DOI:
10.1039/D4RA00001C
(Paper)
RSC Adv., 2024,
14, 18478-18488
A novel μ3-CO3 bridged linear polymeric Cu-complex ([Cu3(DMAP)8(μ3-CO3)2]I2)n·xH2O: synthesis, characterization and catalytic applications in the synthesis of phenoxypyrimidines and arylthiopyrimidines via C–O and C–S cross-coupling reactions†
Received
1st January 2024
, Accepted 25th May 2024
First published on 10th June 2024
Abstract
This manuscript reports on the synthesis and characterization of a new polymeric copper complex ([Cu3(DMAP)8(μ3-CO3)2]I2)n·xH2O and its successful application in C–O and C–S cross coupling reactions for the synthesis of biologically important phenoxypyrimidine and arylthiopyrimidine scaffolds. In an attempt to synthesize [Cu(DMAP)4I]I by adopting a procedure reported by Roy et al. with slight modification, the authors discovered a new polymeric Cu-complex that contains μ3-CO3 bridges. The polymeric linear structure of the complex was established using single crystal X-ray analysis. FT-IR, UV-vis and DSC studies were also performed on the polymeric complex. This novel polymeric Cu-complex was found to efficiently catalyse C–O/C–S cross coupling reactions between chloropyrimidines and phenols/thiophenols in an aqueous medium within a short reaction time, delivering their corresponding phenoxypyrimidines and arylthiopyrimidines. Using this protocol, 22 phenoxypyrimidines and 6 arylthiopyrimidines were successfully synthesized. The synthesized novel compounds were well characterized using 1H and 13C NMR spectroscopy and HRMS analysis and were screened for their drug-likeness properties using the SwissADME webtool.
Introduction
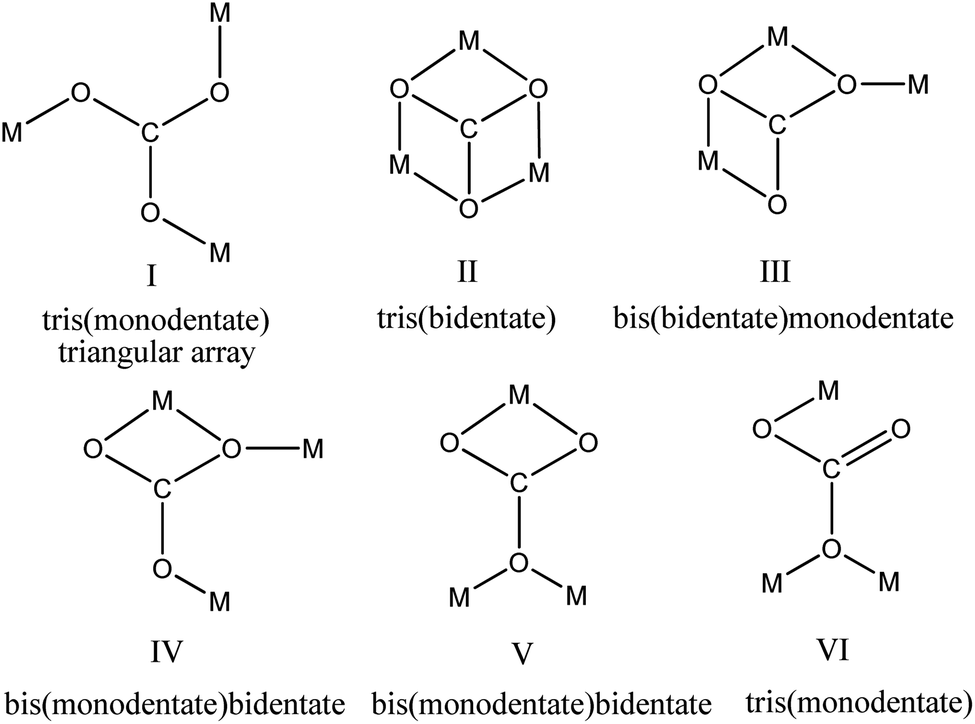
Transition metals are known for their ability to form complexes with organic ligands that result in metal–organic frameworks. Thus, copper can make complexes with different types of N-ligands such as bipyridine, 1,4-dimethylaminopyridine (1,4-DMAP),1 phenanthroline,2 pyrazine,3 pyrazole,4 N-(2-pyridylmethyl)-glycine and N-(2-pyridylmethyl)-glycine.5 Copper can form polynuclear complexes and coordination polymers through bridging ligands such as SCN−, OCN−, CO32−, N3−, N(CN)2−, OH− or heterocyclic organic molecules.6–10 The planar carbonate anion (CO32−) in its metal complexes can adopt several coordination bonding modes to bridge multiple metal centres to afford complexes with dinuclear μ2-CO3, trinuclear μ3-CO3, and tetranuclear μ4-CO3, enabling a wide variety of nuclearity and dimensionality.11,12 Fig. 1 depicts a few different (but not the only) coordination modes (I–VI) of the bridging μ3-CO3 group in metal complexes. While a triangular array (I) is the most commonly reported coordination pattern of the μ3-CO3 ligand,12–15 tris(bidentate) (II),16,17 bis(bidentate)monodentate (III)18 and bis(monodentate)bidentate (IV)19 coordination modes are also observed.
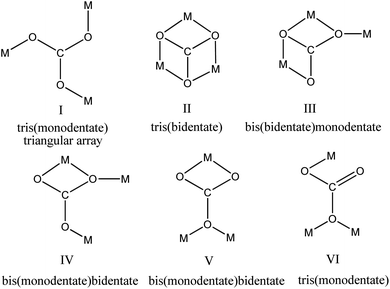 |
| | Fig. 1 Different coordination modes of the μ3-CO3 ligand. | |
Copper complexes enjoy wide applications such as single molecular magnets,20 single chain magnets,21 solar energy conversion and storage,22 drugs for the treatment of cancer,23 and nonsteroidal anti-inflammatory agents.24 Copper as a catalyst has always held immense importance owing to its lower cost (in comparison to Pd, Ru, Rh species) and applicability in diverse organic transformations. A copper–bipyridyl complex was utilised in Ullmann ether synthesis for the formation of carbon heteroatom bonds.25 A Chan–Lam type coupling reaction was also reported using a diamine copper complex between arylboronic acids and imidazoles.26 Roy et al. reported the applications of a square pyramidal [Cu(DMAP)4I]I complex in quick Chan–Lam C–N and C–S cross coupling reactions at room temperature.27 There are numerous reports on alcohol oxidation in the presence of Cu(II)-complexes.28,29
Phenoxypyrimidines and arylthiopyrimidines are essential classes of organic compounds because of their immense biological significances. Phenoxypyrimidines are found to have antidiabetic,30 anti-inflammatory,31,32 epidermal growth factor receptor (EGFR) inhibitory,33 focal adhesion tyrosine kinase (FAK) inhibitory,34 antimicrobial,35 FMS-like tyrosine kinase 3 inhibitory,36,37 herbicidal,38 HIV-1 non-nucleoside reverse transcriptase inhibitory (NNRTI),39 human protein interacting with NIMA1 (Pin1) inhibitory,40 and p38α kinase inhibitory41 activity, among others. Similarly, arylthiopyrimidines are found to possess activities such as human immunodeficiency virus type 1 (HIV-1) inhibitory,42,43 Hsp70 (heat shock protein 70) inhibitory,44 and herbicidal activity.45,46 Apart from their biological significances, phenoxypyrimidines and arylthiopyrimidines have acquired wide applications as substrates for various organic transformations including the ortho C–H functionalization of phenols and the preparation of other valued products.47–51
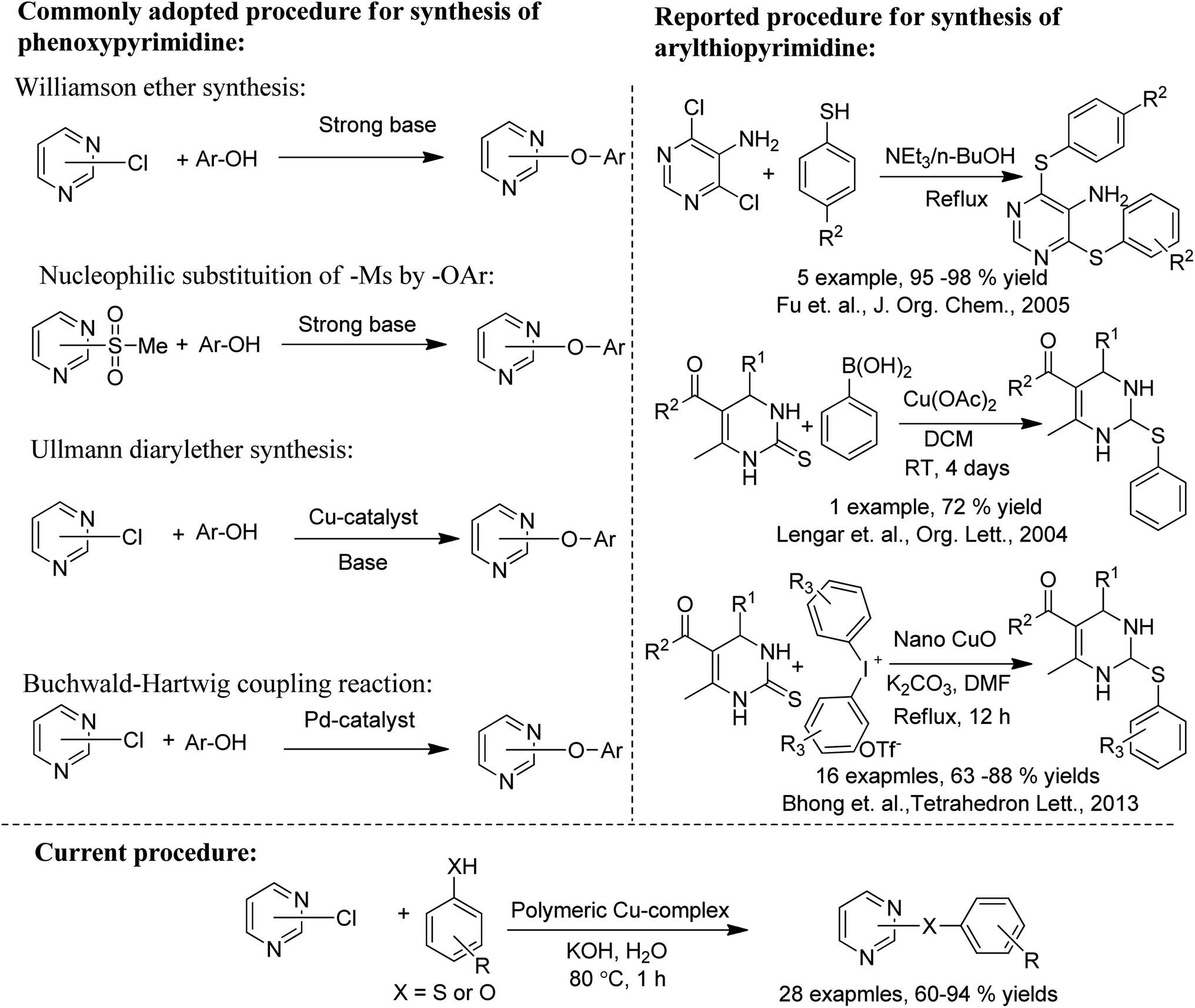
Phenoxypyrimidines are commonly synthesized by the nucleophilic substitution reaction, which includes Williamson ether synthesis30,32–34,40,52–55 and the removal of -Ms ion by phenolate ion.31,41,56,57 These processes are non-catalytic in nature and often demand a long reaction time. There are only a limited number of reports that have adopted catalytic pathways for their synthesis. For example, we have found only three reports of successful use of Ullmann coupling30,37,58 and one report of Buchwald–Hartwig coupling59 in the synthesis of phenoxypyrimidines. Similarly, the synthesis of arylthiopyrimidines is also observed to be largely dependent on non-catalytic processes.52,60,61 However, two reports employing copper-based catalytic procedures for the synthesis of arylthiopyrimidines are found in the literature.62,63
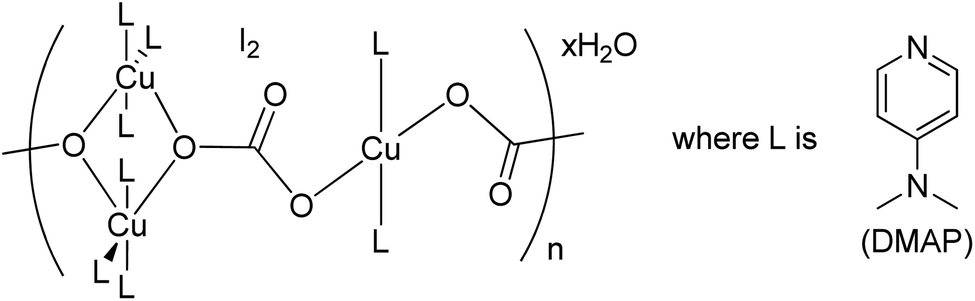
Herein, we are reporting an easy synthetic procedure for the synthesis of a novel μ3-CO3 bridged linear polymeric Cu-complex ([Cu3(DMAP)8(CO3)2]I2)n·xH2O (Fig. 2), which has been characterized using different analytical techniques including single crystal X-ray analysis. The crystalline material was checked for its catalytic activity in C–O and C–S cross-coupling reaction and was successful in synthesizing a good number of phenoxypyrimidines and arylthiopyrimidines in aqueous medium in a short duration of time in good to excellent yield. To the best of our knowledge, Cu-catalysed C–O and C–S cross coupling reactions in aqueous solution have been performed for the first time to synthesize arylthiopyrimidines. Scheme 1 draws the comparison of existing procedures of phenoxypyrimidines and arylthiopyrimidines with the current one.
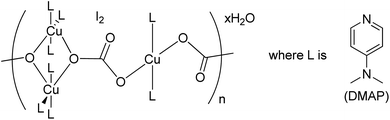 |
| | Fig. 2 Structure of the μ3-CO3 bridged linear polymeric Cu-complex ([Cu3(DMAP)8(CO3)2]I2)n·xH2O. | |
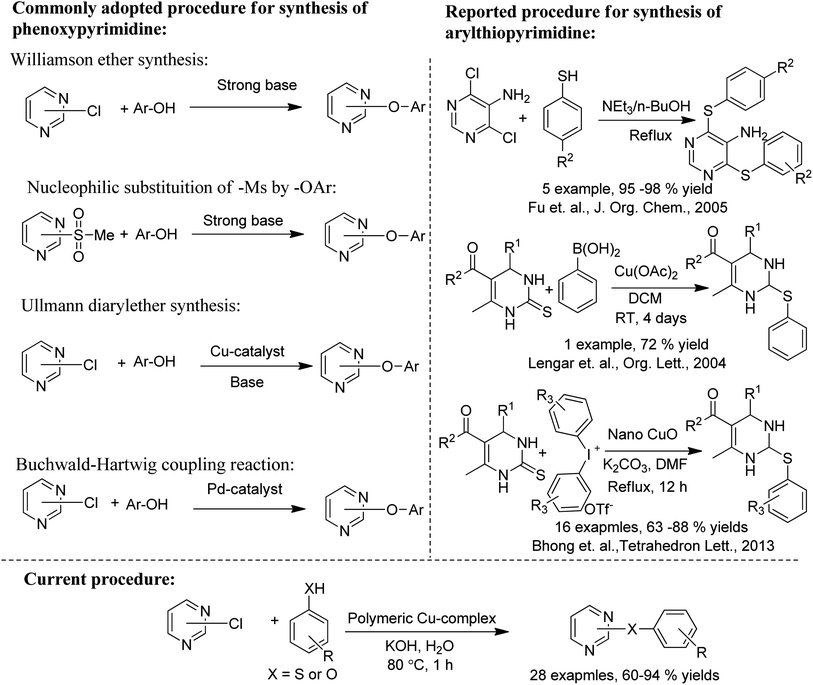 |
| | Scheme 1 Comparison between the reported and current protocol for the synthesis of phenoxypyrimidines and arylthiopyrimidines. | |
Results and discussion
Synthesis and characterization of ([Cu3(DMAP)8(CO3)2]I2)n·xH2O
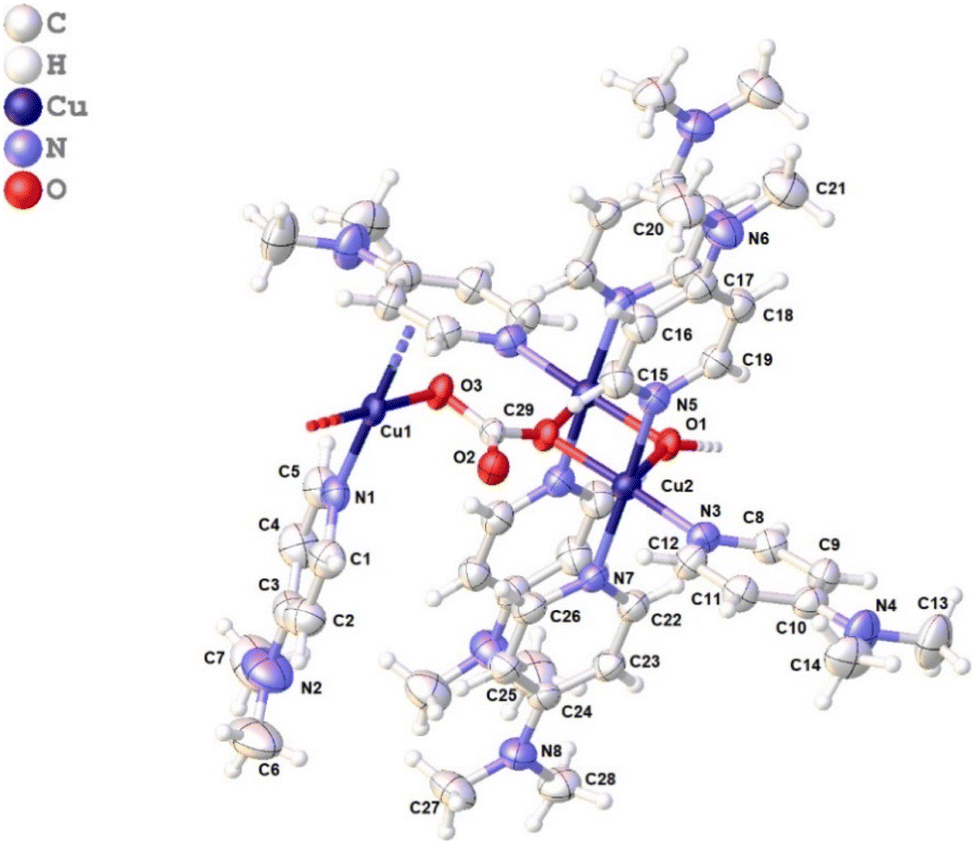
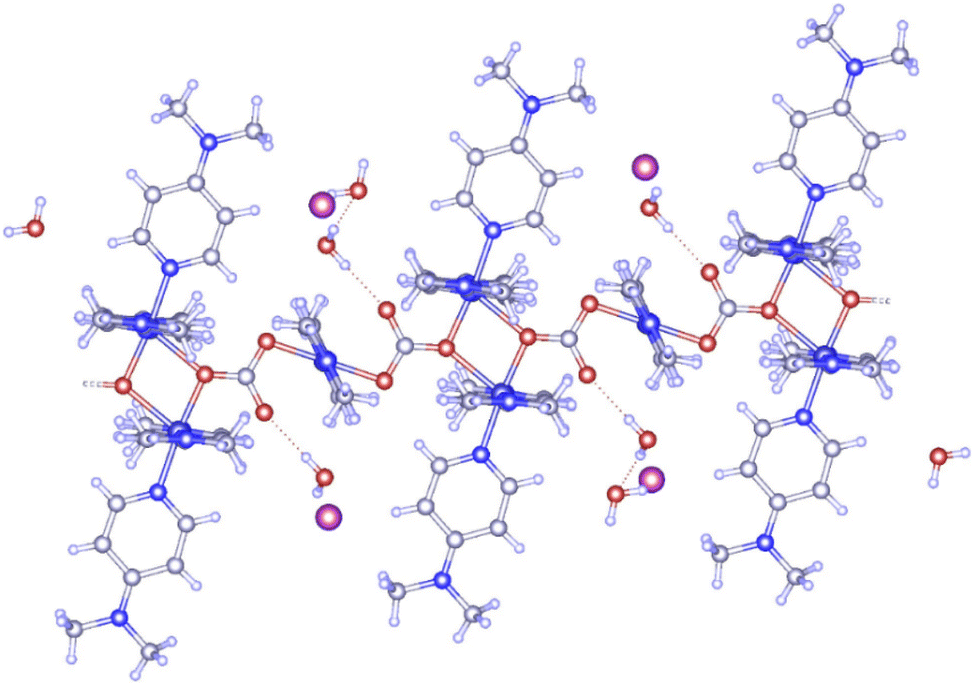
We started with an attempt to synthesize [Cu(DMAP)4I]I in aqueous medium using a procedure similar to that reported by Roy et al.27 Specifically, CuI (4 mmol) and 4-DMAP (16.4 mmol) were refluxed in H2O (8 mL) for 4 h, filtered in a beaker (25 mL) and then kept undisturbed for the possible crystallization of the product. Unlike in the work of Roy et al., the solute did not crystalize out even in 4 weeks. However, some needle-like crystals were observed in the 5th week, which were later collected in the 8th week after accumulation in a good amount. While the structure of the complex was confirmed using single crystal X-ray analysis (CCDC deposition no. 2311902), a few other analyses were carried out to acquire other information. Fig. 3 presents the asymmetric unit of the polymeric complex. It features a one-dimensional coordination polymer network along the a axes, where the asymmetric unit is composed of three Cu(II) centers: one mono and a dimeric center.
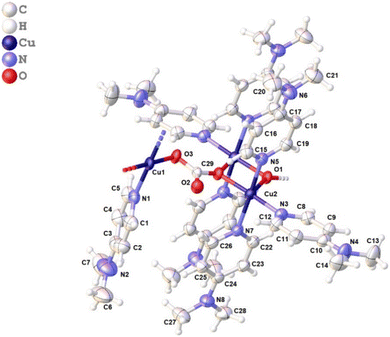 |
| | Fig. 3 Asymmetric unit of the polymeric complex (for a clear view, lattice water and iodide are omitted). | |
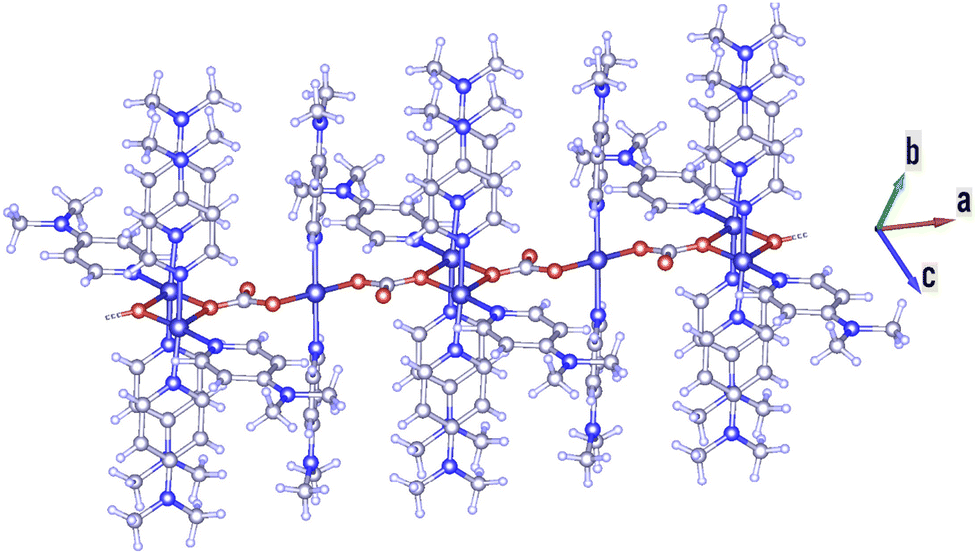
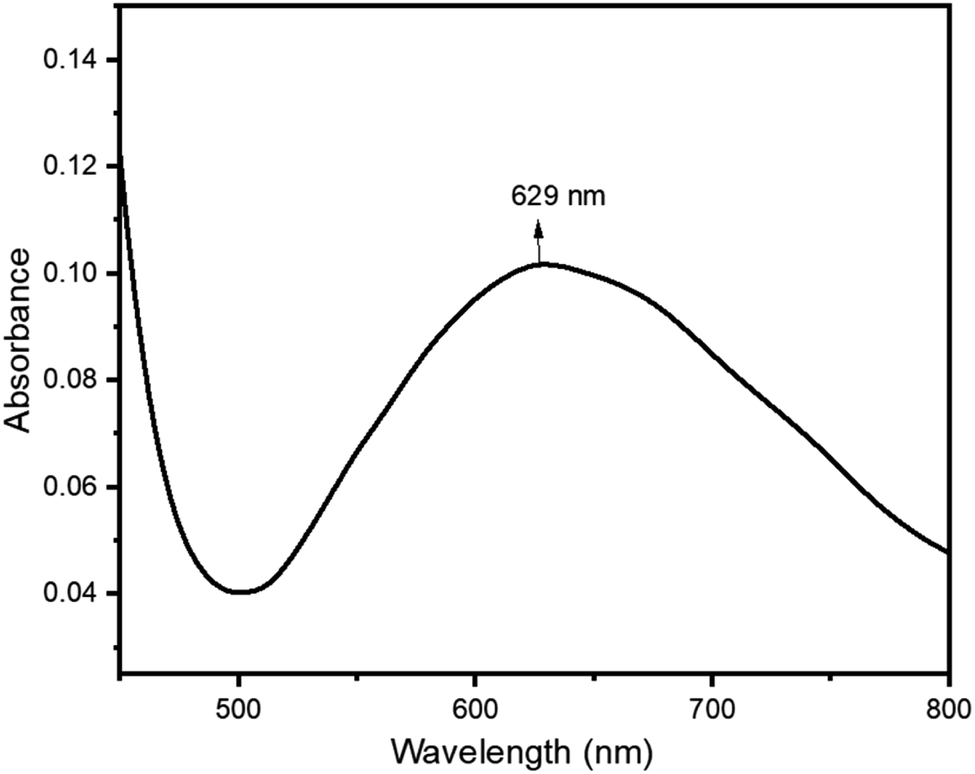
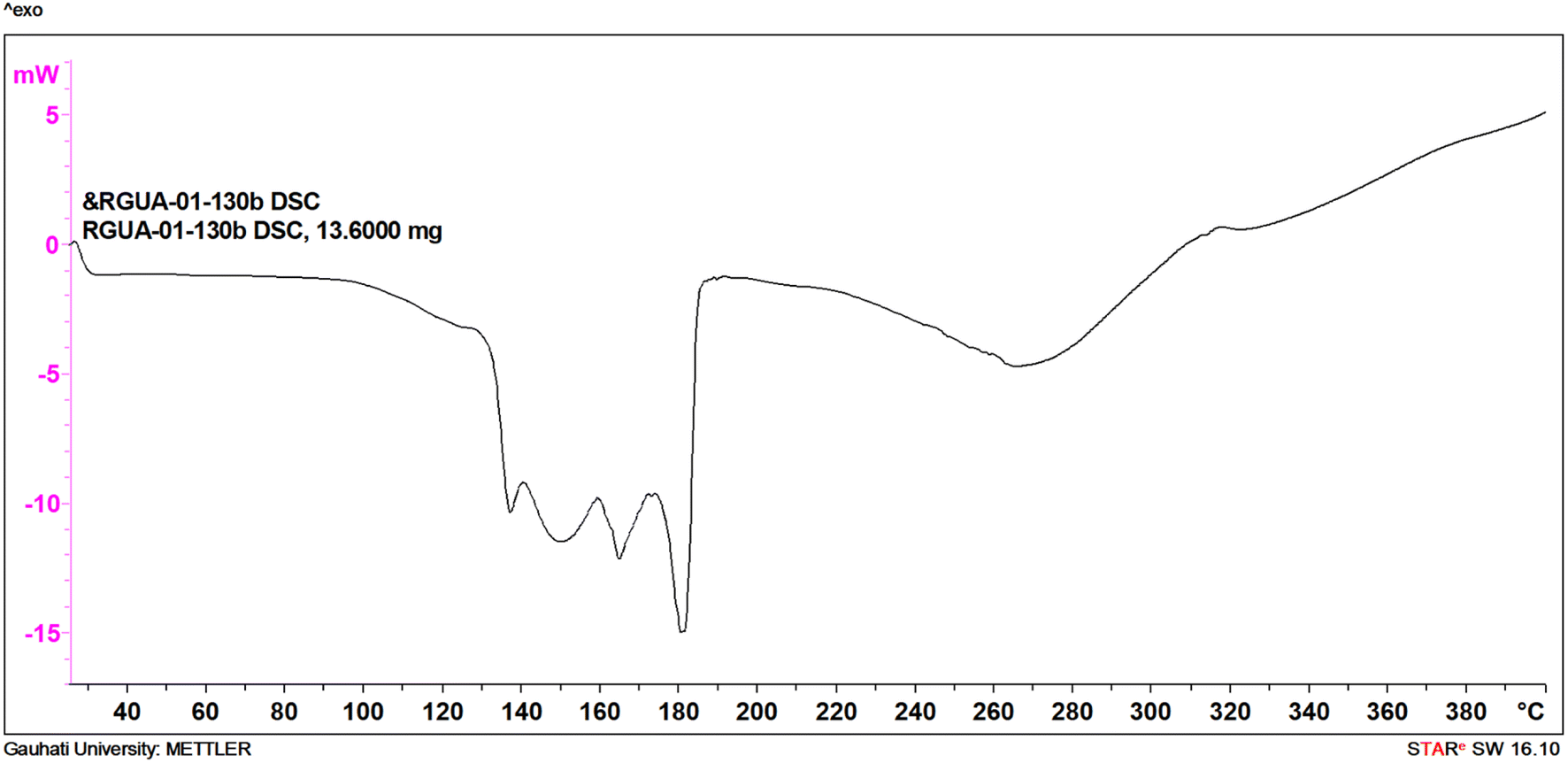
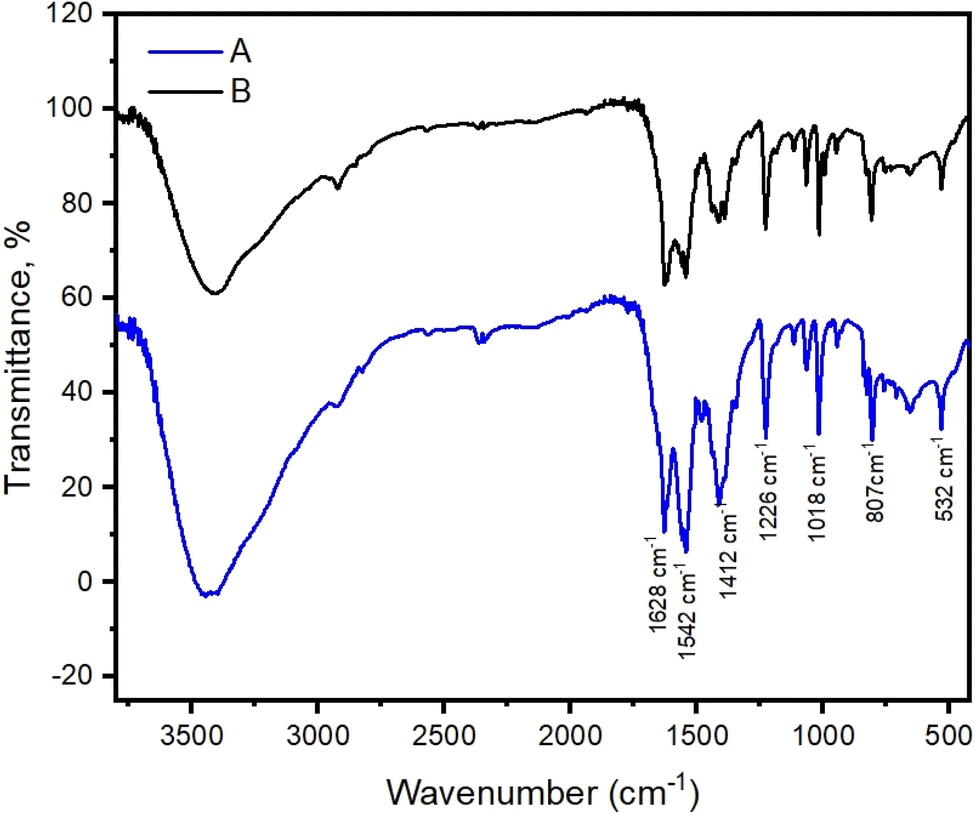
Cu-unit bridges via μ3-CO3 in the tris(monodentate) (VI in Fig. 1) coordination mode of the carbonate bridging ligand. The four co-ordinated Cu(II) center has a square planar geometry with the DMAP ligand bonded in an axial fashion (∠N1–Cu1–N1 = 180°) while two μ3:η1-CO32− bridges (∠O3–Cu1–O3 = 180°) complete the geometry (Fig. 2 and 3). The dimeric unit has a distorted trigonal bipyramidal geometry around the metal centre (∠N5–Cu2–N7 = 172.18(7)°, ∠O1–Cu2–O1 = 74.51(3)°) where the M⋯M distance is found to be 3.393(6) Å. The dangling O atom of the carbonate ligand is involved in intramolecular H-bonding with the lattice water (O2–H⋯O4 = 2.808 Å, O4–H⋯O5 = 2.822 Å) (Fig. 4). Fig. 5 depicts the one-dimensional polymeric growth of the Cu-complex. The bond valence calculation confirms that all the copper centres are in the +2 oxidation state. In UV-Vis spectroscopy, a broad peak at 629 nm was observed, which further confirmed the presence of copper in the Cu2+ state (Fig. 6). From DSC analysis, it was found that the compound is stable up to 137 °C (Fig. 7). In the IR spectra (Fig. 8), characteristics peaks at 1629 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric stretching), 1410 cm−1 (CO symmetric stretching), 1018 cm−1 (C–N–C stretching), 807 cm−1 (N–C–H wagging), and 532 cm−1 (Cu–O wagging) were observed.
O asymmetric stretching), 1410 cm−1 (CO symmetric stretching), 1018 cm−1 (C–N–C stretching), 807 cm−1 (N–C–H wagging), and 532 cm−1 (Cu–O wagging) were observed.
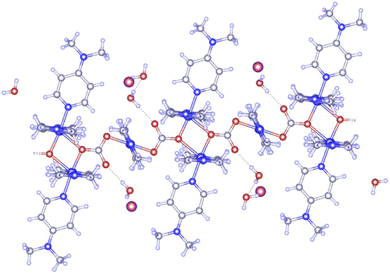 |
| | Fig. 4 μ3-CO3 coordination mode of the carbonate bridging ligand in the polymeric complex. | |
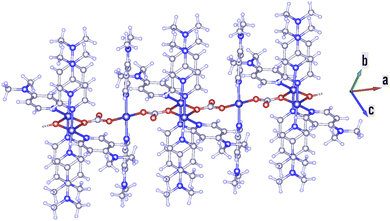 |
| | Fig. 5 Polymeric growth of the Cu-complex in the a direction. | |
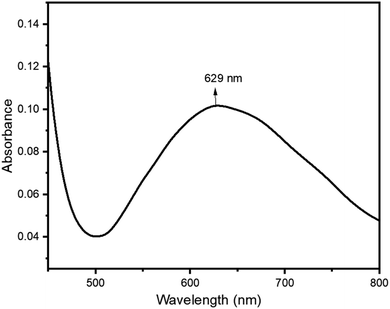 |
| | Fig. 6 UV-vis spectrum of ([Cu3(DMAP)8(CO3)2]I2)n·xH2O. | |
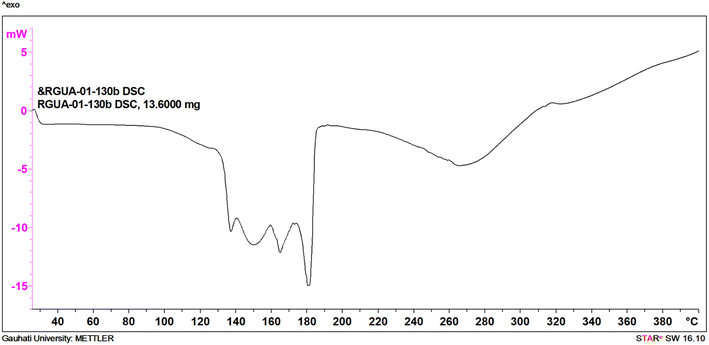 |
| | Fig. 7 DSC curve of ([Cu3(DMAP)8(CO3)2]I2)n·xH2O. | |
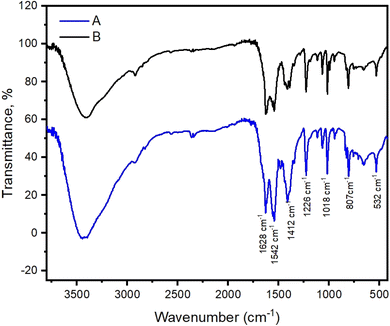 |
| | Fig. 8 IR spectra of the polymeric Cu-complex synthesized in the presence of K2CO3 (A) and synthesized in the absence of K2CO3 (B). | |
The μ3-CO3 bridge in the polymeric complex ([Cu3(DMAP)8(CO3)2]I2)n·xH2O, as confirmed by the single crystal X-ray analysis, compelled us to think what might be the possible source of CO32− in the complex as no CO32− source was used during the synthesis of the complex. We carried out the reaction in the presence of K2CO3 (as CO32− source), and we observed the formation of the same product, which was supported by IR spectral analyses (Fig. 8). The product was obtained in reduced time when the reaction was carried out in the presence of K2CO3. These observations coupled with the structure of the polymeric complex ([Cu3(DMAP)8(CO3)2]I2)n·xH2O suggested atmospheric CO2 fixation as the possible source of CO32− in our initial synthesis of the polymeric Cu-complex in the absence of the added CO32− source. There are literature reports available supporting atmospheric CO2 fixation as a possible source of CO32− in the synthesis of carbonato-bridged copper(II) complexes.11,64
After the complete characterization of the synthesized polymeric Cu-complex, we checked its catalytic activity in the synthesis of phenoxypyrimidines via C–O cross-coupling reactions. For that, we took a model reaction between 6-chlorouracil (1a) and phenol (2a) (Scheme 2) and carried out a series of reactions in the presence of ([Cu3(DMAP)8(CO3)2]I2)n·xH2O in different solvents and base combinations. We started out by carrying out the reactions in DMF at 100 °C (Table 1). DMF was used as the solvent as there are many reports available in the literature demonstrating its suitability in Ulmann coupling reactions for the synthesis of various organic compounds including phenoxypyrimidines.58,65 Our initial attempt using 10 wt% of the Cu-polymer and 3 equivalent Cs2CO3 as the base resulted in 6-phenoxypyrimidine in 71% yield in 4 h (entry 1, Table 1). When catalyst loading was varied keeping other parameters constant, it was observed that 10 wt% catalyst loading was sufficient for the purpose. While the lowering of the catalyst loading to 5 wt% led to a significant decrease in the yield (entry 3, Table 1), an increase in the catalyst loading to 15 wt% did not improve the yield (entry 2, Table 1). Then, a few other bases such as Na2CO3, K2CO3, K3PO4, NaOH and KOH were checked. Among all the bases tested, hydroxide bases were observed to furnish better results (entries 7–8 vs. entries 1, 4–6, Table 1); thus, these two hydroxide bases, i.e., NaOH and KOH, were taken for further investigation. Next, we tested the effect of various solvents on the synthesis of 6-phenoxypyrimidine (Table 2). Among the tested solvents, DMF and DMSO resulted in moderate yields in the presence of either of the bases (entries 1–2, 5–6, Table 2). Methanol offered much lower yields (entries 3 & 7, Table 2) while H2O emerged as the best solvent under the reaction conditions where the reactions were completed within a short period of time (1 h) with an increased amount of yield (entries 4 & 8, Table 2). Among the two hydroxide bases, KOH was observed to be offer a slightly better yield (entries 4 & 8, Table 2). The use of 3 equivalents of KOH was found optimal for the purpose (entries 8–10, Table 2). Then, we checked the effect of temperature on the reaction (entries 8 & 11–13, Table 2). It was found that the reaction was sluggish at room temperature, leading to low yield (60%) even in much longer duration (entry 11, Table 2). Lowering the temperature to 60 °C demanded longer time duration (8 h) (entry 12, Table 2). A reaction temperature of 80 °C was found optimal, furnishing the product in good yield (86%) within 1 h. Finally, we checked whether a stoichiometry of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 instead of 1:1.2 for 1a and 2a could be sufficient, and the result was positive (entry 14, Table 2).
:1.1 instead of 1:1.2 for 1a and 2a could be sufficient, and the result was positive (entry 14, Table 2).
 |
| | Scheme 2 Model reaction for the synthesis of phenoxypyrimidine. | |
Table 1 Effect of a base on the Cu-polymer-catalyzed synthesis of 6-phenoxypyrimidinea
|
| Entry |
Base |
Time (h) |
Yieldb [%] |
| Reaction conditions: 1a (1 mmol), 2a (1.2 mmol), Cu-polymer (10 wt%), base (3 eq.), DMF (3 mL), 100 °C (silicon oil bath). Isolated yield. Cu-polymer (15 wt%). Cu-polymer (5 wt%). Without a catalyst. |
| 1 |
Cs2CO3 |
4 |
71% |
| 2 |
Cs2CO3 |
4 |
71%c |
| 3 |
Cs2CO3 |
4 |
62%d |
| 4 |
Na2CO3 |
4 |
64% |
| 5 |
K2CO3 |
6 |
68% |
| 6 |
K3PO4 |
24 |
40% |
| 7 |
NaOH |
3 |
76% |
| 8 |
KOH |
2 |
78% |
| 9 |
KOH |
24 |
24%e |
Table 2 Effect of a solvent on the synthesis of 6-phenoxypyrimidinea
|
| Entry |
Base |
Solvent |
Time |
Yieldb [%] |
| Reaction conditions: 1a (1 mmol), 2a (1.2 mmol), Cu-polymer (10 wt%), base (3 eq.), solvent (3 mL), 100 °C (silicon oil bath). Isolated yield. KOH (2 eq.). KOH (3.5 eq.). rt. 60 °C. 80 °C. 2a (1.1 mmol), (80 °C). |
| 1 |
NaOH |
DMF |
3 h |
76 |
| 2 |
DMSO |
72 |
| 3 |
MeOH |
6 h |
60 |
| 4 |
H2O |
1 h |
81 |
| 5 |
KOH |
DMF |
2 h |
78 |
| 6 |
DMSO |
74 |
| 7 |
MeOH |
6 h |
61 |
| 8 |
H2O |
1 h |
84 |
| 9 |
KOH |
H2O |
1 h |
78c |
| 10 |
KOH |
H2O |
1 h |
85d |
| 11 |
KOH |
H2O |
18 h |
60e |
| 12 |
KOH |
H2O |
8 h |
80f |
| 13 |
KOH |
H2O |
1 h |
86g |
| 14 |
KOH |
H2O |
1 h |
86h |
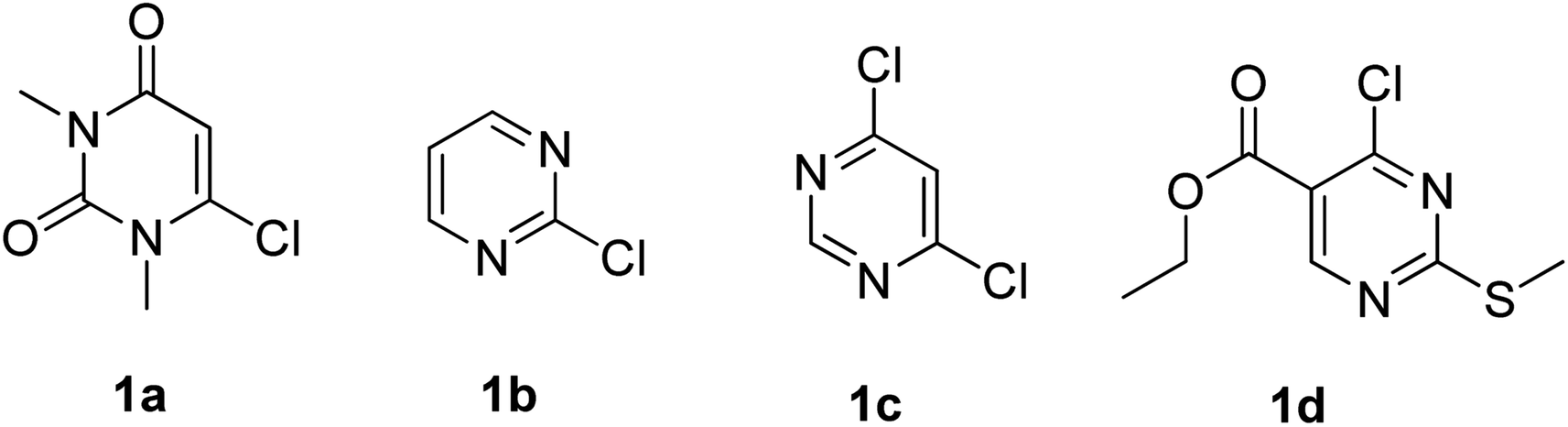
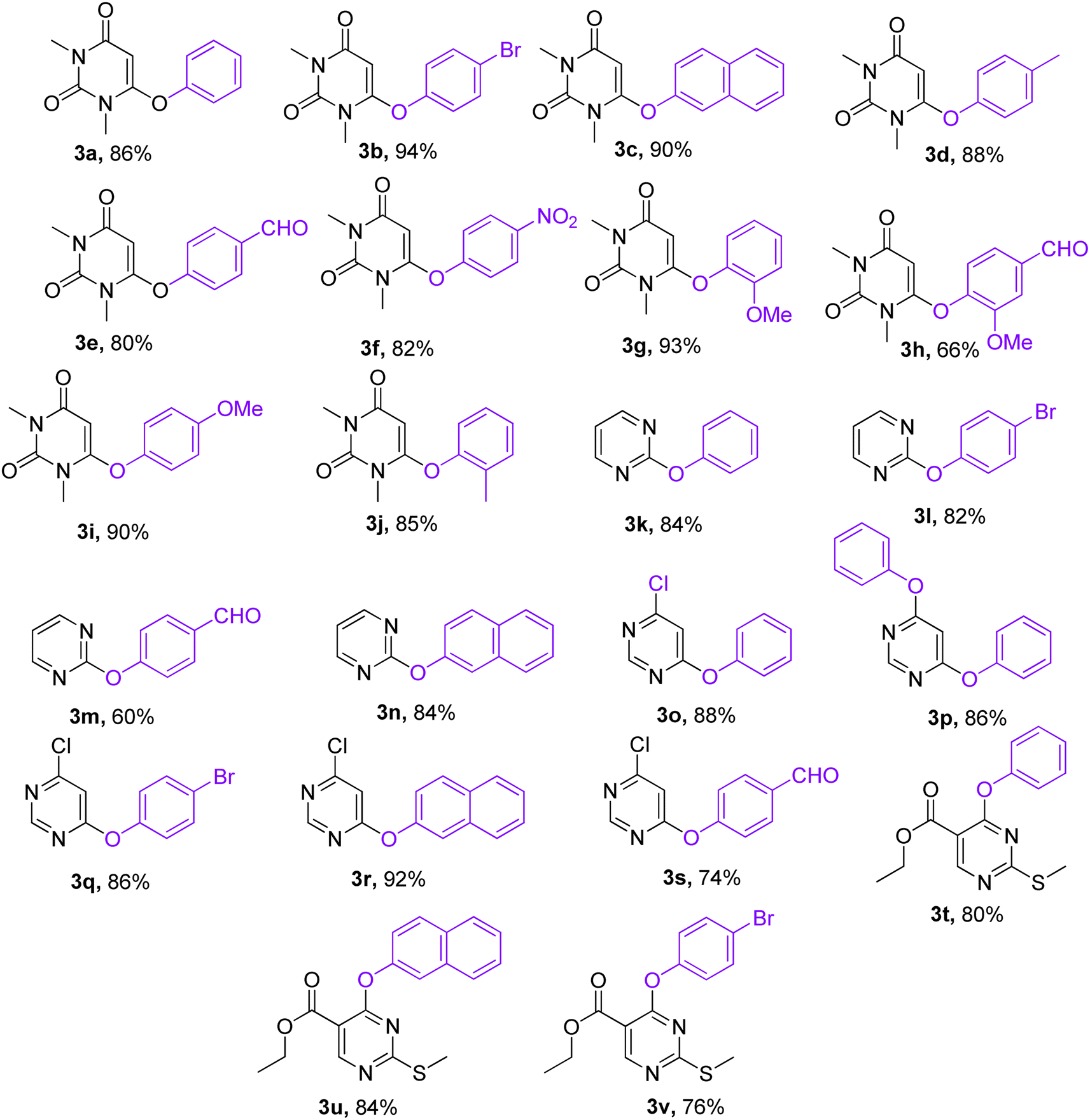
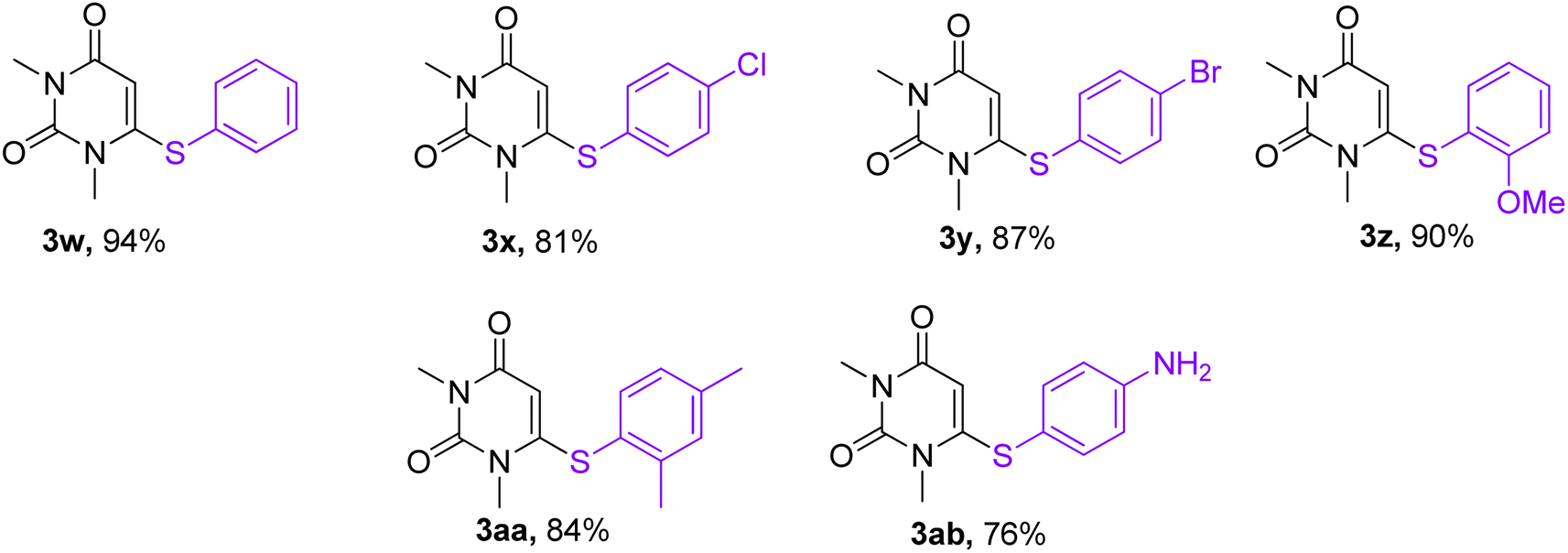
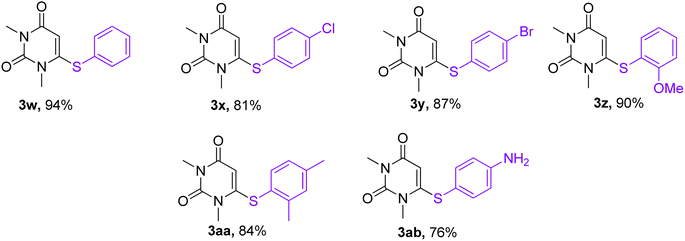
With the optimized conditions in hand, we went for synthesizing an array of phenoxypyrimidines starting from four number of chloropyrimidines (1a–1d, Table 3) and a series of phenols, and the results have been summarized in Table 4. The optimized reaction condition was tolerant to various functional group such as –Br, –Cl, –Me, –NO2, –CHO, –OMe, –SMe, –COOEt, –COCH3, and –NH2 irrespective of its presence either on the pyrimidine or phenyl ring. 10 numbers of 1,3-dimethyl-6-aryloxypyrimidine-2,4(1H,3H)-diones were synthesized from 6-chloro-1,3-dimethyluracil (6-chloro-1,3-dimethylpyrimidine-2,4(1H,3H)-dione) (1a) with good to excellent yield (80–94%, except in 3h) within a short span of time (1 h) by varying the phenols. Phenols with strong electron withdrawing groups such as –NO2 and –CHO were observed to deliver products comparatively in lower yields (3e, 3f & 3h). Both p-methoxy and o-methoxyphenol delivered their corresponding products in excellent yields (3g & 3i), and thus the steric factor was not observed to be very influential in the reaction. Fused ring systems such as 2-naphthol responded to the reaction quite well (90%, 3c). Completely aromatic chloropyrimidines (1b–1d) were also found to be very effective as substrates in delivering their corresponding aryloxypyrimidines (3k–3v). Here too, phenols with strongly electron withdrawing groups were found to deliver products in lower yields (3m & 3s). 4,6-dichloropyrimidine (1c), under the reaction conditions, primarily delivered the mono substituted products (3o–3s). However, the disubstituted product could be prepared as the major product when the reaction was carried out in the presence of 2.5 equivalents of phenol (3p). Highly substituted pyrimidine, ethyl-4-chloro-2-(methylthio)pyrimidine-5-carboxylate (1d), also responded to the reaction protocol well, and both –SMe and –COOEt were found to be tolerant to the reaction conditions, delivering products in very good yields (76–84%, 3t–3v). We intended then to check and extend the reaction protocol for thiophenols, and to our delight, thiophenols reacted well with 6-chloro-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (1a) under the reaction conditions to generate 1,3-dimethyl-6-(aryllthio)pyrimidine-2,4(1H,3H)-diones in good to excellent (3w–3ab, 76–94%) yield within 1 h (Table 5). Interestingly, 4-aminothiophenol, when allowed to react with 1a under the reaction conditions, delivered a single product (3ab) through C–S bond formation without any disturbance to the –NH2 group.
Table 3 Chloropyrimidines used as substrates in this study
Table 4 Library synthesis of phenoxypyrimidinesa
| Reaction conditions: 1a (1 mmol), 2a (1.1 mmol), Cu-polymer (10 wt%), KOH (3 eq.), solvent (3 mL), 80 °C (silicon oil bath). |
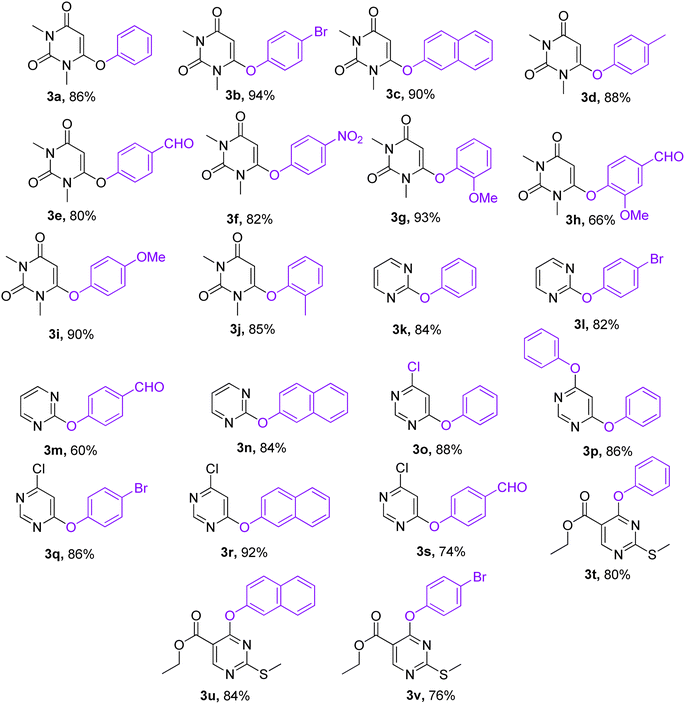 |
Table 5 Synthesis of 1,3-dimethyl-6-(arylthio)pyrimidine-2,4(1H,3H)-dionesa
| Reaction conditions: 1a (1 mmol), 2a (1.1 mmol), Cu-polymer (10 wt%), base (3 eq.), solvent (3 mL), 80 °C (silicon oil bath). |
 |
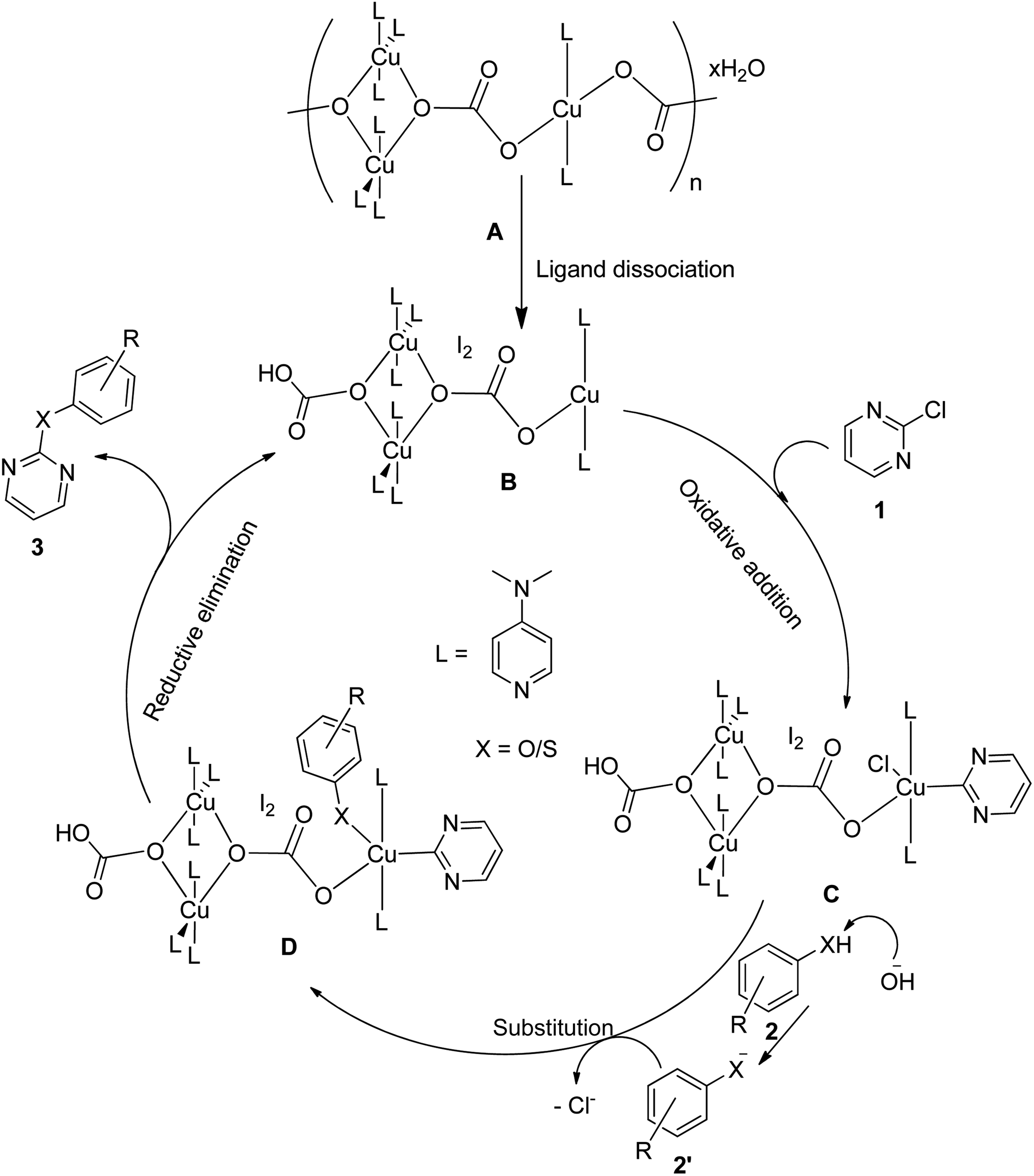
Based on literature reports66,67 and our knowledge on the reaction mechanisms, a plausible reaction mechanism for the C–O and C–S coupling reaction using the polymeric Cu-complex has been proposed, as shown in Fig. 9. Cu(II) centers (A) are expected to undergo –CO3– ligand dissociation to generate the Cu(I) centers (B), which then undergo subsequent oxidative addition reactions with chloropyrimidines (1) to give Cu(III) species (C). Phenols/thiols (2) in alkaline medium undergo ligand substitution reactions with C to deliver D, which finally undergo reductive elimination to furnish the product (3) regenerating the active catalyst (B).
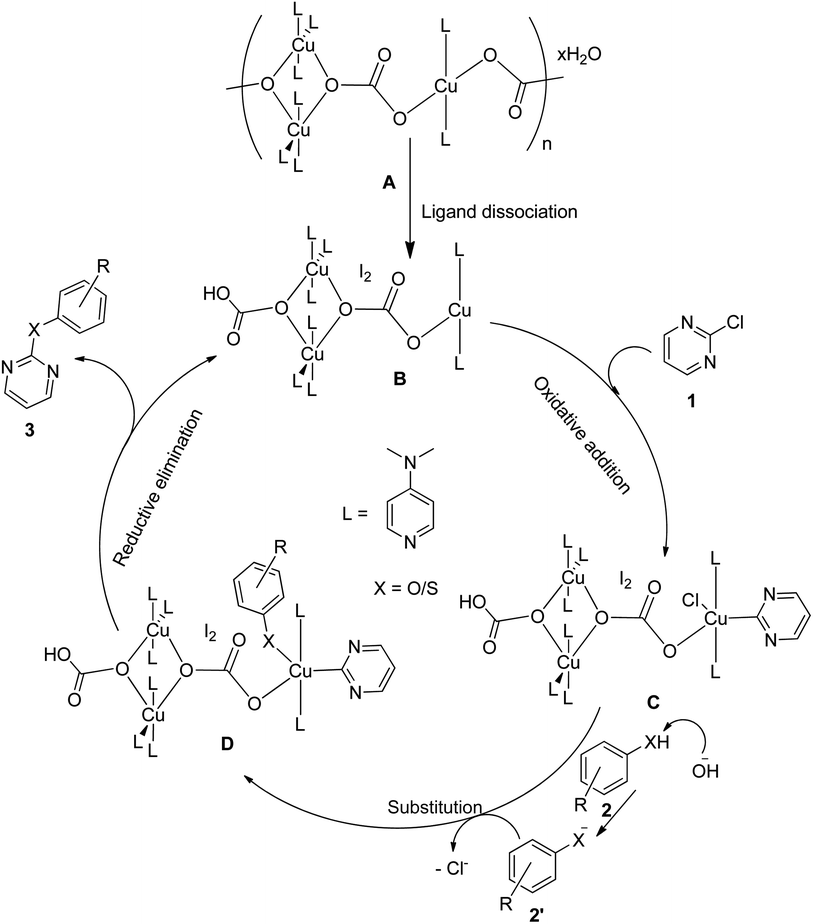 |
| | Fig. 9 Plausible reaction mechanism for the C–O and C–S coupling reaction. | |
In silico investigation of druglikeness properties of the synthesized compounds
All the synthesized 28 compounds (22 aryloxypyrimidines and 6 arylthiopyrimidines) were screened for drug-likeliness properties using the SwissADME prediction tool.68 Drug-likeliness properties of compounds were examined in silico based on the criteria set by Lipinski, Ghose, Veber, Egan and Muggee.69 Only one compound (3k) failed to follow the criterion set by Muggee (molecular weight < 200). All the compounds were observed to possess good pharmacokinetic properties, as reflected by their bioavailability score, which was found to be 0.55 for all. Among the tested compounds, arylthiopyrimidine 3aa was found to be completely fitting in the bioavailability aspect. No PAINS alert was observed for any of the synthesized compounds, while 6 compounds among the tested 28 compounds were found to have Brenk's alert. 14 of the synthesized compounds were found to not violate the criteria for leadlikeness and can be potential leads for further study. From the BOILED-egg structure of the compounds (Fig. S76 & S77 in ESI†), it was observed that all of our synthesized aryloxypyrimidines and arythiopyrimidines have high gastrointestinal (GI) absorption. 16 aryloxypyrimidines and 3 arythiopyrimidines were found to have blood–brain barrier (BBB) penetration ability.
Conclusion
In conclusion, a μ3-CO3 bridged linear polymeric Cu-complex ([Cu3(DMAP)8(μ3-CO3)2]I2)n·xH2O has been synthesized from CuI and DMAP through the fixation of atmospheric CO2. The same complex can be synthesized from the same set of substrates CuI and DMAP by carrying out the reaction in the presence of K2CO3 as the CO32− source. The structure of the polymeric Cu-complex was confirmed by single crystal X-ray analysis and other spectroscopic technique. The catalytic activity of the polymeric Cu-complex was checked in the synthesis of phenoxypyrimidines and arylthiopyrimidines via C–O and C–S cross-coupling reactions and found to be very effective in delivering products in good to excellent yields, starting from chloropyrimidines and phenol/thiophenol. The reaction conditions were tolerant to functional groups such as –Br, –Cl, –Me, –NO2, –CHO, –OMe, –SMe, –COOEt, –COCH3, and –NH2. The developed protocol has been successfully employed for the synthesis of 22 number of phenoxypyrimidines and 6 number of arylthiopyrimidines. In silico study using the SwissADME webtool showed that 14 of the synthesized products were not violating the criteria for leadlikeness and can be potential lead molecules.
Experimental
General information
One of the starting materials (1a) was prepared by the previously described procedure.70 All other starting materials (1b–1d & 2) and chemicals were of analytical reagent grade (AR grade), purchased from Sigma-Aldrich, Merck and TCI, and used without further purification. NMR spectra were recorded using tetramethylsilane (TMS) as the internal standard with a 400 MHz spectrophotometer and expressing chemical shift in ppm. Silica gel60F254 (Merck) over aluminium sheets as a thin-layer chromatography plate were used to monitor the progress of the reactions and UV light as the visualizer for the purpose. HRMS data were collected using electrospray ionization technique with a Q-TOF mass analyzer. SCXRD data were collected with an AXS SMART APEX-I single crystal XRD machine of Bruker, Germany.
Synthetic procedure for ([Cu3(DMAP)8(μ-CO3)2]I2)n·xH2O
CuI (4 mmol) and 4-DMAP (16.4 mmol) were refluxed in H2O (8 mL) for 4 h, filtered hot using a Whatman No. 1 paper in a beaker (25 mL), and kept undisturbed for 8 weeks. Needle-like crystals of ([Cu3(DMAP)8(CO3)2]I2)n·xH2O accumulated in the beaker were collected in 88% yield.
General reaction procedure for the synthesis of phenoxypyrimidines and arylthiopyrimidines
Chloropyrimidine (1 mmol), phenol/thiophenol (1.1 mmol) and KOH (3 mmol) were taken in a round bottom flask. ([Cu3(DMAP)8(CO3)2]I2)n·xH2O (10 wt%) and 3 mL of H2O were added to the reaction mixture. The resulting mixture was subjected to heating at 80 °C under conventional heating using a silicon oil bath. The progress of the reaction was monitored using thin-layer chromatography technique. After the completion of the reaction (as indicated by TLC), the reaction mixture was allowed to cool to room temperature. The organic compounds were extracted using ethyl acetate (2 × 30 mL), followed by washing the organic layer with brine (1 × 30 mL). The resulting organic layer was dried over anhydrous Na2SO4, evaporated under vacuum and finally the crude product was purified by performing column chromatography (100–200 mesh silica gel as the stationary phase and ethyl acetate–hexane as the eluent).
Author contributions
AJB: conceptualization, methodology, formal analysis, writing – original draft; PPN: data collection and analysis; SJB: crystallography; LS: conceptualization, methodology, investigation, funding acquisition, writing – final draft.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by DST, India through DST PURSE project grant (SR/PURSE/2021/58). AJB is thankful to DST, India for financial aid in terms of DST-IINSPIRE fellowship (IF170498). Authors are thankful to SAIC, Tezpur University, Assam and North East Centre for Biological Sciences and Healthcare Engineering (NECBH), IIT Guwahati and Department of Biotechnology (DBT), Govt. of India for providing instrumentation facilities.
References
- E. Liu, Y. Z. Zhang, L. Li, C. Yang, J. C. Fettinger and G. Zhang, Polyhedron, 2015, 99, 223–229 CrossRef CAS
 .
. - L.-G. Zhu and G.-Q. Cai, Chin. J. Chem., 2002, 20, 990–995 CrossRef CAS .
- M. Zhu, J. Peng, H.-J. Pang, P.-P. Zhang, Y. Chen, D.-D. Wang, M.-G. Liu and Y.-H. Wang, Inorg. Chim. Acta, 2011, 370, 260–264 CrossRef CAS .
- A. Beheshti, A. Lalegani, A. Noshadian, G. Bruno and H. A. Rudbari, Polyhedron, 2015, 85, 690–696 CrossRef CAS .
- X. Wang, J. D. Ranford and J. J. Vittal, J. Mol. Struct., 2006, 796, 28–35 CrossRef CAS .
- A. Dehno Khalaji, M. Weil, H. Hadadzadeh and M. Daryanavard, Inorg. Chim. Acta, 2009, 362, 4837–4842 CrossRef CAS .
- A. Chakraborty, S. R. Lingampalli, A. Kumari, J. Ribas, J. Ribas-Arino and T. K. Maji, Inorg. Chem., 2014, 53, 11991–12001 CrossRef CAS PubMed .
- A. D. Khalaji and H. Stoekli-Evans, Polyhedron, 2009, 28, 3769–3773 CrossRef CAS .
- M. Casarin, A. Cingolani, C. Di Nicola, D. Falcomer, M. Monari, L. Pandolfo and C. Pettinari, Cryst. Growth Des., 2007, 7, 676–685 CrossRef CAS .
- R. Bikas, P. Aleshkevych, H. Hosseini-Monfared, J. Sanchiz, R. Szymczak and T. Lis, Dalton Trans., 2015, 44, 1782–1789 RSC .
- S. S. Massoud, F. R. Louka, M. A. Al-Hasan, R. Vicente and F. A. Mautner, New J. Chem., 2015, 39, 5944–5952 RSC .
- S. Youngme, N. Wannarit, T. Remsungnen, N. Chaichit, G. A. van Albada and J. Reedijk, Inorg. Chem. Commun., 2008, 11, 179–185 CrossRef CAS .
- S. Youngme, N. Wannarit, T. Remsungnen, C. Pakawatchai, N. Chaichit, C. Engkagul, G. A. van Albada and J. Reedijk, Inorg. Chem. Commun., 2008, 11, 427–432 CrossRef CAS .
- S. Khan, S. Roy, K. Bhar, R. K. Kumar, T. K. Maji and B. K. Ghosh, Polyhedron, 2012, 32, 54–59 CrossRef CAS .
- A. Majumder, C. R. Choudhury, S. Mitra, G. M. Rosair, M. S. El Fallah and J. Ribas, Chem. Commun., 2005, 16, 2158–2160 RSC .
- P. Mukherjee, M. G. B. Drew, M. Estrader and A. Ghosh, Inorg. Chem., 2008, 47, 7784–7791 CrossRef CAS PubMed .
- P. Bag, S. Dutta, P. Biswas, S. K. Maji, U. Flörke and K. Nag, Dalton Trans., 2012, 41, 3414–3423 RSC .
- A. Escuer, R. Vicente, S. B. Kumar, X. Solans, M. Font-Bardía and A. Caneschi, Inorg. Chem., 1996, 35, 3094–3098 CrossRef CAS PubMed .
- H. Fu, W.-L. Chen, D.-G. Fu, M.-L. Tong, X.-M. Chen, L.-N. Ji and Z.-W. Mao, Inorg. Chem. Commun., 2004, 7, 1285–1288 CrossRef CAS .
- J.-P. Costes, F. Dahan and W. Wernsdorfer, Inorg. Chem., 2006, 45, 5–7 CrossRef CAS PubMed .
- J.-P. Costes, J. M. Clemente-Juan, F. Dahan and J. Milon, Inorg. Chem., 2004, 43, 8200–8202 CrossRef CAS PubMed .
- I. Benesperi, R. Singh and M. Freitag, Energies, 2020, 13, 2198 CrossRef CAS .
- Y. Sun, D.-J. Xing, J.-J. Wei, W.-Z. Ding and S.-R. Cheng, Polym. Bull., 2023, 80, 607–619 CrossRef CAS .
- A. Hussain, M. F. AlAjmi, M. T. Rehman, S. Amir, F. M. Husain, A. Alsalme, M. A. Siddiqui, A. A. AlKhedhairy and R. A. Khan, Sci. Rep., 2019, 9, 5237 CrossRef PubMed .
- J. Niu, H. Zhou, Z. Li, J. Xu and S. Hu, J. Org. Chem., 2008, 73, 7814–7817 CrossRef CAS PubMed .
- J. P. Collman and M. Zhong, Org. Lett., 2000, 2, 1233–1236 CrossRef CAS PubMed .
- S. Roy, M. J. Sarma, B. Kashyap and P. Phukan, Chem. Commun., 2016, 52, 1170–1173 RSC .
- C. E. Koning, F. J. Viersen, G. Challa and J. Reedijk, J. Mol. Catal., 1988, 44, 245–257 CrossRef CAS .
- G. Zhang, Y. Z. Zhang, W.-F. Lo, J. Jiang, J. A. Golen and A. L. Rheingold, Polyhedron, 2016, 103, 227–234 CrossRef CAS .
- H. Duan, M. Ning, X. Chen, Q. Zou, L. Zhang, Y. Feng, L. Zhang, Y. Leng and J. Shen, J. Med. Chem., 2012, 55, 10475–10489 CrossRef CAS PubMed .
- T. D. Venu, S. A. Khanum, A. Firdouse, B. K. Manuprasad, S. Shashikanth, R. Mohamed and B. S. Vishwanth, Bioorg. Med. Chem. Lett., 2008, 18, 4409–4412 CrossRef CAS PubMed .
- A. K. Farag, A. Elkamhawy, A. M. Londhe, K.-T. Lee, A. N. Pae and E. J. Roh, Eur. J. Med. Chem., 2017, 141, 657–675 CrossRef CAS PubMed .
- A. Elkamhawy, S. Paik, A. H. E. Hassan, Y. S. Lee and E. J. Roh, Bioorg. Chem., 2017, 75, 393–405 CrossRef CAS PubMed .
- J. Popow, H. Arnhof, G. Bader, H. Berger, A. Ciulli, D. Covini, C. Dank, T. Gmaschitz, P. Greb, J. Karolyi-Özguer, M. Koegl, D. B. McConnell, M. Pearson, M. Rieger, J. Rinnenthal, V. Roessler, A. Schrenk, M. Spina, S. Steurer, N. Trainor, E. Traxler, C. Wieshofer, A. Zoephel and P. Ettmayer, J. Med. Chem., 2019, 62, 2508–2520 CrossRef CAS PubMed .
- N. M. Goudgaon and B. U. Sheshikant, J. Pharm. Res., 2013, 7, 75–79 CAS .
- Y. Zhong, R.-Z. Qiu, S.-L. Sun, C. Zhao, T.-Y. Fan, M. Chen, N.-G. Li and Z.-H. Shi, J. Med. Chem., 2020, 63, 12403–12428 CrossRef CAS PubMed .
- X. Liang, B. Wang, C. Chen, A. Wang, C. Hu, F. Zou, K. Yu, Q. Liu, F. Li, Z. Hu, T. Lu, J. Wang, L. Wang, E. L. Weisberg, L. Li, R. Xia, W. Wang, T. Ren, J. Ge, J. Liu and Q. Liu, J. Med. Chem., 2019, 62, 875–892 CrossRef CAS PubMed .
- T. Jojima and S. Tamura, Agric. Biol. Chem., 1966, 30, 896–905 CAS .
- H. Zhang, Y. Tian, D. Kang, Z. Huo, Z. Zhou, H. Liu, E. De Clercq, C. Pannecouque, P. Zhan and X. Liu, Eur. J. Med. Chem., 2017, 130, 209–222 CrossRef CAS PubMed .
- G. Cui, J. Jin, H. Chen, R. Cao, X. Chen and B. Xu, Bioorg. Med. Chem., 2018, 26, 2186–2197 CrossRef CAS PubMed .
- J. C. Boehm, M. J. Bower, T. F. Gallagher, S. Kassis, S. R. Johnson and J. L. Adams, Bioorg. Med. Chem. Lett., 2001, 11, 1123–1126 CrossRef CAS PubMed .
- M. Baba, M. Ito, S. Shigeta, H. Tanaka, T. Miyasaka, M. Ubasawa, K. Umezu, R. T. Walker and E. De Clercq, Antimicrob. Agents Chemother., 1991, 35, 1430–1433 CrossRef CAS PubMed .
- I. W. Althaus, K.-C. Chou, R. J. Lemay, K. M. Franks, M. R. Deibel, F. J. Kezdy, L. Resnick, M. E. Busso, A. G. So, K. M. Downey, D. L. Romero, R. C. Thomas, P. A. Aristoff, W. G. Tarpley and F. Reusser, Biochem. Pharmacol., 1996, 51, 743–750 CrossRef CAS PubMed .
- Y. Kang, T. Taldone, H. J. Patel, P. D. Patel, A. Rodina, A. Gozman, R. Maharaj, C. C. Clement, M. R. Patel, J. L. Brodsky, J. C. Young and G. Chiosis, J. Med. Chem., 2014, 57, 1188–1207 CrossRef CAS PubMed .
- Y. Nezu, N. Wada, F. Yoshida, T. Miyazawa, T. Shimizu and T. Fujita, Pestic. Sci., 1998, 52, 343–353 CrossRef .
- A. K. Sharma, L. Wen, L. R. Hall, J. G. Allan and B. J. Clark, J. Agric. Food Chem., 2016, 64, 5793–5802 CrossRef CAS PubMed .
- S. Gu, C. Chen and W. Chen, J. Org. Chem., 2009, 74, 7203–7206 CrossRef CAS PubMed .
- K. Raghuvanshi, K. Rauch and L. Ackermann, Chem.–Eur. J., 2015, 21, 1790–1794 CrossRef CAS PubMed .
- M. Ravi, S. Allu and K. C. K. Swamy, J. Org. Chem., 2017, 82, 2355–2363 CrossRef CAS PubMed .
- Y. Oh, J. Lee, H. Shin and J.-H. Sohn, Tetrahedron Lett., 2019, 60, 2074–2077 CrossRef CAS .
- S. Senda, K. Hirota and M. Takahashi, J. Chem. Soc., Perkin Trans. 1, 1975,(5), 503–507 RSC .
- Y.-J. Cherng, Tetrahedron, 2002, 58, 887–890 CrossRef CAS .
- S. Li, C. Guo, H. Zhao, Y. Tang and M. Lan, Bioorg. Med. Chem., 2012, 20, 877–885 CrossRef CAS PubMed .
- Q. Liu, Z. Lu, W. Ren, K. Shen, Y. Wang and Q. Xu, Chin. J. Chem., 2013, 31, 764–772 CrossRef CAS .
- K. Walsh, H. F. Sneddon and C. J. Moody, RSC Adv., 2014, 4, 28072–28077 RSC .
- S. K. Laughlin, M. P. Clark, J. F. Djung, A. Golebiowski, T. A. Brugel, M. Sabat, R. G. Bookland, M. J. Laufersweiler, J. C. VanRens, J. A. Townes, B. De, L. C. Hsieh, S. C. Xu, R. L. Walter, M. J. Mekel and M. J. Janusz, Bioorg. Med. Chem. Lett., 2005, 15, 2399–2403 CrossRef CAS PubMed .
- M. Matloobi and C. O. Kappe, J. Comb. Chem., 2007, 9, 275–284 CrossRef CAS PubMed .
- N. D. D'Angelo, J. J. Peterson, S. K. Booker, I. Fellows, C. Dominguez, R. Hungate, P. J. Reider and T.-S. Kim, Tetrahedron Lett., 2006, 47, 5045–5048 CrossRef .
- M. Platon, L. Cui, S. Mom, P. Richard, M. Saeys and J.-C. Hierso, Adv. Synth. Catal., 2011, 353, 3403–3414 CrossRef CAS .
- B. Roth and G. H. Hitchings, J. Org. Chem., 1961, 26, 2770–2778 CrossRef CAS .
- R. Fu, X. Xu, Q. Dang and X. Bai, J. Org. Chem., 2005, 70, 10810–10816 CrossRef CAS PubMed .
- A. Lengar and C. O. Kappe, Org. Lett., 2004, 6, 771–774 CrossRef CAS PubMed .
- B. Y. Bhong, A. V. Shelke and N. N. Karade, Tetrahedron Lett., 2013, 54, 739–743 CrossRef CAS .
- G. A. van Albada, I. Mutikainen, O. S. Roubeau, U. Turpeinen and J. Reedijk, Eur. J. Inorg. Chem., 2000, 2000, 2179–2184 CrossRef .
- Z. Chen, Y. Jiang, L. Zhang, Y. Guo and D. Ma, J. Am. Chem. Soc., 2019, 141, 3541–3549 CrossRef CAS PubMed .
- E. Sperotto, G. P. M. van Klink, G. van Koten and J. G. de Vries, Dalton Trans., 2010, 39, 10338–10351 RSC .
- A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301–2318 RSC .
- A. Daina and V. Zoete, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed .
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed .
- W. Pfleiderer and K.-H. Schündehütte, Justus Liebigs Ann. Chem., 1958, 612, 158–163 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Partha Pratim Nathbc,
Sourav Jyoti Bharali
a,
Partha Pratim Nathbc,
Sourav Jyoti Bharali