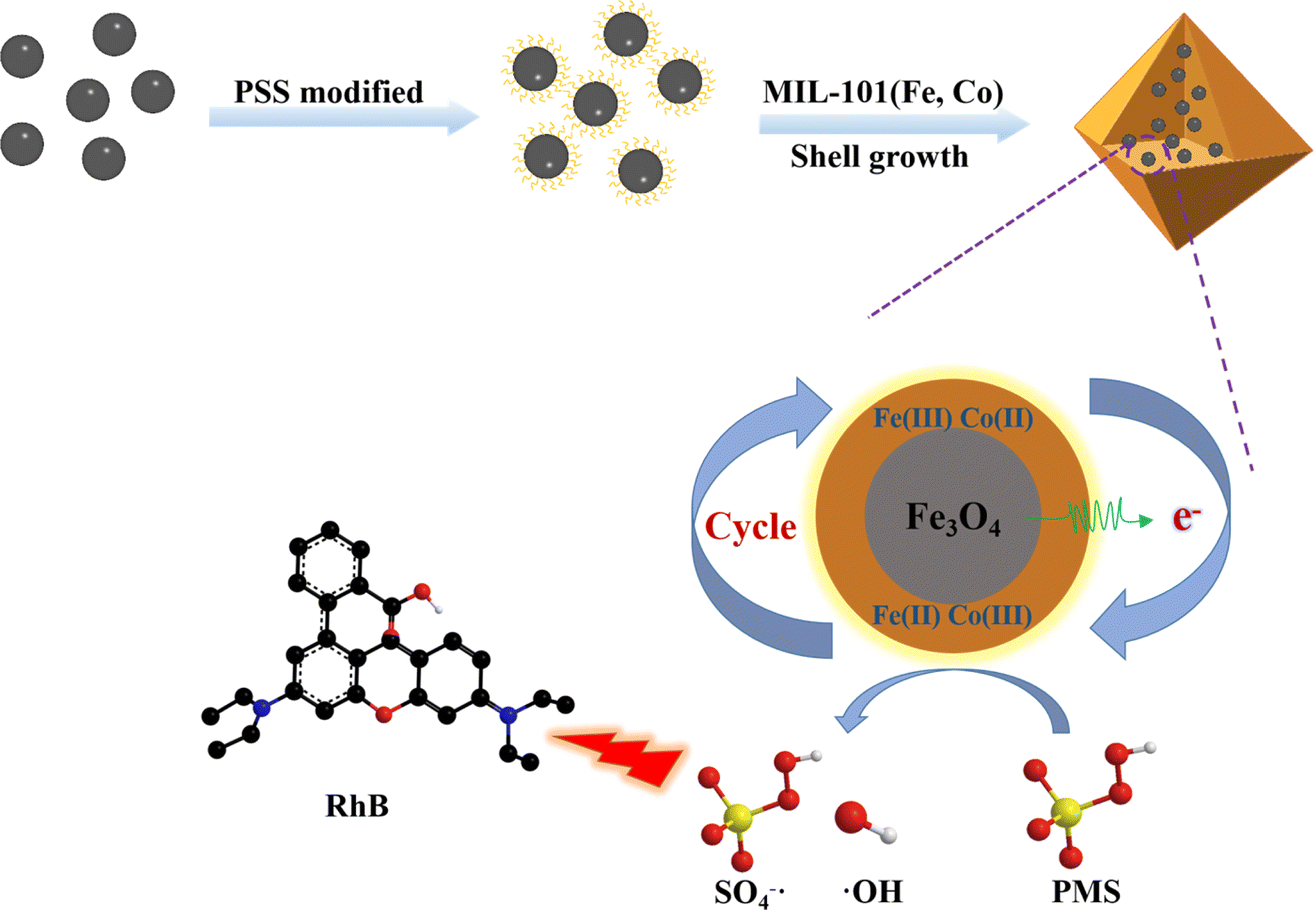 | ||
| Scheme 1 Schematic illustration of synthetic procedure of Fe3O4@MIL-101(Fe, Co) for PMS activation. | ||
2. Experimental section
2.1. Chemicals and materials
All chemicals used were of at least analytical grade. Hexahydrate ferric chloride, anhydrous ethanol, N,N-dimethylformamide (DMF), methylene blue (MB), malachite green (MG), amaranth red (E123), rhodamine B (RhB), methanol (MeOH), and tert-butanol (TBA) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). FeCl3·6H2O, Co(NO3)2·6H2O, terephthalic acid (H2BDC), sodium polystyrene sulfonate (PSS), and peroxomonosulfate (PMS) were provided by Aladdin (Shanghai, China). Fresh double-distilled water was used in all experiments.2.2. Preparation of catalysts
Fe3O4@MIL-101(Fe, Co) was prepared according to the reported literature, with some modifications.34 Initially, 0.01 g of Fe3O4 was dispersed in a 30 mL aqueous solution containing 0.09 g of sodium polystyrene sulfonate (PSS) and subjected to ultrasonic treatment for 30 minutes. The resulting mixture was recovered by an external magnetic field and washed several times. Subsequently, the material was dispersed in a 15 mL DMF solution containing FeCl3·6H2O and Co(NO3)2·6H2O, and stirred for 1 hour. Then, 0.206 g of H2BDC was dissolved in the above solution and stirred for an additional hour. The reaction mixture was then transferred to a Teflon autoclave and heated at 120 °C for 24 hours. After cooling, the final product was recovered using a magnet, washed several times, and dried, resulting in the formation of Fe3O4@MIL-101(Fe, Co).2.3. Characterization
The chemical compositions of the prepared catalysts were analyzed using X-ray diffraction (XRD, D8 ADVANCE Bruker, Cu-Kα radiation), Fourier-transform infrared spectroscopy (FT-IR, Nicolet Is10, Thermo), and X-ray Photoelectron Spectroscopy (XPS, ESCALAB Xi+, Thermo). The morphologies of the synthesized catalysts were characterized using scanning electron microscopy (SEM, JSM6510LV, JEOL) and high resolution transmission electron microscopy (HRTEM FEI talos F200). Absorbance measurements were obtained at 664 nm, 619 nm, 520 nm, and 552 nm using a UV-visible spectrophotometer (UV-Vis, 752 N, INESA), from which the concentration of the degraded dye was calculated.2.4. Degradation experiments
The catalytic performance of Fe3O4@MIL-101(Fe, Co) was evaluated by activating PMS for RhB degradation using a UV-visible spectrophotometer. All degradation experiments were conducted in a 50 mL beaker equipped with a mechanical stirrer, and a dark environment was maintained throughout. Typically, 4 mg of Fe3O4@MIL-101(Fe, Co) and 8 mg of PMS were added to a 20 mL dye solution with a concentration of 10 mg L−1 prepared from deionized water with a pH of 7. At preset reaction times, samples of the dye solution were taken and quenched with 0.1 mL of methanol to determine the residual dye concentration at that time. The samples were then filtered using a 0.22 μm filter, and the degree of RhB degradation was assessed by measuring the absorbance of the filtrate at 554 nm on a UV-visible spectrophotometer.In experiments with optimized conditions, the concentration of Fe3O4@MIL-101(Fe, Co), PMS, and dye, as well as the initial pH of the solution, and the type of dye, were varied. The same steps as described above were then followed.
3. Results and discussion
3.1. Characterization of materials
The crystalline phases of Fe3O4, MIL-101(Fe, Co), Fe3O4@MIL-101(Fe, Co) were characterized by XRD. Fig. 1a showed the XRD pattern of Fe3O4@MIL-101(Fe, Co). It was obvious that the peaks of 30.13°, 35.52°, 43.14°, 53.64°, 57.03° and 62.64° in the XRD pattern of Fe3O4 correspond to (220), (311), (400), (422), (511) and (440).35 Additionally, peaks at 5.2°, 5.8°, 9.0°, and 16.4° were observed, indicating the presence of MIL-101(Fe, Co). The coexistence of characteristic peaks of Fe3O4 and MIL-101(Fe, Co) in the XRD pattern of the complex indicated successful growth of MIL-101(Fe, Co) on the surface of Fe3O4, albeit with reduced crystallinity compared to MIL-101(Fe, Co). In Fig. 1b, the Fourier-transform infrared (FT-IR) absorption spectrum of Fe3O4@MIL-101(Fe, Co) showed characteristic peaks at 3390 cm−1 and 1631.5 cm−1, corresponding to the stretching vibrations of –OH and Fe–O in Fe3O4,34 respectively. Besides, the bands in the range of 3300–3500 cm−1 were assigned to the stretching vibration of –OH, and the bands in the range of 1400–1600 cm−1 were attributed to the asymmetric and symmetric stretching of organic ligand O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C, which were the characteristic peaks of MIL-101(Fe, Co).32 Additionally, for Fe3O4@MIL-101(Fe, Co), it was equipped with characteristic peaks of MIL-101(Fe, Co), and the peak at 577 cm−1 belonged to the stretching vibration of Fe–O, it can be seen that there was an extraordinary apparent shift in Fe3O4@MIL-101(Fe, Co) in contrast with the peak in Fe3O4, Fig. S1a† showed the X-ray photoelectron spectroscopy (XPS) spectrum of the material, and the XPS peaks for Co2p, Fe 2p, O 1s, and C 1s were observed apparently. High-resolution X-ray photoelectron spectroscopy (XPS) spectra of Fe2p and Co2p were presented in Fig. S1b and c† respectively, revealing primary peaks positioned at binding energies of 712.3 eV and 725.7 eV, corresponding to the Fe 2p3/2 and Fe 2p1/2. Moreover, the differing oxidation states of iron within Fe3O4 lead to the splitting of both the Fe 2p3/2 and Fe 2p1/2 signals into separate peaks attributed to Fe(III) and Fe(II).36,37 Besides, the Co 2p peak contained 2p3/2 and 2p1/2 at 781.6 and 797.5 eV as well as satellite peaks (Sat.) at 786.5 and 803.4 eV.38 In a word, successful formation of Fe3O4@MIL-101(Fe, Co) was further proved.
C–C, which were the characteristic peaks of MIL-101(Fe, Co).32 Additionally, for Fe3O4@MIL-101(Fe, Co), it was equipped with characteristic peaks of MIL-101(Fe, Co), and the peak at 577 cm−1 belonged to the stretching vibration of Fe–O, it can be seen that there was an extraordinary apparent shift in Fe3O4@MIL-101(Fe, Co) in contrast with the peak in Fe3O4, Fig. S1a† showed the X-ray photoelectron spectroscopy (XPS) spectrum of the material, and the XPS peaks for Co2p, Fe 2p, O 1s, and C 1s were observed apparently. High-resolution X-ray photoelectron spectroscopy (XPS) spectra of Fe2p and Co2p were presented in Fig. S1b and c† respectively, revealing primary peaks positioned at binding energies of 712.3 eV and 725.7 eV, corresponding to the Fe 2p3/2 and Fe 2p1/2. Moreover, the differing oxidation states of iron within Fe3O4 lead to the splitting of both the Fe 2p3/2 and Fe 2p1/2 signals into separate peaks attributed to Fe(III) and Fe(II).36,37 Besides, the Co 2p peak contained 2p3/2 and 2p1/2 at 781.6 and 797.5 eV as well as satellite peaks (Sat.) at 786.5 and 803.4 eV.38 In a word, successful formation of Fe3O4@MIL-101(Fe, Co) was further proved.
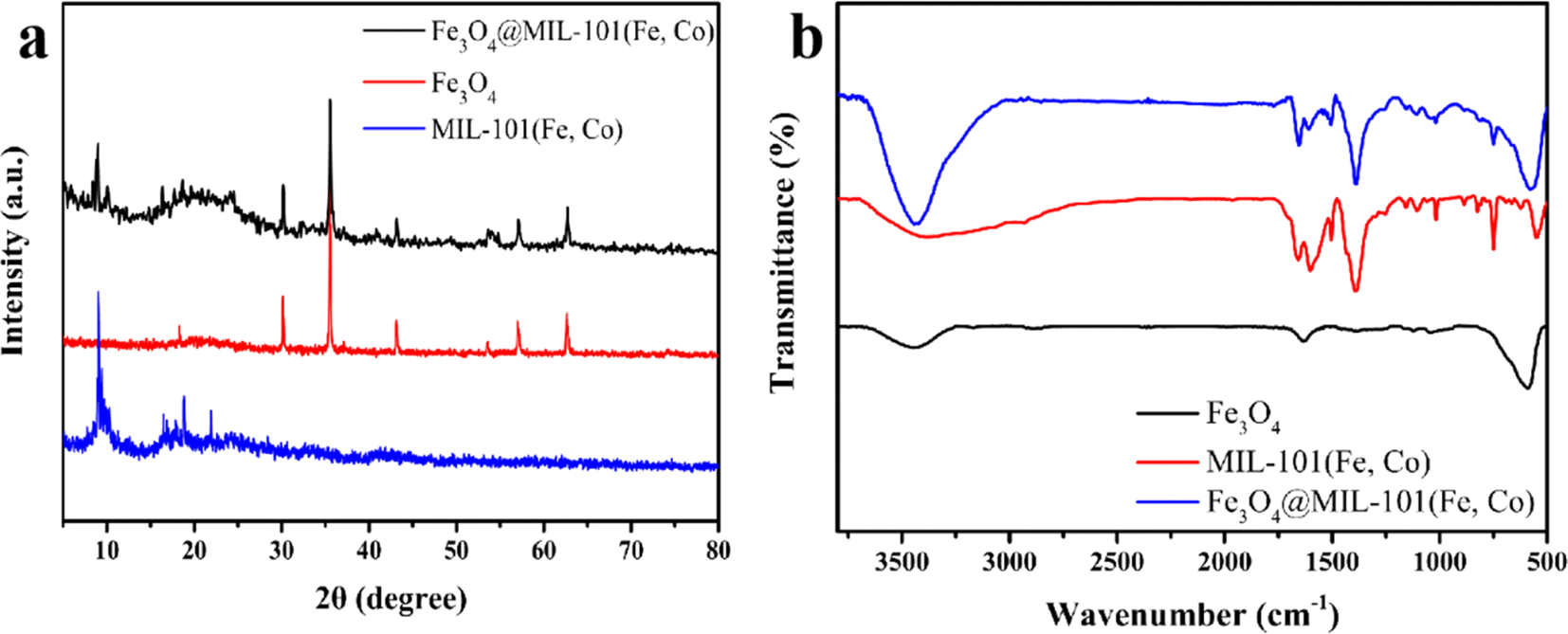 | ||
| Fig. 1 (a) XRD of Fe3O4@MIL-101(Fe, Co), MIL-101(Fe, Co) and Fe3O4; (b) FT-IR of Fe3O4@MIL-101(Fe, Co), MIL-101(Fe, Co) and Fe3O4. | ||
SEM, HRTEM and EDS were conducted to confirm structure of Fe3O4@MIL-101(Fe, Co) and the results were exhibited in Fig. 2. In the scanning electron microscope (SEM) images (Fig. 2a and b), Fe3O4@MIL-101(Fe, Co) exhibited a uniform morphology with slight aggregation. Additionally, the high-resolution transmission electron microscopy (HRTEM) image (Fig. 2d–f) revealed an obvious core–shell structure of Fe3O4@MIL-101(Fe, Co), with a diameter of approximately 150 nm and a shell length of about 20 nm. Furthermore, the lattice spacing width was measured to be 0.21 nm, corresponding to the (400) crystal plane of Fe3O4.39 Energy dispersive spectrometer (EDS) analysis (Fig. 2c) confirmed the presence of elements C, O, Fe, and Co in Fe3O4@MIL-101(Fe, Co), indicating the existence of bimetallic MOFs coated on the surface of Fe3O4 and the successful synthesis of Fe3O4@MIL-101(Fe, Co). The magnetic properties of Fe3O4@MIL-101(Fe, Co) were analyzed by applying an external magnetic field (as shown in Fig. 2g), demonstrating that the material can be easily separated from the dispersion by the action of a magnet, thus confirming its magnetic recoverability. Despite being coated with MIL-101(Fe, Co), Fe3O4@MIL-101(Fe, Co) still exhibited strong magnetic properties.
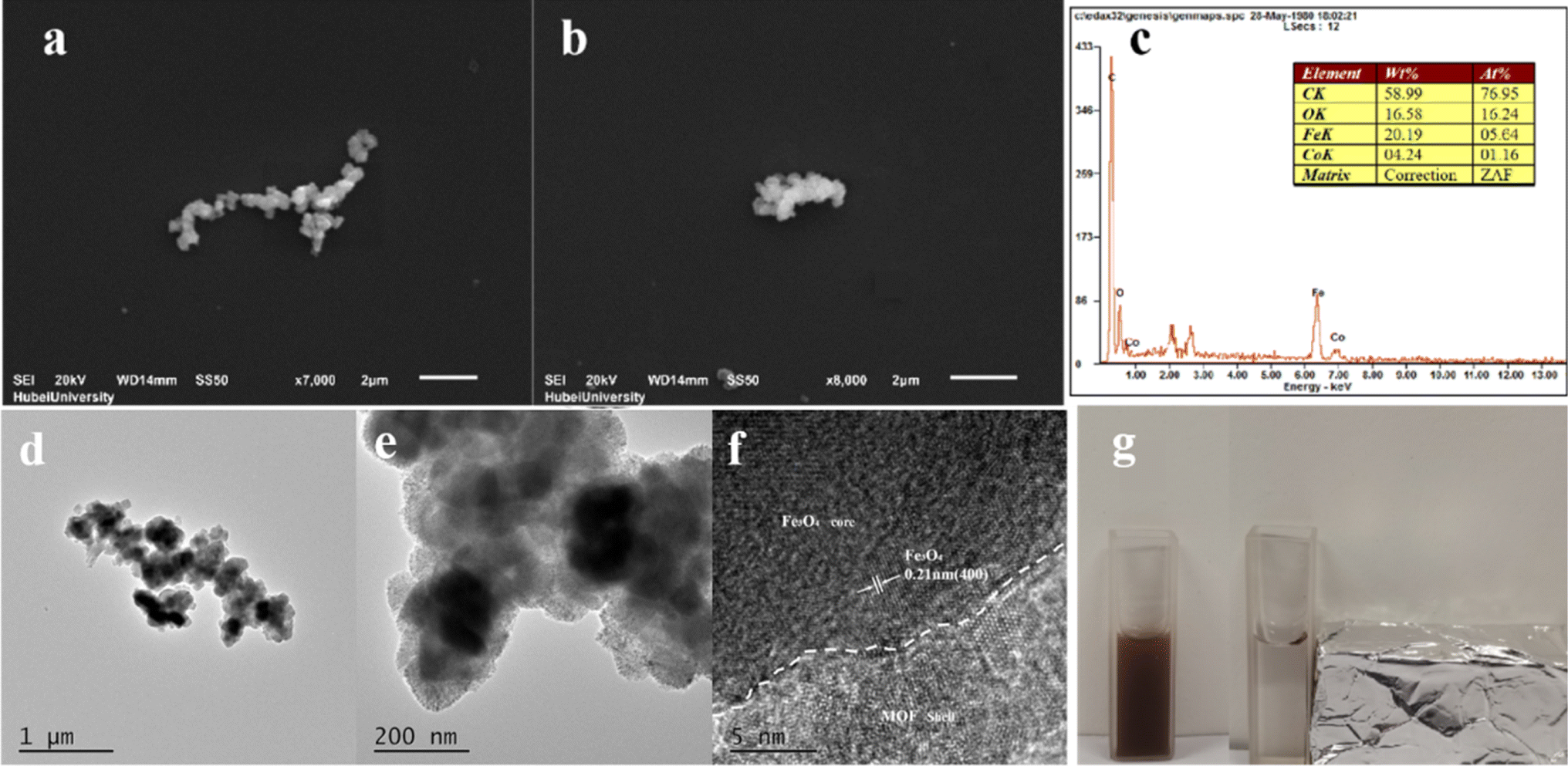 | ||
| Fig. 2 SEM images of Fe3O4@MIL-101(Fe, Co) (a and b); the EDX spectrum of Fe3O4@MIL-101(Fe, Co) (c) HRTEM image of Fe3O4@MIL-101(Fe, Co) (d–f); changes of Fe3O4@MIL-101(Fe, Co) before and after magnetic field application (g). | ||
3.2. Catalytic performance
In this study, we aimed to evaluate the catalytic activity of Fe3O4@MIL-101(Fe, Co) in activating PMS for the degradation of a model pollutant, rhodamine B (RhB). As shown in Fig. 3a, Fe3O4 alone and Fe3O4@MIL-101(Fe, Co) exhibited minimal removal effect on RhB, suggesting negligible adsorption of RhB by these materials. However, direct oxidation of RhB by PMS alone was limited, resulting in no significant change in RhB concentration. Combining these catalysts with PMS led to varying degrees of enhancement in RhB removal rate, indicating that Fe3O4, MIL-101(Fe, Co), and Fe3O4@MIL-101(Fe, Co) can serve as heterogeneous catalysts for Fenton-like reactions. Notably, the Fe3O4/PMS system achieved only 29.3% RhB degradation within 15 minutes, while the Fe3O4@MIL-101(Fe, Co)/PMS system reported in this study achieved 99% removal of RhB within 10 minutes. Both MIL-101(Fe, Co) and Fe3O4@MIL-101(Fe, Co) achieved over 99% RhB removal, with Fe3O4@MIL-101(Fe, Co) demonstrating enhanced catalytic performance.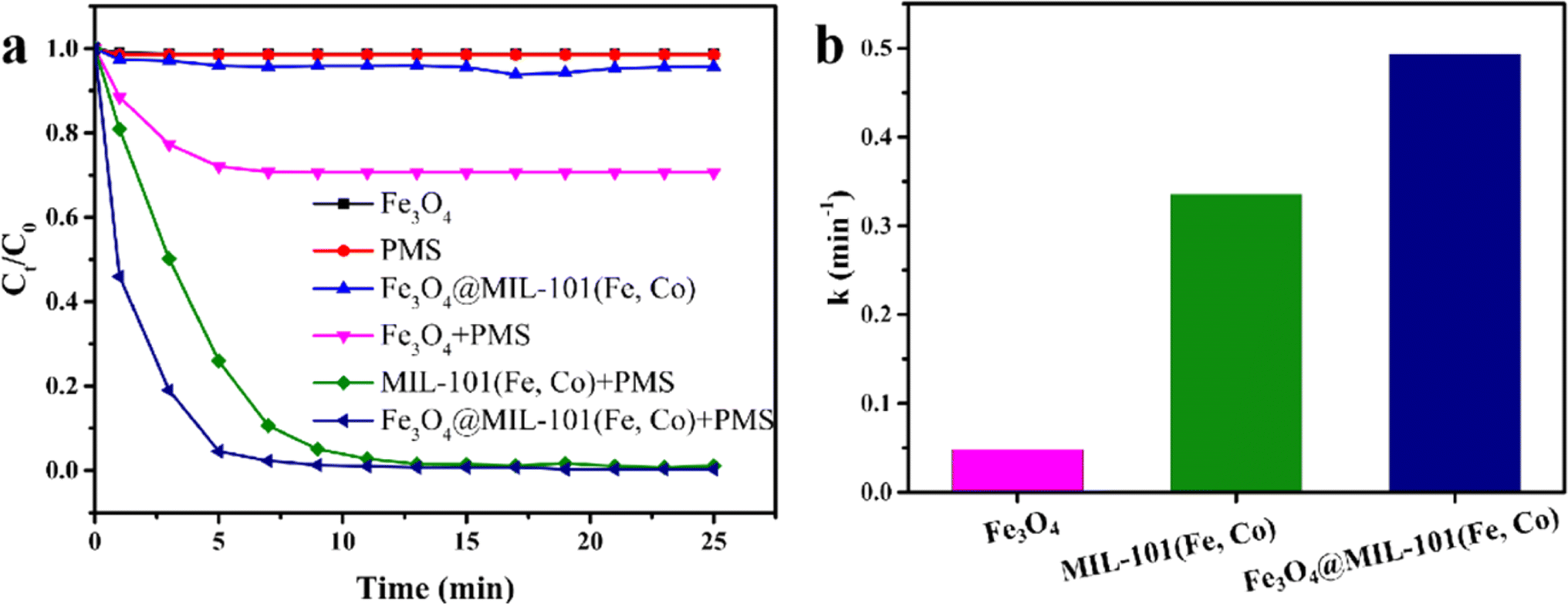 | ||
| Fig. 3 The degradation of RhB under different reaction conditions (a). Concentration: [RhB] = 10 mg L−1, [PMS] = 0.4 g L−1, [catalyst] = 0.2 g L−1, volume: 20 mL; temperature: 25 °C; initial solution pH: 7.0; pseudo-first order kinetics of different materials (b). | ||
Furthermore, as illustrated in Fig. 3b, pseudo first-order kinetic curves were fitted with the kinetic data of the three catalysts, and the apparent kinetic rate constants (k, min−1) of Fe3O4, MIL-101(Fe, Co), and Fe3O4@MIL-101(Fe, Co) were calculated as 0.0482 min−1, 0.3359 min−1, and 0.4934 min−1, respectively. These results underscored the synergistic effect between Fe3O4 and MIL-101(Fe, Co) enhancing the removal efficiency of RhB. Additionally, as shown in Table 1, the pseudo-first-order kinetics of the Fe3O4@MIL-101(Fe, Co)/PMS system reported in this study were much faster than those reported in other literature on AOPs.
| Catalysts | Reaction condition | Degradation rate (%) | Rate constant (k) (min−1) | Ref. |
|---|---|---|---|---|
| Co-tib(Co-MOFs) | [RhB] = 10.0 mg L−1, [PMS] = 0.15 mM, [catalyst] = 0.2 g L−1, 15 min | 100 | 0.20525 | 40 |
| Fe2(MoO4)3 | [RhB] = 10.0 mg L−1, [PMS] = 0.2 g L−1 [catalyst] = 0.1 g L−1, 30 min | 98.4 | 0.1149 | 41 |
| CNTS-Fe-Mn-0.5 | [RhB] = 15.0 mg L−1, [PMS] = 0.4 g L−1 [catalyst] = 0.1 g L−1, 60 min | 95 | 0.042 | 42 |
| Mn2O3 | [RhB] = 10.0 mg L−1, [PMS] = 0.5 g L−1 [catalyst] = 0.4 g L−1, 70 min | 98.2 | 0.0521 | 43 |
| Fe–Co–Co | [RhB] = 15.0 mg L−1, [PMS] = 0.1 g L−1 [catalyst] = 0.4 g L−1, 60 min | 94.3 | 0.042 | 44 |
| PBA@PmPDs | ||||
| Mg/Fe2O3 | [RhB] = 15.0 mg L−1, [PMS] = 0.2 g L−1 [catalyst] = 0.5 g L−1, 120 min | 96 | 0.0343 | 45 |
| Fe3O4@MIL-101(Fe, Co) | [RhB] = 1.0 mg L−1, [PMS] = 0.4 g L−1 [catalyst] = 0.2 g L−1, 10 min | 99 | 0.4934 | This work |
3.3. Catalytic performance of catalysts
As the Fenton-like reaction is influenced by the dosage of catalyst and PMS, as well as the initial pH of the solution, understanding the versatility of Fe3O4@MIL-101(Fe, Co) under varied conditions is crucial for their practical applications. To further optimize the operational conditions, these effects were investigated. Fig. 4a illustrated the influence of the concentration of Fe3O4@MIL-101(Fe, Co) on the degradation of RhB. When the catalyst concentration was 0.1 g L−1, the removal rate of RhB reached 98.2% within 23 minutes. With the same dosage of PMS, the time to reach degradation equilibrium of RhB decreased as the catalyst concentration increased. This effect can be attributed to the provision of more activation sites for PMS with higher catalyst concentrations.46 However, increasing the catalyst concentration from 0.2 g L−1 to 0.8 g L−1 did not significantly improve the removal efficiency of RhB. Considering economic feasibility, a catalyst concentration of 0.2 g L−1 was selected for subsequent experiments. As depicted in Fig. 4b, the effect of PMS dosage on the removal rate of RhB during the reaction was investigated. Under the catalysis of Fe3O4@MIL-101(Fe, Co), RhB was degraded by 76.8% within 10 minutes with the addition of 0.1 g per L PMS. However, when the dosage of PMS was in the range of 0.1 g L−1 to 0.4 g L−1, the degradation rate of RhB showed an upward trend with the increase of PMS concentration. This trend can be attributed to the fact that more PMS promotes the production of additional free radicals, enhancing the degradation efficiency.47 However, when the dosage of PMS increased from 0.4 g L−1 to 0.6 g L−1, the degradation rate of RhB exhibited a decreasing trend. This phenomenon is attributed to the self-quenching of free radicals caused by excessive PMS,48 which ultimately hinders the degradation efficiency. Therefore, a concentration of 0.4 g L−1 was chosen as the optimal PMS concentration for subsequent experiments.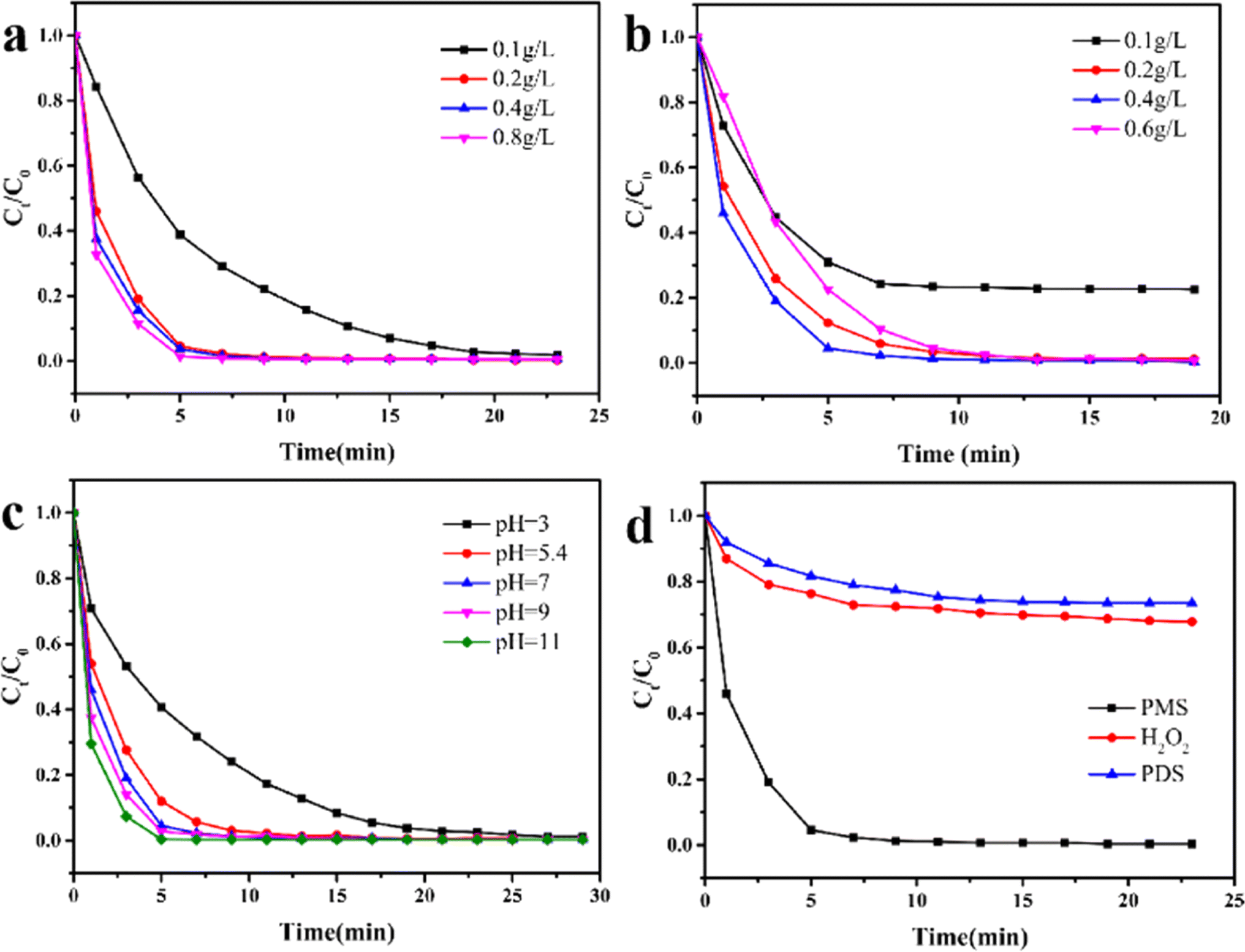 | ||
| Fig. 4 The influence of catalyst (a) and PMS (b) dosage, initial pH (c) in RhB removal and the efficiencies of different oxidants in Fe3O4@MIL-101(Fe, Co)/PMS system (d). Concentration: [dyes] = 10 mg L−1 (for a–d), [PMS] = 0.4 g L−1 (for a, c, d), [catalyst] = 0.2 g L−1 (for b–d), volume: 20 mL; temperature: 25; initial solution pH: 7.0. | ||
The effect of initial pH was further evaluated by varying the pH value adjusted using NaOH and HCl. As illustrated in Fig. 4c, the degradation efficiency of RhB gradually improved with increasing pH value. For instance, 98.8% of RhB was slowly degraded within 27 minutes under the condition of pH = 3. This low degradation efficiency at low pH values can be attributed to two main reasons: firstly, SO4−˙ and ˙OH radicals are captured by H+ ions and transformed into HSO4− and H2O, and secondly, an overly acidic environment may lead to the collapse of the material structure, resulting in a high ion leaching rate and a reduction in active sites.49 Furthermore, the degradation rate exceeded 99% within 12 minutes in the pH range of 5.4–11. Interestingly, the degradation efficiency was slightly enhanced under alkaline conditions. Previous studies have suggested that alkaline conditions are favorable for converting PMS into HSO5−˙, which is easily activated by metal active sites.50 These results demonstrate that Fe3O4@MIL-101(Fe, Co) exhibits remarkable catalytic ability over a wide pH range (pH = 3–11).
The effects of different oxidants on the degradation of RhB were also investigated. As shown in Fig. 4d, degradation equilibrium was reached within 15 minutes with all three oxidants. However, compared to PMS, the catalyst exhibited poor ability to activate H2O2 and PDS to generate free radicals. When PMS was used as the oxidant, 99% of RhB was degraded within 10 minutes when catalyzed by Fe3O4@MIL-101(Fe, Co). This indicates that PMS is a suitable oxidant for Fe3O4@MIL-101(Fe, Co) activation.
To investigate the potential application for removing toxic pollutants in complex water bodies, the catalytic performance of Fe3O4@MIL-101(Fe, Co) for RhB degradation was evaluated by adding certain anions that may affect the stability of the catalyst/PMS system. Specifically, two common water environment anions (Cl− and HCO3−) were chosen to test the anti-interference ability of the Fe3O4@MIL-101(Fe, Co)/PMS system. The effects of anions at concentrations of 1 mM, 5 mM, and 10 mM on the degradation of RhB were examined.
Fig. 5a demonstrated that the presence of Cl− can enhance the degradation of RhB to some extent, and this enhancement increases with the concentration of Cl−. This phenomenon was attributed to the formation of reactive chlorine species (HOCl, Cl˙, and Cl2, etc.) resulting from the reaction between Cl− and SO4−˙ and ˙OH. These reactive chlorine species, with certain oxidation ability, participate in the degradation of RhB.51 In addition, it can be seen from Fig. 5b that when the concentration of HCO3− was 1 mM, the reaction rate was accelerated, which may be due to the formation of weakly alkaline environment due to the existence of a small amount of HCO3−, while the degradation rate of Fe3O4@MIL-101(Fe, Co)/PMS system was increased under weakly alkaline conditions. HCO3− exhibited inhibitory effects on the RhB removal when the concentration were 5 mM and 10 mM, the removal of RhB was slightly restrained, and the higher the concentration, the more obvious the inhibition effect, which may owing to the lower active by-products (˙HCO3) produced from the reaction between HCO3− and active free radical, thus consuming the amount of active free radicals.52 But 89.5% of RhB can still be degraded within 15 min, in a word, Fe3O4@MIL-101(Fe, Co)/PMS system displayed excellent anti-anion interference ability.
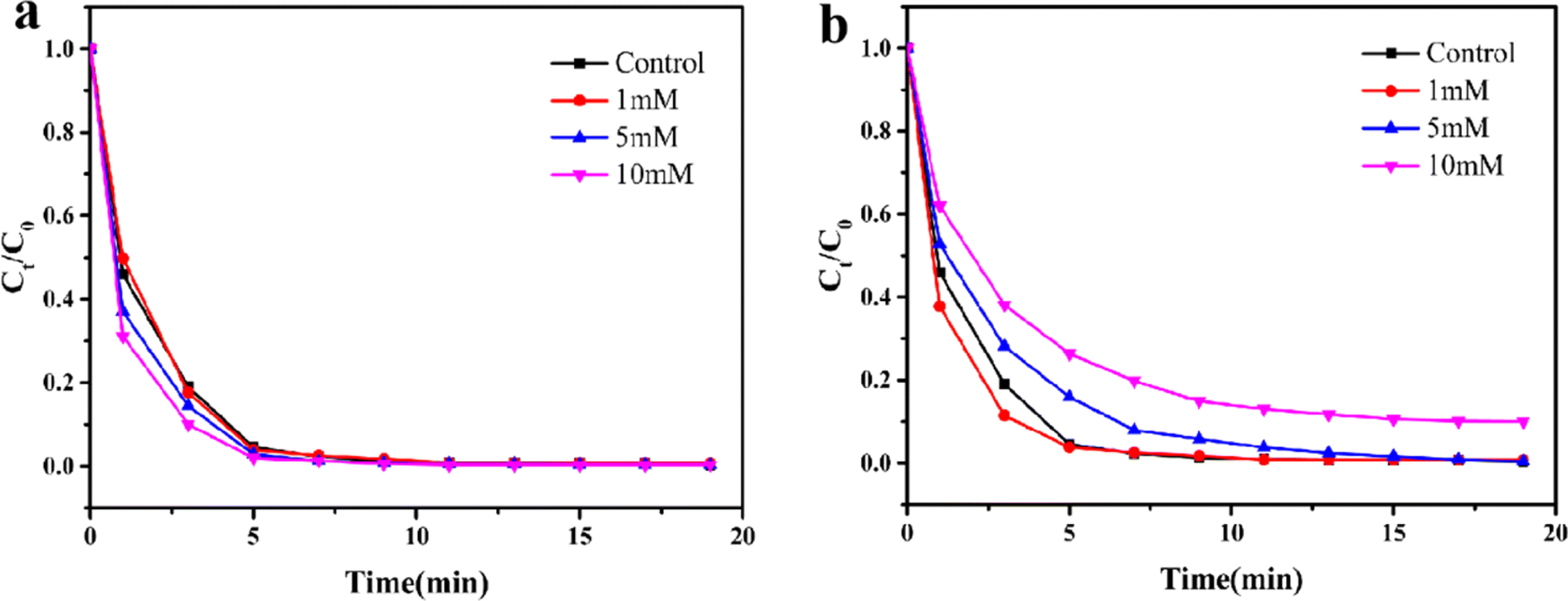 | ||
| Fig. 5 Effects of chloride (a) and bicarbonate (b) on RhB degradation in Fe3O4@MIL-101(Fe, Co)/PMS system. Concentration: [RhB] = 10 mg L−1, [PMS] = 0.4 g L−1, [catalyst] = 0.2 g L−1, volume: 20 mL; temperature: 25 °C; initial solution pH: 7.0. | ||
The impact of varying stirring rates on the catalytic reaction rate was investigated, Fig. S2a† demonstrated that under several different stirring velocities, the catalytic degradation performance of the material towards Rhodamine B (RhB) remained virtually unchanged, suggesting that the stirring speed did not significantly affect the material's catalytic properties, what's more, the effect of temperature variations on the reaction rate was studied, as presented in Fig. S3.† With the increase in temperature, the catalytic reaction rate steadily escalated. At a temperature of T = 30 °C, the catalytic degradation rate reached its peak, achieving a 99% removal of RhB within just 7 min. Furthermore, a first-order kinetic curve was fitted for the first 7 min at different temperatures, and the outcomes are depicted in Fig. S4.† At T = 30 °C, the apparent first-order rate constant (kobs) was calculated to be 0.74692 min−1, suggesting that elevated temperatures were conducive to enhancing the progress of the catalytic reaction. Besides, the catalytic degradation of methylene blue (MB), malachite green (MG) and amaranth red (E123) dyes by the material was illustrated in the Fig. S2b,† within 10 minutes, the material achieved removal rates exceeding 99% for all four dyes, signifying that the material possesses exceptional removal capabilities for these particular dyes.
3.4. Catalyst stability
The stability of Fe3O4@MIL-101(Fe, Co) in solution was investigated by measuring the ion leaching rate after the reaction. As depicted in Fig. S5a,† following the Fenton-like reaction, the concentrations of leached Fe and Co ions were found to be 0.6986 mg L−1 and 0.2972 mg L−1, respectively. These concentrations were significantly lower than the wastewater discharge standards of China, which specify maximum allowable concentrations of Fe (2 mg L−1) and Co (1 mg L−1) (GB 13456-2012 and GB 25467-2010, respectively).53 Subsequently, the material underwent recycling degradation experiments over five cycles under the same conditions, as depicted in Fig. S5b.† The cycle test demonstrated that even after five cycles, the degradation rate of RhB remained above 91%. Additionally, leaching of metal ions from the material results in the loss of active components, directly impacting catalytic performance. During the cyclic reaction process, the gradual decline in catalytic activity may be attributed to the deposition of dye degradation products or other byproducts within the pores of the catalyst, impeding access of dye molecules to active sites and thereby leading to reduced degradation efficiency. Notably, the loss rate of materials was greatly reduced compared to previous work due to the magnetic recovery performance of the materials. Furthermore, further evidence of excellent stability was obtained by comparing the FT-IR and XRD patterns of Fe3O4@MIL-101(Fe, Co) before and after the reaction. Clearly, the peak positions in both the FT-IR diagram (Fig. S6a†) and the XRD diagram (Fig. S6b†) of the material showed no significant differences between fresh and used materials, confirming the stability of its structure.3.5. Possible mechanism
In order to explore the degradation mechanism of RhB, free radicals involved in Fenton-like reactions were distinguished through free radical quenching experiments. Methanol (MeOH) was chosen to quench SO4−˙, while tert-butyl alcohol (TBA) was selected to scavenge ˙OH. Ethanol (EtOH) can quench both SO4−˙ and ˙OH. As depicted in Fig. S7,† in the absence of radical quenchers, the Fe3O4@MIL-101(Fe, Co)/PMS system removed 99.3% of RhB within 15 minutes. However, when 50 mM TBA or methanol was added, the degradation rate of RhB decreased to 90.8% and 35.3%, respectively. Adding 100 mM ethanol resulted in a degradation rate of 37.4%. These results indicate the participation of both ˙OH and SO4−˙ in the Fenton-like reaction, with a significant reduction in RhB removal rate observed upon methanol addition, suggesting that SO4−˙ was the primary free radical involved in RhB degradation.Based on these findings, we proposed a possible degradation mechanism in the Fe3O4@MIL-101(Fe, Co)/PMS system. Fe(II) on the surface of Fe3O4and Co(II) on the MOFs effectively react with HSO5− to generate Fe(III), Co(III), ˙OH, and SO4−˙ (eqn (1)). Simultaneously, Fe(III) and Co(III) on the MOFs receive electrons transferred from Fe3O4, leading to the generation of Fe(II) and Co(II) (eqn (2)). This establishes the Fenton-like cycle reactions of Fe(II)/Fe(III) and Co(II)/Co(III). Furthermore, considering the difference in standard redox potential, the remaining Fe(II) can further react with Co(III) to form Co(II) with high PMS activation ability (eqn (3)), while Co(III) and Fe(III) can also react with the remaining PMS to form Co(II) and Fe(II) (eqn (4)). The resulting strongly oxidizing radicals contact RhB and oxidize it into carbon dioxide and water27 (eqn (7)). In summary, the synergistic effect between MIL-101(Fe, Co) and Fe3O4 plays a significant role in the removal of organic dyes.
| Co(II)/Fe(II) + HSO5− → Co(III)/Fe(III) + SO4−˙ + ˙OH | (1) |
| Co(III)/Fe(III) + e− → Co(II)/Fe(II) | (2) |
| Co(III) + Fe(II) → Co(II) + Fe(III) | (3) |
| Co(III)/Fe(III) + HSO5− → Co(II)/Fe(II) + SO5−˙ + H+ | (4) |
| SO5−˙ → SO4−˙ + O2 | (5) |
| SO4−˙ + H2O → SO42− + ˙OH + H+ | (6) |
| SO4−˙ + ˙OH + dye → degradation products | (7) |
4. Conclusions
In summary, the core–shell structured material Fe3O4@MIL-101(Fe, Co) was successfully synthesized via a solvothermal method. The findings demonstrate that Fe3O4@MIL-101(Fe, Co) exhibits superior efficiency in activating PMS for RhB degradation compared to Fe3O4and MIL-101(Fe, Co) alone. Moreover, the catalytic enhancement can be attributed to the synergistic effect between Fe3O4 and MIL-101(Fe, Co), facilitating electron transfer and redox cycling between Fe(III)/Fe(II) and Co(II)/Co(III), thereby promoting active free radical generation. Additionally, the material exhibits excellent pH adaptability and stability. Its magnetic recovery capabilities result in minimal loss and high recycling rates, rendering it a promising candidate for practical applications in water treatment.Conflicts of interest
There are no conflicts to declare.References
- A. G. Varghese, S. A. Paul and M. S. Latha, Environ. Chem. Lett., 2019, 17, 867–877 CrossRef CAS.
- G. Sriram, A. Bendre, E. Mariappan, T. Altalhi, M. Kigga, Y. C. Ching, H.-Y. Jung, B. Bhaduri and M. Kurkuri, Sustainable Mater. Technol., 2022, 31, e00378 CrossRef CAS.
- S. Jorfi, S. Pourfadakari and B. Kakavandi, Chem. Eng. J., 2018, 343, 95–107 CrossRef CAS.
- X. Ma, S. Zhao, Z. Tian, G. Duan, H. Pan, Y. Yue, S. Li, S. Jian, W. Yang, K. Liu, S. He and S. Jiang, Chem. Eng. J., 2022, 446, 136851 CrossRef CAS.
- Q. Lin, G. Zeng, G. Yan, J. Luo, X. Cheng, Z. Zhao and H. Li, Chem. Eng. J., 2022, 427, 131668 CrossRef CAS.
- X. Wu, J. Xiong, S. Liu, J.-H. Cheng, M.-H. Zong and W.-Y. Lou, J. Hazard. Mater., 2021, 417, 126011 CrossRef CAS PubMed.
- F. R. Furlan, L. G. de Melo da Silva, A. F. Morgado, A. A. U. de Souza and S. M. A. Guelli Ulson de Souza, Resour., Conserv. Recycl., 2010, 54, 283–290 CrossRef.
- D. Olivo-Alanís, A. García-González, M. A. Mueses and R. B. García-Reyes, Appl. Catal., B, 2022, 317, 121804 CrossRef.
- Y. Zheng, X. Wu, Y. Zhang, Y. Li, W. Shao, J. Fu, Q. Lin, J. Tan, S. Gao, W. Ye and H. Huang, Chem. Eng. J., 2023, 453, 139919 CrossRef CAS.
- S. Wang, W. An, J. Lu, L. Liu, J. Hu, Y. Liang and W. Cui, Chem. Eng. J., 2022, 441, 135944 CrossRef CAS.
- W. Tang, H. Zhang, X. Yang, Z. Dai, Y. Sun, H. Liu, Z. Hu and X. Zheng, Appl. Catal., B, 2023, 320, 121952 CrossRef CAS.
- Y. Yuan, W. Yin, Y. Huang, A. Feng, T. Chen, L. Qiao, H. Cheng, W. Liu, Z. Li, C. Ding, F. Chen and A. Wang, Chem. Eng. J., 2023, 451, 138732 CrossRef CAS.
- F. Yang, M. Du, K. Yin, Z. Qiu, J. Zhao, C. Liu, G. Zhang, Y. Gao and H. Pang, Small, 2022, 18, 2105715 CrossRef CAS PubMed.
- J. Zdarta, A. S. Meyer, T. Jesionowski and M. Pinelo, Biotechnol. Adv., 2019, 37, 107401 CrossRef CAS PubMed.
- M. Zhu, D. Guan, Z. Hu, H.-J. Lin, C.-T. Chen, H.-S. Sheu, S. Wang, J. Zhou, W. Zhou and Z. Shao, Appl. Catal., B, 2021, 297, 120463 CrossRef CAS.
- N. Song, S. Ren, Y. Zhang, C. Wang and X. Lu, Adv. Funct. Mater., 2022, 32, 2204751 CrossRef CAS.
- X. Du and M. Zhou, Chem. Eng. J., 2021, 403, 126346 CrossRef CAS.
- Z. Liu, Z. Gao and Q. Wu, Chem. Eng. J., 2021, 423, 130283 CrossRef CAS.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- C. Wang, H. Wang, R. Luo, C. Liu, J. Li, X. Sun, J. Shen, W. Han and L. Wang, Chem. Eng. J., 2017, 330, 262–271 CrossRef CAS.
- Y. Qian, F. Zhang and H. Pang, Adv. Funct. Mater., 2021, 31, 2104231 CrossRef CAS.
- C. Gao, S. Chen, X. Quan, H. Yu and Y. Zhang, J. Catal., 2017, 356, 125–132 CrossRef CAS.
- X. Li, W. Guo, Z. Liu, R. Wang and H. Liu, Appl. Surf. Sci., 2016, 369, 130–136 CrossRef CAS.
- J. Wang, J. Wan, Y. Ma, Y. Wang, M. Pu and Z. Guan, RSC Adv., 2016, 6, 112502–112511 RSC.
- H. Li, J. Zhang, Y. Yao, X. Miao, J. Chen and J. Tang, Environ. Pollut., 2019, 255, 113337 CrossRef CAS PubMed.
- H. Liang, R. Liu, X. An, C. Hu, X. Zhang and H. Liu, Chem. Eng. J., 2021, 414, 128669 CrossRef CAS.
- Z. Xiao, R. Wu, T. Shu, Y. Wang and L. Li, Sep. Purif. Technol., 2023, 304, 122300 CrossRef CAS.
- L. Dong, Y. Li, X. Chen, D. Zhang and Y. Guan, Adv. Mater. Interfaces, 2021, 8, 2100178 CrossRef CAS.
- X. Jia, X. Chen, Y. Liu, B. Zhang, H. Zhang and Q. Zhang, Appl. Organomet. Chem., 2019, 33, e4826 CrossRef.
- K. Wang, E. Ma and H. Wang, Adv. Mater. Interfaces, 2022, 9, 2200271 CrossRef CAS.
- H. Alamgholiloo, N. N. Pesyan, R. Mohammadi, S. Rostamnia and M. Shokouhimehr, J. Environ. Chem. Eng., 2021, 9, 105486 CrossRef CAS.
- X. Yue, W. Guo, X. Li, H. Zhou and R. Wang, Environ. Sci. Pollut. Res., 2016, 23, 15218–15226 CrossRef CAS PubMed.
- H. Lv, H. Zhao, T. Cao, L. Qian, Y. Wang and G. Zhao, J. Mol. Catal. A: Chem., 2015, 400, 81–89 CrossRef CAS.
- T. Zhang, X. Zhang, X. Yan, L. Kong, G. Zhang, H. Liu, J. Qiu and K. L. Yeung, Chem. Eng. J., 2013, 228, 398–404 CrossRef CAS.
- K. M. L. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef CAS PubMed.
- Q. Gao, C. Dong, X. Hu, J. Zhang, J. Xue, Y. Zhao and X. Wang, Mol. Catal., 2023, 547, 113308 CrossRef CAS.
- S. Eslaminejad, R. Rahimi and M. Fayazi, J. Taiwan Inst. Chem. Eng., 2023, 152, 105181 CrossRef CAS.
- F.-X. Wang, Z.-C. Zhang, X.-H. Yi, C.-C. Wang, P. Wang, C.-Y. Wang and B. Yu, CrystEngComm, 2022, 24, 5557–5561 RSC.
- X. Hao, Z. Hu, D. Xiang and Z. Jin, Mol. Catal., 2022, 528, 112417 CrossRef CAS.
- M. Wang, F. Wang, P. Wang, H. Chu, H. Fu, C. Zhao, C.-C. Wang and Y. Zhao, Sep. Purif. Technol., 2023, 326, 124806 CrossRef CAS.
- L. Chang, X. Xie, X. Zhang, H. Chai and Y. Huang, Sep. Purif. Technol., 2023, 322, 124360 CrossRef CAS.
- X. Tian and L. Xiao, J. Colloid Interface Sci., 2020, 580, 803–813 CrossRef CAS PubMed.
- V. D. Thao, N. T. Dung, N. D. Duc, T. T. Trang, D. T. M. Thanh, N. T. Phuong, N. T. Thuy, K.-Y. A. Lin and N. N. Huy, Int. J. Environ. Sci. Technol., 2023, 20, 10163–10178 CrossRef CAS.
- L. Zeng, L. Xiao, X. Shi, M. Wei, J. Cao and Y. Long, J. Colloid Interface Sci., 2019, 534, 586–594 CrossRef CAS PubMed.
- S. Guo, M. Liu, L. You, G. Cheng, J. Li and K. Zhou, Chemosphere, 2021, 279, 130482 CrossRef CAS PubMed.
- M. Nie, Y. Deng, S. Nie, C. Yan, M. Ding, W. Dong, Y. Dai and Y. Zhang, Chem. Eng. J., 2019, 369, 35–45 CrossRef CAS.
- Z. Wu, Y. Wang, Z. Xiong, Z. Ao, S. Pu, G. Yao and B. Lai, Appl. Catal., B, 2020, 277, 119136 CrossRef CAS.
- Y. Mao, J. Liang, F. Ji, H. Dong, L. Jiang, Q. Shen and Q. Zhang, Sci. Total Environ., 2021, 787, 147584 CrossRef CAS PubMed.
- J. Yu, J. Cao, Z. Yang, W. Xiong, Z. Xu, P. Song, M. Jia, S. Sun, Y. Zhang and J. Zhu, J. Colloid Interface Sci., 2020, 580, 470–479 CrossRef CAS PubMed.
- Z. Xiao, Y. Li, L. Fan, Y. Wang and L. Li, J. Colloid Interface Sci., 2021, 589, 298–307 CrossRef CAS PubMed.
- Y. Yang, J. J. Pignatello, J. Ma and W. A. Mitch, Environ. Sci. Technol., 2014, 48, 2344–2351 CrossRef CAS PubMed.
- C. Guan, J. Jiang, S. Pang, X. Chen, R. D. Webster and T.-T. Lim, Chem. Eng. J., 2020, 387, 123726 CrossRef CAS.
- H. Liang, R. Liu, C. Hu, X. An, X. Zhang, H. Liu and J. Qu, J. Hazard. Mater., 2021, 406, 124692 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08768a |
| ‡ First authors. |
| This journal is © The Royal Society of Chemistry 2024 |